Endoplasmic Reticulum Detergent-Resistant Membranes Accommodate Hepatitis C Virus Proteins for Viral Assembly

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Cell Lysis
2.2. Antibodies
2.3. Membrane Fractionation
2.4. Immunoprecipitation Assay
2.5. Electron Microscopy Analysis of the Ultrastructure of the Infected Cells
3. Results
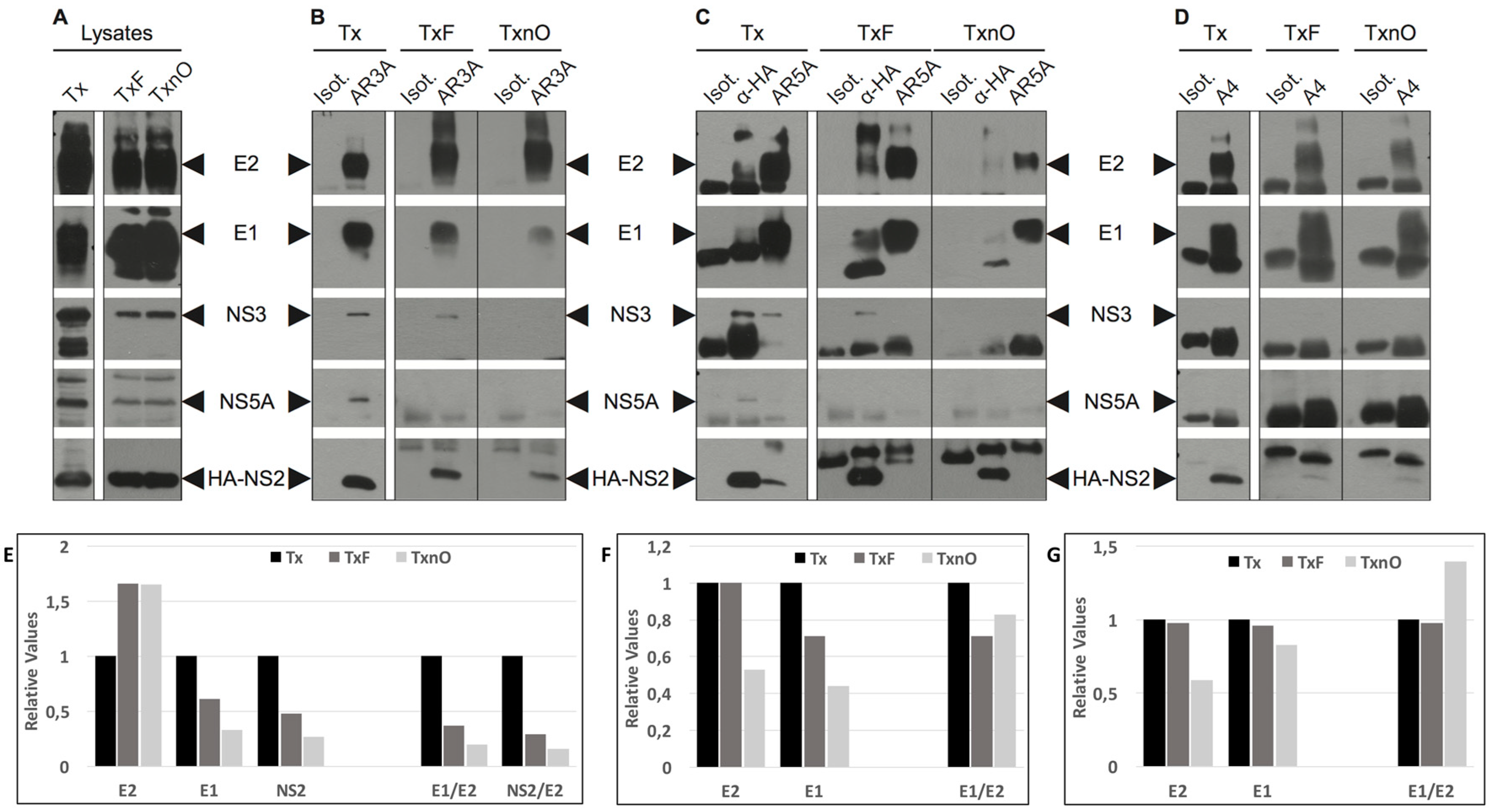
3.1. DRMs Can Be Solubilized in A Combination of Triton X-100 and n-Octyl-β-d-glucopyranoside
3.2. The Tx, TxF, and TxnO Buffers Do Not Alter Specific Protein–Protein Interactions
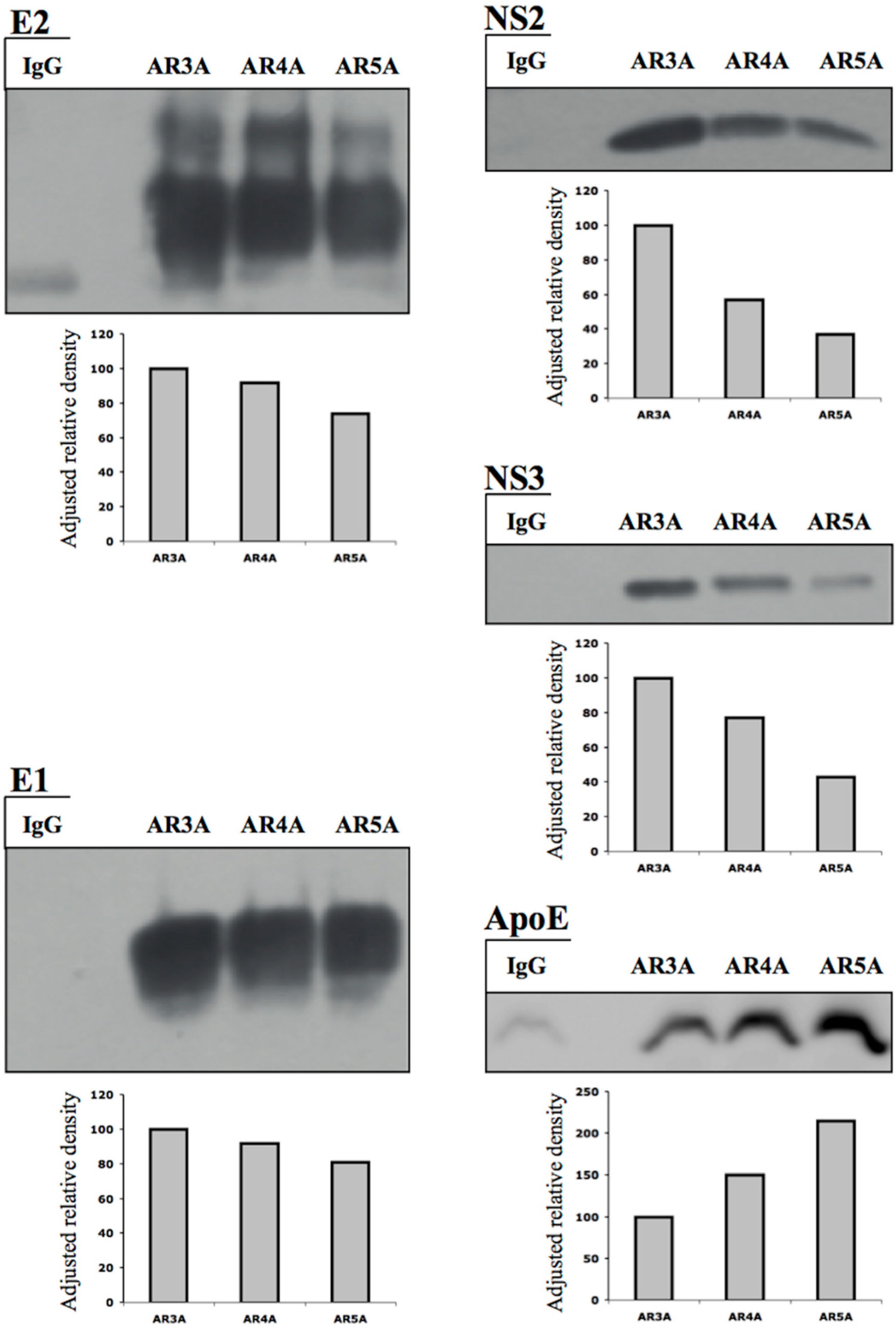
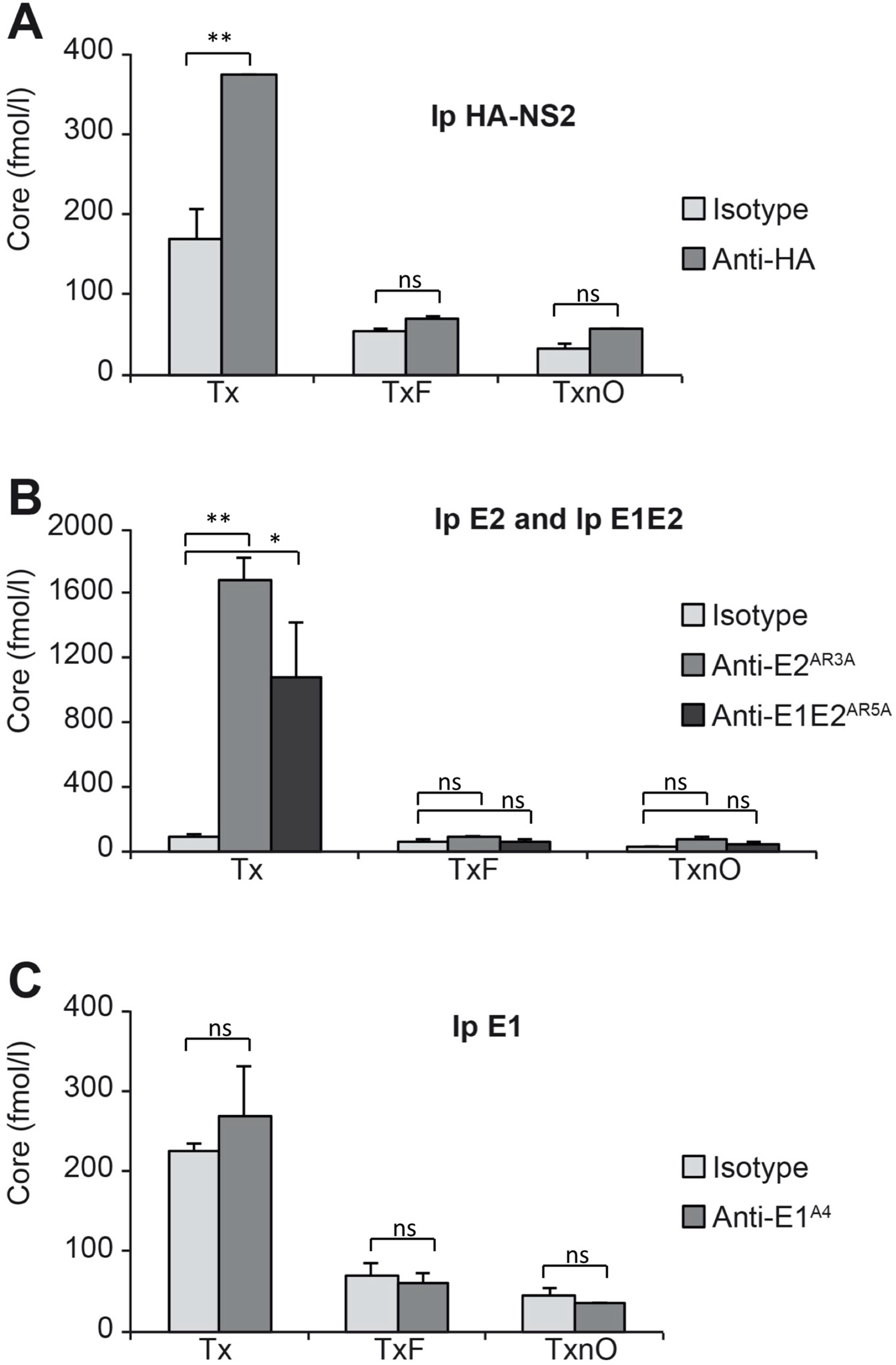
3.3. HCV NS2 Associates with Glycoproteins E1 and E2 through Direct Protein–Protein Interactions
3.4. HCV NS2 Associates with NS3 and NS5A through DRMs
3.5. The Native E1E2 Complex Associates Efficiently with ApoE, but Much Less Efficiently with NS2 and NS3
3.6. HCV Core Associates with NS2, Well-Folded E2, and the E1E2 Complex through Less Stable Domains of DRMs
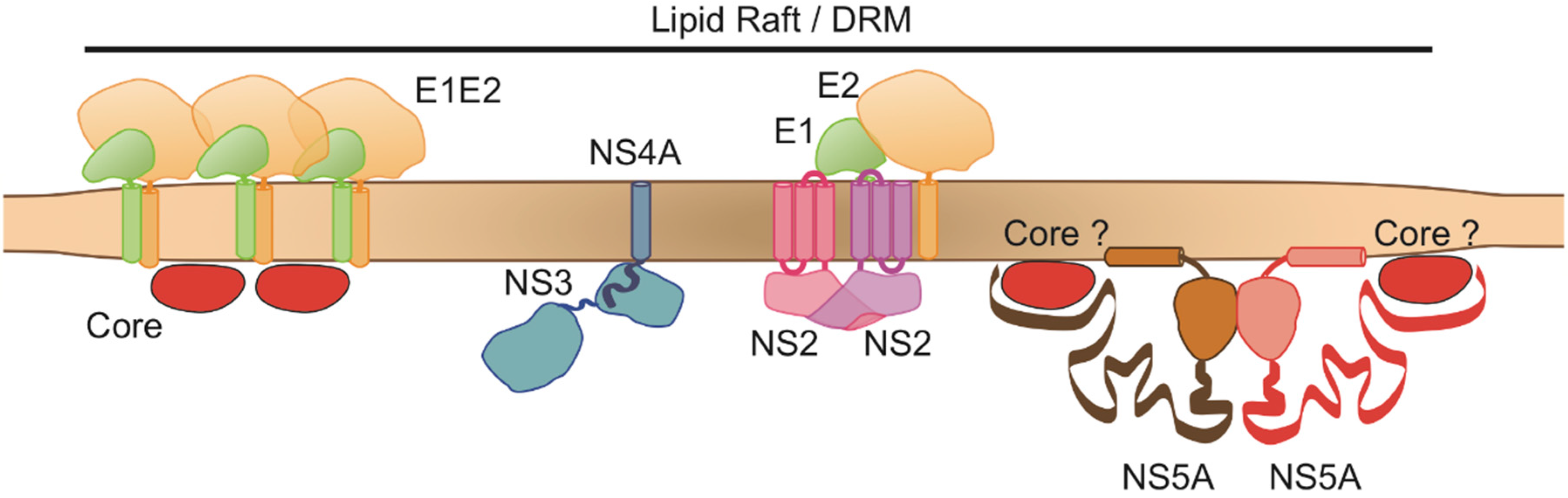
3.7. HCV NS2, E2, NS5A and the E1E2 Complex Are Colocalized at the Same Membranes of the Membranous Web
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Andre, P.; Komurian-Pradel, F.; Deforges, S.; Perret, M.; Berland, J.L.; Sodoyer, M.; Pol, S.; Brechot, C.; Paranhos-Baccala, G.; Lotteau, V. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol. 2002, 76, 6919–6928. [Google Scholar] [CrossRef]
