Transcription Factors Indirectly Regulate Genes through Nuclear Colocalization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Preparation
2.2. Randomized Experiments
2.3. Statistical Method
3. Results
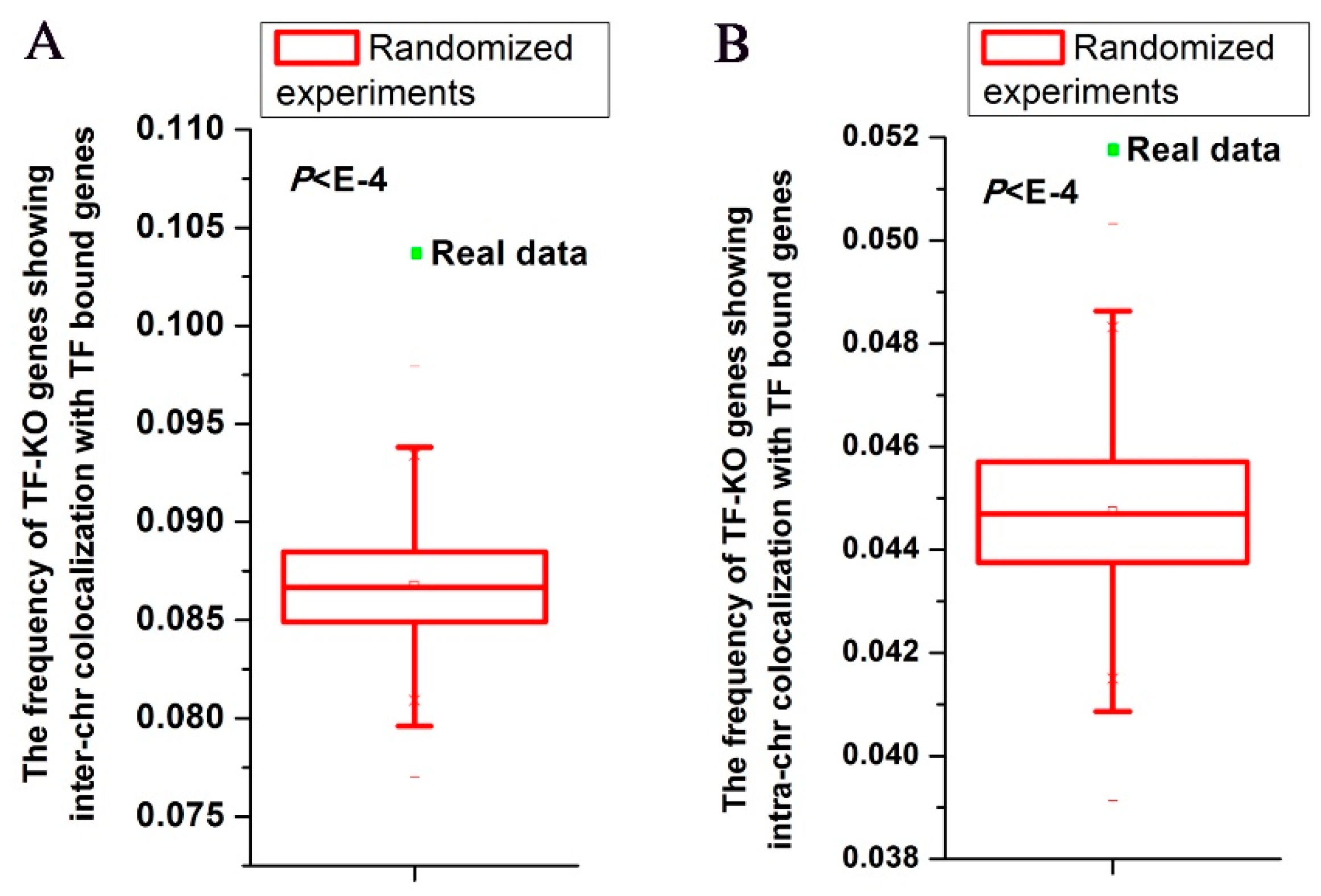
3.1. TF-KO Genes Show Nuclear Colocalization with Genes Bound by the Corresponding TF
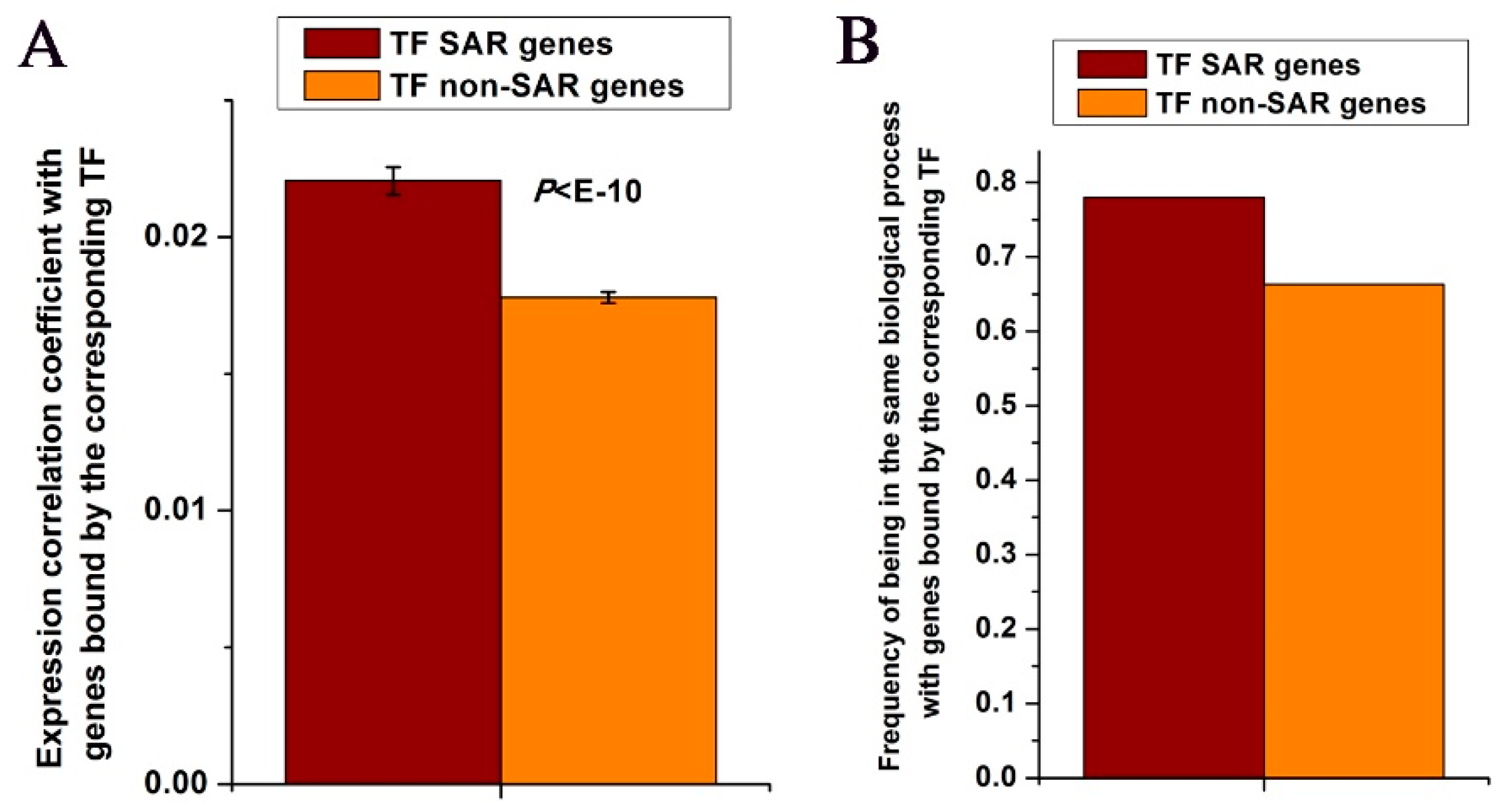
3.2. TFs Tend to Be in the Same Cellular Component with Binding TFs of their SAR Genes
3.3. TF SAR Genes Show Co-expression with Genes Bound by the Corresponding TF
3.4. TF SAR Genes Lack DNA Motifs of the Corresponding TF
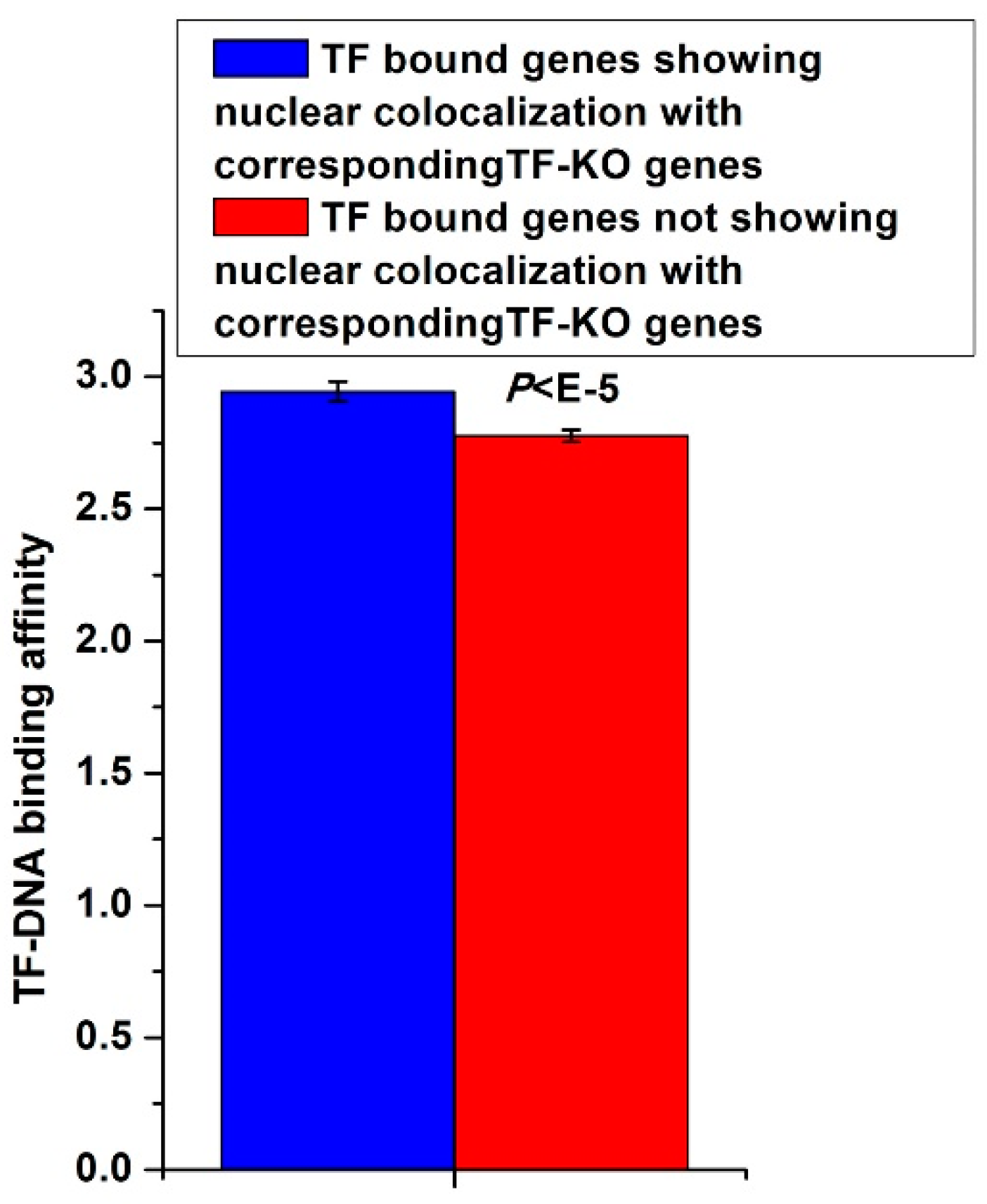
3.5. TF-KO Genes Show Nuclear Colocalization with TF Bound Genes that Have High Corresponding TF-DNA Binding Affinities
4. Discussion
Supplementary Materials
Funding
Conflicts of Interest
Abbreviations
| TF-KO genes | TF knockout affected genes |
| inter-chr colocalization | inter-chromosomal colocalization |
| intra-chr colocalization | intra-chromosomal colocalization |
| TF SAR genes | TF spatially adjacent regulated genes |
| SAR TFs | spatially adjacent regulatory TFs |
References
- Lee, T.I.; Rinaldi, N.J.; Robert, F.; Odom, D.T.; Bar-Joseph, Z.; Gerber, G.K.; Hannett, N.M.; Harbison, C.T.; Thompson, C.M.; Simon, I.; et al. Transcriptional regulatory networks in saccharomyces cerevisiae. Science 2002, 298, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Ren, B.D. Genome-wide analysis of protein-DNA interactions. Annu. Rev. Genom. Hum. Genet. 2006, 7, 81–102. [Google Scholar] [CrossRef] [PubMed]
- Lieb, J.D. Genome-wide mapping of protein-DNA interactions by chromatin immunoprecipitation and DNA microarray hybridization. Methods Mol. Biol. 2003, 224, 99–109. [Google Scholar] [PubMed]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science 2007, 316, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.; Hirst, M.; Bainbridge, M.; Bilenky, M.; Zhao, Y.; Zeng, T.; Euskirchen, G.; Bernier, B.; Varhol, R.; Delaney, A.; et