CPT1A Supports Castration-Resistant Prostate Cancer in Androgen-Deprived Conditions

 ,
, 

Abstract
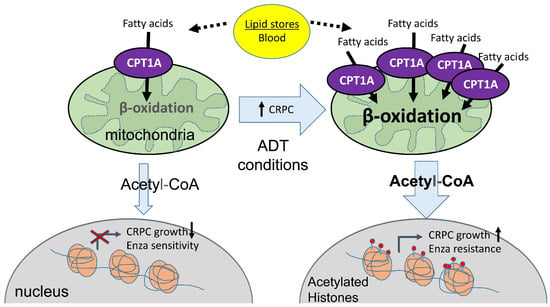
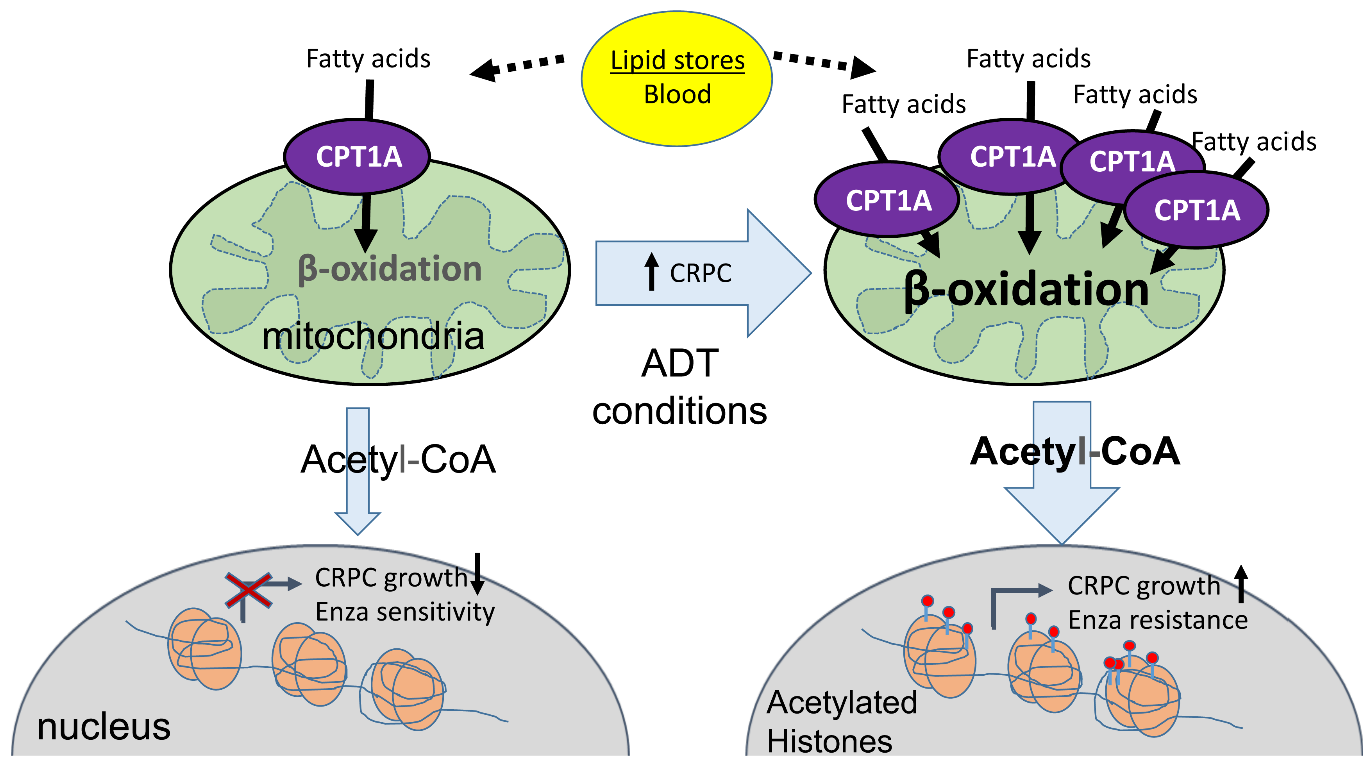{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
