Targeting IKKβ in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKβ Inhibitors

Signalling Laboratory, The Babraham Institute, Babraham Research Campus, Cambridge CB22 3AT, UK
*
Authors to whom correspondence should be addressed.
Cells 2018, 7(9), 115; https://doi.org/10.3390/cells7090115
Submission received: 16 July 2018
/
Revised: 15 August 2018
/
Accepted: 19 August 2018
/
Published: 23 August 2018
(This article belongs to the Special Issue NF-κB in Cancer)
Abstract
:Deregulated NF-κB signalling is implicated in the pathogenesis of numerous human inflammatory disorders and malignancies. Consequently, the NF-κB pathway has attracted attention as an attractive therapeutic target for drug discovery. As the primary, druggable mediator of canonical NF-κB signalling the IKKβ protein kinase has been the historical focus of drug development pipelines. Thousands of compounds with activity against IKKβ have been characterised, with many demonstrating promising efficacy in pre-clinical models of cancer and inflammatory disease. However, severe on-target toxicities and other safety concerns associated with systemic IKKβ inhibition have thus far prevented the clinical approval of any IKKβ inhibitors. This review will discuss the potential reasons for the lack of clinical success of IKKβ inhibitors to date, the challenges associated with their therapeutic use, realistic opportunities for their future utilisation, and the alternative strategies to inhibit NF-κB signalling that may overcome some of the limitations associated with IKKβ inhibition.
1. Introduction
There are five members of the nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells (NF-κB) transcription factor family in mammals: RelA (p65), RelB, c-Rel, NF-κB1 (p50, initially synthesised as a larger precursor, p105) and NF-κB2 (p52, initially synthesised as a larger precursor, p100). All members share a conserved Rel homology domain (RHD) that enables them to associate with each other to form a diverse array of transcriptionally active homo- and hetero-dimeric complexes. In normal unstimulated cells, NF-κB dimers are tightly bound by members of the inhibitor of kappa B (IκB) family of proteins, which maintain the cytoplasmic steady-state localisation of NF-κB dimers and inhibit their DNA-binding activity. The precursor proteins p100 and p105 also contain ankyrin repeat domains, which are cleaved upon processing to p52/p50, such that they comprise internal inhibitors of NF-κB dimers.
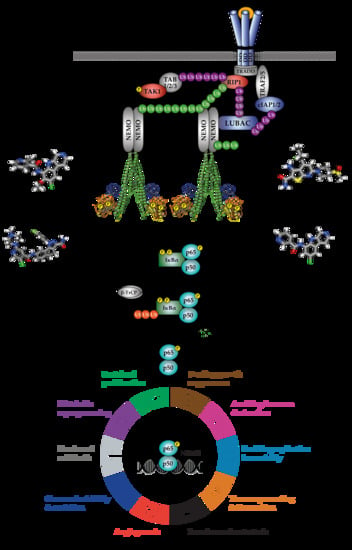
The transcriptional activity of NF-κB dimers is regulated by several distinct pathways (Figure 1). The first is the canonical pathway, which is induced by pro-inflammatory cytokines, such as tumour necrosis factor-alpha (TNF-α) and interleukin-1 (IL-1), engagement of antigen receptors, such as the T- and B-cell receptor (T/B-CR), pathogen-associated molecules such as lipopolysaccharides (LPS) and certain growth factors (Figure 1A–D; [1]). Engagement of the canonical NF-κB pathways triggers signalling cascades that converge on activation of the IκB kinase (IKK) complex, which is formed by the kinase subunits, IKKα and IKKβ, and a regulatory subunit, IKKγ (also known as NEMO, NF-κB essential modifier). The non-canonical pathway, meanwhile, is stimulated by a more restricted set of cytokines all belonging to the TNF superfamily, including BAFF (B-cell activating factor) and lymphotoxin β [2]. Non-canonical NF-κB signalling requires IKKα, but not NEMO or IKKβ activity, and so will not be discussed in detail here. Several atypical pathways are also capable of activating the IKK complex in response to diverse stresses, such as DNA damage (Figure 1E; [3]). These distinct NF-κB pathways regulate different subsets of target genes, and hence different biological functions.
IKKα and IKKβ are ubiquitously expressed serine/threonine kinases with 52% sequence identity and 70% homology [4]. They also share highly similar domain organisation and tertiary structure, as demonstrated by the recent X-ray crystal structures of human IKKα and IKKβ (Figure 2; [5,6,7]. Activation of IKKα and IKKβ kinase activity requires the phosphorylation of specific residues in the activation loop of their active sites: serine-176 (S176) and serine-180 (S180) for IKKα and S177, and S181 for IKKβ [8,9]. The precise sequence of molecular events involved in IKK activation remain to be fully determined. However, recent evidence for higher-order IKK complexes [10], combined with X-ray crystal structures of IKKβ dimers in catalytically active conformations [6], has led to the proposal of a model for IKK activation involving oligomerization-mediated trans-autophosphorylation of IKK subunits. Indeed, in the case of IKKβ activation downstream of IL-1 and TNFα in mouse embryonic fibroblasts (MEFs) and TLR ligands in macrophages, TAK1 has been proposed to phosphorylate IKKβ at S177, which primes subsequent IKKβ-catalysed autophosphorylation of S181 [11]. Subsequently, the activated IKK complex phosphorylates IκB proteins (at S32 and S36 in the case of IκBα), promoting their K48-linked ubiquitylation by the S phase kinase-associated protein 1 (SKP1)-cullin 1-F-box protein (SCF)/beta-transducing repeat-containing protein (β-TrCP) E3 ubiquitin ligase complex, which targets them for proteasomal degradation [12,13,14,15,16]. In the context of canonical NF-κB signalling, this enables RelA- and c-Rel-containing NF-κB dimers to accumulate in the nucleus where they coordinate the expression of genes involved in diverse biological process including cell proliferation/survival, immune and inflammatory responses, and other host defence mechanisms [17].
The activity of NF-κB subunits is also regulated through direct post-translational modification including phosphorylation, ubiquitination, sumoylation, acetylation and nitrosylation [18,19]. In many cases, these modifications are mediated by IKKα and IKKβ, as well as components of heterologous signalling pathways. This provides an additional layer of fine control through which NF-κB transcriptional activity can be modulated and represents a key site of cross-talk within the wider signalling network [3].
NF-κB signalling pathways are normally tightly controlled by multiple regulatory mechanisms to ensure minimal basal activation [20]. However, given its critical role in regulating the expression of genes involved in cell-survival, proliferation, angiogenesis, metabolism, inflammation and cell adhesion/migration, it is unsurprising that a wide range of inflammatory diseases and cancers have been shown to exhibit deregulated NF-κB signalling that results in constitutive pathway activation [21]. Indeed, the ability of NF-κB to induce inflammation places it as one of the crucial links between chronic inflammation and cancer [22]. However, aberrant NF-κB activation is also capable of promoting tumorigenesis in cancers whose early progression isn’t typically associated with inflammation through contributions of NF-κB target genes to almost all the hallmarks of cancer [23,24].
