Serine Protease HtrA2/Omi Deficiency Impairs Mitochondrial Homeostasis and Promotes Hepatic Fibrogenesis via Activation of Hepatic Stellate Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Samples and Animal Studies
2.2. Histological Analysis and Immunohistochemistry
2.3. TUNEL Assay
2.4. Isolation of Mouse Primary Hepatocytes and Cell Culture
2.5. Western Blot Analysis
2.6. Transmission Electron Microscopy (TEM)
2.7. Immunofluorescence Staining
2.8. Serum Aminotransferase Activity and Hydroxyproline Determination
2.9. Lentiviral Vector Transduction
2.10. Quantitative Real Time-PCR-Based Gene Expression
2.11. Mitochondrial Fractionation and mtDNA Extraction
2.12. Genomic DNA Extraction and Quantitative PCR (qPCR)
2.13. Mitochondrial Membrane Potential and ROS Production
2.14. Measurement of Mitochondrial Respiration
2.15. Serine Protease Activity Assy
2.16. Statistical Analysis
3. Results
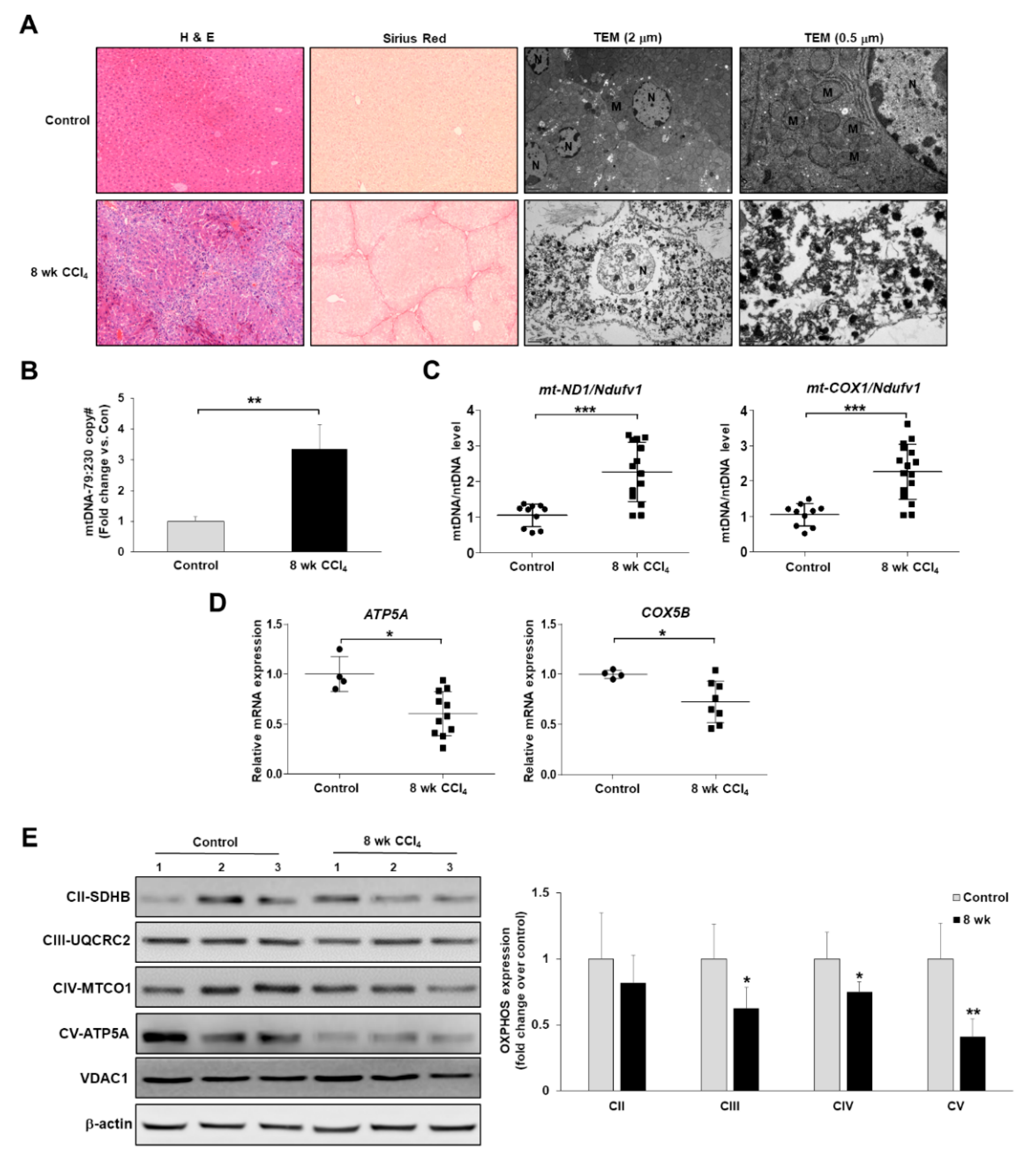
3.1. Mitochondrial Dysfunction Is Present in the CCl4-Induced Mouse Model of Liver Fibrosis
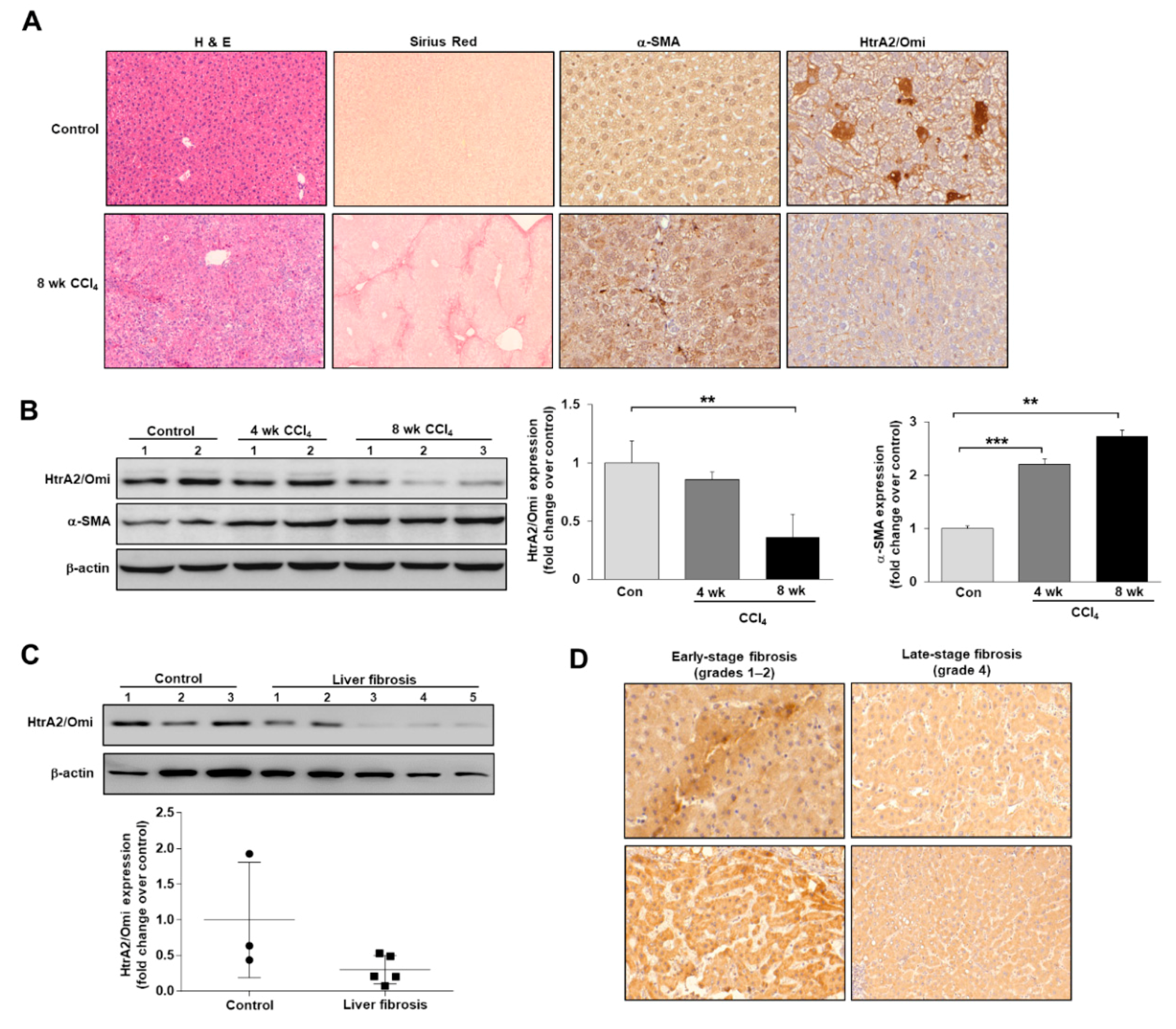
3.2. HtrA2/Omi Expression Is Decreased in CCl4-Induced Fibrotic Mice and Patients with Liver Fibrosis
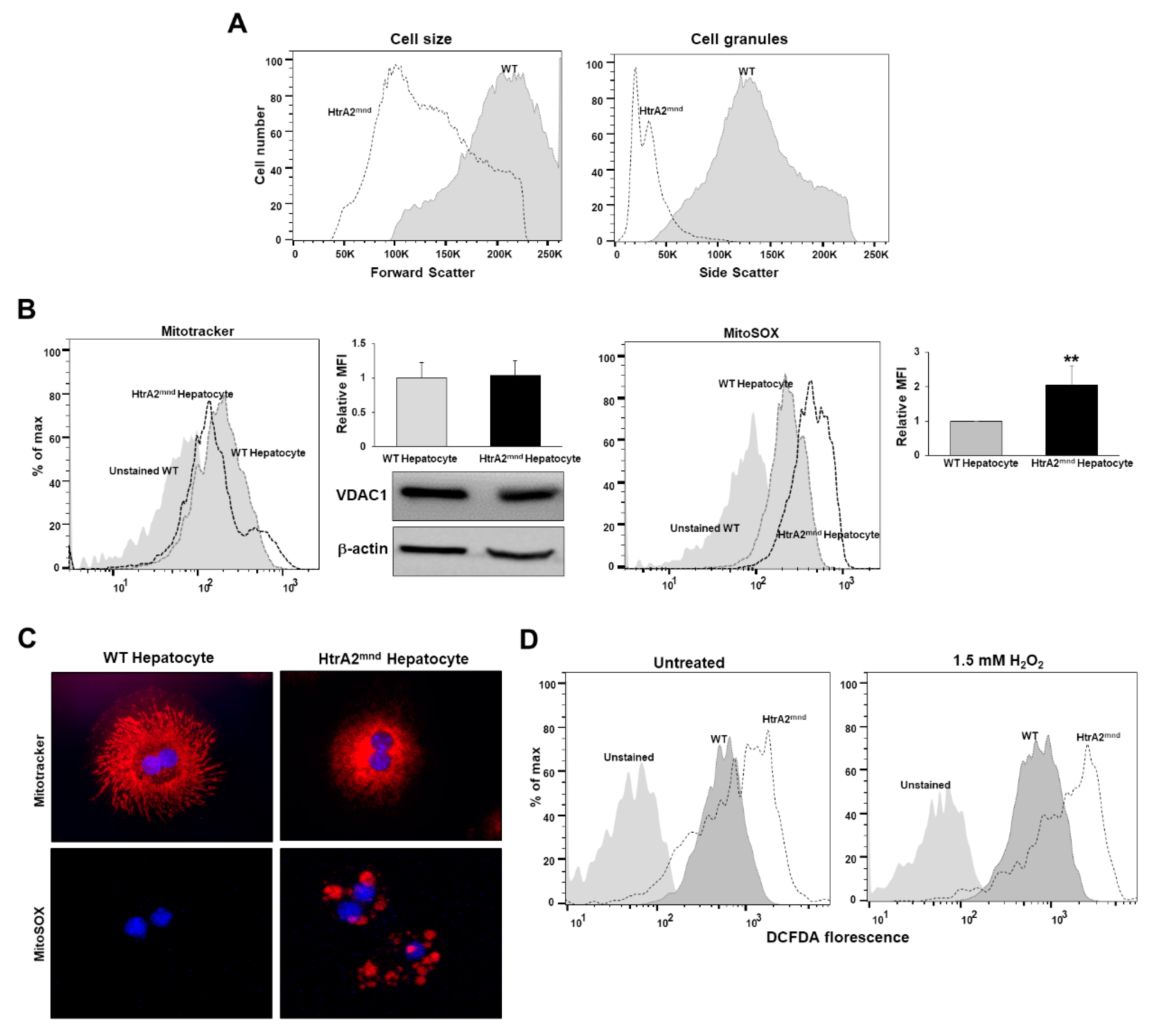
3.3. HtrA2/Omi-Deficient Hepatocytes Cause Mitochondrial Accumulation and Structural Anomalies
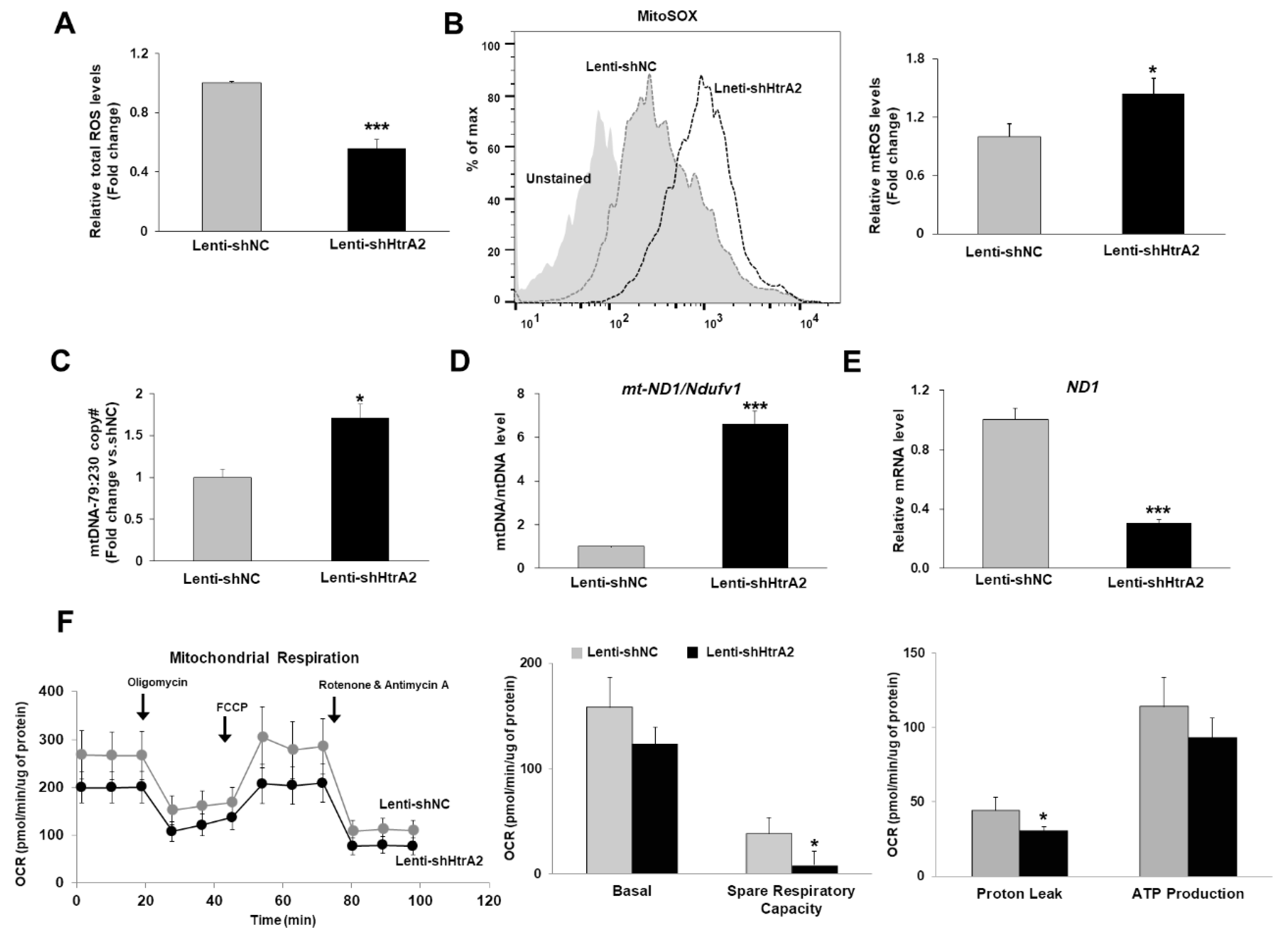
3.4. Loss of HtrA2/Omi Protease Activity in Hepatocytes Results in the Accumulation of Dysfunctional Mitochondria and Oxidative Stress
3.5. Loss of HtrA2/Omi Mitochondrial Protease Activity in mnd2-Mutant Mice Promotes Liver Fibrosis
