A 2-Min Transient Ischemia Confers Cerebral Ischemic Tolerance in Non-Obese Gerbils, but Results in Neuronal Death in Obese Gerbils by Increasing Abnormal mTOR Activation-Mediated Oxidative Stress and Neuroinflammation

 , , , and
, , , and
Abstract
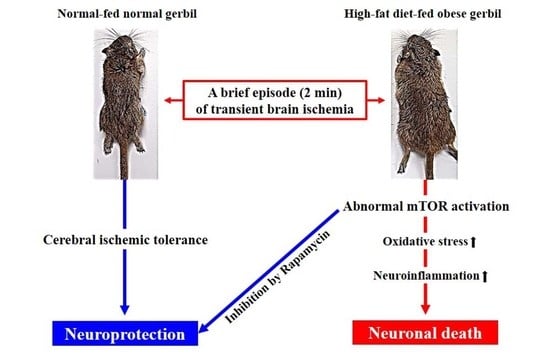
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
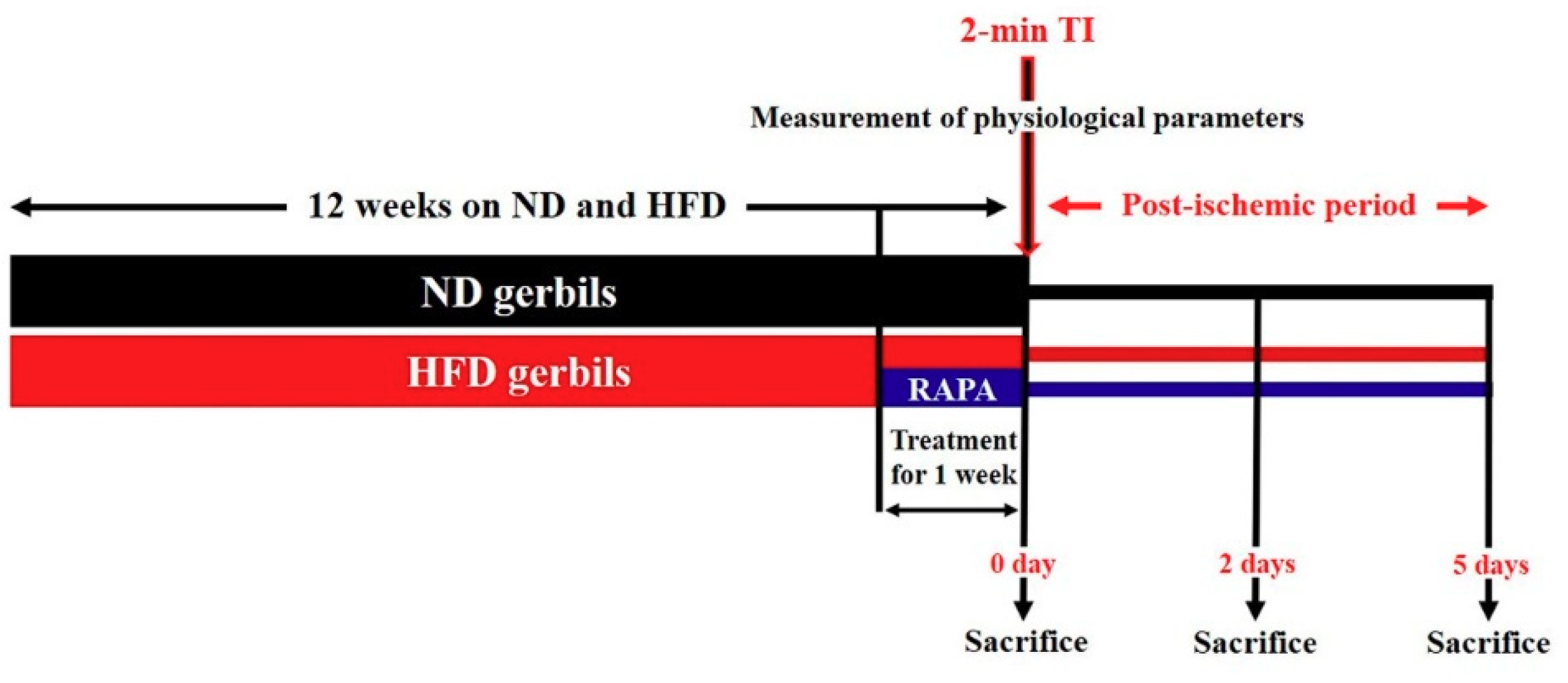
2.1. Experimental Animals and Diets
2.2. Treatment with RAPA
2.3. Analyses of Glucose Levels, and Lipid and Leptin Profiles
2.4. Induction of 2-min TI
2.5. Tissue Section for Histology
2.6. Cresyl Violet Staining
2.7. NeuN Immunofluorescence and F-J B Histofluorescence Staining
2.8. Immunohistochemistry
2.9. Dihydroethidium Fluorescence Staining
2.10. Western Blot Analysis
2.11. Data Analyses
2.12. Statistical Analysis
3. Results
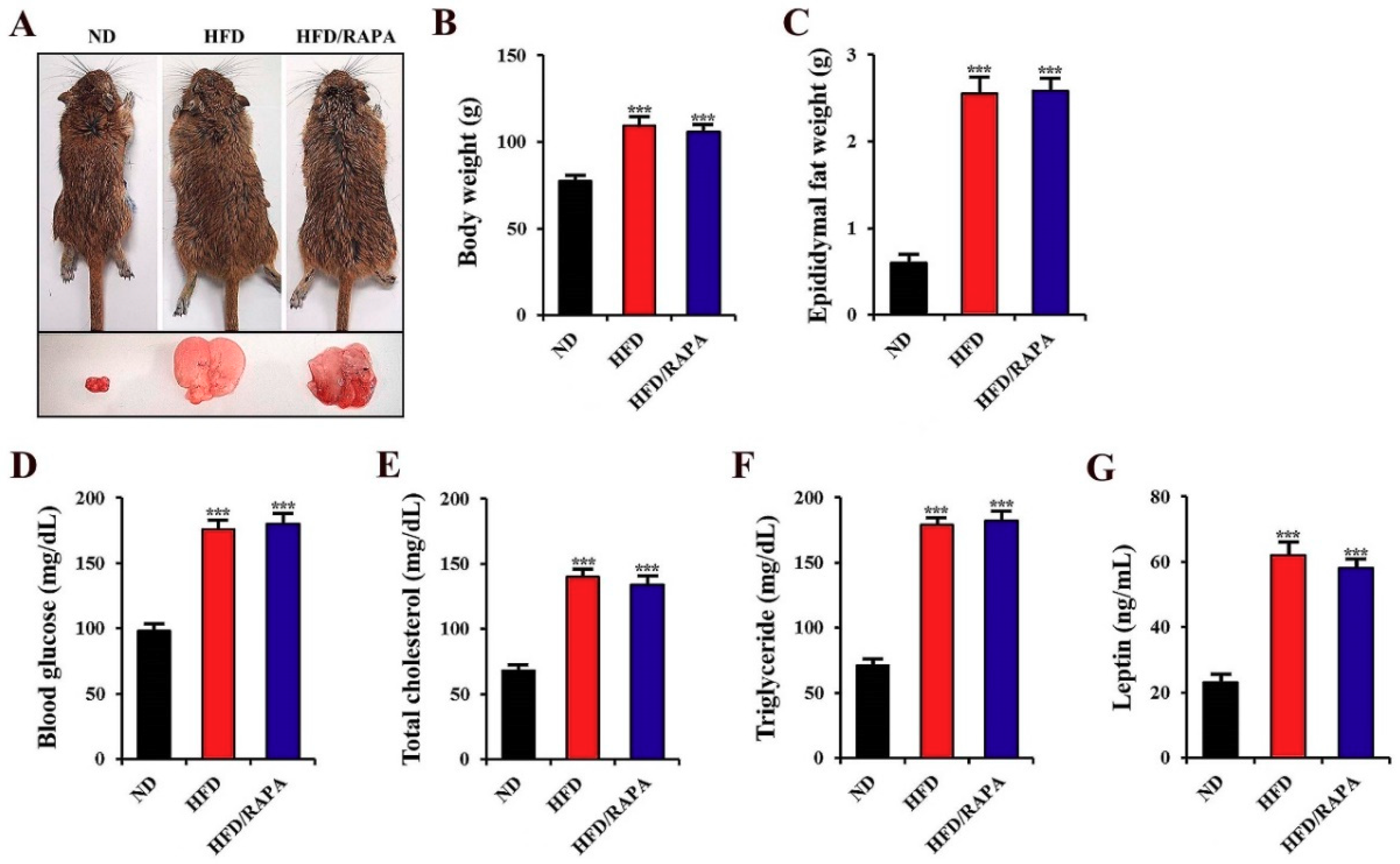
3.1. Changes in Physiological Parameters
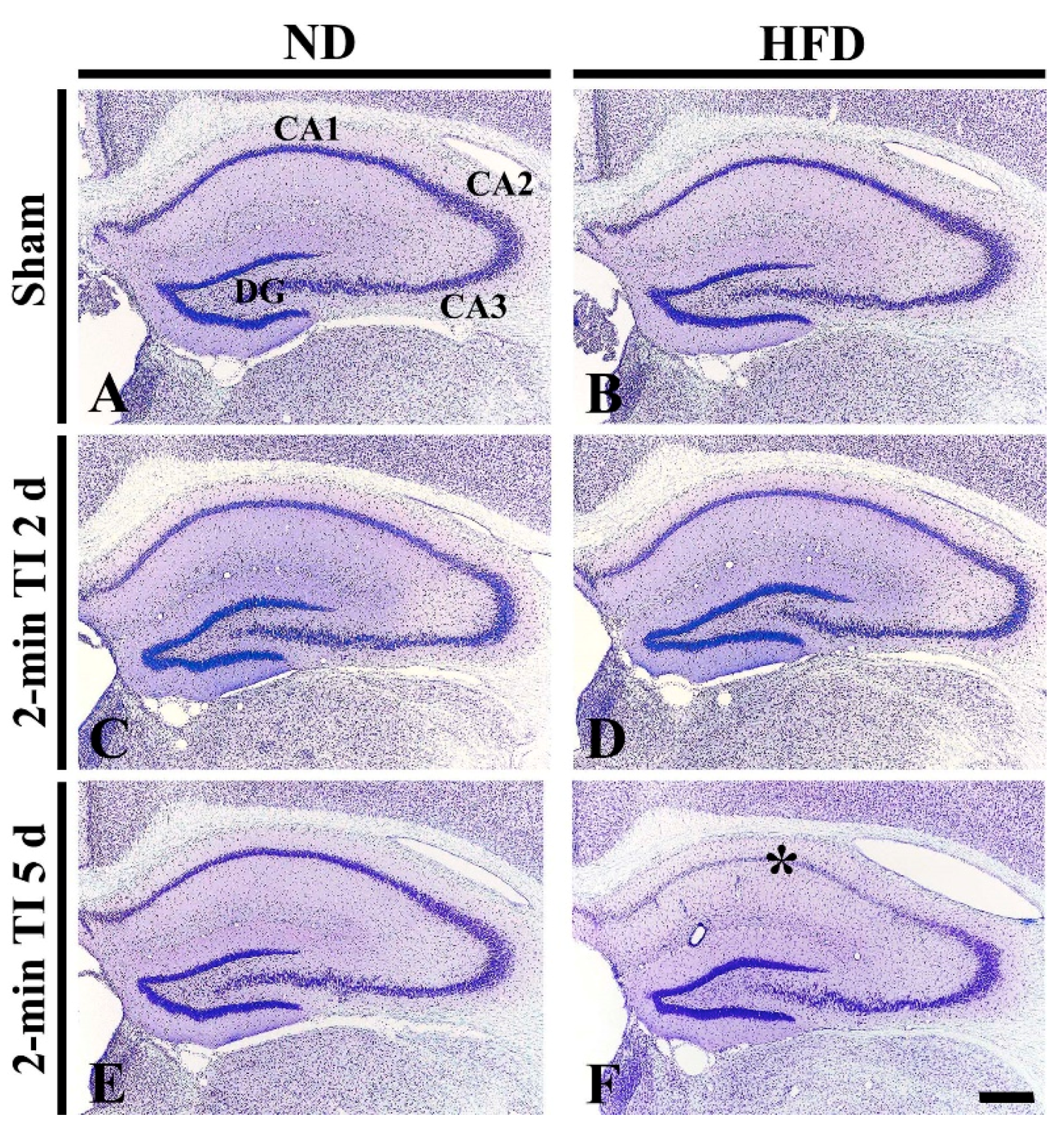
3.2. Neuronal Death by 2 Min of TI in HFD-Fed, Obese Gerbils
CV+ Cells
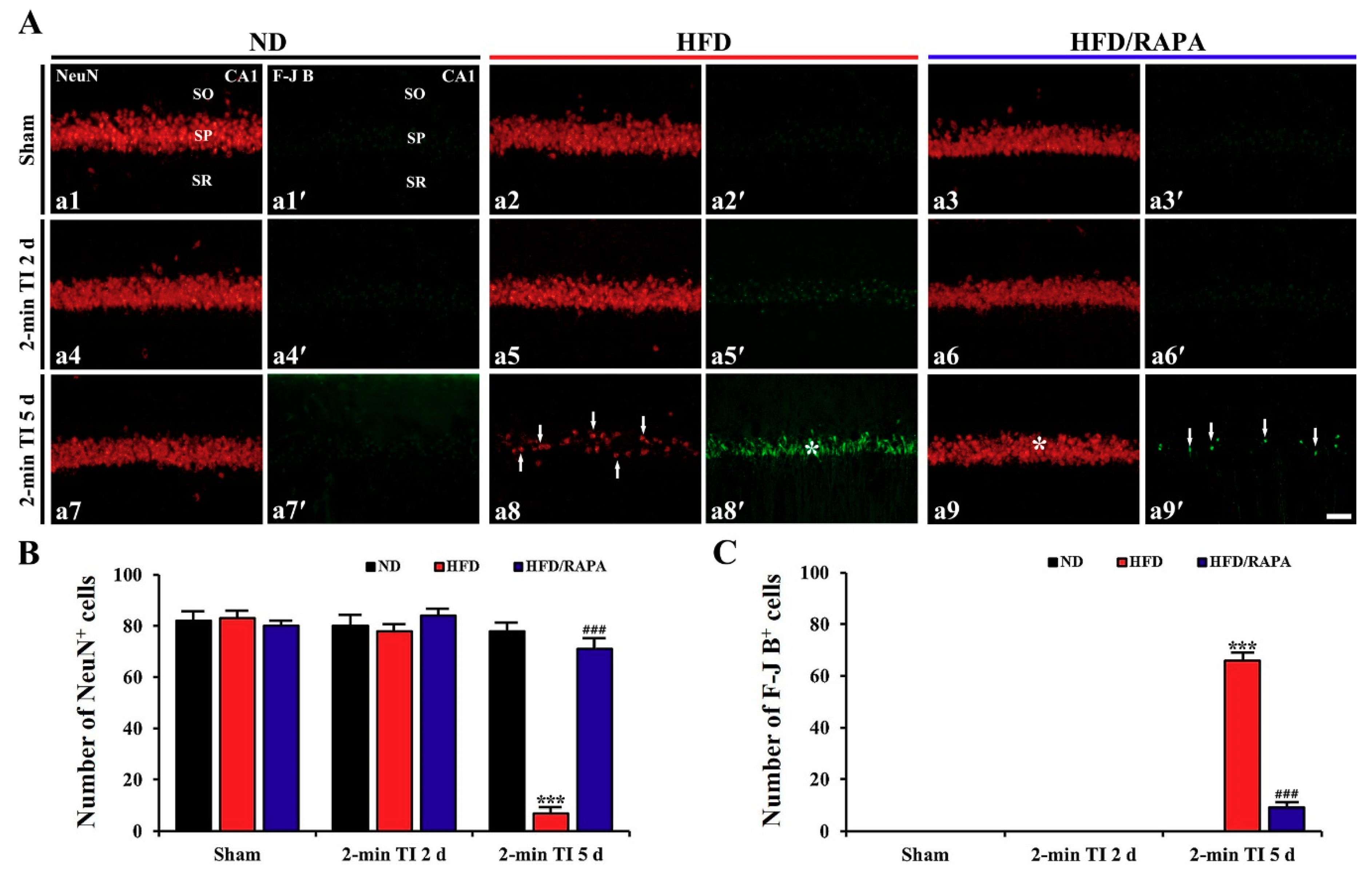
3.3. Neuroprotection by RAPA in HFD-Fed, Obese Gerbils
NeuN+ and F-J B+ Cells
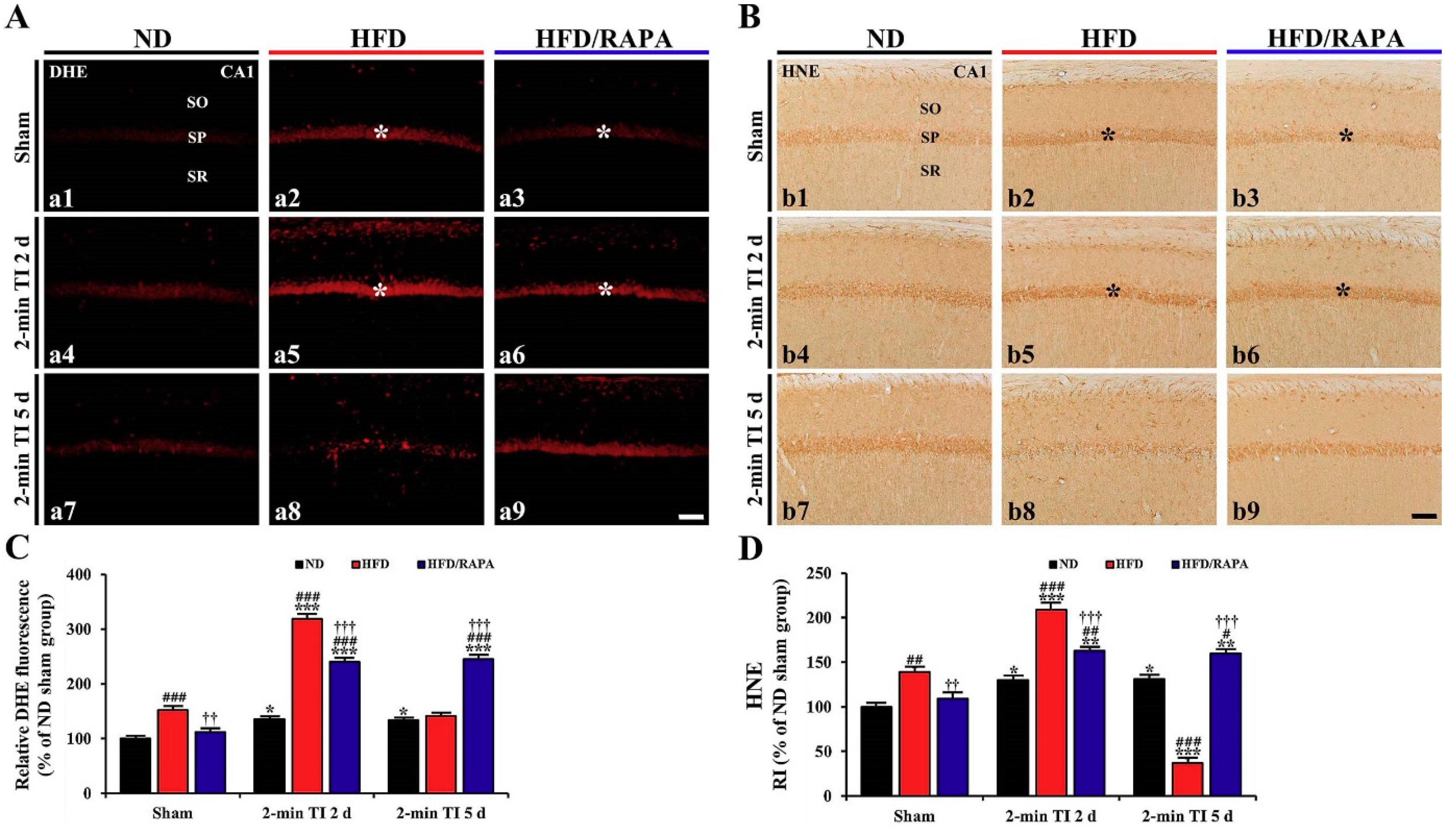
3.4. Attenuation of Oxidative Stress by RAPA in HFD-fed Group
3.4.1. DHE Fluorescence
3.4.2. HNE Immunoreactivity
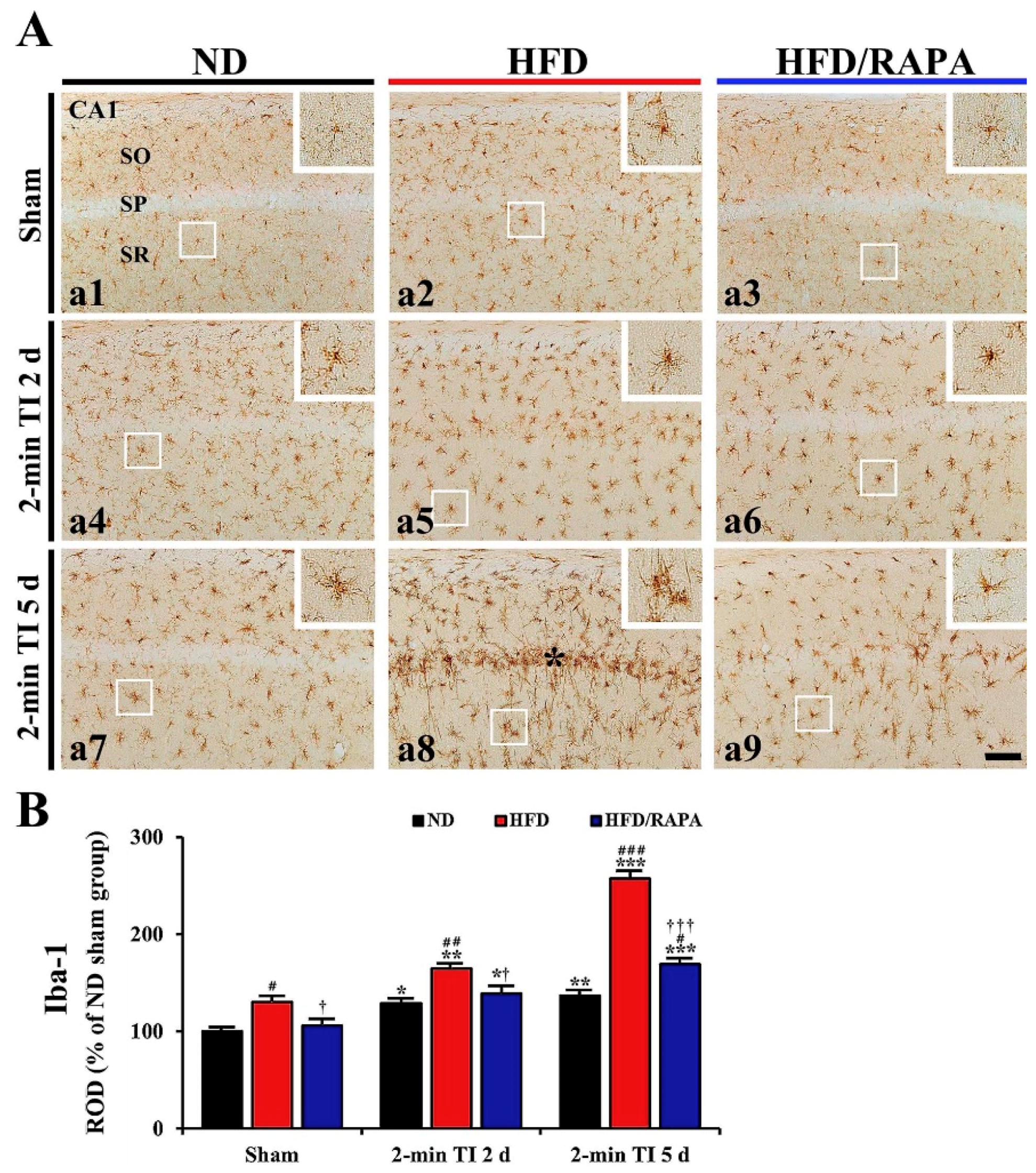
3.5. Reduction of Neuroinflammation by RAPA in HFD-fed Group
3.5.1. Iba-1+ Microglia
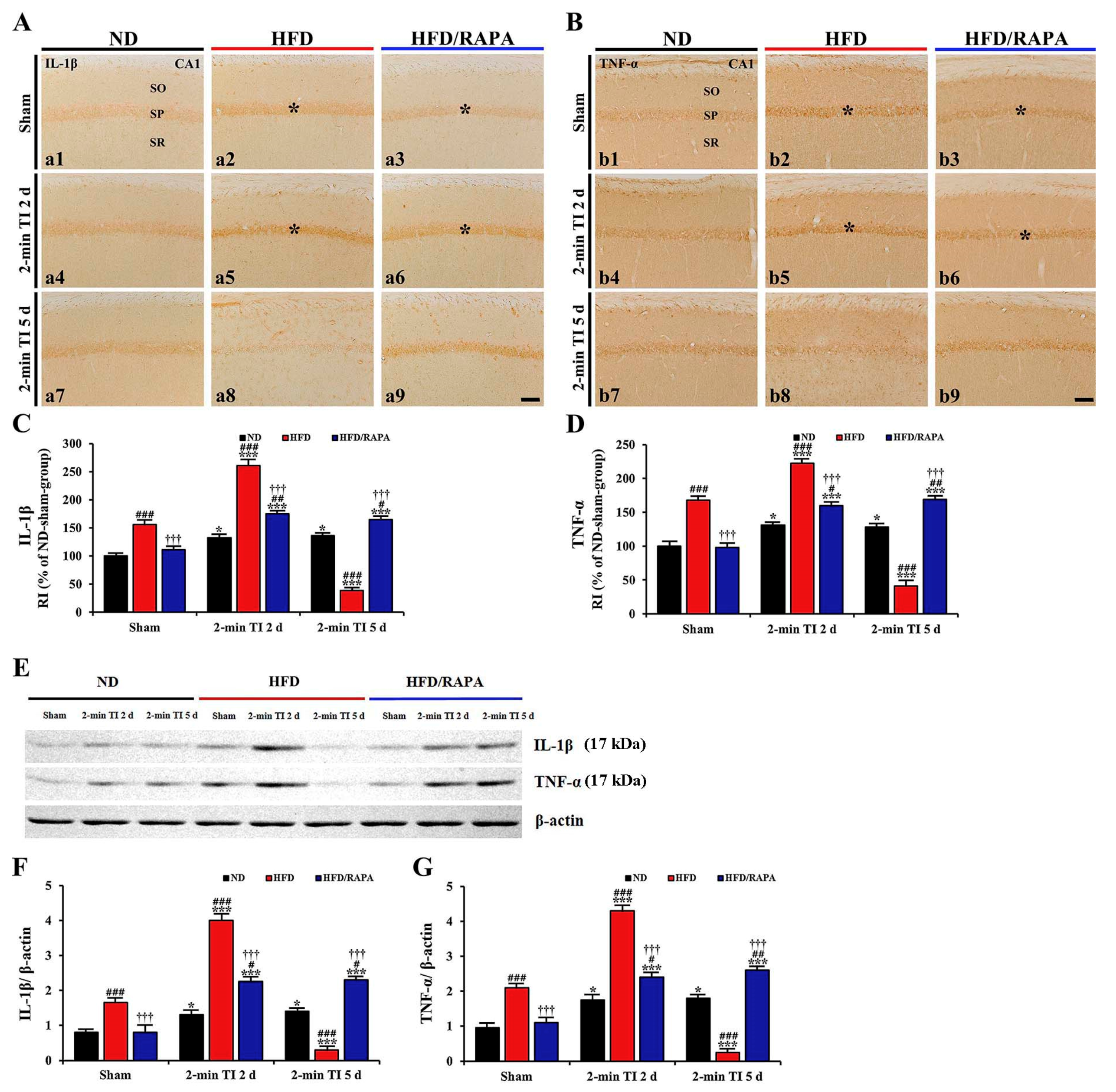
3.5.2. IL-1β and TNF-α Immunoreactivity
3.5.3. IL-1β and TNF-α Protein Levels
3.6. Inhibition of Abnormal mTOR Activation by RAPA in HFD-fed Group
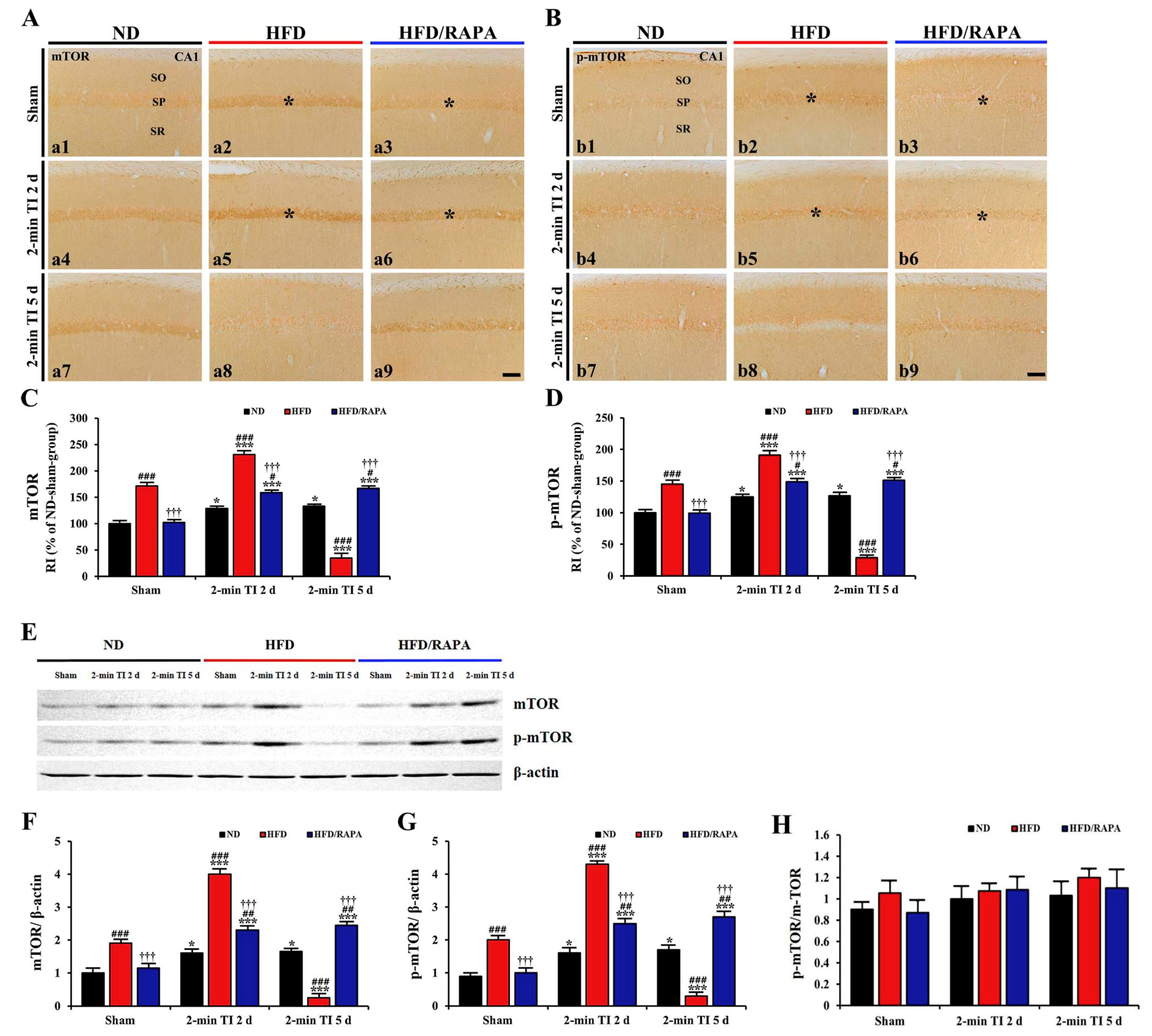
3.6.1. mTOR and p-mTOR Immunoreactivity
3.6.2. mTOR and p-mTOR Protein Levels
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Globus, M.Y.; Busto, R.; Martinez, E.; Valdes, I.; Dietrich, W.D.; Ginsberg, M.D. Comparative effect of transient global ischemia on extracellular levels of glutamate, glycine, and gamma-aminobutyric acid in vulnerable and nonvulnerable brain regions in the rat. J. Neurochem. 1991, 57, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Polsky, K.; Nadler, J.V.; Crain, B.J. Selective neocortical and thalamic cell death in the gerbil after transient ischemia. Neuroscience 1990, 35, 289–299. [Google Scholar] [CrossRef]