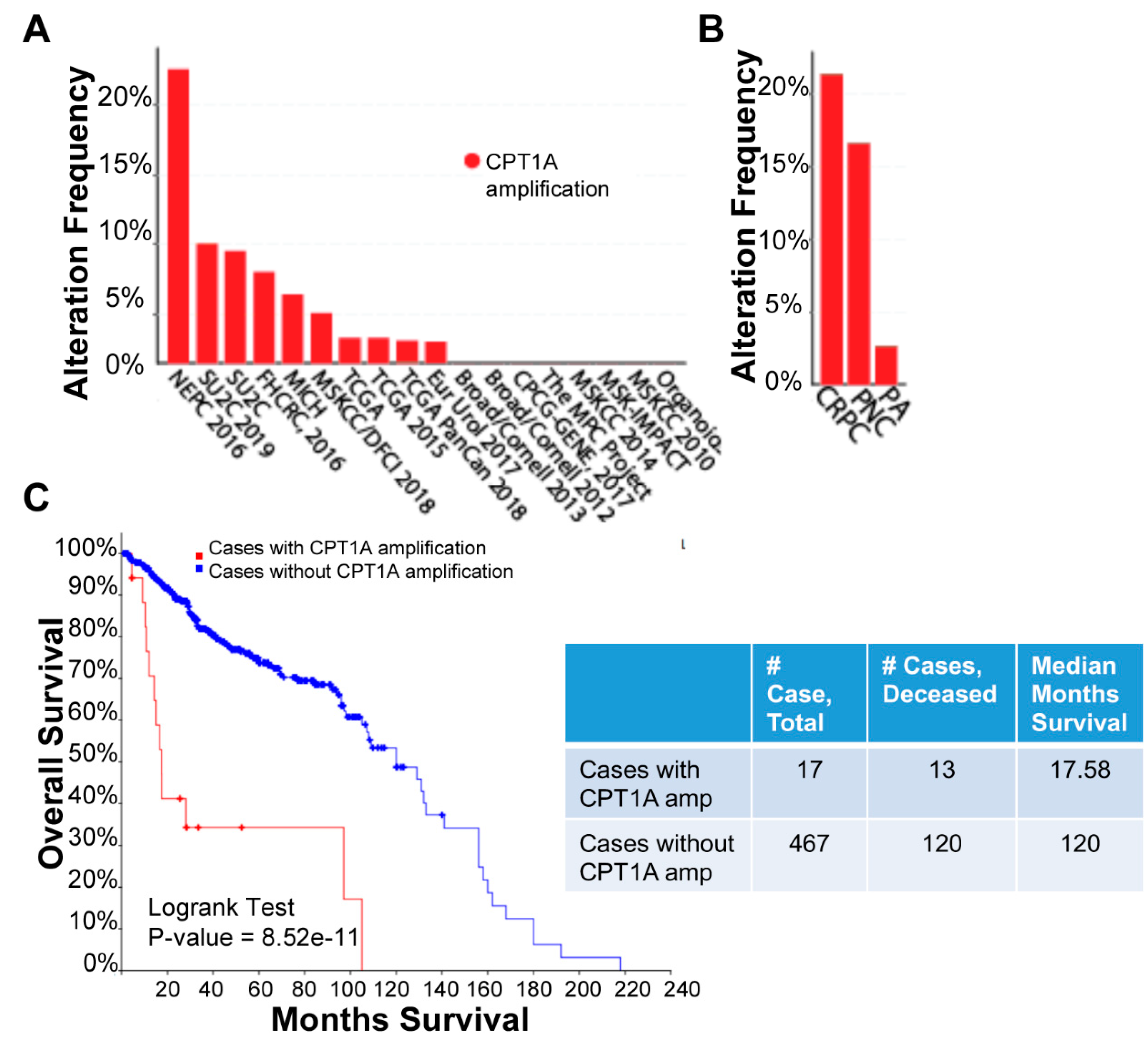{kind=link}
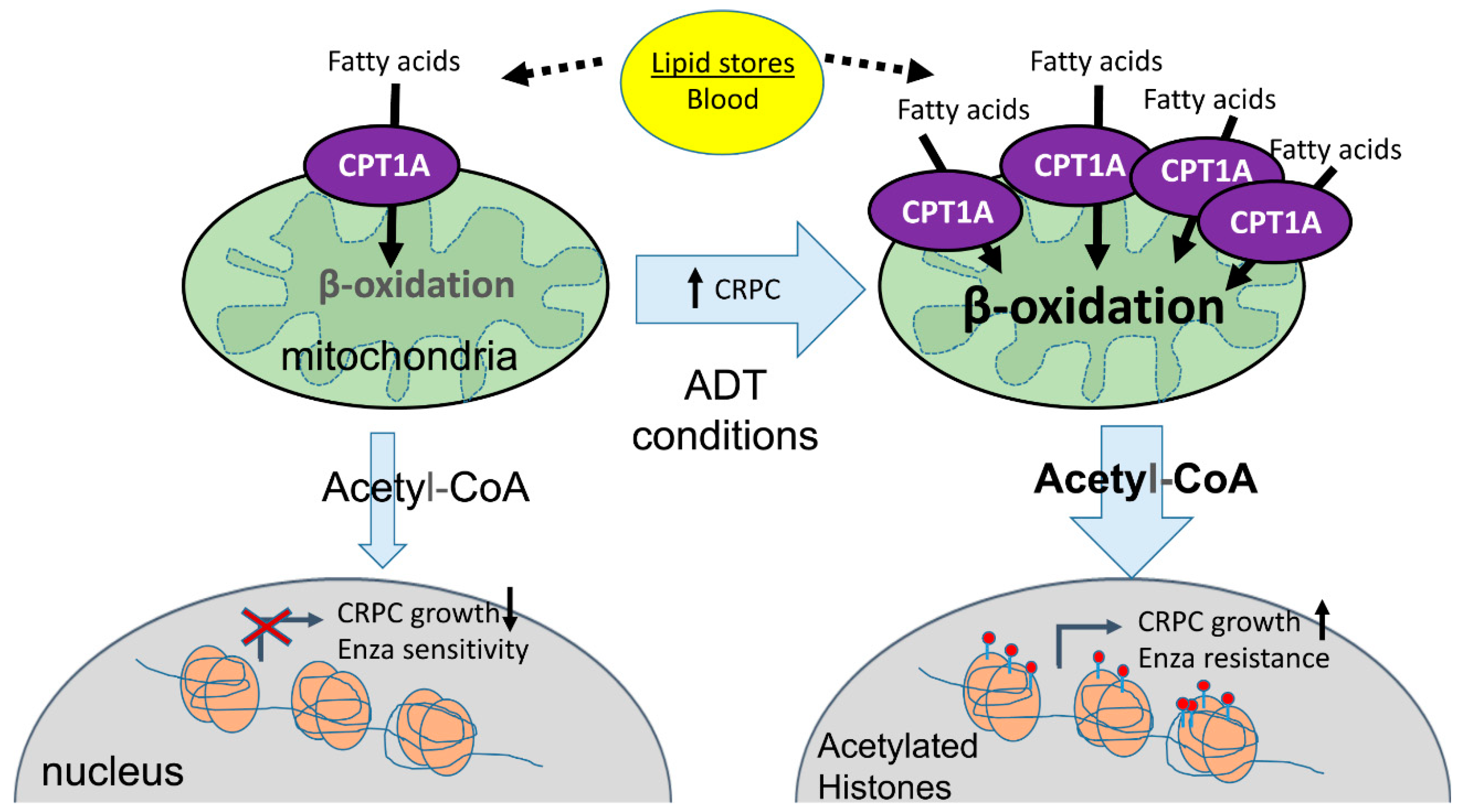{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Clonogenic Growth, Migration, and Growth in Suspension Assays