al. Genome-wide profiles of stat1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods 2007, 4, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Neph, S.; Vierstra, J.; Stergachis, A.B.; Reynolds, A.P.; Haugen, E.; Vernot, B.; Thurman, R.E.; John, S.; Sandstrom, R.; Johnson, A.K.; et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 2012, 489, 83–90. [Google Scholar] [CrossRef]
- Song, L.; Crawford, G.E. Dnase-seq: A high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. Atac-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar]
- Hesselberth, J.R.; Chen, X.; Zhang, Z.; Sabo, P.J.; Sandstrom, R.; Reynolds, A.P.; Thurman, R.E.; Neph, S.; Kuehn, M.S.; Noble, W.S.; et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat. Methods 2009, 6, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Schulz, M.H.; Look, T.; Begemann, M.; Zenke, M.; Costa, I.G. Identification of transcription factor binding sites using atac-seq. Genome Biol. 2019, 20, 45. [Google Scholar] [CrossRef]
- Giresi, P.G.; Kim, J.; McDaniell, R.M.; Iyer, V.R.; Lieb, J.D. Faire (formaldehyde-assisted isolation of regulatory elements) isolates active regulatory elements from human chromatin. Genome Res. 2007, 17, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.M.; Giresi, P.G.; Davis, I.J.; Lieb, J.D. Using formaldehyde-assisted isolation of regulatory elements (faire) to isolate active regulatory DNA. Nat. Protoc. 2012, 7, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.R.; Marton, M.J.; Jones, A.R.; Roberts, C.J.; Stoughton, R.; Armour, C.D.; Bennett, H.A.; Coffey, E.; Dai, H.; He, Y.D.; et al. Functional discovery via a compendium of expression profiles. Cell 2000, 102, 109–126. [Google Scholar] [CrossRef]