- Lee, J.C.; Park, J.H.; Ahn, J.H.; Kim, I.H.; Cho, J.H.; Choi, J.H.; Yoo, K.Y.; Lee, C.H.; Hwang, I.K.; Cho, J.H.; et al. New GABAergic Neurogenesis in the Hippocampal CA1 Region of a Gerbil Model of Long-Term Survival after Transient Cerebral Ischemic Injury. Brain Pathol. 2016, 26, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Nitatori, T.; Sato, N.; Waguri, S.; Karasawa, Y.; Araki, H.; Shibanai, K.; Kominami, E.; Uchiyama, Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J. Neurosci. 1995, 15, 1001–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakhan, S.E.; Kirchgessner, A.; Hofer, M. Inflammatory mechanisms in ischemic stroke: Therapeutic approaches. J. Transl. Med. 2009, 7, 97. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Park, J.H.; Kim, I.H.; Cho, G.S.; Ahn, J.H.; Tae, H.J.; Choi, S.Y.; Cho, J.H.; Kim, D.W.; Kwon, Y.G.; et al. Neuroprotection of ischemic preconditioning is mediated by thioredoxin 2 in the hippocampal CA1 region following a subsequent transient cerebral ischemia. Brain Pathol. 2017, 27, 276–291. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.P.; Shi, Y.W.; Tang, M.; Zhang, X.C.; Gu, Y.; Liang, X.M.; Wang, Z.W.; Ding, F. Isoquercetin Ameliorates Cerebral Impairment in Focal Ischemia Through Anti-Oxidative, Anti-Inflammatory, and Anti-Apoptotic Effects in Primary Culture of Rat Hippocampal Neurons and Hippocampal CA1 Region of Rats. Mol. Neurobiol. 2017, 54, 2126–2142. [Google Scholar] [CrossRef] [PubMed]
- Dirnagl, U.; Becker, K.; Meisel, A. Preconditioning and tolerance against cerebral ischaemia: From experimental strategies to clinical use. Lancet Neurol. 2009, 8, 398–412. [Google Scholar] [CrossRef]
- Wegener, S.; Gottschalk, B.; Jovanovic, V.; Knab, R.; Fiebach, J.B.; Schellinger, P.D.; Kucinski, T.; Jungehulsing, G.J.; Brunecker, P.; Muller, B.; et al. Transient ischemic attacks before ischemic stroke: Preconditioning the human brain? A multicenter magnetic resonance imaging study. Stroke 2004, 35, 616–621. [Google Scholar] [CrossRef]
- Choi, Y.S.; Cho, K.O.; Kim, E.J.; Sung, K.W.; Kim, S.Y. Ischemic preconditioning in the rat hippocampus increases antioxidant activities but does not affect the level of hydroxyl radicals during subsequent severe ischemia. Exp. Mol. Med. 2007, 39, 556–563. [Google Scholar] [CrossRef]