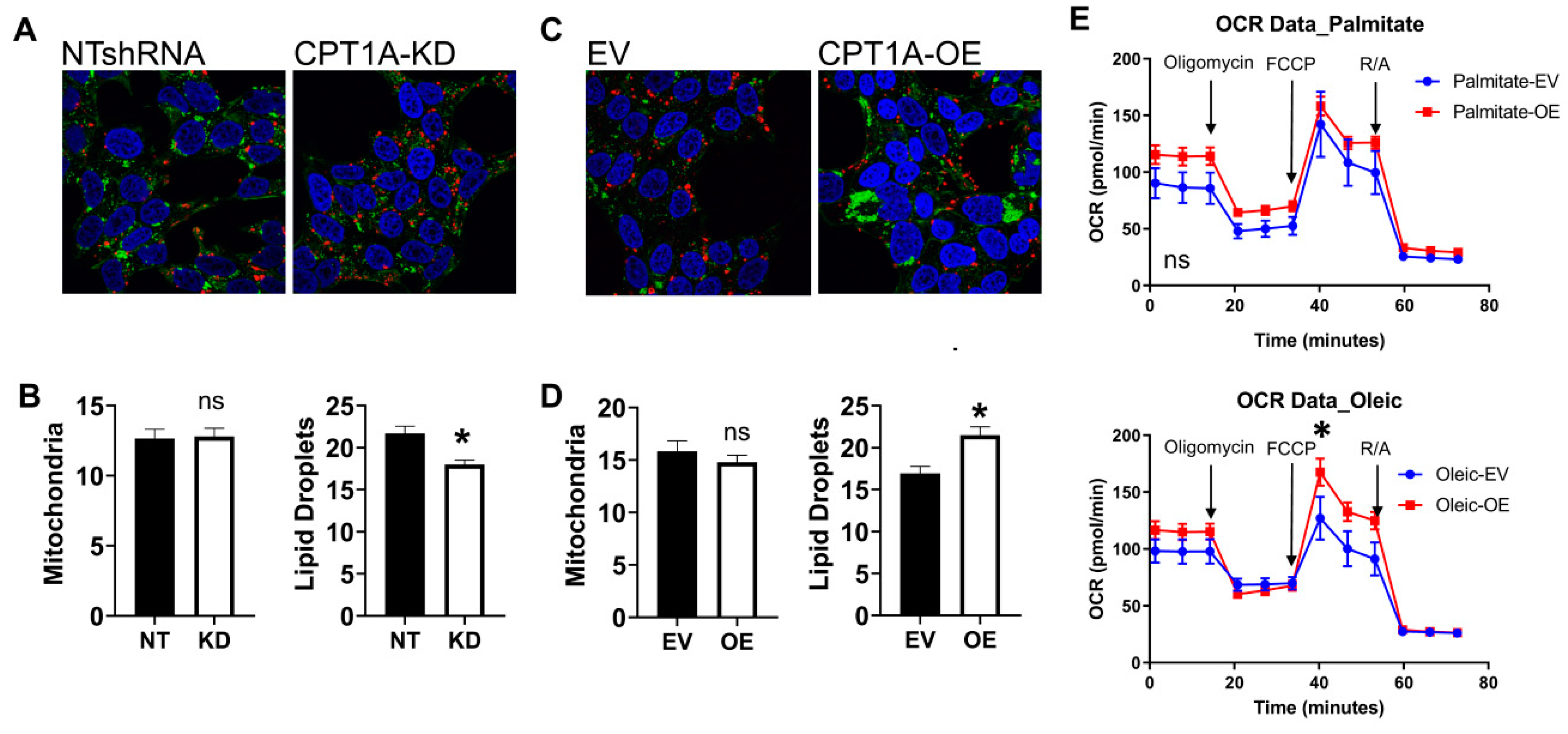
2.3. Seahorse Metabolic Flux Analysis
2.4. Immunofluorescence and Microscopy
2.5. Lentivirus Preparation and Transfection
2.6. Reverse-Transcriptase-PCR
2.7. Mouse Xenograft Studies
2.8. Western Blot Analysis
2.9. Histone Isolation and Mass Spectrometric Analysis
2.10. Statistics
2.11. Clinical Data Analysis
3. Results
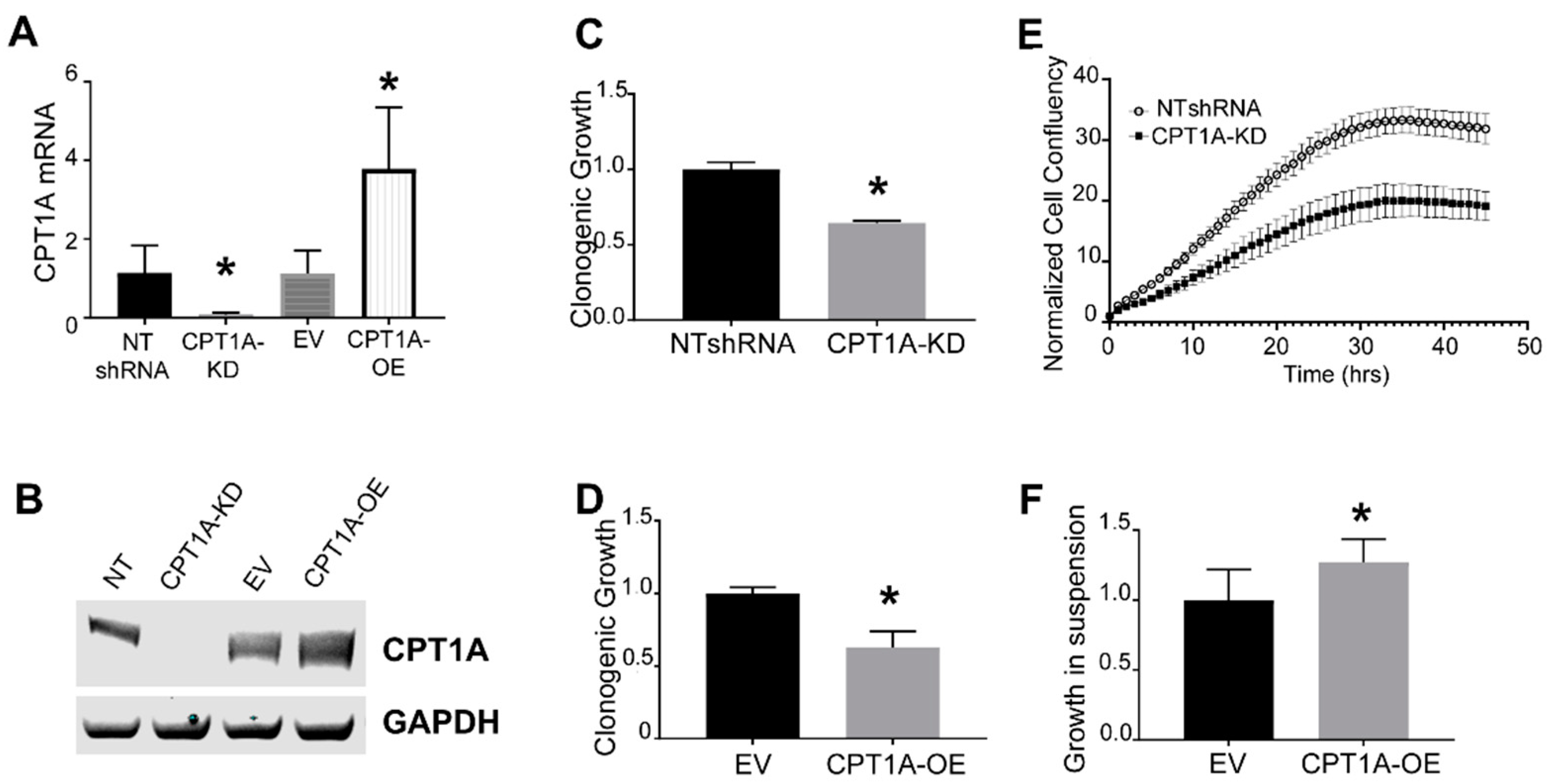
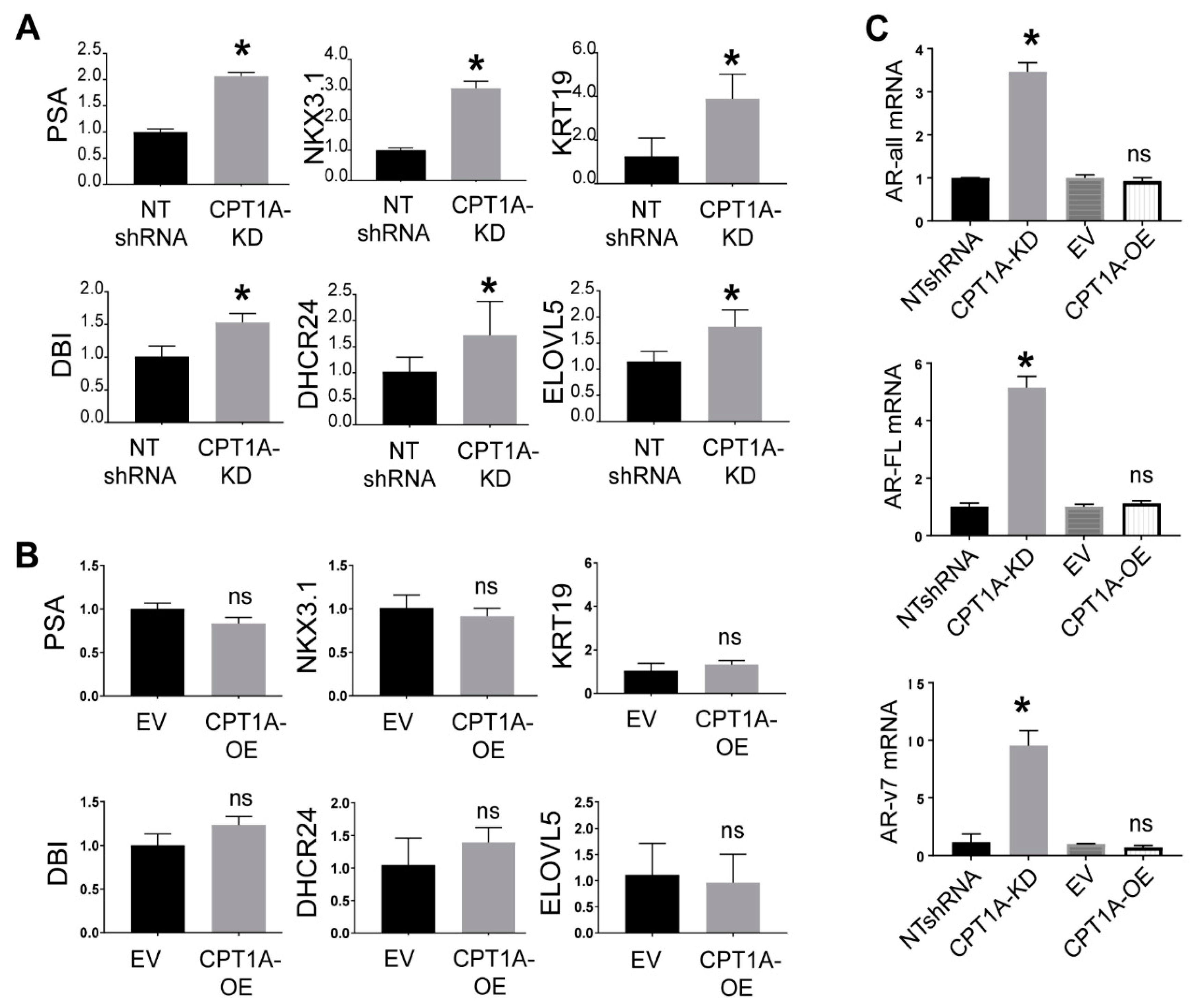
3.1. Characterization of CRPC C4-2 Cells with CPT1A Knock-Down (KD) or Overexpression (OE)
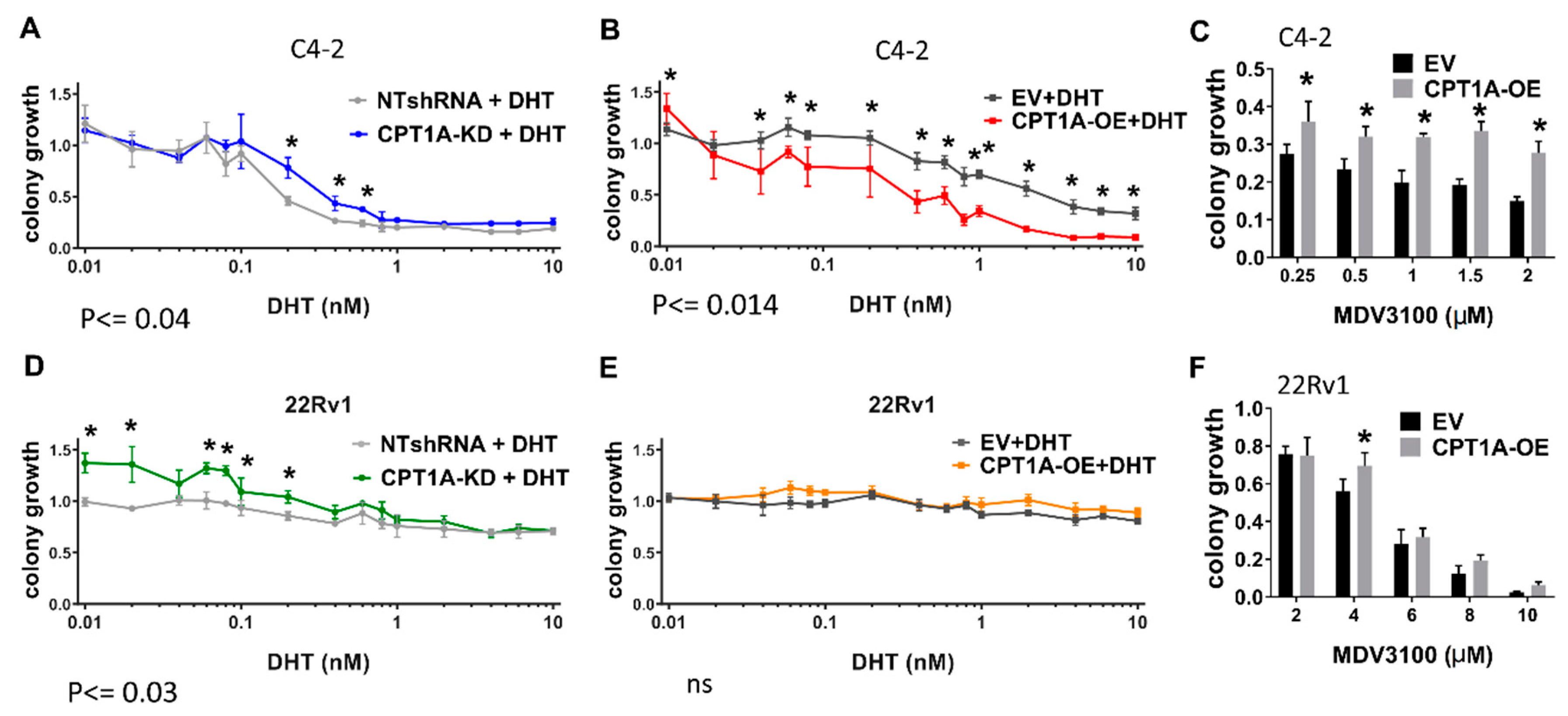
3.2. The CPT1A KD cells Show Increased Response to Androgens, While CPT1A OE Cells Show a More AR-Independent Phenotype
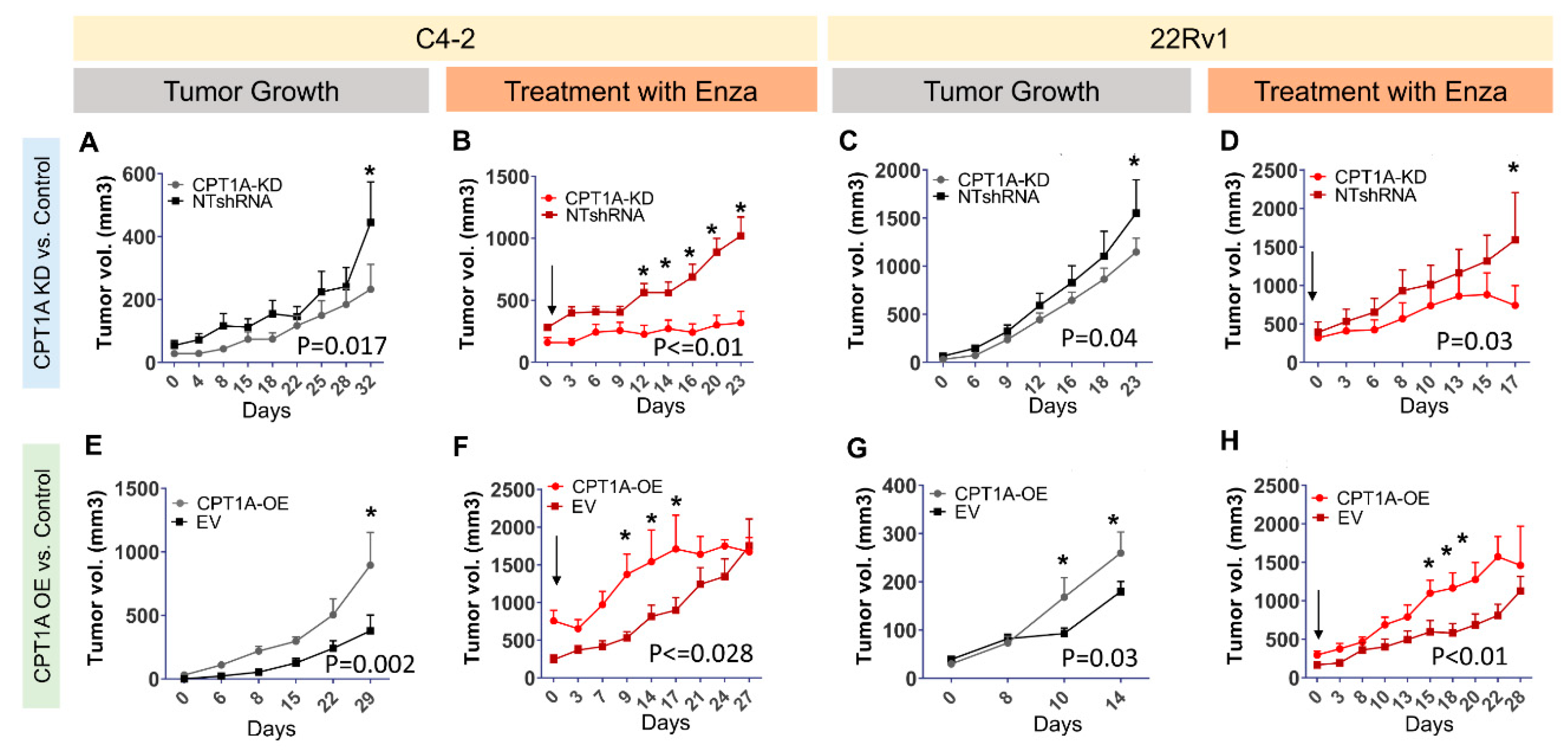
3.3. CPT1A Expression in Tumors Modulates Growth and Response to Enzalutamide in Castrated Mice
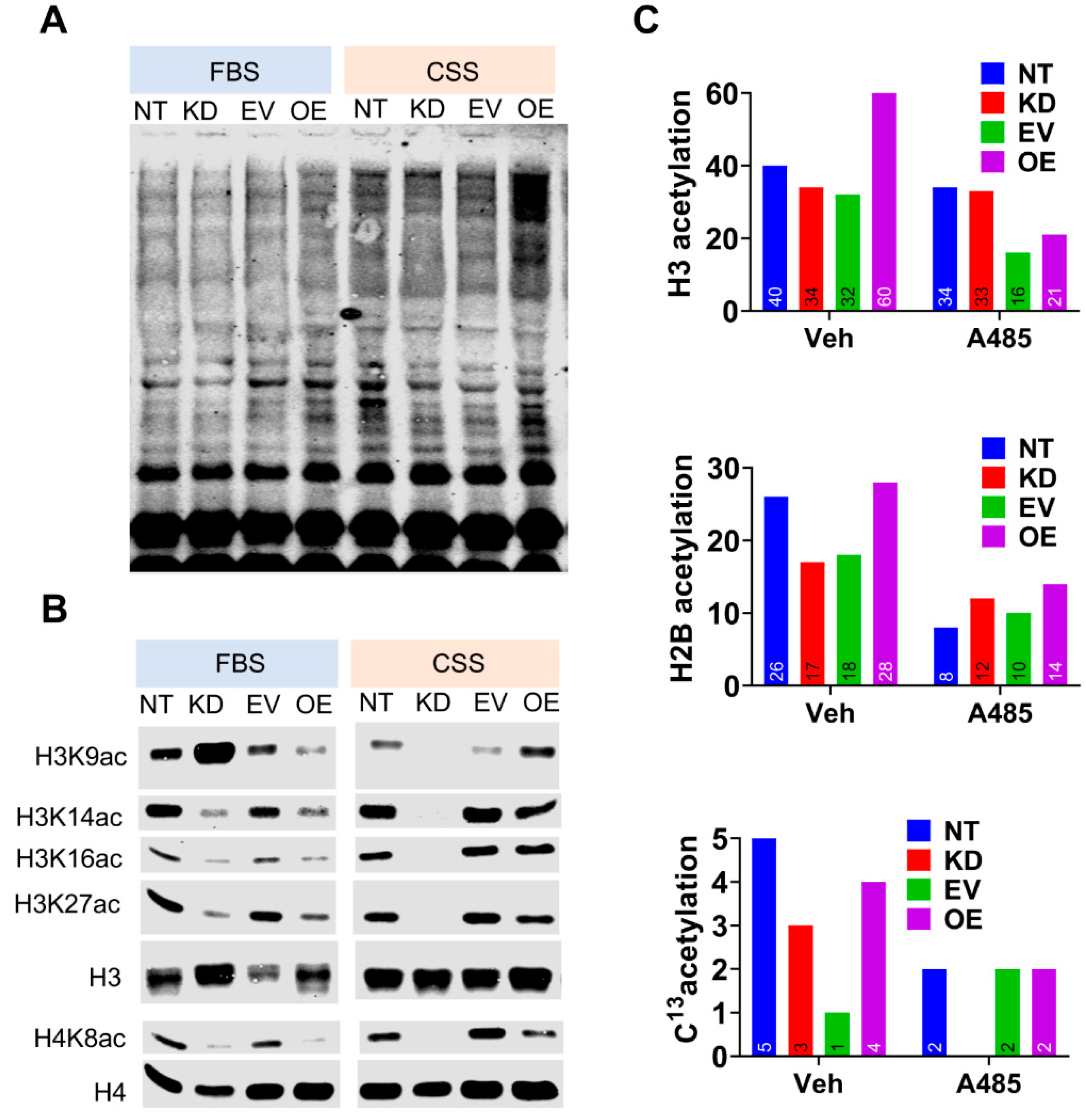
3.4. CPT1A Expression Modulates Histone Acetylation in An Androgen-Dependent Manner.
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Litwin, M.S.; Tan, H.-J. The Diagnosis and Treatment of Prostate Cancer. JAMA 2017, 317, 2532–2542. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Parker, C.; Eeles, R.A.; Schröder, F.; Tomlins, S.A.; Tannock, I.; Drake, C.G.; De Bono, J.S. Prostate cancer. Lancet 2016, 387, 70–82. [Google Scholar] [CrossRef]
- Davies, A.; Conteduca, V.; Zoubeidi, A.; Beltran, H. Biological Evolution of Castration-resistant Prostate Cancer. Eur. Urol. Focus 2019, 5, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M.; Tindall, D.J. Alternatively Spliced Androgen Receptor Variants. Endocr.-Relat. Cancer 2011, 18, R183–R196. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- De Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B.; Saad, F.; et al. Abiraterone and Increased Survival in Metastatic Prostate Cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef]
- Fizazi, K.; Taplin, M.-E.; Sternberg, C.N.; Miller, K.; Flaig, T.W.; Hirmand, M.; Selby, B.; Scher, H.I.; Saad, F.; De Wit, R.; et al. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar]