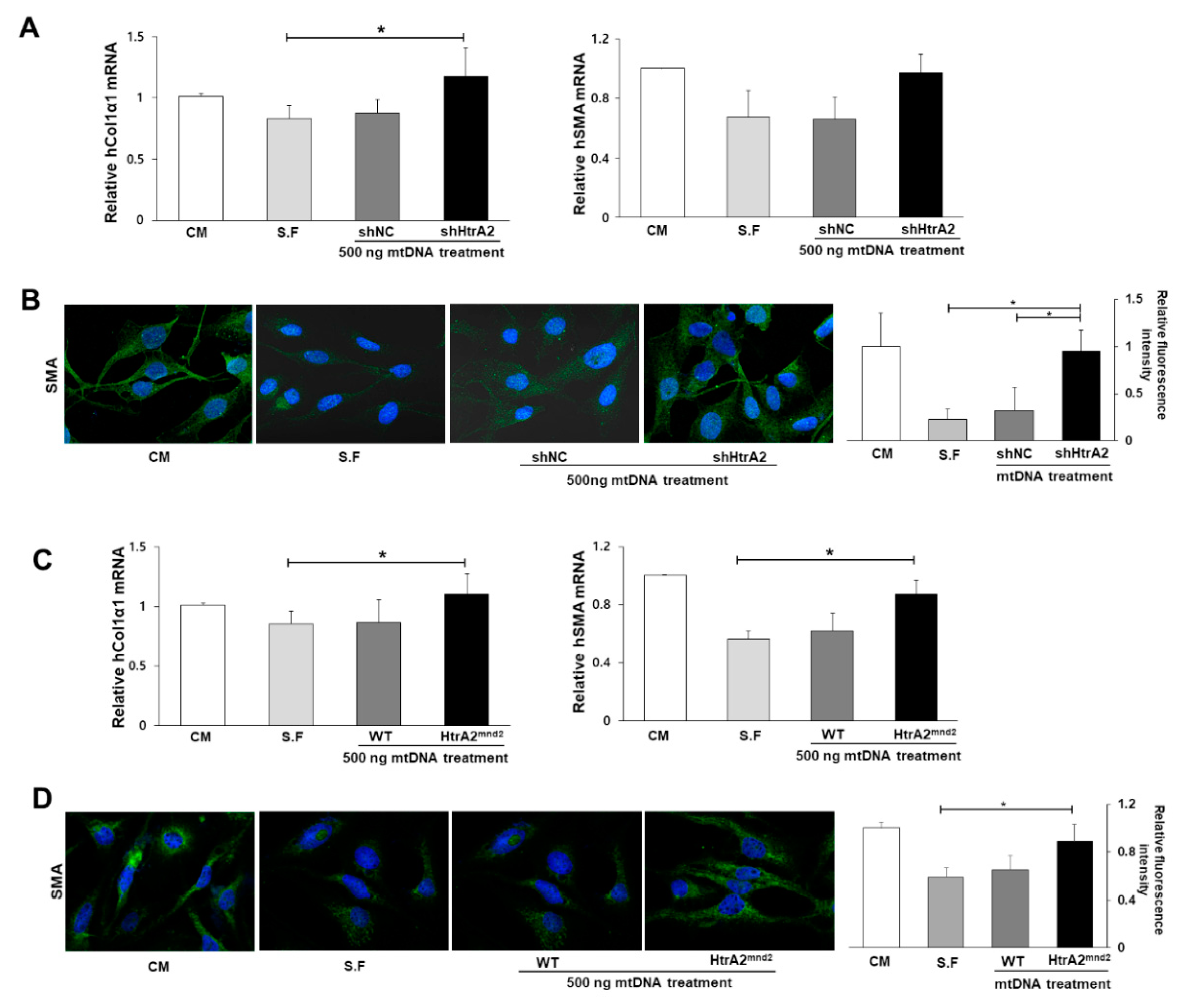
3.6. HtrA2/Omi Deficient Hepatocyte Derived-mtDNA Induces Liver Fibrogenesis
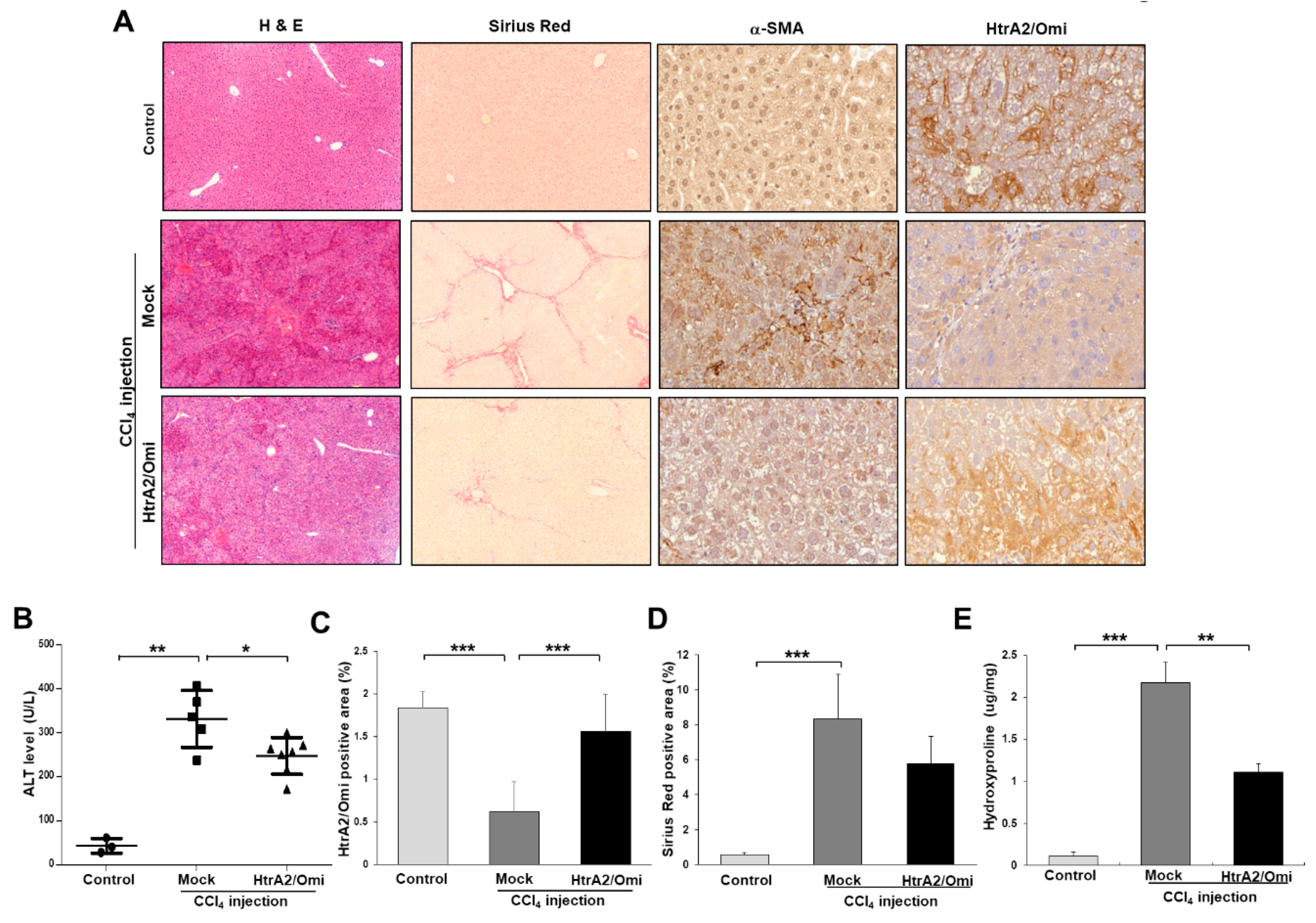
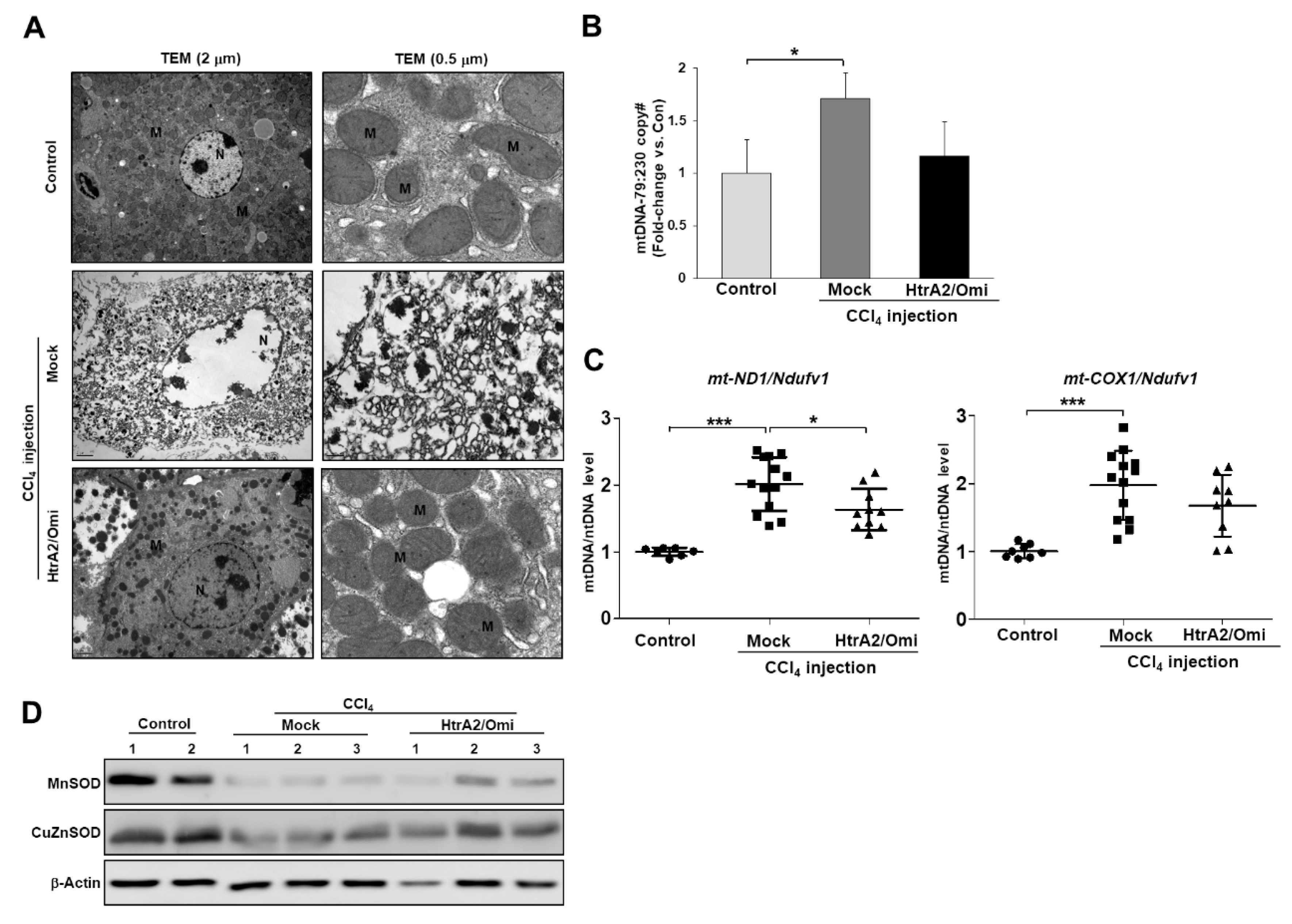
3.7. Restoration of HtrA2/Omi Expression Rescues CCl4-Induced Liver Fibrosis and Reverses Mitochondrial Dysfunction in Hepatocyte
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Friedman, S.L. Evolving challenges in hepatic fibrosis. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Schwabe, R.F. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Poelstra, K.; Schuppan, D. Targeted therapy of liver fibrosis/cirrhosis and its complications. J. Hepatol. 2011, 55, 726–728. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Massart, J.; Robin, M.A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef] [PubMed]
- Auger, C.; Alhasawi, A.; Contavadoo, M.; Appanna, V.D. Dysfunctional mitochondrial bioenergetics and the pathogenesis of hepatic disorders. Front. Cell Dev. Biol. 2015, 3, 40. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, E.L.; Best, J.; Dolle, L.; Najimi, M.; Sokal, E.; van Grunsven, L.A. Mitochondrial uncouplers inhibit hepatic stellate cell activation. Bmc Gastroenterol. 2012, 12, 68. [Google Scholar] [CrossRef] [PubMed]
- De Minicis, S.; Seki, E.; Oesterreicher, C.; Schnabl, B.; Schwabe, R.F.; Brenner, D.A. Reduced nicotinamide adenine dinucleotide phosphate oxidase mediates fibrotic and inflammatory effects of leptin on hepatic stellate cells. Hepatology 2008, 48, 2016–2026. [Google Scholar] [CrossRef]