- Nakajima, T.; Hata, R.; Kondo, T.; Takenaka, S. Proteomic analysis of the hippocampus in naive and ischemic-preconditioned rat. J. Neurol. Sci. 2015, 358, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Cai, Y.; Zhang, X.; Huang, H.; Wang, J.; Wang, Y.; Tong, X.; Wang, J.; Wu, J. Ischemic Preconditioning Mediates Neuroprotection against Ischemia in Mouse Hippocampal CA1 Neurons by Inducing Autophagy. PLoS ONE 2015, 10, e0137146. [Google Scholar] [CrossRef] [PubMed]
- Duszczyk, M.; Ziembowicz, A.; Gadamski, R.; Wieronska, J.M.; Smialowska, M.; Lazarewicz, J.W. Changes in the NPY immunoreactivity in gerbil hippocampus after hypoxic and ischemic preconditioning. Neuropeptides 2009, 43, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Tae, H.J.; Kim, I.H.; Cho, J.H.; Lee, T.K.; Park, J.H.; Ahn, J.H.; Choi, S.Y.; Bai, H.C.; Shin, B.N.; et al. Roles of HIF-1alpha, VEGF, and NF-kappaB in Ischemic Preconditioning-Mediated Neuroprotection of Hippocampal CA1 Pyramidal Neurons Against a Subsequent Transient Cerebral Ischemia. Mol. Neurobiol. 2017, 54, 6984–6998. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.L.; Bayraktutan, U. Risk factors for ischaemic stroke. Int. J. Stroke 2008, 3, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef]
- Bas, D.F.; Ozdemir, A.O. The effect of metabolic syndrome and obesity on outcomes of acute ischemic stroke patients treated with systemic thrombolysis. J. Neurol. Sci. 2017, 383, 1–4. [Google Scholar] [CrossRef]
- Langdon, K.D.; Clarke, J.; Corbett, D. Long-term exposure to high fat diet is bad for your brain: Exacerbation of focal ischemic brain injury. Neuroscience 2011, 182, 82–87. [Google Scholar] [CrossRef]
- Li, W.; Prakash, R.; Chawla, D.; Du, W.; Didion, S.P.; Filosa, J.A.; Zhang, Q.; Brann, D.W.; Lima, V.V.; Tostes, R.C.; et al. Early effects of high-fat diet on neurovascular function and focal ischemic brain injury. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R1001–R1008. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Tolhurst, A.T.; Cho, S. Deregulation of inflammatory response in the diabetic condition is associated with increased ischemic brain injury. J. Neuroinflamm. 2014, 11, 83. [Google Scholar] [CrossRef]