- Karantanos, T.; Evans, C.P.; Tombal, B.; Thompson, T.C.; Montironi, R.; Isaacs, W.B. Understanding the Mechanisms of Androgen Deprivation Resistance in Prostate Cancer at the Molecular Level. Eur. Urol. 2015, 67, 470–479. [Google Scholar] [CrossRef]
- Saylor, P.J.; Karoly, E.D.; Smith, M.R. Prospective study of changes in the metabolomic profiles of men during their first three months of androgen deprivation therapy for prostate cancer. Clin. Cancer Res. 2012, 18, 3677–3685. [Google Scholar] [CrossRef]
- Smith, M.R.; Finkelstein, J.S.; McGovern, F.J.; Zietman, A.L.; Fallon, M.A.; Schoenfeld, D.A.; Kantoff, P.W. Changes in Body Composition during Androgen Deprivation Therapy for Prostate Cancer. J. Clin. Endocrinol. Metab. 2002, 87, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Suburu, J.; Chen, Y.Q. Lipids and Prostate Cancer. Prostaglandins Other Lipid Mediat. 2012, 98, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, J.V.; Brusselmans, K.; Verhoeven, G. Increased lipogenesis in cancer cells: New players, novel targets. Curr. Opin. Clin. Nutr. Metab Care 2006, 9, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, J.V.; Van Veldhoven, P.P.; Esquenet, M.; Heyns, W.; Verhoeven, G. Androgens markedly stimulate the accumulation of neutral lipids in the human prostatic adenocarcinoma cell line LNCaP. Endocrinology 1996, 137, 4468–4474. [Google Scholar] [CrossRef] [PubMed]
- Stoykova, G.E.; Schlaepfer, I.R. Lipid Metabolism and Endocrine Resistance in Prostate Cancer, and New Opportunities for Therapy. Int. J. Mol. Sci. 2019, 20, 2626. [Google Scholar] [CrossRef] [PubMed]
- Zadra, G.; Ribeiro, C.F.; Chetta, P.; Ho, Y.; Cacciatore, S.; Gao, X.; Syamala, S.; Bango, C.; Photopoulos, C.; Huang, Y.; et al. Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2018, 116, 631–640. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D.; Leatherman, G.F.; Foster, D.W. Carnitine palmitoyltransferase I. The site of inhibition of hepatic fatty acid oxidation by malonyl-CoA. J. Biol. Chem. 1978, 253, 4128–4136. [Google Scholar]
