Zileuton, a 5-Lipoxygenase Inhibitor, Exerts Anti-Angiogenic Effect by Inducing Apoptosis of HUVEC via BK Channel Activation




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animal and Ethics Statement
2.3. Cell Cultures
2.4. Cell Viability Assay
2.5. BrdU Incorporation Assay
2.6. Scratch Wound Healing Assays
2.7. Tube Formation Assay
2.8. Matrigel Plug Assay and Analysis of Hemoglobin Content
2.9. Measurement of Nitric Oxide (NO) Production
2.10. BK Channel Small Interfering RNA (siRNA) Transfection
2.11. Assay of LTB4, PGE2, PGI2, and Caspase-3 Activity
2.12. RNA Preparation and Real-Time PCR
2.13. Western Blot Analysis
2.14. Whole-Cell Patch Clamp Recordings
2.15. Measurement of Intracellular Ca2+
2.16. Statistical Analysis
3. Results
3.1. Zileuton Inhibits VEGF-Induced Angiogenesis In Vitro
3.2. Zileuton Inhibits Angiogenesis In Vivo
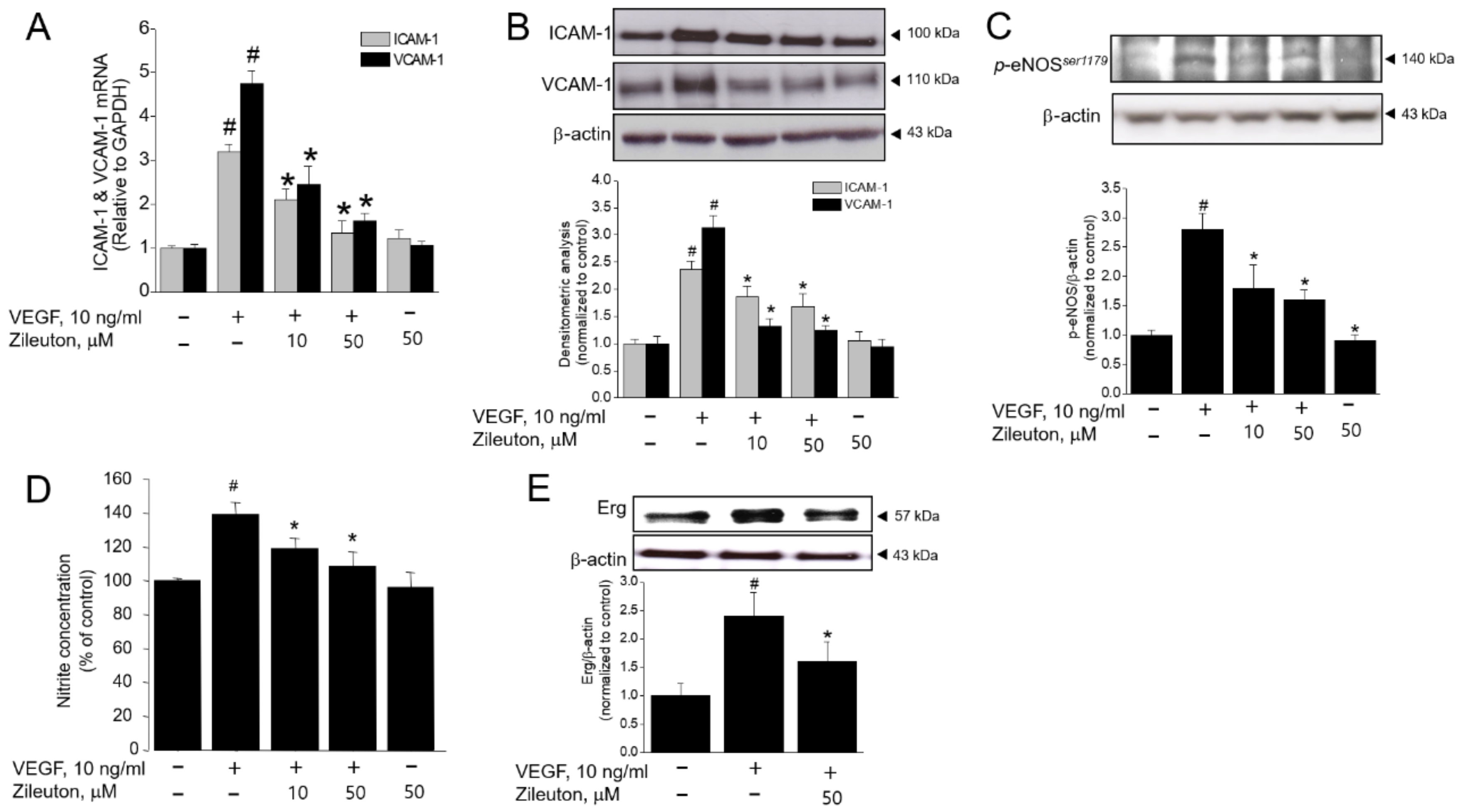
3.3. Zileuton Inhibits VEGF-Induced Adhesion Molecules, Erg, and Production of NO
3.4. Zileuton Augments Whole-Cell K+ Outward Currents in HUVECs
3.5. Zileuton Attenuates VEGF-Induced Angiogenesis and Proliferation via BK Channel Opening
3.6. Effect of BK Channel Knockdown on Zileuton-Induced Antiangiogenic and Anti-Proliferative Activity
3.7. Zileuton Induces Apoptosis Through BK Channel in VEGF-Induced Angiogenenic HUVECs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ferrara, N.; Kerbel, R.S. Angiogenesis as a therapeutic target. Nature 2005, 438, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Chiodoni, C.; Colombo, M.P.; Sangaletti, S. Matricellular proteins: From homeostasis to inflammation, cancer, and metastasis. Cancer Metastasis Rev. 2010, 29, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef]
- Heinzman, J.M.; Brower, S.L.; Bush, J.E. Comparison of angiogenesis-related factor expression in primary tumor cultures under normal and hypoxic growth conditions. Cancer Cell Int. 2008, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Ben-Baruch, A. Host microenvironment in breast cancer development: Inflammatory cells, cytokines and chemokines in breast cancer progression: Reciprocal tumor-microenvironment interactions. Breast Cancer Res. 2003, 5, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef]
- Wu, S.N.; Lo, Y.K.; Li, H.F.; Shen, A.Y. Functional coupling of voltage-dependent L-type Ca2+ current to Ca2+-activated K+ current in pituitary GH3 cells. Chin. J. Physiol. 2001, 44, 161–167. [Google Scholar]
- Parihar, A.S.; Coghlan, M.J.; Gopalakrishnan, M.; Shieh, C.C. Effects of intermediate-conductance Ca2+-activated K+ channel modulators on human prostate cancer cell proliferation. Eur. J. Pharmacol. 2003, 471, 157–164. [Google Scholar] [CrossRef]
- Bloch, M.; Ousingsawat, J.; Simon, R.; Schraml, P.; Gasser, T.C.; Mihatsch, M.J.; Kunzelmann, K.; Bubendorf, L. KCNMA1 gene amplification promotes tumor cell proliferation in human prostate cancer. Oncogene 2007, 26, 2525–2534. [Google Scholar] [CrossRef]