- Maysami, S.; Haley, M.J.; Gorenkova, N.; Krishnan, S.; McColl, B.W.; Lawrence, C.B. Prolonged diet-induced obesity in mice modifies the inflammatory response and leads to worse outcome after stroke. J. Neuroinflamm. 2015, 12, 140. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, D.S.; Kang, S.; Kwon, D.Y. Ischemic hippocampal cell death induces glucose dysregulation by attenuating glucose-stimulated insulin secretion which is exacerbated by a high fat diet. Life Sci. 2011, 88, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Gao, X.X.; Chen, L.; You, X.Q. Rapamycin suppresses Abeta25-35-or LPS-induced neuronal inflammation via modulation of NF-kappaB signaling. Neuroscience 2017, 355, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Yang, J.; Yang, L. Insights for Oxidative Stress and mTOR Signaling in Myocardial Ischemia/Reperfusion Injury under Diabetes. Oxid. Med. Cell. Longev. 2017, 2017, 6437467. [Google Scholar] [CrossRef] [PubMed]
- Catania, C.; Binder, E.; Cota, D. mTORC1 signaling in energy balance and metabolic disease. Int. J. Obes. (Lond.) 2011, 35, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Wong, M. Mammalian target of rapamycin (mTOR) pathways in neurological diseases. Biomed. J. 2013, 36, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, M.D.; Busto, R. Rodent models of cerebral ischemia. Stroke 1989, 20, 1627–1642. [Google Scholar] [CrossRef] [PubMed]
- Hei, C.; Liu, P.; Yang, X.; Niu, J.; Li, P.A. Inhibition of mTOR signaling Confers Protection against Cerebral Ischemic Injury in Acute Hyperglycemic Rats. Int. J. Biol. Sci. 2017, 13, 878–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.; Ahn, J.H.; Kim, H.; Kim, D.W.; Lee, T.K.; Lee, J.C.; Kim, Y.M.; Lee, C.H.; Hwang, I.K.; Yan, B.C.; et al. Chronic high-fat diet-induced obesity in gerbils increases pro-inflammatory cytokines and mTOR activation, and elicits neuronal death in the striatum following brief transient ischemia. Neurochem. Int. 2018, 121, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Park, C.W.; Ahn, J.H.; Choi, S.Y.; Shin, M.C.; Cho, J.H.; Lee, T.K.; Kim, I.H.; Cho, J.H.; Lee, J.C.; et al. Neuroprotection and reduced gliosis by pre-and post-treatments of hydroquinone in a gerbil model of transient cerebral ischemia. Chem. Biol. Interact. 2017, 278, 230–238. [Google Scholar] [CrossRef]