- Mitchell, C.; Robin, M.A.; Mayeuf, A.; Mahrouf-Yorgov, M.; Mansouri, A.; Hamard, M.; Couton, D.; Fromenty, B.; Gilgenkrantz, H. Protection against hepatocyte mitochondrial dysfunction delays fibrosis progression in mice. Am. J. Pathol. 2009, 175, 1929–1937. [Google Scholar] [CrossRef]
- Vande Walle, L.; Lamkanfi, M.; Vandenabeele, P. The mitochondrial serine protease HtrA2/Omi: An overview. Cell Death Differ. 2008, 15, 453–460. [Google Scholar] [CrossRef]
- Kang, S.; Louboutin, J.P.; Datta, P.; Landel, C.P.; Martinez, D.; Zervos, A.S.; Strayer, D.S.; Fernandes-Alnemri, T.; Alnemri, E.S. Loss of HtrA2/Omi activity in non-neuronal tissues of adult mice causes premature aging. Cell Death Differ. 2013, 20, 259–269. [Google Scholar] [CrossRef]
- Kang, S.; Fernandes-Alnemri, T.; Alnemri, E.S. A novel role for the mitochondrial HTRA2/OMI protease in aging. Autophagy 2013, 9, 420–421. [Google Scholar] [CrossRef] [PubMed]
- Goo, H.G.; Jung, M.K.; Han, S.S.; Rhim, H.; Kang, S. HtrA2/Omi deficiency causes damage and mutation of mitochondrial DNA. Biochim. Biophys. Acta 2013, 1833, 1866–1875. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Sung, P.S.; Lee, E.B.; Hur, W.; Park, D.J.; Shin, E.C.; Windisch, M.P.; Yoon, S.K. GRIM-19 Restricts HCV Replication by Attenuating Intracellular Lipid Accumulation. Front. Microbiol. 2017, 8, 576. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.A.; Kim, G.Y.; Nam, M.K.; Goo, H.G.; Kang, S.; Rhim, H. A Simple and Accurate Genotype Analysis of the motor neuron degeneration 2 (mnd2) Mice: An Easy-to-Follow Guideline and Standard Protocol Applicable to Mutant Mouse Model. Interdiscip. Bio Cent. 2012, 4, 1–7. [Google Scholar] [CrossRef]