- Lang, F.; Foller, M.; Lang, K.S.; Lang, P.A.; Ritter, M.; Gulbins, E.; Vereninov, A.; Huber, S.M. Ion channels in cell proliferation and apoptotic cell death. J. Membr. Biol. 2005, 205, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Stegen, B.; Butz, L.; Klumpp, L.; Zips, D.; Dittmann, K.; Ruth, P.; Huber, S.M. Ca2+-Activated IK K+ Channel Blockade Radiosensitizes Glioblastoma Cells. Mol. Cancer Res. 2015, 13, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Sontheimer, H. An unexpected role for ion channels in brain tumor metastasis. Exp. Biol. Med. (Maywood) 2008, 233, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels and the hallmarks of cancer. Trends Mol. Med. 2010, 16, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Khaitan, D.; Sankpal, U.T.; Weksler, B.; Meister, E.A.; Romero, I.A.; Couraud, P.O.; Ningaraj, N.S. Role of KCNMA1 gene in breast cancer invasion and metastasis to brain. BMC Cancer 2009, 9, 258. [Google Scholar] [CrossRef] [PubMed]
- Oeggerli, M.; Tian, Y.; Ruiz, C.; Wijker, B.; Sauter, G.; Obermann, E.; Guth, U.; Zlobec, I.; Sausbier, M.; Kunzelmann, K.; et al. Role of KCNMA1 in breast cancer. PLoS ONE 2012, 7, e41664. [Google Scholar] [CrossRef] [PubMed]
- Goda, A.A.; Siddique, A.B.; Mohyeldin, M.; Ayoub, N.M.; El Sayed, K.A. The Maxi-K (BK) Channel Antagonist Penitrem A as a Novel Breast Cancer-Targeted Therapeutic. Mar. Drugs 2018, 16, 157. [Google Scholar] [CrossRef]
- Han, X.; Xi, L.; Wang, H.; Huang, X.; Ma, X.; Han, Z.; Wu, P.; Ma, X.; Lu, Y.; Wang, G.; et al. The potassium ion channel opener NS1619 inhibits proliferation and induces apoptosis in A2780 ovarian cancer cells. Biochem. Biophys. Res. Commun. 2008, 375, 205–209. [Google Scholar] [CrossRef]
- Kim, K.Y.; Cheon, H.G. Antiangiogenic effect of rosiglitazone is mediated via peroxisome proliferator-activated receptor gamma-activated maxi-K channel opening in human umbilical vein endothelial cells. J. Biol. Chem. 2006, 281, 13503–13512. [Google Scholar] [CrossRef]
- Cai, R.P.; Xue, Y.X.; Huang, J.; Wang, J.H.; Wang, J.H.; Zhao, S.Y.; Guan, T.T.; Zhang, Z.; Gu, Y.T. NS1619 regulates the expression of caveolin-1 protein in a time-dependent manner via ROS/PI3K/PKB/FoxO1 signaling pathway in brain tumor microvascular endothelial cells. J. Neurol. Sci. 2016, 369, 109–118. [Google Scholar] [CrossRef]
- Barlow, R.S.; El-Mowafy, A.M.; White, R.E. H(2)O(2) opens BK(Ca) channels via the PLA(2)-arachidonic acid signaling cascade in coronary artery smooth muscle. Am. J. Physiol. Heart. Circ. Physiol. 2000, 279, H475–H483. [Google Scholar] [CrossRef] [PubMed]