- Radtke-Schuller, S.; Schuller, G.; Angenstein, F.; Grosser, O.S.; Goldschmidt, J.; Budinger, E. Brain atlas of the Mongolian gerbil (Meriones unguiculatus) in CT/MRI-aided stereotaxic coordinates. Brain Struct. Funct. 2016, 221, 1–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, K.B.; Lemberg, L. Obesity and the metabolic syndrome. Am. J. Crit. Care 2003, 12, 167–170. [Google Scholar] [PubMed]
- Moon, S.M.; Choi, G.M.; Yoo, D.Y.; Jung, H.Y.; Yim, H.S.; Kim, D.W.; Hwang, I.K.; Cho, B.M.; Chang, I.B.; Cho, S.M.; et al. Differential Effects of Pioglitazone in the Hippocampal CA1 Region Following Transient Forebrain Ischemia in Low-and High-Fat Diet-Fed Gerbils. Neurochem. Res. 2015, 40, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Ventura, L.L.; Fortes, N.C.; Santiago, H.C.; Caliari, M.V.; Gomes, M.A.; Oliveira, D.R. Obesity-induced diet leads to weight gain, systemic metabolic alterations, adipose tissue inflammation, hepatic steatosis, and oxidative stress in gerbils (Meriones unguiculatus). PeerJ 2017, 5, e2967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, M.G., Jr.; Leibel, R.L.; Seeley, R.J.; Schwartz, M.W. Obesity and leptin resistance: Distinguishing cause from effect. Trends Endocrinol. Metab. 2010, 21, 643–651. [Google Scholar] [CrossRef]
- Deutsch, C.; Portik-Dobos, V.; Smith, A.D.; Ergul, A.; Dorrance, A.M. Diet-induced obesity causes cerebral vessel remodeling and increases the damage caused by ischemic stroke. Microvasc. Res. 2009, 78, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Zhang, J.; Feng, C.; Xiong, L.; Zuo, Z. Critical role of matrix metalloprotease-9 in chronic high fat diet-induced cerebral vascular remodelling and increase of ischaemic brain injury in micedagger. Cardiovasc. Res. 2014, 103, 473–484. [Google Scholar] [CrossRef]
- ElAli, A.; Doeppner, T.R.; Zechariah, A.; Hermann, D.M. Increased blood-brain barrier permeability and brain edema after focal cerebral ischemia induced by hyperlipidemia: Role of lipid peroxidation and calpain-1/2, matrix metalloproteinase-2/9, and RhoA overactivation. Stroke 2011, 42, 3238–3244. [Google Scholar] [CrossRef]