Outside of their direct role in the NF-κB signalling pathway, both IKKα and IKKβ have been proposed to phosphorylate an ever-growing list of ‘non-classical’ substrates involved in diverse biological processes [25]. Whilst, many of these ‘non-classical’ phosphorylation events and the associated NF-κB-independent functions of IKKs await thorough validation, it is clear that the IKK complex is a central point of cross-talk between NF-κB and other signalling pathways. Many of the substrates phosphorylated by IKKα and IKKβ, such as FOXO3a and TSC1, are involved in proliferative and pro-survival pathways, and so these NF-κB-independent functions of the IKKs may also contribute to tumorigenesis [26,27].
2. IKKβ Inhibitors
The identification of NF-κB signalling pathways as important drivers of human disease and the essential role of the IKK complex as the ubiquitous signal integration hub for NF-κB activation pathways led to a concerted effort by the pharmaceutical industry to identify small molecule inhibitors of the IKKs via high-throughput screening programmes. The primary focus of efforts to inhibit canonical NF-κB signalling has been the druggable IKKβ kinase due to foundational mouse studies that indicated IKKβ and NEMO are essential for activation of this pathway, while IKKα is largely dispensable [28,29,30]. Instead, IKKα, but not IKKβ or NEMO, is indispensable for non-canonical NF-κB activation [31,32,33,34].
A vast drug-discovery effort has identified numerous synthetic small molecules with activity against the IKKs that typically exhibit selectivity for IKKβ over IKKα. Indeed, the first IKKα-selective inhibitor series was only very recently reported [35] and IKKα-selective inhibitors are described in another review within this special issue by Pepper & Mackay. In addition, a range of natural products, such as wedelactone, reportedly inhibit IKKβ [36,37]. However, these compounds tend to have pleiotropic effects, and with a few notable exceptions, their mechanism of IKK/NF-κB inhibition has not been fully characterised. Collectively, the number of compounds with reported activity against IKKβ continues to grow [38,39]. A selection of the best-characterised, commercially-available IKKβ inhibitors is shown in Table 1.
To date, chemical IKKβ inhibitors with four mechanisms of action have been characterised (see Table 1). The clear majority are ATP analogues that exhibit reversible, ATP-competitive activity, with some degree of selectivity for IKKβ over IKKα and other kinases. Due to the structural similarity of protein kinase ATP-binding sites, ATP mimetics often exhibit activity against other kinases, resulting in ‘off-target’ effects at or near concentrations of drug required to inhibit its primary target in cells [96]. Indeed, several widely-used ‘selective’ IKKβ inhibitors have recently been shown to exhibit significant off-target effects. For example, the compound Bay 11-7082, which inhibits IκBα phosphorylation and NF-κB transcriptional activity in cells, has been used to study IKK and NF-κB function in >350 publications [97]. However, a recent report showed that Bay 11-7082 inhibits NF-κB not through direct inhibition of IKK activity, but through irreversible covalent inactivation of the E2-conjugating enzymes Ubc (ubiquitin conjugating) 13 and UbcH7, and the E3-ligase LUBAC (linear ubiquitin assembly complex) [98]. In addition, the widely used compound TPCA-1 was recently shown to inhibit STAT3 signalling through direct binding to the STAT3 Src Homology 2 (SH2) domain, in addition to its activity against IKKβ [51]. Of great concern is the continued use of these non-selective inhibitors to make inferences about IKK function. The current best-in-class, ATP-competitive IKKβ inhibitors are MLN-120B and BI605906, which exhibit >50- and >300-fold selectivity for IKKβ over IKKα, respectively [40,41,99]. The selectivity of BI605906 for IKKβ over 100 other serine/threonine and tyrosine kinases has also been confirmed [40], while MLN-120B exhibited a highly favourable selectivity profile when tested against a panel of 442 kinases [99], making these inhibitors the preferred tools for dissecting IKKβ-dependent functions [100].
Meanwhile, BMS-345541 acts as an allosteric inhibitor of the IKKs, displaying moderate (13-fold) selectivity for IKKβ over IKKα [83]. Curiously, BMS-345541 binds to IKKβ in a mutually exclusive manner with respect to phosphorylated IκBα substrate and in a non-mutually exclusive manner with respect to ADP. Binding to IKKα has the opposite effect, leading to a proposed binding model whereby BMS-345541 binds to similar allosteric sites on IKKα and IKKβ but affects the active sites of the subunits differently.
A third class of IKKβ inhibitors constitute thiol-reactive compounds that interact with key cysteine residues in IKKβ. For example, berberine [101], nimbolide [102] and withaferin A [92] have been proposed to inhibit IKKβ activity through covalent modification of cysteine 179 (C179), a residue that may promote phosphorylation of ser-177/Ser-181 and, in turn, kinase activity [103]. However, the involvement of C179 modification in the mechanism of action of withaferin A has been questioned recently by a study that found it to be a poor direct inhibitor of IKKβ in an in vitro kinase assay; instead it was proposed to inhibit IKKβ indirectly through blockade of signal-induced NEMO reorganization into ubiquitin-based signalling foci [89]. Other molecules, such as the epoxyquinoid derivatives, manumycin A [104] and jesterone dimer [105], are thought to inhibit IKKβ activity through covalent cross-linking of IKKβ monomers via C179, which disrupts the essential interaction between IKKβ and NEMO. Modification of C179 may also block S-glutathionylation at this residue, which is thought to be important for the kinase activity of IKKβ [106]. The natural product ainsliadimer A, meanwhile, covalently modifies the conserved residue, cysteine-46, in both IKKα and IKKβ, to inhibit ATP-binding and kinase activity via a putative allosteric mechanism [95]. A fourth mechanism of action for IKKβ inhibitors has been proposed for the benzoxathiole derivate, BOT-64, which inhibited IKKβ kinase activity via an apparent direct interaction with S177 and/or S181 residues in the activation loop of IKKβ [94], although 10 years on from the primary paper further work is required to validate this.
In addition, IKKβ kinase activity may be inhibited indirectly through blockade of the essential interaction between IKKβ and NEMO. For example, cell-permeable peptides corresponding to the NEMO-binding domain (NBD) of IKKα/β display non-selective inhibitory activity against IKKα and IKKβ [107]. These molecules will be discussed further in Section 7.2.
Multiple clinically approved non-steroidal anti-inflammatory agents (NSAIDs), including sodium salicylate (aspirin), sulindac sulphide and exisulind, have also been proposed to inhibit the NF-κB pathway at the level of IKKβ. The primary anti-inflammatory mechanism of these compounds is via inhibition of the cyclooxygenase enzymes, COX1 and COX2 [108]. However, these compounds have also been reported to inhibit NF-κB activation by inhibiting IκBα phosphorylation [109,110,111,112], while at higher concentrations aspirin has been proposed to act as an ATP-competitive inhibitor of IKK-β (in vitro IC50 ~ 80µM) [110,112]. However, these findings should be interpreted with extreme caution as more recent kinase profiling studies suggest that aspirin inhibits vast numbers of other kinases at least as potently as it inhibits IKKβ [113]. Furthermore, some studies have suggested that the direct inhibition of IKKβ activity by aspirin in vitro does not reflect its inhibitory mechanism in vivo, where aspirin is proposed to inhibit TNFα-induced IκBα phosphorylation and degradation indirectly through activation of p38 kinase [114,115]. In addition, more recent studies have proposed that while short-term treatment (1–2 h) with NSAIDs may block stimuli-induced NF-κB activation in cells, prolonged exposure with pharmacologically relevant doses of NSAIDs, in fact, stimulates the NF-κB pathway both in vitro and in vivo ([116,117,118,119,120]. Furthermore, this activation has been causally associated with pro-apoptotic effects of these agents in cancer cells and may involve NSAID-induced nucleolar translocation of RelA [121].