- Radmark, O.; Samuelsson, B. Regulation of 5-lipoxygenase enzyme activity. Biochem. Biophys. Res. Commun. 2005, 338, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Werz, O.; Steinhilber, D. Development of 5-lipoxygenase inhibitors—Lessons from cellular enzyme regulation. Biochem. Pharmacol. 2005, 70, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Braeckman, R.A.; Granneman, G.R.; Locke, C.S.; Machinist, J.M.; Cavannaugh, J.H.; Awni, W.M. The pharmacokinetics of zileuton in healthy young and elderly volunteers. Clin. Pharmacokinet. 1995, 29 (Suppl. 2), 42–48. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, Y.; Zhang, H.; Peng, C.; Li, S. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat. Genet. 2009, 41, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Ihara, A.; Wada, K.; Yoneda, M.; Fujisawa, N.; Takahashi, H.; Nakajima, A. Blockade of leukotriene B4 signaling pathway induces apoptosis and suppresses cell proliferation in colon cancer. J. Pharmacol. Sci. 2007, 103, 24–32. [Google Scholar] [CrossRef]
- Yektaei-Karin, E.; Zovko, A.; Nilsson, A.; Nasman-Glaser, B.; Kanter, L.; Radmark, O.; Wallvik, J.; Ekblom, M.; Dolinska, M.; Qian, H.; et al. Modulation of leukotriene signaling inhibiting cell growth in chronic myeloid leukemia. Leuk Lymphoma. 2017, 58, 1903–1913. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis and apoptosis. Semin. Cancer. Biol. 2003, 13, 159–167. [Google Scholar] [CrossRef]
- Huang, X.; Sun, J.; Chen, G.; Niu, C.; Wang, Y.; Zhao, C.; Sun, J.; Huang, H.; Huang, S.; Liang, Y.; et al. Resveratrol Promotes Diabetic Wound Healing via SIRT1-FOXO1-c-Myc Signaling Pathway-Mediated Angiogenesis. Front. Pharmacol. 2019, 10, 421. [Google Scholar] [CrossRef]
- Zhu, H.; Gao, M.; Gao, X.; Tong, Y. Vascular endothelial growth factor-B: Impact on physiology and pathology. Cell. Adh. Migr. 2018, 12, 215–227. [Google Scholar] [CrossRef]
- Kwak, H.J.; Park, K.M.; Choi, H.E.; Chung, K.S.; Lim, H.J.; Park, H.Y. PDE4 inhibitor, roflumilast protects cardiomyocytes against NO-induced apoptosis via activation of PKA and Epac dual pathways. Cell Signal 2008, 20, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, K.S.; Lim, H.J.; Kim, H.; Kwak, H.J.; Park, H.Y. The PPARdelta ligand L-165041 inhibits VEGF-induced angiogenesis, but the antiangiogenic effect is not related to PPARdelta. J. Cell. Biochem. 2012, 113, 1947–1954. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.D.; Meininger, C.J.; Ziche, M.; Granger, H.J. VEGF upregulates ecNOS message, protein, and NO production in human endothelial cells. Am J Physiol 1998, 274, H1054–H1058. [Google Scholar] [CrossRef] [PubMed]
- Tamarat, R.; Silvestre, J.S.; Kubis, N.; Benessiano, J.; Duriez, M.; de Gasparo, M.; Henrion, D.; Levy, B.I. Endothelial nitric oxide synthase lies downstream from angiotensin II-induced angiogenesis in ischemic hindlimb. Hypertension 2002, 39, 830–835. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, X.; Wang, S.; Wu, N.; Sood, S.; Wang, P.; Jin, Z.; Beer, D.G.; Giordano, T.J.; Lin, Y.; Shih, W.C.; et al. Overexpression of 5-lipoxygenase in rat and human esophageal adenocarcinoma and inhibitory effects of zileuton and celecoxib on carcinogenesis. Clin Cancer Res 2004, 10, 6703–6709. [Google Scholar] [CrossRef] [PubMed]