- Piver, E.; Boyer, A.; Gaillard, J.; Bull, A.; Beaumont, E.; Roingeard, P.; Meunier, J.C. Ultrastructural Organization of Hepatitis C Virus From the Bloodstream of Infected Patients Revealed by Electron Microscopy After Specific Immunocapture. Gut 2017, 66, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Prince, A.M.; Huima-Byron, T.; Parker, T.S.; Levine, D.M. Visualization of hepatitis C virions and putative defective interfering particles isolated from low-density lipoproteins. J. Viral Hepat. 1996, 3, 11–17. [Google Scholar] [CrossRef]
- Thomssen, R.; Bonk, S.; Propfe, C.; Heermann, K.H.; Kochel, H.G.; Uy, A. Association of hepatitis C virus in human sera with beta-lipoprotein. Med. Microbiol. Immunol. (Berl) 1992, 181, 293–300. [Google Scholar] [CrossRef]
- Jirasko, V.; Montserret, R.; Appel, N.; Janvier, A.; Eustachi, L.; Brohm, C.; Steinmann, E.; Pietschmann, T.; Penin, F.; Bartenschlager, R. Structural and functional characterization of nonstructural protein 2 for its role in hepatitis C virus assembly. J. Biol. Chem. 2008, 283, 28546–28562. [Google Scholar] [CrossRef]
- Jirasko, V.; Montserret, R.; Lee, J.Y.; Gouttenoire, J.; Moradpour, D.; Penin, F.; Bartenschlager, R. Structural and functional studies of nonstructural protein 2 of the hepatitis C virus reveal its key role as organizer of virion assembly. PLoS Pathog. 2010, 6, e1001233. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Anantpadma, M.; Timpe, J.M.; Shanmugam, S.; Singh, S.M.; Lemon, S.M.; Yi, M. Hepatitis C virus NS2 protein serves as a scaffold for virus assembly by interacting with both structural and nonstructural proteins. J. Virol. 2011, 85, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Popescu, C.I.; Callens, N.; Trinel, D.; Roingeard, P.; Moradpour, D.; Descamps, V.; Duverlie, G.; Penin, F.; Heliot, L.; Rouille, Y.; et al. NS2 protein of hepatitis C virus interacts with structural and non-structural proteins towards virus assembly. PLoS Pathog. 2011, 7, e1001278. [Google Scholar] [CrossRef] [PubMed]