3. Pre-clinical Development of IKKβ Inhibitors
IKKβ inhibitors have demonstrated efficacy in various pre-clinical models of cancer and inflammatory disease (see Table 1). For instance, MLN-120B inhibited multiple myeloma (MM) cell growth in a clinically relevant severe combined immunodeficient (SCID)-hu mouse model [42] and exhibited significant therapeutic efficacy in a rat model of rheumatoid arthritis (RA) [43]. However, clinical use of these inhibitors has not yet been reported. Indeed, only a handful of phase I/II clinical trials with IKK inhibitors have been performed. The earliest example was the ATP-competitive IKKβ inhibitor, MLN-0415, which failed in phase I human trials for inflammatory disorders due to an unfavourable safety profile [122]. It is now being tested in dogs with high-grade lymphomas. The Institute of Medicinal Molecular Design Inc (IMMD) has several compounds in current or completed phase I/II trials, for which results have yet to be published [122]. For example, IMD-2560 (a pro-drug of IMD-0560) [123] underwent a phase I trial for the treatment of rheumatoid arthritis (RA), rheumatic osteoporosis and osteoarthritis, IMD-0354 [124] underwent a phase I trial for the topical treatment of atopic dermatitis and IMD-0354 (and its pro-drug IMD-1041) underwent a proof-of-concept (POC) study for the treatment of chronic obstructive pulmonary disease (COPD; Identifier: NCT00883584). However, questions surrounding the true molecular target of these compounds has been raised by the lack of conclusive biochemical evidence for IKKβ antagonism. Indeed, IMD-0354 was recently shown to exhibit no activity against IKKβ or IKKα in an ATP-based kinase assay [125].
The IKKβ-selective compound, SAR-113945 [126], has progressed the furthest through clinical development. Multiple phase I trials demonstrated its safety/tolerability following intra-articular injection in patients with knee osteoarthritis (Identifier: NCT01113333/NCT01463488/ NCT01511549). Positive efficacy trends in these studies motivated the undertaking of a Phase IIa POC trial (Identifier: NCT01598415). However, SAR-113945 failed to show efficacy in this larger patient sample size [127].
The ATP-competitive IKKβ inhibitor, SPC-839 (also known as AS602868; [128,129]), also progressed into phase I trials for haematological malignancies, but the trial was prematurely terminated due to portfolio repositioning. This reflects a common trend within the pharmaceutical industry; interest in the clinical development of IKKβ-selective inhibitors has significantly diminished in the last 10 years.
4. Potential Reasons for the Lack of Clinical Success of IKKβ Inhibitors
Evidently, the potential of IKKβ inhibition as a therapeutic strategy remains unrealized and previous optimism for IKKβ as a therapeutic target has significantly cooled. There are several plausible reasons for this lack of clinical success. The simplest is that IKKβ inhibitors developed to date may not exhibit the combination of properties required to achieve success during preclinical development, including but not limited to: nanomolar-range potency, high selectivity over IKKα/other kinases, and clinically relevant pharmacokinetics/pharmacodynamics. As mentioned earlier, several ‘selective’ IKKβ inhibitors that continue to be used by the research community have been shown to be anything but selective. IKKβ inhibitors described to date were identified following hit-to-lead development and characterization of structure–activity relationships in the absence of resolved crystal structures for the IKKs. However, the recent reports of human IKKβ X-ray crystal structures have revealed many new structural details at a sufficiently high resolution that will hopefully facilitate the structure-guided design of next-generation IKKβ inhibitors with enhanced potency and selectivity (Figure 2 and Figure 3; [5,6]).
These structures identified opportunities to rationally design highly selective non-ATP competitive inhibitors. For example, Liu et al. captured an asymmetric dimer of human IKKβ at a resolution of 2.8A, with one protomer in an active and the other in an inactive conformation, each with phosphorylated and unphosphorylated S177/S181 residues, respectively [5]. The binding mode of an inhibitor within the ATP-binding site of IKKβ was essentially identical regardless of the activation state of the kinase domain (KD), suggesting that ATP-competitive compounds are unlikely to selectively capture the inactive conformation. However, elsewhere the protein conformations were distinct, highlighting the potential for selective, non-ATP competitive inhibitors. Indeed, a recent study utilised a potential allosteric site identified at the KD-ubiquitin-like domain (ULD) interface (Figure 3A) to perform a virtual screen for allosteric inhibitors [130]. They identified a lead compound (3,4-dichloro-2-ethoxy-N-(2,2,6,6-tetramethylpiperidin-4-yl) benzenesulfonamide), which inhibited TNFα-induced NF-κB transcriptional activity through selective capture of the inactive conformation and hence blockade of IKKβ S177/S181 phosphorylation. The selectivity of this compound over IKKα was, unfortunately, not assessed. However, it is interesting to note that X-ray and cryo-EM structures of human IKKα demonstrated pronounced differences between IKKα and IKKβ in the orientation of the KD relative to the α-helical scaffold/dimerization domain (SDD) and its stably associated ULD, suggesting that the KD-ULD interfaces of IKKβ and IKKα may be sufficiently unique to facilitate the design of highly selective IKK inhibitors [7]. Meanwhile, Polley et al. demonstrated that the introduction of mutations within the two oligomerization interfaces identified in their constitutively active (S177E/S181E) human IKKβ X-ray crystal structure—the SDD-SDD interface of dimeric IKKβ (Figure 3B,C), and the KD-KD interface of oligomeric IKKβ (Figure 3D,E)—was sufficient to interfere with IKKβ activation/catalytic activity in vitro, indicating that small molecules designed to interfere with IKKβ oligomerization through these interfaces may function as highly selective inhibitors of IKKβ [6].
Another reason for the lack of success of IKKβ inhibitors in pre-clinical development may be their inappropriate therapeutic application. In the simplest sense this could reflect use of the wrong dosing strategy (drug concentration/dosing schedule, etc.) for effective target inhibition. More challenging is the appropriate selection of patient subgroups to achieve the desired therapeutic efficacy. For example, the proof of concept study for SAR113945 failed to show any effect in the overall group of recruited study participants for the primary endpoint; however, post-study analysis demonstrated a statistically significant difference in a patient subgroup that had presented with synovial effusion at baseline [127]. Stratification of patient subgroups to identify those with a clear NF-κB-driven, inflammatory phenotype that would benefit from IKKβ inhibition may be one solution to this problem.
A further underappreciated factor that may have impacted the clinical success of IKKβ inhibitors is the relative contribution of IKKα and/or IKKβ to the disease state being targeted. As already discussed, IKKβ has historically been considered the primary viable target to inhibit pathogenic canonical NF-κB signalling due to seminal knock-out experiments in murine cells. However, this isoform-specific delineation of function has turned out to be overly simplistic. Increasingly, it appears that in certain human cell-types IKKα may play a substantial role, alongside IKKβ, in activation of canonical NF-κB signalling, both under physiological or pathological conditions [133,134,135] and as an adaptive response to inhibition of IKKβ [136]. The relative contribution of IKKα and IKKβ to disease-associated canonical NF-κB signalling should therefore be assessed prior to any decisions to apply IKKβ-, IKKα- or dual-selective inhibitors. For example, dual siRNA-mediated knockdown of IKKα and IKKβ or dual IKKα/β inhibition had a greater suppressive effect on canonical NF-κB activation and proliferation, survival and migration of head and neck squamous cell carcinoma (HNSCC) cells than knockdown or inhibition of each IKK individually [134].