- Cheon, S.H.; Yan, B.C.; Chen, B.H.; Park, J.H.; Ahn, J.H.; Kim, I.H.; Lee, J.C.; Park, Y.S.; Kim, M.J.; Lee, Y.L.; et al. Accelerated and exacerbated effects of high dietary fat on neuronal damage induced by transient cerebral ischemia in the gerbil septum. Endocrinol. Metab. (Seoul) 2014, 29, 328–335. [Google Scholar] [CrossRef]
- Yan, B.C.; Park, J.H.; Ahn, J.H.; Kim, I.H.; Lee, J.C.; Yoo, K.Y.; Choi, J.H.; Hwang, I.K.; Cho, J.H.; Kwon, Y.G.; et al. Effects of high-fat diet on neuronal damage, gliosis, inflammatory process and oxidative stress in the hippocampus induced by transient cerebral ischemia. Neurochem. Res. 2014, 39, 2465–2478. [Google Scholar] [CrossRef] [PubMed]
- Hye Kim, I.; Lee, J.C.; Ha Park, J.; Hyeon Ahn, J.; Cho, J.H.; Hui Chen, B.; Na Shin, B.; Chun Yan, B.; Rueol Ryu, D.; Hong, S.; et al. Time interval after ischaemic preconditioning affects neuroprotection and gliosis in the gerbil hippocampal CA1 region induced by transient cerebral ischaemia. Neurol. Res. 2016, 38, 210–219. [Google Scholar] [CrossRef]
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The science of stroke: Mechanisms in search of treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef] [PubMed]
- Chu, K.; Yin, B.; Wang, J.; Peng, G.; Liang, H.; Xu, Z.; Du, Y.; Fang, M.; Xia, Q.; Luo, B. Inhibition of P2X7 receptor ameliorates transient global cerebral ischemia/reperfusion injury via modulating inflammatory responses in the rat hippocampus. J. Neuroinflamm. 2012, 9, 69. [Google Scholar] [CrossRef]
- Leak, R.K.; Li, P.; Zhang, F.; Sulaiman, H.H.; Weng, Z.; Wang, G.; Stetler, R.A.; Shi, Y.; Cao, G.; Gao, Y.; et al. Apurinic/apyrimidinic endonuclease 1 upregulation reduces oxidative DNA damage and protects hippocampal neurons from ischemic injury. Antioxid. Redox Signal. 2015, 22, 135–148. [Google Scholar] [CrossRef]
- Ohtsuki, T.; Matsumoto, M.; Kuwabara, K.; Kitagawa, K.; Suzuki, K.; Taniguchi, N.; Kamada, T. Influence of oxidative stress on induced tolerance to ischemia in gerbil hippocampal neurons. Brain Res. 1992, 599, 246–252. [Google Scholar] [CrossRef]