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijón, M.A.; Pac, C.T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glode, L.M.; Eckel, R.H.; et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol. Cancer Ther. 2014, 13, 2361–2371. [Google Scholar] [CrossRef]
- Flaig, T.W.; Salzmann-Sullivan, M.; Su, L.-J.; Zhang, Z.; Joshi, M.; Gijón, M.A.; Kim, J.; Arcaroli, J.J.; Van Bokhoven, A.; Lucia, M.S.; et al. Lipid catabolism inhibition sensitizes prostate cancer cells to antiandrogen blockade. Oncotarget 2017, 8, 56051–56065. [Google Scholar] [CrossRef]
- Itkonen, H.M.; Brown, M.; Urbanucci, A.; Tredwell, G.; Lau, C.H.; Barfeld, S.; Hart, C.; Guldvik, I.J.; Takhar, M.; Heemers, H.V.; et al. Lipid degradation promotes prostate cancer cell survival. Oncotarget 2017, 8, 38264–38275. [Google Scholar] [CrossRef]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-N.; Zeng, Z.-L.; Lu, J.; Wang, Y.; Liu, Z.-X.; He, M.-M.; Zhao, Q.; Wang, Z.-X.; Li, T.; Lu, Y.-X.; et al. CPT1A-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis. Oncogene 2018, 37, 6025–6040. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.-L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Regulation of Oncogenic Properties in Triple Negative Breast Cancer. Cell Rep. 2016, 14, 2154–2165. [Google Scholar] [CrossRef] [PubMed]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, E.; Crown, S.B.; Fox, D.B.; Kitir, B.; Ilkayeva, O.R.; Olsen, C.A.; Grimsrud, P.A.; Hirschey, M.D. Lipids reprogram metabolism to become a major carbon source for histone acetylation. Cell Rep. 2016, 17, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.W.; Wang, X.; Zecchin, A.; Thienpont, B.; Cornelissen, I.; Kalucka, J.; Garcia-Caballero, M.; Missiaen, R.; Huang, H.; Bruning, U.; et al. The role of fatty acid beta-oxidation in lymphangiogenesis. Nature 2017, 542, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Thalmann, G.N.; Anezinis, P.E.; Chang, S.M.; Zhau, H.E.; Kim, E.E.; Hopwood, V.L.; Pathak, S.; Von Eschenbach, A.C.; Chung, L.W.K. Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res. 1994, 54, 2577–2581. [Google Scholar] [PubMed]