Many of these issues are likely compounded by the lack of widespread use of isoform-specific readouts of cellular IKK activity in pre-clinical studies. Non-canonical NF-κB stimuli/FBS-induced phosphorylation of human p100 at S866/870 may be used as a cellular readout of IKKα-induced non-canonical NF-κB signalling [137]. Meanwhile, the relative contributions of IKKα and IKKβ to canonical NF-κB signalling (typically assessed by measurement of phosphorylation of IκBα at S32/36 and p65 at S468 and/or S536) may be assessed using a combination of highly selective IKKβ inhibitors (such as BI605906) and siRNA or CRISPR-Cas9-mediated genetic ablation of the IKKs. IKKα-selective inhibitors may also be commercially available in the near future [35].
5. Safety Concerns Surrounding the Therapeutic Use of IKKβ Inhibitors
Irrespective of the reasons for the lack of clinical success of IKKβ inhibitors to date, there are several real and perceived concerns surrounding the safety of systemic administration of IKKβ inhibitors that have contributed to a significant decrease in interest in their clinical development.
One of the first observations to raise concerns about the safety of systemic IKKβ/NF-κB inhibition was the marked increase in susceptibility to apoptosis that accompanied genetic ablation of NF-κB pathway components. For example, ikkb-/- and rela-/- mice exhibit embryonic lethality due to severe liver apoptosis, which results from an absence of survival signalling in response to TNFα stimulation [28,29,138,139]. Meanwhile, enterocyte-specific ablation of IKKβ in a mouse model of gut ischemia-reperfusion resulted in severe apoptotic damage to the intestinal mucosa, highlighting a role for NF-κB signalling in maintaining tissue homeostasis in the adult mouse [140]. Curiously, however, the consequences of a lack of IKKβ in humans are strikingly different; homozygous deletion of the IKBKB gene is not embryonic lethal—at least in patients examined to date—but leads to a lack of regulatory (Treg) and γδ T cells, defects in T- and B-cell activation and to SCID associated with early infections with various pathogens [141,142,143,144]. These differences could relate to compensatory IKKα-dependent canonical NF-κB signalling in humans in certain cell types, or other species variations, and imply there may be broader differences in the response of mice and humans to systemic IKKβ inhibition. Nevertheless, these genetic studies highlight another concern with systemic IKKβ inhibition: the potential for increased susceptibility to infection due to the vital role for NF-κB signalling in the host defence system [145,146].
Additional safety concerns surrounding systemic IKKβ inhibition are largely related to the complexity of function of IKK/NF-κB signalling in inflammation and, in turn, inflammatory diseases and cancer; NF-κB signalling is often described as a ‘double-edged sword’, having pro- and anti-inflammatory functions in different contexts. Further complexity is introduced by the context-dependent roles of the NF-κB-mediated inflammatory response itself in tumorigenesis. As an example, immune-cell infiltration of tumours can have a dual role: either leading to an anti-tumour response, or immune evasion and active promotion of tumorigenesis. For a comprehensive review of this complex subject, see [147,148].
Chronic NF-κB activity has been implicated in the pathogenesis of many inflammation-related diseases [149,150] and genetic mutations that lead to increased NF-κB activity often trigger chronic inflammation and associated pathologies [151,152,153]. Indeed, NF-κB is one of the key links between chronic inflammation and cancer, as shown by the critical role of NF-κB in inflammation-driven, colitis-associated cancer (CAC) and hepatocellular carcinoma (HCC) [154,155]. Beyond maintaining a chronic inflammatory microenvironment, NF-κB has established tumour promoting roles in various cancers through the aberrant regulation of genes that influence all of the hallmarks of cancer. Many studies have confirmed that NF-κB inhibition through genetic ablation or small-molecule inhibition of IKKβ has anti-inflammatory and/or anti-tumorigenic effects in vivo [154,156,157].
However, several studies have surprisingly shown that IKKβ inhibition in certain cells/tissues triggers the spontaneous development of severe inflammatory conditions, and in some cases promotes malignant development, indicating anti-inflammatory and tumour suppressor roles for NF-κB; it appears, therefore, that the consequence of IKKβ deletion and NF-κB inhibition is highly cell-type and context dependent [158]. For example, a series of seminal studies demonstrated a role for NF-κB signalling in the negative control of inflammasome-dependent IL-1β secretion [159,160]. Hematopoietic-cell specific ablation of IKKβ or prolonged, systemic IKKβ inhibition in mice resulted in enhanced inflammasome-dependent, caspase-1-mediated IL-1β production and hyper-susceptibility to septic shock-induced endotoxin challenge or bacterial infection [159]. In turn, the enhanced IL-1β secretion following systemic IKKβ inhibition was found to promote the proliferation of granulocytic progenitors and increase the survival of mature neutrophils leading to neutrophilia and inflammatory destruction of tissues [160]. The mechanism for this NF-κB-dependent inhibition of the inflammasome remained obscure until recently when it was proposed that NF-κB promotes the expression of sequestosome-1/p62 (SQSTM1, here referred to as p62) in macrophages to facilitate p62-dependent elimination of damaged mitochondria, which act as cell-intrinsic inflammasome activating signals [161]. The clinical relevance of these observations is supported by the observation of enhanced inflammation and neutrophilia during human phase I trials of IKKβ inhibitors. Furthermore, IL-1β protein is significantly increased in the plasma of advanced non-small-cell lung carcinoma (NSCLC) patients treated with Bortezomib (trial protocol: NCT01633645) [162]. While IKKβΔ IL-1βR1-/- mice do not display neutrophil-driven inflammation they exhibit severely compromised innate immunity and susceptibility to bacterial infection [160]. Furthermore, IL-1β is required for immunogenic cell death (ICD)-mediated dendritic cell (DC) maturation and antigen presentation as part of the adaptive immune response [163]. Combined inhibition of IKKβ and IL-1β signalling is, therefore, unlikely to be viable, necessitating an alternative solution to the toxicity issues of systemic IKKβ inhibition, unless coupled with intense medical supervision and strong antibiotics.
In addition, there are concerns that systemic IKKβ inhibition might promote malignant development in tissues/contexts where IKKβ/NF-κB activity plays a dominant tumour suppressor role. For example, IKKβ has been shown to act as a tumour suppressor in cancer-associated fibroblasts (CAFs) during intestinal tumorigenesis [164]. However, these findings directly contrast with other studies where IKKβ ablation in mesenchymal cells protected against inflammation-induced intestinal carcinogenesis [165,166]. The reasons for these differences are unclear but may be due to temporal differences in IKKβ inactivation and/or the targeting of different mesenchymal cell subpopulations in each model. The outcome of IKKβ ablation often depends on the targeted cell type. For example, in melanoma IKK has both tumour-promoting activity in melanocytes [156] and tumour-suppressive activity in myeloid cells [167]. Furthermore, IKKβ doesn’t always act as a ‘real’ tumour suppressor. For example, in models of chemically-induced HCC (which is not accompanied by chronic inflammation) NF-κB inactivation through liver-targeted deletion of IKKβ strongly enhances diethylnitrosamine (DEN)-induced carcinogenesis [168]. However, IKKβ inhibition only potentiates HCC under conditions of elevated hepatocyte injury, indicating that IKKβ is not a true tumour suppressor in this context. Indeed, mice with hepatocyte-specific expression of constitutively active IKKβ exhibit enhanced HCC development [169].