- Kim, S.M.; Choi, J.E.; Hur, W.; Kim, J.H.; Hong, S.W.; Lee, E.B.; Lee, J.H.; Li, T.Z.; Sung, P.S.; Yoon, S.K. RAR-Related Orphan Receptor Gamma (ROR-gamma) Mediates Epithelial-Mesenchymal Transition Of Hepatocytes During Hepatic Fibrosis. J. Cell Biochem. 2017, 118, 2026–2036. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Song, Y.; Liu, D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene 1999, 6, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Sun, R.; Huang, Q.; Tian, Z. Technical Improvement and Application of Hydrodynamic Gene Delivery in Study of Liver Diseases. Front. Pharm. 2017, 8, 591. [Google Scholar] [CrossRef] [PubMed]
- Nam, M.K.; Seong, Y.M.; Park, H.J.; Choi, J.Y.; Kang, S.; Rhim, H. The homotrimeric structure of HtrA2 is indispensable for executing its serine protease activity. Exp. Mol. Med. 2006, 38, 36–43. [Google Scholar] [CrossRef][Green Version]
- Xu, L.; Hui, A.Y.; Albanis, E.; Arthur, M.J.; O’Byrne, S.M.; Blaner, W.S.; Mukherjee, P.; Friedman, S.L.; Eng, F.J. Human hepatic stellate cell lines, LX-1 and LX-2: New tools for analysis of hepatic fibrosis. Gut 2005, 54, 142–151. [Google Scholar] [CrossRef]
- Higuchi, Y.; Linn, S. Purification of all forms of HeLa cell mitochondrial DNA and assessment of damage to it caused by hydrogen peroxide treatment of mitochondria or cells. J. Biol. Chem. 1995, 270, 7950–7956. [Google Scholar] [CrossRef]
- Sadikot, R.T.; Bedi, B.; Li, J.; Yeligar, S.M. Alcohol-induced mitochondrial DNA damage promotes injurious crosstalk between alveolar epithelial cells and alveolar macrophages. Alcohol 2018. pii: S0741–8329, 30181–30188. [Google Scholar] [CrossRef]