- Chua, G.; Morris, Q.D.; Sopko, R.; Robinson, M.D.; Ryan, O.; Chan, E.T.; Frey, B.J.; Andrews, B.J.; Boone, C.; Hughes, T.D. Identifying transcription factor functions and targets by phenotypic activation. Proc. Natl. Acad. Sci. USA 2006, 103, 12045–12050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Killion, P.J.; Iyer, V.D. Genetic reconstruction of a functional transcriptional regulatory network. Nat. Genet. 2007, 39, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Harbison, C.T.; Gordon, D.B.; Lee, T.I.; Rinaldi, N.J.; Macisaac, K.D.; Danford, T.W.; Hannett, N.M.; Tagne, J.B.; Reynolds, D.B.; Yoo, J.; et al. Transcriptional regulatory code of a eukaryotic genome. Nature 2004, 431, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Reimand, J.; Vaquerizas, J.M.; Todd, A.E.; Vilo, J.; Luscombe, N.D. Comprehensive reanalysis of transcription factor knockout expression data in saccharomyces cerevisiae reveals many new targets. Nucleic Acids Res. 2010, 38, 4768–4777. [Google Scholar] [CrossRef]
- Duan, Z.; Andronescu, M.; Schutz, K.; McIlwain, S.; Kim, Y.J.; Lee, C.; Shendure, J.; Fields, S.; Blau, C.A.; Noble, W.D. A three-dimensional model of the yeast genome. Nature 2010, 465, 363–367. [Google Scholar] [CrossRef]
- Jin, F.; Li, Y.; Dixon, J.R.; Selvaraj, S.; Ye, Z.; Lee, A.Y.; Yen, C.A.; Schmitt, A.D.; Espinoza, C.A.; Ren, B.D. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 2013, 503, 290–294. [Google Scholar] [CrossRef] [Green Version]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Sexton, T.; Yaffe, E.; Kenigsberg, E.; Bantignies, F.; Leblanc, B.; Hoichman, M.; Parrinello, H.; Tanay, A.; Cavalli, G. Three-dimensional folding and functional organization principles of the drosophila genome. Cell 2012, 148, 458–472. [Google Scholar] [CrossRef]
- Nagano, T.; Lubling, Y.; Stevens, T.J.; Schoenfelder, S.; Yaffe, E.; Dean, W.; Laue, E.D.; Tanay, A.; Fraser, P. Single-cell hi-c reveals cell-to-cell variability in chromosome structure. Nature 2013, 502, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Flyamer, I.M.; Gassler, J.; Imakaev, M.; Brandao, H.B.; Ulianov, S.V.; Abdennur, N.; Razin, S.V.; Mirny, L.A.; Tachibana-Konwalski, K. Single-nucleus hi-c reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 2017, 544, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Stevens, T.J.; Lando, D.; Basu, S.; Atkinson, L.P.; Cao, Y.; Lee, S.F.; Leeb, M.; Wohlfahrt, K.J.; Boucher, W.; O’Shaughnessy-Kirwan, A.; et al. 3D structures of individual mammalian genomes studied by single-cell hi-c. Nature 2017, 544, 59–64. [Google Scholar] [CrossRef]
- Nagano, T.; Lubling, Y.; Varnai, C.; Dudley, C.; Leung, W.; Baran, Y.; Mendelson Cohen, N.; Wingett, S.; Fraser, P.; Tanay, A. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 2017, 547, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.; Xing, D.; Chang, C.H.; Li, H.; Xie, X.S. Three-dimensional genome structures of single diploid human cells. Science 2018, 361, 924–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3d map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Boettiger, A.N.; Bintu, B.; Moffitt, J.R.; Wang, S.; Beliveau, B.J.; Fudenberg, G.; Imakaev, M.; Mirny, L.A.; Wu, C.T.; Zhuang, X. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 2016, 529, 418–422. [Google Scholar] [CrossRef] [Green Version]
- Dai, Z.; Dai, X.D. Nuclear colocalization of transcription factor target genes strengthens coregulation in yeast. Nucleic Acids Res. 2012, 40, 27–36. [Google Scholar] [CrossRef]
- Fanucchi, S.; Shibayama, Y.; Burd, S.; Weinberg, M.S.; Mhlanga, M.M. Chromosomal contact permits transcription between coregulated genes. Cell 2013, 155, 606–620. [Google Scholar] [CrossRef] [PubMed]
- Rieder, D.; Ploner, C.; Krogsdam, A.M.; Stocker, G.; Fischer, M.; Scheideler, M.; Dani, C.; Amri, E.Z.; Muller, W.G.; McNally, J.G.; et al. Co-expressed genes prepositioned in spatial neighborhoods stochastically associate with sc35 speckles and rna polymerase ii factories. Cell. Mol. Life Sci. 2014, 71, 1741–1759. [Google Scholar] [CrossRef] [PubMed]