These examples highlight that the biological determinants influencing the pro- or anti-inflammatory/tumorigenic roles of IKKβ are complex and ill-defined. Further work is needed, both in animal models and patient tumoral samples, to unravel the context-dependencies of IKKβ functions in inflammation/cancer to better define the circumstances where the therapeutic benefits of systemic IKKβ inhibition may outweigh the risk of potential side-effects.
6. Recent Therapeutic Opportunities to Target IKKβ
Despite shifts in the priorities of the pharmaceutical industry away from the development of IKKβ inhibitors there are several active research areas where IKKβ remains a highly attractive clinical target. Furthermore, therapeutic strategies such as rational combination therapies and targeted delivery may be able to mitigate some of the host toxicity observed following systemic delivery of IKKβ inhibitors. Here we discuss some of these recent therapeutic opportunities to target IKKβ.
6.1. Cancers Exhibiting Clear ‘Addiction’ to Canonical NF-κB Signalling
While the inflammatory side-effects associated with prolonged IKKβ inhibition may limit systemic application of IKKβ inhibitors in the treatment of chronic inflammatory and autoimmune diseases, a partial and/or short-lived inhibition of IKKβ, which is unlikely to trigger widespread neutrophilia, could still find utility in the treatment of cancers in which NF-κB plays a clear initiating/driving role in tumorigenesis. The priority would be tumours bearing oncogenic, NF-κB-activating lesions, followed by tumours with constitutive NF-κB activation due to factors within the tumour microenvironment (TME). In either case, NF-κB activation must be strongly correlated with poor prognosis. In other words, identification of cancers exhibiting a wide therapeutic window of opportunity may enable partial/short-term IKKβ inhibition to have a preferential effect on malignant cells relative to normal host cells. In this context, it is important that tumour subtypes exhibiting addiction to the NF-κB pathway are identified. This can be achieved using whole genome sequencing/copy number analysis to identify relevant NF-κB component lesions alongside expression profiling of NF-κB target genes to identify gene signatures diagnostic of NF-κB pathway addiction [170,171]. A full account of the link between NF-κB and cancer is beyond the scope of this review, and readers are directed elsewhere [22,148]. However, a few illustrative examples will be discussed to highlight those cancers for which IKKβ inhibition offers the greatest therapeutic potential.
Both cell-intrinsic and -extrinsic factors contribute to aberrant NF-κB activity. Enhanced NF-κB activity can be a direct consequence of mutations of NF-κB pathway components and/or upstream oncogenes that activate NF-κB. On the other hand, a tumour can acquire elevated NF-κB activity through interaction with the inflammatory milieu of the TME. Lymphoid malignancies, particularly B-cell lymphomas, frequently exhibit direct mutations of NF-κB signalling genes [172]. For example, the pathogenesis of activated B cell-like Diffuse large B-cell lymphoma (ABC DLBCL) involves oncogenic activation of various upstream NF-κB pathway components, including the CD79B subunit of the BCR, CARD11 and MyD88, which drive cancer cell proliferation/survival through canonical NF-κB activity [171,173]. Oncogenic addiction of ABC DLBCL cells to high NF-κB activity has been demonstrated by the selective cytotoxicity of IKKβ inhibitors, providing a clear rational for therapeutic strategies targeting IKKβ/NF-κB [62,170].
In contrast, with a few exceptions [174,175,176], solid malignancies rarely exhibit direct oncogenic mutations of NF-κB pathway components. In most solid tumours NF-κB is constitutively activated due to chronic pro-inflammatory signalling within the TME. However, certain oncogenes can also drive downstream NF-κB activity. For example, studies of mouse models of Kirsten rat sarcoma viral oncogene homolog (KRAS; G12D)-induced lung cancer have demonstrated that KRAS activates NF-κB in lung tumours in situ [177,178,179]. Various mechanisms have been proposed for KRAS-mediated activation of NF-κB, including cell-autonomous feed-forward activation of PI3K-AKT, MEK-ERK and DNA-damage response signalling pathways and feed-forward autocrine signalling [157,179]. Concomitant loss of p53 activity dramatically enhances the activation of NF-κB in lung cancer cells [177,179]. Significantly, IKKβ-dependent NF-κB activation in these models has been shown to drive lung tumourigenesis, primarily through enhanced cancer cell proliferation [65,178,179]. Furthermore, KRAS-driven NF-κB activation has been associated with feedforward amplification of RAS signalling, drug resistance, and tumour stemness [180,181]. These studies collectively identify IKKβ inhibition as a promising therapeutic strategy in KRAS-driven lung cancer with altered p53 activity [65]. It should be noted, however, that several studies have also linked KRAS-driven IKKα and TBK1 activation to pathogenic NF-κB activity [181,182,183,184], suggesting that IKKβ inhibition alone may not be an optimal therapeutic strategy.
A potentially crucial aspect of IKKβ inhibition as a cancer therapy is the appropriate timing of treatment with respect to the stage of cancer progression. Given the potential toxicity of IKKβ inhibition towards T cells, such treatment is likely to be undesirable during the early tumour-eliminating phase of the immune system, when cytotoxic T lymphocytes (CTLs) target transformed cells. Rather, based on current knowledge, IKKβ inhibition is more likely to have positive therapeutic effects in the chronic inflammatory phase of tumour progression [22]. However, this generalisation may be an oversimplification (see Section 6.3) and may not apply to all cancers. Indeed, given the context-dependency of the pro- and anti-inflammatory effects of IKKβ inhibition, it remains to be determined what the net effect systemic, pharmacological IKKβ inhibition may have in different cancers.
6.2. Use of IKKβ Inhibitors in Combination Therapies to Combat Chemoresistance
The consensus from pre-clinical studies is that IKKβ inhibitors are unlikely to achieve broad clinical success as single agents in cancer therapy, except perhaps for certain types of lymphoma/leukaemia. However, there is significant optimism that they might yet enter the clinic as part of combination therapies with conventional therapeutics (chemo- and radiotherapeutics), and certain targeted therapies, in cancers where NF-κB signalling has been associated with chemoresistance [185]. Use of synergistic combinations may enable lower concentrations of IKKβ inhibitors to be employed to achieve a desired therapeutic effect, thus reducing systemic toxicity.