- Wu, Z.; Conaway, M.; Gioeli, D.; Weber, M.; Theodorescu, D. Conditional expression of PTEN alters the androgen responsiveness of prostate cancer cells. Prostate 2006, 66, 1114–1123. [Google Scholar] [CrossRef]
- Schlaepfer, I.R.; Glode, L.M.; Hitz, C.A.; Pac, C.T.; Boyle, K.E.; Maroni, P.; Deep, G.; Agarwal, R.; Lucia, S.M.; Cramer, S.D.; et al. Inhibition of Lipid Oxidation Increases Glucose Metabolism and Enhances 2-Deoxy-2-[18F]Fluoro-d-Glucose Uptake in Prostate Cancer Mouse Xenografts. Mol. Imaging Boil. 2015, 17, 529–538. [Google Scholar] [CrossRef]
- Sramkoski, R.M.; Pretlow, T.G.; Giaconia, J.M.; Pretlow, T.P.; Schwartz, S.; Sy, M.-S.; Marengo, S.R.; Rhim, J.S.; Zhang, D.; Jacobberger, J.W. A new human prostate carcinoma cell line, 22Rv1. Vitr. Cell. Dev. Boil.-Anim. 1999, 35, 403–409. [Google Scholar] [CrossRef]
- Toren, P.; Kim, S.; Johnson, F.; Zoubeidi, A. Combined AKT and MEK Pathway Blockade in Pre-Clinical Models of Enzalutamide-Resistant Prostate Cancer. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Sedelaar, J.M.; Isaacs, J.T. Tissue Culture Media Supplemented with 10% Fetal Calf Serum Contains a Castrate level of Testosterone. Prostate 2009, 69, 1724–1729. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Chung, M.K.; Fan, J.; Rabinowitz, J.D. Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer Metab. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; Digiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Gao, S.; Barrett, D.; Ahmed, M.; Han, D.; Macoska, J.A.; He, H.H.; Cai, C. Reactivation of androgen receptor-regulated lipid biosynthesis drives the progression of castration-resistant prostate cancer. Oncogene 2018, 37, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Zeng, F.; Liu, X.; Wang, Q.J.; Deng, F. Fatty acid oxidation and carnitine palmitoyltransferase I: Emerging therapeutic targets in cancer. Cell Death Dis. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.S.; Smift, A.L.; Croteau, N.J.; Ferrick, D.A.; Wu, M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim. Biophys. Acta 2011, 1807, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.J.; Clark, A.K.; Selth, L.A.; Haynes, V.R.; Lister, N.; Rebello, R.; Porter, L.H.; Niranjan, B.; Whitby, S.T.; Lo, J.; et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Kuhajda, F.P.; Date, O.; Katsura, M.; Ishida, M.; Yoshihara, T.; Kinomura, A.; Sueda, T.; Miyagawa, K. Fatty Acid Synthase and Cancer: New Application of an Old Pathway: Figure 1. Cancer Res. 2006, 66, 5977–5980. [Google Scholar] [CrossRef]
- Little, J.L.; Wheeler, F.B.; Fels, D.R.; Koumenis, C.; Kridel, S.J. Inhibition of Fatty Acid Synthase Induces Endoplasmic Reticulum Stress in Tumor Cells. Cancer Res. 2007, 67, 1262–1269. [Google Scholar] [CrossRef]
- Migita, T.; Ruiz, S.; Fornari, A.; Fiorentino, M.; Priolo, C.; Zadra, G.; Inazuka, F.; Grisanzio, C.; Palescandolo, E.; Shin, E.; et al. Fatty Acid Synthase: A Metabolic Enzyme and Candidate Oncogene in Prostate Cancer. J. Natl. Cancer Inst. 2009, 101, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Dart, D.A.; Brooke, G.N.; Sita-Lumsden, A.; Waxman, J.; Bevan, C.L. Reducing prohibitin increases histone acetylation, and promotes androgen independence in prostate tumours by increasing androgen receptor activation by adrenal androgens. Oncogene 2012, 31, 4588–4598. [Google Scholar] [CrossRef] [PubMed]
- Mostaghel, E.A.; Solomon, K.R.; Pelton, K.; Freeman, M.R.; Montgomery, R.B. Impact of Circulating Cholesterol Levels on Growth and Intratumoral Androgen Concentration of Prostate Tumors. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.-M.; Corey, E. Supraphysiological Testosterone Therapy as Treatment for Castration-Resistant Prostate Cancer. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.-F.; Yao, T.-P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef]