- Thevenin, A.; Ein-Dor, L.; Ozery-Flato, M.; Shamir, R. Functional gene groups are concentrated within chromosomes, among chromosomes and in the nuclear space of the human genome. Nucleic Acids Res. 2014, 42, 9854–9861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Scholer, A.; van Nimwegen, E.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D.; et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Heintzman, N.D.; Ren, B.D. Finding distal regulatory elements in the human genome. Curr. Opin. Genet. Dev. 2009, 19, 541–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spitz, F.; Furlong, E.D. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar]
- Deng, W.; Lee, J.; Wang, H.; Miller, J.; Reik, A.; Gregory, P.D.; Dean, A.; Blobel, G.A. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell 2012, 149, 1233–1244. [Google Scholar] [CrossRef]
- Kalhor, R.; Tjong, H.; Jayathilaka, N.; Alber, F.; Chen, L. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nat. Biotechnol. 2011, 30, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Weintraub, A.S.; Li, C.H.; Zamudio, A.V.; Sigova, A.A.; Hannett, N.M.; Day, D.S.; Abraham, B.J.; Cohen, M.A.; Nabet, B.; Buckley, D.L.; et al. Yy1 is a structural regulator of enhancer-promoter loops. Cell 2017, 171, 1573–1588. [Google Scholar] [CrossRef]
- David, L.; Huber, W.; Granovskaia, M.; Toedling, J.; Palm, C.J.; Bofkin, L.; Jones, T.; Davis, R.W.; Steinmetz, L.M. A high-resolution map of transcription in the yeast genome. Proc. Natl. Acad. Sci. USA 2006, 103, 5320–5325. [Google Scholar] [CrossRef] [Green Version]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. The gene ontology (go) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [PubMed]
- Gasch, A.P.; Spellman, P.T.; Kao, C.M.; Carmel-Harel, O.; Eisen, M.B.; Storz, G.; Botstein, D.; Brown, P.O. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 2000, 11, 4241–4257. [Google Scholar] [CrossRef] [PubMed]