- Stapleford, K.A.; Lindenbach, B.D. Hepatitis C virus NS2 coordinates virus particle assembly through physical interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J. Virol. 2011, 85, 1706–1717. [Google Scholar] [CrossRef]
- Phan, T.; Beran, R.K.; Peters, C.; Lorenz, I.C.; Lindenbach, B.D. Hepatitis C virus NS2 protein contributes to virus particle assembly via opposing epistatic interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J. Virol. 2009, 83, 8379–8395. [Google Scholar] [CrossRef] [PubMed]
- Tedbury, P.; Welbourn, S.; Pause, A.; King, B.; Griffin, S.; Harris, M. The subcellular localization of the hepatitis C virus non-structural protein NS2 is regulated by an ion channel-independent function of the p7 protein. J. Gen. Virol. 2011, 92, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, S.; Saravanabalaji, D.; Yi, M.K. Detergent-Resistant Membrane Association of NS2 and E2 during Hepatitis C Virus Replication. J. Virol. 2015, 89, 4562–4574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aizaki, H.; Lee, K.J.; Sung, V.; Ishiko, H.; Lai, M. Characterization of the hepatitis C virus RNA replication complex associated with lipid rafts. Virology 2004, 324, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Aizaki, H.; Morikawa, K.; Fukasawa, M.; Hara, H.; Inoue, Y.; Tani, H.; Saito, K.; Nishijima, M.; Hanada, K.; Matsuura, Y.; et al. Critical role of virion-associated cholesterol and sphingolipid in hepatitis C virus infection. J. Virol. 2008, 82, 5715–5724. [Google Scholar] [CrossRef]