- Haemmerle, G.; Moustafa, T.; Woelkart, G.; Buttner, S.; Schmidt, A.; van de Weijer, T.; Hesselink, M.; Jaeger, D.; Kienesberger, P.C.; Zierler, K.; et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nat. Med. 2011, 17, 1076–1085. [Google Scholar] [CrossRef]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef]
- Moiseeva, O.; Bourdeau, V.; Roux, A.; Deschenes-Simard, X.; Ferbeyre, G. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol. Cell Biol. 2009, 29, 4495–4507. [Google Scholar] [CrossRef]
- Miettinen, T.P.; Bjorklund, M. Mitochondrial Function and Cell Size: An Allometric Relationship. Trends Cell Biol. 2017, 27, 393–402. [Google Scholar] [CrossRef]
- Mikhed, Y.; Daiber, A.; Steven, S. Mitochondrial Oxidative Stress, Mitochondrial DNA Damage and Their Role in Age-Related Vascular Dysfunction. Int. J. Mol. Sci. 2015, 16, 15918–15953. [Google Scholar] [CrossRef]
- Jones, J.M.; Datta, P.; Srinivasula, S.M.; Ji, W.; Gupta, S.; Zhang, Z.; Davies, E.; Hajnoczky, G.; Saunders, T.L.; Van Keuren, M.L.; et al. Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature 2003, 425, 721–727. [Google Scholar] [CrossRef]
- Martins, L.M.; Morrison, A.; Klupsch, K.; Fedele, V.; Moisoi, N.; Teismann, P.; Abuin, A.; Grau, E.; Geppert, M.; Livi, G.P.; et al. Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol. Cell Biol. 2004, 24, 9848–9862. [Google Scholar] [CrossRef]