- Ohtsuki, T.; Ruetzler, C.A.; Tasaki, K.; Hallenbeck, J.M. Interleukin-1 mediates induction of tolerance to global ischemia in gerbil hippocampal CA1 neurons. J. Cereb. Blood Flow Metab. 1996, 16, 1137–1142. [Google Scholar] [CrossRef]
- Haley, M.J.; Lawrence, C.B. Obesity and stroke: Can we translate from rodents to patients? J. Cereb. Blood Flow Metab. 2016, 36, 2007–2021. [Google Scholar] [CrossRef] [PubMed]
- Savini, I.; Catani, M.V.; Evangelista, D.; Gasperi, V.; Avigliano, L. Obesity-associated oxidative stress: Strategies finalized to improve redox state. Int. J. Mol. Sci. 2013, 14, 10497–10538. [Google Scholar] [CrossRef]
- Hajiluian, G.; Abbasalizad Farhangi, M.; Nameni, G.; Shahabi, P.; Megari-Abbasi, M. Oxidative stress-induced cognitive impairment in obesity can be reversed by vitamin D administration in rats. Nutr. Neurosci. 2018, 21, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Jeon, B.T.; Jeong, E.A.; Shin, H.J.; Lee, Y.; Lee, D.H.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Roh, G.S. Resveratrol attenuates obesity-associated peripheral and central inflammation and improves memory deficit in mice fed a high-fat diet. Diabetes 2012, 61, 1444–1454. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.L.; Du, J.; Zhang, Y.; Yan, J.T.; Hu, X.M. Hyperlipidemia exacerbates cerebral injury through oxidative stress, inflammation and neuronal apoptosis in MCAO/reperfusion rats. Exp. Brain Res. 2015, 233, 2753–2765. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Hei, C.; Liu, P.; Song, Y.; Thomas, T.; Tshimanga, S.; Wang, F.; Niu, J.; Sun, T.; Li, P.A. Inhibition of mTOR Pathway by Rapamycin Reduces Brain Damage in Rats Subjected to Transient Forebrain Ischemia. Int. J. Biol. Sci. 2015, 11, 1424–1435. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, A.; Sharma, U.; Jagannathan, N.R.; Reeta, K.H.; Gupta, Y.K. Rapamycin protects against middle cerebral artery occlusion induced focal cerebral ischemia in rats. Behav. Brain Res. 2011, 225, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Sun, F.; Wang, J.; Mao, X.; Xie, L.; Yang, S.H.; Su, D.M.; Simpkins, J.W.; Greenberg, D.A.; Jin, K. mTOR signaling inhibition modulates macrophage/microglia-mediated neuroinflammation and secondary injury via regulatory T cells after focal ischemia. J. Immunol. 2014, 192, 6009–6019. [Google Scholar] [CrossRef]