- Paul, D.; Hoppe, S.; Saher, G.; Krijnse-Locker, J.; Bartenschlager, R. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J. Virol. 2013, 87, 10612–10627. [Google Scholar] [CrossRef]
- Shi, S.T.; Polyak, S.J.; Tu, H.; Taylor, D.R.; Gretch, D.R.; Lai, M.M. Hepatitis C virus NS5A colocalizes with the core protein on lipid droplets and interacts with apolipoproteins. Virology 2002, 292, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Gerl, M.J. Revitalizing membrane rafts: New tools and insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 688–699. [Google Scholar] [CrossRef]
- Manes, S.; Del Real, G.; Martinez, C. Pathogens: Raft hijackers. Nat. Rev. Immunol. 2003, 3, 557–568. [Google Scholar] [CrossRef]
- Boslem, E.; Weir, J.; MacIntosh, G.; Sue, N.; Cantley, J.; Meikle, P.J.; Biden, T.J. Alteration of endoplasmic reticulum lipid rafts contributes to lipotoxicity in pancreatic β-cells. J. Biol. Chem. 2013, 288, 26569–26582. [Google Scholar] [CrossRef]
- Browman, D.T.; Resek, M.E.; Zajchowski, L.D.; Robbins, S.M. Erlin-1 and erlin-2 are novel members of the prohibitin family of proteins that define lipid-raft-like domains of the ER. J. Cell Sci. 2006, 119, 3149–3160. [Google Scholar] [CrossRef] [Green Version]
- Giang, E.; Dorner, M.; Prentoe, J.C.; Dreux, M.; Evans, M.J.; Bukh, J.; Rice, C.M.; Ploss, A.; Burton, D.R.; Law, M. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc. Natl. Acad. Sci. USA 2012, 109, 6205–6210. [Google Scholar] [CrossRef] [Green Version]