The cytotoxic/cytostatic effects of conventional therapeutics, such as cisplatin and radiotherapy, typically rely on their ability to preferentially induce DNA damage in highly proliferative cancer cells, which triggers cell cycle arrest and, ultimately, cell death or senescence. NF-κB activity is associated with chemoresistance in various cancers through the induction of genes involved in the control of survival, proliferation, inflammation, DNA repair, metabolic reprogramming, angiogenesis, drug uptake/inactivation, etc., that reduce the efficacy of conventional therapeutics [186]. Intrinsic chemoresistance has been correlated with constitutive NF-κB activation in numerous cancers [187,188,189]. Chemoresistance is also commonly acquired or enhanced by therapy-induced NF-κB activation [186,190,191]. For example, genotoxic agents that induce double-strand DNA breaks (DSBs) activate IKK-dependent NF-κB activity via a NEMO-ATM dependent pathway initiated in the nucleus (Figure 1E; [192]). As such, IKKβ inhibitors are under investigation as a route to sensitize cancer cells to conventional genotoxic therapeutics. For example, inhibition of IKKβ with MLN-120B leads to synergistic enhancement of vincristine cytotoxicity in non-Hodgkin’s lymphoma via suppression of vincristine-induced NF-κB activity [193]. Interest in IKK/NF-κB inhibition is likely to enhance as ever greater numbers of studies/clinical trials identify therapy-induced NF-κB activity as a key factor in the relapse response to conventional therapies (Identifier: NCT00280761).
IKKβ inhibitors are also being investigated in combination with certain targeted therapeutics to overcome intrinsic resistance. For example, activation of NF-κB has been strongly linked to intrinsic and acquired resistance to epidermal growth factor receptor (EGFR) inhibitors [181,194,195], while synergistic cytotoxicity has been observed in ABC DLBCL treated with JAK and IKKβ inhibitors [196]. However, it should be noted that, in general, the IKK isoform-dependence of NF-κB-mediated chemoresistance mechanisms has not been characterised. IKKβ inhibition may not always be the most effective means to counteract chemoresistance. Indeed, dual IKKα/β inhibition may be more effective than IKKβ inhibition alone in the counteraction of resistance to EGFR inhibitors in HNSCC [134].
6.3. IKKβ Inhibitors as an Adjunct to Cancer Immunotherapies
Recent reports suggest IKKβ inhibitors may also combine well with certain cancer immunotherapies. Cancer immunotherapy is a general term for treatments that harness or reactivate the body’s own immune system to target and destroy tumours. For instance, this may be a tumour-specific vaccination that enhances the anti-tumour activity of the patients own CTLs. However, CTL-dependent anti-tumour responses are often suppressed by FOXP3+ Tregs that infiltrate tumours. Therefore, several cancer immunotherapies that inhibit or deplete FOXP3+ Treg cells are currently being tested [197]. As described earlier, patients with rare homozygous deletion of the IKBKB gene typically lack Treg cells [141]. Consistent with this, prolonged IKKβ inhibition was recently shown to partially deplete circulating FOXP3+ Treg cells, due to their dependence on NF-κB signalling for survival [198]. Consequently, administration of an IKKβ inhibitor after tumour vaccination in a mouse melanoma model enhanced the CTL-dependent anti-tumour response and delayed tumour growth, identifying IKKβ as a potential druggable immune checkpoint. A large caveat, however, is that the correct dosage or scheduling of IKKβ inhibition is likely to be critical to achieve potentiation, rather than inhibition of the anti-tumour response, due to the importance of NF-κB activity for T cell survival/function [31]. For example, genetic deletion of IKKβ in T-cells abrogated the anti-tumour response in mice with fibrosarcoma [199]. Indeed, Heuser et al. observed that high doses of IKKβ inhibitor suppressed CTL responses in their model [198]. This dosage effect was explained by the fact that Tregs were more sensitive than CTLs to IKKβ inhibition due to a greater reliance on NF-κB signalling for survival. These findings also suggest further caution is warranted in attempts to utilise systemic IKKβ inhibition to treat inflammatory diseases.
A key feature of tumour immune evasion is the increased expression of certain ligands, notably programmed cell death-ligand 1 (PD-L1), at the cell surface of cancer cells and immune cells, such as dendritic cells and macrophages [200,201]. Binding of PD-L1 to its cognate receptor, PD-1, on T cells inhibits their proliferation/survival and effector cytokine secretion to downregulate CTL responses [202,203]. As such, anti-PD-1/PD-L1 therapies have now demonstrated marked success in a wide range of malignancies [204]. However, high expression of surface PD-L1 by solid tumours and tumour-infiltrating myeloid cells appears to be correlated with poor prognosis in certain cancers and may negatively impact the clinical response to PD-1 blockade [205]. The mechanisms regulating PD-L1 expression are, therefore, of clinical interest. Interestingly, NF-κB signalling has been shown to promote the expression of PD-L1 at the transcriptional [206,207] and, more recently, the post-transcriptional level [208]. Lim et al. demonstrated that in an inflammation-enhanced tumour model, macrophage-derived inflammatory cytokines (such as TNFα) enhanced PD-L1 expression in cancer cells through protein stabilization via NF-κB-dependent upregulation of COP9 signalosome 5 (CSN5). Consequently, administration of curcumin (which can inhibit NF-κB and CSN5 activity directly) inhibited CSN5-dependent PDL1 stabilisation and enhanced the efficacy of anti-CTLA4 therapy. Any study that employs curcumin comes with several caveats because of the sheer number of signalling pathways this natural product deregulates, including ERK, JNK, p38 and Akt, as well as NF-κB, in addition to effects on cathepsins. Nonetheless, this approach has received some support by the observation that TNFα-blockade overcomes resistance to anti-PD-1 in experimental melanoma [209]. Whether these strategies might also apply to IKKβ-selective inhibitors warrants further investigation.
Another form of cancer immunotherapy involves the use of oncolytic viruses (OVs), which specifically infect and kill tumour cells. For example, Talimogene laherparepvec (or T-Vec), based on herpes simplex virus 1 (HSV-1), has been clinically approved for the treatment of advanced melanoma [210]. However, there is considerable heterogeneity in the therapeutic response to OV therapy, with resistance largely due to failure of tumour cells to become infected by the virus [211]. Interestingly, dimethyl fumarate (DMF) was recently shown to enhance OV infection of cancer cells and improve therapeutic outcomes in resistant syngeneic and xenograft tumour mouse models through inhibition of NF-κB signalling and, in turn, the antiviral response of cancer cells [212]. IKKβ-selective inhibitors also improved OV infection in vitro [50,212,213], suggesting a combination of oncolytic virotherapy and IKKβ inhibitors may warrant further clinical investigation.
Finally, there are reports that IKKβ inhibition may enhance anti-tumour immunity through modulation of the activity of tumour-associated macrophages (TAM). A simplified model states that TAMs recruited to tumours characterised by non-resolving inflammation switch from a tumour-killing, ‘classically activated’ M1-like phenotype to a tumour-promoting, ‘alternatively activated’ M2 phenotype that stimulates cancer cell proliferation, angiogenesis and metastasis and suppresses immune effector cells [214]. The modulation of TAM survival/polarization is, therefore, an attractive therapeutic target [215]. Inhibition of NF-κB activation in TAMs through macrophage-specific ablation of IKKβ has been proposed to promote polarization to the anti-tumour M1 phenotype, thereby enhancing tumour regression [216]. However, there are other conflicting reports that suggest that M2 immunosuppressive TAMs from established tumours are characterised by defective NF-κB transcriptional responses, due, in part, to overexpression of repressive p50 homodimers [217,218]. In turn, enforced re-activation of NF-κB can polarize M2-like TAMs to an M1-like phenotype and induce their tumour cytotoxicity [219,220,221]. Various explanations have been proposed for these contrasting findings [222]. As discussed earlier in the broader context of cancer therapy, the clinical outcome of pharmacologic targeting of macrophage IKKβ/NF-κB activities may depend on the disease stage; that is, premalignant, malignant or metastatic. This example is also representative of a wider issue: given the complexity of NF-κB function in the immune system it is perhaps prohibitively difficult to predict the outcome of systemic IKKβ inhibition on anti-tumour immunity, thus limiting the potential of IKKβ inhibitors as adjuncts to cancer immunotherapies.