- Spellman, P.T.; Sherlock, G.; Zhang, M.Q.; Iyer, V.R.; Anders, K.; Eisen, M.B.; Brown, P.O.; Botstein, D.; Futcher, B. Comprehensive identification of cell cycle-regulated genes of the yeast saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell 1998, 9, 3273–3297. [Google Scholar] [CrossRef] [PubMed]
- Cho, R.J.; Campbell, M.J.; Winzeler, E.A.; Steinmetz, L.; Conway, A.; Wodicka, L.; Wolfsberg, T.G.; Gabrielian, A.E.; Landsman, D.; Lockhart, D.J.; et al. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol. Cell 1998, 2, 65–73. [Google Scholar] [CrossRef]
- Lee, W.; Tillo, D.; Bray, N.; Morse, R.H.; Davis, R.W.; Hughes, T.R.; Nislow, C. A high-resolution atlas of nucleosome occupancy in yeast. Nat. Genet. 2007, 39, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- MacIsaac, K.D.; Wang, T.; Gordon, D.B.; Gifford, D.K.; Stormo, G.D.; Fraenkel, E. An improved map of conserved regulatory sites for saccharomyces cerevisiae. BMC Bioinform. 2006, 7, 113. [Google Scholar] [CrossRef] [PubMed]
- Ben-Elazar, S.; Yakhini, Z.; Yanai, I. Spatial localization of co-regulated genes exceeds genomic gene clustering in the saccharomyces cerevisiae genome. Nucleic Acids Res. 2013, 41, 2191–2201. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, Z. Transcription Factors Indirectly Regulate Genes through Nuclear Colocalization. Cells 2019, 8, 754. https://doi.org/10.3390/cells8070754
Dai Z. Transcription Factors Indirectly Regulate Genes through Nuclear Colocalization. Cells. 2019; 8(7):754. https://doi.org/10.3390/cells8070754
Chicago/Turabian StyleDai, Zhiming. 2019. "Transcription Factors Indirectly Regulate Genes through Nuclear Colocalization" Cells 8, no. 7: 754. https://doi.org/10.3390/cells8070754
APA StyleDai, Z. (2019). Transcription Factors Indirectly Regulate Genes through Nuclear Colocalization. Cells, 8(7), 754. https://doi.org/10.3390/cells8070754