- Dubuisson, J.; Duvet, S.; Meunier, J.C.; Op De Beeck, A.; Cacan, R.; Wychowski, C.; Cocquerel, L. Glycosylation of the hepatitis C virus envelope protein E1 is dependent on the presence of a downstream sequence on the viral polyprotein. J. Biol. Chem. 2000, 275, 30605–30609. [Google Scholar] [CrossRef]
- Brisson, L.; Gillet, L.; Calaghan, S.; Besson, P.; Le Guennec, J.-Y.; Roger, S.; Gore, J. Na(V)1.5 enhances breast cancer cell invasiveness by increasing NHE1-dependent H(+) efflux in caveolae. Oncogene 2011, 30, 2070–2076. [Google Scholar] [CrossRef]
- Morandat, S.; El Kirat, K. Solubilization of supported lipid membranes by octyl glucoside observed by time-lapse atomic force microscopy. Colloids Surf. B 2007, 55, 179–184. [Google Scholar] [CrossRef]
- Patel, J.; Patel, A.H.; McLauchlan, J. Covalent interactions are not required to permit or stabilize the non-covalent association of hepatitis C virus glycoproteins E1 and E2. J. Gen. Virol. 1999, 80, 1681–1690. [Google Scholar] [CrossRef]
- Vieyres, G.; Thomas, X.; Descamps, V.; Duverlie, G.; Patel, A.H.; Dubuisson, J. Characterization of the envelope glycoproteins associated with infectious hepatitis C virus. J. Virol. 2010, 84, 10159–10168. [Google Scholar] [CrossRef]
- Boyer, A.; Dumans, A.; Beaumont, E.; Etienne, L.; Roingeard, P.; Meunier, J.C. The association of hepatitis C virus glycoproteins with apolipoproteins E and B early in assembly is conserved in lipoviral particles. J. Biol. Chem. 2014, 289, 18904–18913. [Google Scholar] [CrossRef] [PubMed]