- Ohd, J.F.; Nielsen, C.K.; Campbell, J.; Landberg, G.; Lofberg, H.; Sjolander, A. Expression of the leukotriene D4 receptor CysLT1, COX-2, and other cell survival factors in colorectal adenocarcinomas. Gastroenterology 2003, 124, 57–70. [Google Scholar] [CrossRef]
- Khophai, S.; Thanee, M.; Techasen, A.; Namwat, N.; Klanrit, P.; Titapun, A.; Jarearnrat, A.; Sa-Ngiamwibool, P.; Loilome, W. Zileuton suppresses cholangiocarcinoma cell proliferation and migration through inhibition of the Akt signaling pathway. Onco Targets Ther 2018, 11, 7019–7029. [Google Scholar] [CrossRef] [PubMed]
- Steinhilber, D.; Hofmann, B. Recent advances in the search for novel 5-lipoxygenase inhibitors. Basic Clin Pharmacol Toxicol 2014, 114, 70–77. [Google Scholar] [CrossRef]
- Kwak, H.J.; Park, K.M.; Choi, H.E.; Lim, H.J.; Park, J.H.; Park, H.Y. The cardioprotective effects of zileuton, a 5-lipoxygenase inhibitor, are mediated by COX-2 via activation of PKC delta. Cell Signal 2010, 22, 80–87. [Google Scholar] [CrossRef]
- Kwak, H.J.; Choi, H.E.; Cheon, H.G. 5-LO inhibition ameliorates palmitic acid-induced ER stress, oxidative stress and insulin resistance via AMPK activation in murine myotubes. Sci Rep. 2017, 7, 5025. [Google Scholar] [CrossRef]
- Rioux, N.; Castonguay, A. Inhibitors of lipoxygenase: A new class of cancer chemopreventive agents. Carcinogenesis 1998, 19, 1393–1400. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.X.; Ding, X.L.; Wu, S.B.; Zhang, H.F.; Cao, W.; Qu, L.S.; Zhang, H. Inhibition of 5-lipoxygenase triggers apoptosis in pancreatic cancer cells. Oncol Rep. 2015, 33, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.Y.; Lee, J.W.; Cho, S.H.; Seo, J.M.; Kim, J.H. Role of the low-affinity leukotriene B4 receptor BLT2 in VEGF-induced angiogenesis. Arterioscler Thromb Vasc Biol 2009, 29, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Nie, D.; Honn, K.V. Cyclooxygenase, lipoxygenase and tumor angiogenesis. Cell Mol. Life Sci. 2002, 59, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Cao, R.; Yang, Z.; Liu, T.; Wang, Y.; Wang, X. Inhibitor of 5-lipoxygenase, zileuton, suppresses prostate cancer metastasis by upregulating E-cadherin and paxillin. Urology 2013, 82, 1452.e7–1452.e14. [Google Scholar] [CrossRef] [PubMed]
- Butler, C.T.; Reynolds, A.L.; Tosetto, M.; Dillon, E.T.; Guiry, P.J.; Cagney, G.; O’Sullivan, J.; Kennedy, B.N. A Quininib Analogue and Cysteinyl Leukotriene Receptor Antagonist Inhibits Vascular Endothelial Growth Factor (VEGF)-independent Angiogenesis and Exerts an Additive Antiangiogenic Response with Bevacizumab. J. Biol Chem 2017, 292, 3552–3567. [Google Scholar] [CrossRef] [PubMed]