- Miettinen, T.P.; Pessa, H.K.; Caldez, M.J.; Fuhrer, T.; Diril, M.K.; Sauer, U.; Kaldis, P.; Bjorklund, M. Identification of transcriptional and metabolic programs related to mammalian cell size. Curr. Biol. 2014, 24, 598–608. [Google Scholar] [CrossRef]
- Gandhi, C.R. Oxidative Stress and Hepatic Stellate Cells: A PARADOXICAL RELATIONSHIP. Trends Cell Mol. Biol. 2012, 7, 1–10. [Google Scholar]
- Gandhi, C.R. Hepatic stellate cell activation and pro-fibrogenic signals. J. Hepatol. 2017, 67, 1104–1105. [Google Scholar] [CrossRef]
- Shinde, A.B.; Baboota, R.K.; Denis, S.; Loizides-Mangold, U.; Peeters, A.; Espeel, M.; Malheiro, A.R.; Riezman, H.; Vinckier, S.; Vaz, F.M.; et al. Mitochondrial disruption in peroxisome deficient cells is hepatocyte selective but is not mediated by common hepatic peroxisomal metabolites. Mitochondrion 2018, 39, 51–59. [Google Scholar] [CrossRef]
- Satapati, S.; Kucejova, B.; Duarte, J.A.; Fletcher, J.A.; Reynolds, L.; Sunny, N.E.; He, T.; Nair, L.A.; Livingston, K.A.; Fu, X.; et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Clin. Invest. 2015, 125, 4447–4462. [Google Scholar] [CrossRef]
- Khacho, M.; Clark, A.; Svoboda, D.S.; MacLaurin, J.G.; Lagace, D.C.; Park, D.S.; Slack, R.S. Mitochondrial dysfunction underlies cognitive defects as a result of neural stem cell depletion and impaired neurogenesis. Hum. Mol. Genet. 2017, 26, 3327–3341. [Google Scholar] [CrossRef]
- Krick, S.; Shi, S.; Ju, W.; Faul, C.; Tsai, S.Y.; Mundel, P.; Bottinger, E.P. Mpv17l protects against mitochondrial oxidative stress and apoptosis by activation of Omi/HtrA2 protease. Proc. Natl. Acad. Sci. USA 2008, 105, 14106–14111. [Google Scholar] [CrossRef]