- Egger, D.; Wolk, B.; Gosert, R.; Bianchi, L.; Blum, H.E.; Moradpour, D.; Bienz, K. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 2002, 76, 5974–5984. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Penin, F. Hepatitis C virus proteins: From structure to function. Curr. Top. Microbiol. Immunol. 2013, 369, 113–142. [Google Scholar] [PubMed]
- Ruiz-Herrero, T.; Hagan, M.F. Simulations show that virus assembly and budding are facilitated by membrane microdomains. Biophys. J. 2015, 108, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; Suzuki, R.; Murakami, K.; Aizaki, H.; Ishii, K.; Murayama, A.; Date, T.; Matsuura, Y.; Miyamura, T.; Wakita, T.; et al. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J. Virol. 2008, 82, 7964–7976. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boyer, A.; Dreneau, J.; Dumans, A.; Burlaud-Gaillard, J.; Bull-Maurer, A.; Roingeard, P.; Meunier, J.-C. Endoplasmic Reticulum Detergent-Resistant Membranes Accommodate Hepatitis C Virus Proteins for Viral Assembly. Cells 2019, 8, 487. https://doi.org/10.3390/cells8050487
Boyer A, Dreneau J, Dumans A, Burlaud-Gaillard J, Bull-Maurer A, Roingeard P, Meunier J-C. Endoplasmic Reticulum Detergent-Resistant Membranes Accommodate Hepatitis C Virus Proteins for Viral Assembly. Cells. 2019; 8(5):487. https://doi.org/10.3390/cells8050487
Chicago/Turabian StyleBoyer, Audrey, Julie Dreneau, Amélie Dumans, Julien Burlaud-Gaillard, Anne Bull-Maurer, Philippe Roingeard, and Jean-Christophe Meunier. 2019. "Endoplasmic Reticulum Detergent-Resistant Membranes Accommodate Hepatitis C Virus Proteins for Viral Assembly" Cells 8, no. 5: 487. https://doi.org/10.3390/cells8050487