6.4. Targeted Delivery of IKKβ Inhibitors to Specific Tissues
If the safety issues associated with systemic IKKβ inhibition prove insurmountable, a less toxic therapeutic approach may be the topical/local or targeted administration of inhibitors to specific disease areas. The former strategy has been demonstrated in pre-clinical studies investigating the use of IKKβ inhibitors to treat choroid neovascularization (CNV), which is a major pathological change associated with exudative age-related macular degeneration (AMD) [223]. IKKβ-dependent NF-κB signalling plays a significant role in CNV development [53]. Consequently, retrobulbar administration of IKKβ inhibitor (TPCA-1)-loaded poly-lactide-co-glycolide (PLGA) microparticles achieved controlled, durable intraocular release of drug, leading to attenuation of CNV and macrophage recruitment, without systemic toxicity in a laser-induced mouse model of CNV [223].
Furthermore, as discussed in Section 5, numerous studies have demonstrated that the therapeutic outcome of IKK/NF-κB inhibition in a specific cancer model depends on the cell-type(s) selected for genetic ablation of the NF-κB signalling component. For instance, in a mouse melanoma model, ablation of tumour-intrinsic NF-κB activity using cancer cell-targeted expression of an IκBα super-repressor construct led to cytotoxicity-driven tumour regression following doxorubicin treatment [224]. However, myeloid-specific loss of NF-κB signalling resulted in increased host toxicity and mortality without tumour regression due to excessive IL-1β production, consistent with other studies [159]. Results such as this strongly advocate for a targeted delivery approach to IKKβ inhibition to reduce therapy-associated systemic toxicity. αvβ3-ligand tetraiodothyroacetic acid (TET)-modified micelles have been shown to promote selective accumulation of the NF-κB inhibitor, celestrol, in primary tumours and lung metastases and to inhibit tumour growth and metastasis in a breast cancer mouse model to a greater extent than systemic delivery of celestrol alone [225]. Although relative safety profiles were not assessed in this case, this study demonstrates a promising proof-of-principle.
7. Alternative Approaches to Targeting the NF-κB Pathway
As the ubiquitously expressed, primary, druggable mediator of canonical NF-κB signalling, IKKβ was the logical, first-choice target for the development of pharmacological inhibitors of the NF-κB pathway. As we have discussed, IKKβ inhibitors have displayed significant therapeutic potential, and new developments continue to identify IKKβ as an attractive therapeutic target. However, the potential risks of on-target systemic toxicity, immunodeficiency and malignant development arising in contexts where IKKβ/NF-κB activity plays a dominant tumour suppressor role may prove insurmountable and forever undermine research efforts to clinically develop IKKβ-targeting therapeutics. Some, if not all, of these safety concerns may be circumvented by targeting alternative nodes in the NF-κB pathway [226,227]. Indeed, this strategy has seen far greater clinical success than directly targeting IKKβ. A few illustrative examples will be discussed here.
7.1. Targeting Upstream NF-κB Signalling Components
Multiple signalling pathways converge to activate NF-κB. Inhibition of disease-specific upstream NF-κB components may provide a greater degree of tissue- and context-specificity than directly targeting IKKβ, which is activated by all NF-κB-inducing signals. A clinically-relevant example is the specific targeting of BCR-induced NF-κB signalling (see Figure 1D) in B-cells using inhibitors of Burton tyrosine kinase (BTK), such as Ibrutinib, which is approved for the treatment of refractory mantle cell lymphoma (MCL) [228] and marginal zone lymphoma (MZL) [229], chronic lymphomatic leukemia (CLL) [230], small lymphocytic lymphoma (SLL) [230], Waldenström’s macroglobulinemia [231] and chronic graft versus host disease [232] The highly-restricted pattern of BTK expression to B cells means that Ibrutinib has a generally well-tolerated safety profile.
7.2. Targeting IKKα or NEMO
Until recently IKKα has received minimal attention as a target for drug development [35]. However, IKKα is an essential component of the non-canonical NF-κB pathway, making it an attractive therapeutic target in diseases where aberrant activity of this pathway contributes to pathogenesis, such as mucosa-associated lymphoid tissue (MALT) lymphoma and MM [233]. Furthermore, the growing realisation that IKKα, alongside IKKβ, contributes to canonical NF-κB pathway regulation in certain contexts should motivate the investigation of IKKα-selective or IKKα/β dual selectivity inhibitors in diseases where canonical NF-κB signalling plays a role in pathogenesis [134]. Equally, selective targeting of IKKα may enable context-specific inhibition of pathogenic NF-κB signalling, while retaining sufficient ‘physiological’ levels of canonical NF-κB signalling in host tissues to prevent the adverse effects seen with IKKβ inhibitors. IKKα also has numerous NF-κB-independent functions that could be targeted (see Section 7.3).
Significantly more effort has been devoted to development of molecules that target the NEMO scaffold, which is essential for canonical NF-κB signalling [234]. IKKα and IKKβ interact with NEMO via their short, C-terminal NBDs, consisting of the hexapeptide amino acid sequence, LDWSWL. Peptides corresponding to the NBD domain fused to sequences that facilitate intracellular delivery disrupt the association of IKKs with NEMO and block NF-κB transcriptional activation in cells [107]. NBD peptides have exhibited promising therapeutic effects in numerous disease models [235]. For example, a recent phase I trial of systemic administration of NBD peptides for the treatment of dogs with spontaneous ABC DLBCL demonstrated safety and treatment efficacy, offering hope for translation of this therapy to human ABC DLBCL [236]. The safety/toxicity profile of NBD peptides are generally more favourable than IKKβ inhibitors, potentially because they suppress only stimulus-induced rather than basal, IKK activity [107]. They are also less likely to inhibit ‘off-target’ kinases due to the specificity of the protein interaction between NBD peptides and NEMO. Clinical success may be limited, however, by the expense of peptide synthesis, their short half-life, and poor oral bioavailability, requiring intravenous administration. Small-molecule NBD mimetics that could overcome these limitations have recently been described [225].
Recently, NEMO-ubiquitin interaction inhibitors (iNUBs) have been characterised [237]. iNUBs inhibited IKK/NF-κB activation and target gene expression in response to TNFα, but not IL-1β, stimulation. iNUBs, therefore, may offer a strategy to selectively impair NF-κB signalling in response to specific stimuli.
7.3. Targeting NF-κB-Independent Functions of the IKKs
As described in Section 1, the IKKs exert multiple NF-κB-independent functions through the phosphorylation of so-called ‘non-classical’ substrates. Many of these activities have been proposed to contribute to disease progression [238]. Targeting these NF-κB-independent functions may, therefore, offer an opportunity to block IKK-driven pathogenesis without the side-effects associated with systemic NF-κB inhibition. For example, IKKα has been proposed to promote mammalian target of rapamycin complex 1 (mTORC1) and mTORC2 activity via feedforward signalling, and thus could serve as a therapeutic target in mTOR-dependent cancers in a manner independent of potent canonical NF-κB inhibition [239,240]. Meanwhile, IKKβ phosphorylates FOXO3a at Ser644 to promote its nuclear exclusion and ubiquitination-mediated proteasomal degradation to promote breast cancer progression [26]. However, further work is needed to validate many of these targets as bona fide IKK substrates and to understand the importance of these NF-κB-independent functions of the IKKs in disease progression. Characterisation of NF-κB-independent functions is also vital in order to anticipate and explain the potential side effects of targeting IKK kinase activity.