- Wan, J.; Cui, J.; Wang, L.; Wu, K.; Hong, X.; Zou, Y.; Zhao, S.; Ke, H. Excessive mitochondrial fragmentation triggered by erlotinib promotes pancreatic cancer PANC-1 cell apoptosis via activating the mROS-HtrA2/Omi pathways. Cancer Cell Int. 2018, 18, 165. [Google Scholar] [CrossRef]
- Okochi, O.; Hibi, K.; Uemura, T.; Inoue, S.; Takeda, S.; Kaneko, T.; Nakao, A. Detection of mitochondrial DNA alterations in the serum of hepatocellular carcinoma patients. Clin. Cancer Res. 2002, 8, 2875–2878. [Google Scholar]
- Yu, M. Circulating cell-free mitochondrial DNA as a novel cancer biomarker: Opportunities and challenges. Mitochondrial Dna 2012, 23, 329–332. [Google Scholar] [CrossRef]
- Boyapati, R.K.; Tamborska, A.; Dorward, D.A.; Ho, G.T. Advances in the understanding of mitochondrial DNA as a pathogenic factor in inflammatory diseases. F1000Res 2017, 6, 169. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- McGill, M.R.; Staggs, V.S.; Sharpe, M.R.; Lee, W.M.; Jaeschke, H.; Acute Liver Failure Study, G. Serum mitochondrial biomarkers and damage-associated molecular patterns are higher in acetaminophen overdose patients with poor outcome. Hepatology 2014, 60, 1336–1345. [Google Scholar] [CrossRef]
- Marques, P.E.; Amaral, S.S.; Pires, D.A.; Nogueira, L.L.; Soriani, F.M.; Lima, B.H.; Lopes, G.A.; Russo, R.C.; Avila, T.V.; Melgaco, J.G.; et al. Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology 2012, 56, 1971–1982. [Google Scholar] [CrossRef]
- McGill, M.R.; Sharpe, M.R.; Williams, C.D.; Taha, M.; Curry, S.C.; Jaeschke, H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J. Clin. Invest. 2012, 122, 1574–1583. [Google Scholar] [CrossRef]