- Duah, E.; Teegala, L.R.; Kondeti, V.; Adapala, R.K.; Keshamouni, V.G.; Kanaoka, Y.; Austen, K.F.; Thodeti, C.K.; Paruchuri, S. Cysteinyl leukotriene 2 receptor promotes endothelial permeability, tumor angiogenesis, and metastasis. Proc. Natl. Acad. Sci. USA 2019, 116, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Tsujii, M.; Kawano, S.; Tsuji, S.; Sawaoka, H.; Hori, M.; DuBois, R.N. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell 1998, 94, 273. [Google Scholar] [CrossRef]
- Kawabe, J.-i.; Ushikubi, F.; Hasebe, N. Prostacyclin in Vascular Diseases-Recent Insights and Future Perspectives. Circulation J. 2010, 74, 836–843. [Google Scholar] [CrossRef]
- Hernandez, G.L.; Volpert, O.V.; Iniguez, M.A. Selective inhibition of vascular endothelial growth factor mediated angiogenesis by cyclosporin A: Roles of the nuclear factor of activated T cells and cyclooxygenase 2. J. Exp. Med. 2001, 193, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Neagoe, P.E.; Lemieux, C.; Sirois, M.G. Vascular endothelial growth factor (VEGF)-A156 induced prostacyclin synthesis requries the activation of VEGF receptor-1 and -2 heterodimer. J. Biol. Chem. 2005, 280, 9904–9912. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Acquaviva, A.M.; Iuliano, F.; Di Paola, R.; Cuzzocrea, S.; Sautebin, L. Up-regulation of prostaglandin biosynthesis by leukotriene C4 in elicited mice peritoneal macrophages activated with lipopolysaccharide/interferon-{gamma}. J. Leukoc. Biol. 2005, 78, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Pergola, C.; Koeberle, A.; Hoffmann, M.; Dehm, F.; Bramanti, P.; Cuzzocrea, S.; Werz, O.; Sautebin, L. The 5-lipoxygenase inhibitor, zileuton, suppresses prostaglandin biosynthesis by inhibition of arachidonic acid release in macrophages. Br. J. Pharmacol. 2010, 161, 555–570. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Sood, S.; Li, N.; Ramji, D.; Yang, P.; Newman, R.A.; Yang, C.S.; Chen, X. Involvement of the 5-lipoxygenase/leukotriene A4 hydrolase pathway in 7,12-dimethylbenz[a]anthracene (DMBA)-induced oral carcinogenesis in hamster cheek pouch, and inhibition of carcinogenesis by its inhibitors. Carcinogenesis 2006, 27, 1902–1908. [Google Scholar] [CrossRef] [PubMed]
- Kohler, R.; Kaistha, B.P.; Wulff, H. Vascular KCa-channels as therapeutic targets in hypertension and restenosis disease. Expert Opin. Ther. Targets 2010, 14, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Striano, P.; Striano, S. New and investigational antiepileptic drugs. Expert. Opin. Investig. Drugs 2009, 18, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Wickenden, A.D.; McNaughton-Smith, G. Kv7 channels as targets for the treatment of pain. Curr. Pharm. Des. 2009, 15, 1773–1798. [Google Scholar] [CrossRef]