- Zhou, L.X.; Li, T.; Huang, Y.R.; Sha, J.J.; Sun, P.; Li, D. Application of histone modification in the risk prediction of the biochemical recurrence after radical prostatectomy. Asian J. Androl. 2010, 12, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Welsbie, D.S.; Xu, J.; Chen, Y.; Borsu, L.; Scher, H.I.; Rosen, N.; Sawyers, C.L. Histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer. Cancer Res. 2009, 69, 958–966. [Google Scholar] [CrossRef]
- Lee, J.-H.; Yang, B.; Lindahl, A.J.; Damaschke, N.; Boersma, M.D.; Huang, W.; Corey, E.; Jarrard, D.F.; Denu, J.M. Identifying Dysregulated Epigenetic Enzyme Activity in Castrate-Resistant Prostate Cancer Development. ACS Chem. Boil. 2017, 12, 2804–2814. [Google Scholar] [CrossRef]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef]
- Higano, C.S.; Armstrong, A.J.; Sartor, A.O.; Vogelzang, N.J.; Kantoff, P.W.; McLeod, D.G.; Pieczonka, C.M.; Penson, D.F.; Shore, N.D.; Vacirca, J.; et al. Real-world outcomes of sipuleucel-T treatment in PROCEED, a prospective registry of men with metastatic castration-resistant prostate cancer. Cancer 2019. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joshi, M.; Stoykova, G.E.; Salzmann-Sullivan, M.; Dzieciatkowska, M.; Liebman, L.N.; Deep, G.; Schlaepfer, I.R. CPT1A Supports Castration-Resistant Prostate Cancer in Androgen-Deprived Conditions. Cells 2019, 8, 1115. https://doi.org/10.3390/cells8101115
Joshi M, Stoykova GE, Salzmann-Sullivan M, Dzieciatkowska M, Liebman LN, Deep G, Schlaepfer IR. CPT1A Supports Castration-Resistant Prostate Cancer in Androgen-Deprived Conditions. Cells. 2019; 8(10):1115. https://doi.org/10.3390/cells8101115
Chicago/Turabian StyleJoshi, Molishree, Gergana E. Stoykova, Maren Salzmann-Sullivan, Monika Dzieciatkowska, Lauren N. Liebman, Gagan Deep, and Isabel R. Schlaepfer. 2019. "CPT1A Supports Castration-Resistant Prostate Cancer in Androgen-Deprived Conditions" Cells 8, no. 10: 1115. https://doi.org/10.3390/cells8101115
APA StyleJoshi, M., Stoykova, G. E., Salzmann-Sullivan, M., Dzieciatkowska, M., Liebman, L. N., Deep, G., & Schlaepfer, I. R. (2019). CPT1A Supports Castration-Resistant Prostate Cancer in Androgen-Deprived Conditions. Cells, 8(10), 1115. https://doi.org/10.3390/cells8101115