- Rodrigue-Gervais, I.G.; Doiron, K.; Champagne, C.; Mayes, L.; Leiva-Torres, G.A.; Vanie, P., Jr.; Douglas, T.; Vidal, S.M.; Alnemri, E.S.; Saleh, M. The mitochondrial protease HtrA2 restricts the NLRP3 and AIM2 inflammasomes. Sci. Rep. 2018, 8, 8446. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hur, W.; Kang, B.Y.; Kim, S.M.; Lee, G.W.; Kim, J.-H.; Nam, M.-K.; Rhim, H.; Yoon, S.K. Serine Protease HtrA2/Omi Deficiency Impairs Mitochondrial Homeostasis and Promotes Hepatic Fibrogenesis via Activation of Hepatic Stellate Cells. Cells 2019, 8, 1119. https://doi.org/10.3390/cells8101119
Hur W, Kang BY, Kim SM, Lee GW, Kim J-H, Nam M-K, Rhim H, Yoon SK. Serine Protease HtrA2/Omi Deficiency Impairs Mitochondrial Homeostasis and Promotes Hepatic Fibrogenesis via Activation of Hepatic Stellate Cells. Cells. 2019; 8(10):1119. https://doi.org/10.3390/cells8101119
Chicago/Turabian StyleHur, Wonhee, Byung Yoon Kang, Sung Min Kim, Gil Won Lee, Jung-Hee Kim, Min-Kyung Nam, Hyangshuk Rhim, and Seung Kew Yoon. 2019. "Serine Protease HtrA2/Omi Deficiency Impairs Mitochondrial Homeostasis and Promotes Hepatic Fibrogenesis via Activation of Hepatic Stellate Cells" Cells 8, no. 10: 1119. https://doi.org/10.3390/cells8101119
APA StyleHur, W., Kang, B. Y., Kim, S. M., Lee, G. W., Kim, J.-H., Nam, M.-K., Rhim, H., & Yoon, S. K. (2019). Serine Protease HtrA2/Omi Deficiency Impairs Mitochondrial Homeostasis and Promotes Hepatic Fibrogenesis via Activation of Hepatic Stellate Cells. Cells, 8(10), 1119. https://doi.org/10.3390/cells8101119