- Xing, J.; Lu, J. Effects of mTOR on Neurological Deficits after Transient Global Ischemia. Transl. Neurosci. 2017, 8, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Yoshimoto, K.; Nghiemphu, P.; Brown, K.; Dang, J.; Zhu, S.; Hsueh, T.; Chen, Y.; Wang, W.; Youngkin, D.; et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008, 5, e8. [Google Scholar] [CrossRef]
- Thellung, S.; Corsaro, A.; Nizzari, M.; Barbieri, F.; Florio, T. Autophagy activator drugs: A new opportunity in neuroprotection from misfolded protein toxicity. Int. J. Mol. Sci. 2019, 20, 901. [Google Scholar] [CrossRef]
- Liu, P.; Yang, X.; Hei, C.; Meli, Y.; Niu, J.; Sun, T.; Li, P.A. Rapamycin Reduced Ischemic Brain Damage in Diabetic Animals Is Associated with Suppressions of mTOR and ERK1/2 Signaling. Int. J. Biol. Sci. 2016, 12, 1032–1040. [Google Scholar] [CrossRef] [Green Version]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.H.; Ahn, J.H.; Song, M.; Kim, H.; Park, C.W.; Park, Y.E.; Lee, T.-K.; Lee, J.-C.; Kim, D.W.; Lee, C.-H.; et al. A 2-Min Transient Ischemia Confers Cerebral Ischemic Tolerance in Non-Obese Gerbils, but Results in Neuronal Death in Obese Gerbils by Increasing Abnormal mTOR Activation-Mediated Oxidative Stress and Neuroinflammation. Cells 2019, 8, 1126. https://doi.org/10.3390/cells8101126
Park JH, Ahn JH, Song M, Kim H, Park CW, Park YE, Lee T-K, Lee J-C, Kim DW, Lee C-H, et al. A 2-Min Transient Ischemia Confers Cerebral Ischemic Tolerance in Non-Obese Gerbils, but Results in Neuronal Death in Obese Gerbils by Increasing Abnormal mTOR Activation-Mediated Oxidative Stress and Neuroinflammation. Cells. 2019; 8(10):1126. https://doi.org/10.3390/cells8101126
Chicago/Turabian StylePark, Joon Ha, Ji Hyeon Ahn, Minah Song, Hyunjung Kim, Cheol Woo Park, Young Eun Park, Tae-Kyeong Lee, Jae-Chul Lee, Dae Won Kim, Choong-Hyun Lee, and et al. 2019. "A 2-Min Transient Ischemia Confers Cerebral Ischemic Tolerance in Non-Obese Gerbils, but Results in Neuronal Death in Obese Gerbils by Increasing Abnormal mTOR Activation-Mediated Oxidative Stress and Neuroinflammation" Cells 8, no. 10: 1126. https://doi.org/10.3390/cells8101126