- Kim, I.; Moon, S.O.; Kim, S.H.; Kim, H.J.; Koh, Y.S.; Koh, G.Y. Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-kappa B activation in endothelial cells. J. Biol. Chem. 2001, 276, 7614–7620. [Google Scholar] [CrossRef]
- Garmy-Susini, B.; Jin, H.; Zhu, Y.; Sung, R.J.; Hwang, R.; Varner, J. Integrin α4β1-VCAM-1-mediated adhesion between endothelial and mural cells is required for blood vessel maturation. J. Clin. Investig. 2005, 115, 1542–1551. [Google Scholar] [CrossRef]
- Fukushi, J.; Ono, M.; Morikawa, W.; Iwamoto, Y.; Kuwano, M. The activity of soluble VCAM-1 in angiogenesis stimulated by IL-4 and IL-13. J. Immunol. 2000, 165, 2818–2823. [Google Scholar] [CrossRef]
- Fukumura, D.; Gohongi, T.; Kadambi, A.; Izumi, Y.; Ang, J.; Yun, C.O.; Buerk, D.G.; Huang, P.L.; Jain, R.K. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc. Natl. Acad. Sci. USA 2001, 98, 2604–2609. [Google Scholar] [CrossRef] [PubMed]
- Pham, V.N.; Lawson, N.D.; Mugford, J.W.; Dye, L.; Castranova, D.; Lo, B.; Weinstein, B.M. Combinatorial function of ETS transcription factors in the developing vasculature. Dev. Biol. 2007, 303, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Sperone, A.; Dryden, N.H.; Birdsey, G.M.; Madden, L.; Johns, M.; Evans, P.C.; Mason, J.C.; Haskard, D.O.; Boyle, J.J.; Paleolog, E.M.; et al. The transcription factor Erg inhibits vascular inflammation by repressing NF-kappaB activation and pro-inflammatory gene expression in endothelial cells. Arterioscler Thromb. Vasc. Biol. 2011, 31, 142–150. [Google Scholar] [CrossRef]
- Birdsey, G.M.; Dryden, N.H.; Amsellem, V.; Gebhardt, F.; Sahnan, K.; Haskard, D.O.; Dejana, E.; Mason, J.C.; Randi, A.M. Transcription factor Erg regulates angiogenesis and endothelial apoptosis through VE-cadherin. Blood 2008, 111, 3498–3506. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, H.-J.; Park, J.; Um, J.-Y.; Lee, S.-S.; Kwak, H.-J. Zileuton, a 5-Lipoxygenase Inhibitor, Exerts Anti-Angiogenic Effect by Inducing Apoptosis of HUVEC via BK Channel Activation. Cells 2019, 8, 1182. https://doi.org/10.3390/cells8101182
Lim H-J, Park J, Um J-Y, Lee S-S, Kwak H-J. Zileuton, a 5-Lipoxygenase Inhibitor, Exerts Anti-Angiogenic Effect by Inducing Apoptosis of HUVEC via BK Channel Activation. Cells. 2019; 8(10):1182. https://doi.org/10.3390/cells8101182
Chicago/Turabian StyleLim, Hyun-Joung, Jinbong Park, Jae-Young Um, Sang-Seob Lee, and Hyun-Jeong Kwak. 2019. "Zileuton, a 5-Lipoxygenase Inhibitor, Exerts Anti-Angiogenic Effect by Inducing Apoptosis of HUVEC via BK Channel Activation" Cells 8, no. 10: 1182. https://doi.org/10.3390/cells8101182
APA StyleLim, H.-J., Park, J., Um, J.-Y., Lee, S.-S., & Kwak, H.-J. (2019). Zileuton, a 5-Lipoxygenase Inhibitor, Exerts Anti-Angiogenic Effect by Inducing Apoptosis of HUVEC via BK Channel Activation. Cells, 8(10), 1182. https://doi.org/10.3390/cells8101182