7.4. Targeting IκBα Degradation
The ubiquitination and proteasomal degradation of IκB proteins represents an essential step in the activation of NF-κB pathways. Inhibitors of the ubiquitin-proteasome system (UPS), therefore, inhibit NF-κB activity by stabilising IκB proteins. A prominent example is the first-generation proteasome inhibitor, bortezomib, which has achieved significant clinical successes, particularly in relapsed and/or refractory MM and MCL [241,242]. However, despite these successes, therapeutic proteasome inhibitors have several limitations, including dose-limiting toxicity, the rapid onset of secondary drug resistance and broad cellular activities, which often lead to unpredictable outcomes. It is also unclear to what extent the clinical response to proteasome inhibitors in lymphomas results from the inhibition of NF-κB signalling [243]. Proteasome inhibitors will impinge on a legion of critical cell regulatory pathways, so the notion that they can be used as a strategy to selectively target NF-κB pathways is, at best, fanciful. Indeed, malignant plasma cells are thought to be more sensitive to proteasome inhibition than non-malignant cells because their increased rate of immunoglobulin protein production leads to enhanced cellular accumulation of undegraded, polyubiquitinated proteins in the presence of proteasome inhibitors, which activates the unfolded protein response (UPR), thereby accelerating tumour-cell apoptosis [244].
7.5. Targeting NF-κB Activity Directly
The activity of the NF-κB transcription factors themselves could theoretically be inhibited by several different mechanisms including blockade of nuclear translocation, dimerization and DNA binding, and inhibition of protein interactions with other essential cofactors. The reality is that drug development success in this area has been limited, mostly due to the generalised difficulty of targeting nuclear transport and of developing inhibitors of protein-protein interactions. Peptidomimetics, such as SN-50, which blocks nuclear import of p50-containing NF-κB dimers, have been widely used as research tools, but are currently unsuitable for clinical development due to their vast non-specific effects [245]. However, a collection of recent studies has reinvigorated interest in directly targeting NF-κB subunits. Conditional deletion of p65 and c-Rel in developing and mature Treg cells demonstrated their unique but partially overlapping roles in Treg cell development [246]. Subsequently, targeting c-Rel with an FDA-approved xanthine derivative, pentoxifylline (PTXF; [247]), improved checkpoint-targeting immunotherapy protocols (e.g., anti-PD1 therapy) to enhance anti-tumour immunity in a melanoma model [248]. In contrast to IKKβ inhibition [198], minimal adverse effects were observed. Because the critical biological function of c-Rel appears to be mainly restricted to the adaptive immune system, it may represent a more suitable target than IKKβ/p65 to enhance checkpoint-targeting immunotherapies [249]. In addition, IKKβ deletion in mouse bone-marrow derived hematopoietic cells results in significant defects in haematopoiesis not seen upon p65 deletion, suggesting NF-κB-independent pathways may mediate some of the hematopoietic defects associated with IKKβ deletion/inhibition [250]. This provides further rationale for targeting NF-κB subunits directly.
7.6. Targeting Downstream Effectors of NF-κB-Dependent Pathogenesis
It is the aberrant NF-κB-dependent regulation of specific gene expression profiles that primarily contributes to disease progression. These transcriptional programmes are often highly stimulus and tissue specific. Therefore, an attractive alternative to IKKβ inhibition is the targeting of non-redundant, disease-specific downstream mediators of pathogenic NF-κB activity. In theory, this approach should identify safer, context-specific therapeutics that preserve the multiple critical physiological functions of NF-κB. A few studies have demonstrated the exciting potential of this strategy. For example, TIMP-1 is a key downstream modulator of NF-κB-dependent tumour growth in mouse KRAS-driven lung cancer models and represents a potentially safer therapeutic target than IKKβ [179]. Another approach is to target subsets of NF-κB-controlled genes based on their dependence on specific regulatory mechanisms that are not involved in the activation of other genes. For example, the selective blockade of pro-inflammatory NF-κB-dependent transcriptional responses using inhibitors of the epigenetic modifiers bromodomain and extra-terminal (BET) proteins, such as JQ1, exhibited synergistic effects alongside JAK/STAT inhibition in myeloproliferative neoplasms [251,252].
8. Conclusions
As our appreciation of the fundamental role of dysregulated NF-κB signalling in the pathogenesis of inflammatory disease and cancer continues to grow so the resolve of the pharmaceutical industry to pharmacologically target NF-κB pathway components strengthens. Only a small cross-section of recent developments in the therapeutic application of IKKβ inhibitors have been discussed in this review. The number of conditions where IKKβ inhibitors are being pursued as a treatment option was too great to cover here but includes: obesity-associated metabolic disease [253], atherosclerosis [254], multiple sclerosis [255], COPD [256], muscular dystrophy [257], Parkinson’s disease [258], inflammatory bowel disease [259] and chronic arthritis [260]. However, as has often been the case in studies of the role of IKKβ in cancer progression, there have been conflicting reports of the importance of IKKβ activity in the progression of other inflammatory disorders. In the context of atherosclerosis, for example, aberrant vascular endothelial cell-specific canonical NF-κB pathway activation has been shown, quite convincingly, to promote monocyte recruitment and atherosclerotic plaque formation [261,262]. Indeed, endothelial cell-specific NEMO ablation or expression of dominant-negative IκBα protects mice from atherosclerosis [263]. The role of myeloid-specific IKKβ activity in atherosclerosis is less clear, however, as myeloid-specific IKKβ deletion has been shown to both attenuate [264] and enhance [265] atherosclerosis severity in LDL receptor-deficient mice in separate studies. These discrepancies again raise questions about whether a favourable clinical outcome could be achieved following systemic IKKβ inhibition in this disease context.
Despite promising pre-clinical results for IKKβ inhibitors in the aforementioned disease settings, there is little optimism that IKKβ inhibitors will soon broadly enter the clinic. IKKβ is ubiquitously expressed and carries out many critical physiological roles. Furthermore, the complex context- and tissue-specific functions of IKK/NF-κB signalling have made it difficult to predict the net effect and outcome of systemic intervention, and safety concerns currently appear too great a barrier to overcome. As discussed, the most compelling rationale for clinical use of IKKβ inhibitors appears to be in combination therapies and/or as part of a targeted delivery approach, particularly in cancers exhibiting clear addiction to constitutive NF-κB signalling.
If the safety concerns associated with IKKβ inhibitors are to be overcome and they are to offer lasting health benefits as a cancer therapy, research into intrinsic and acquired resistance mechanisms will also be necessary. Resistant tumours eventually develop in a mouse model of KRAS-driven, p53 mutant NSCLC treated with IKKβ inhibitors after a prolonged period of tumour-free survival [266], but no mechanisms of resistance to IKK/NF-κB inhibitors have been defined to-date. Furthermore, further work is needed to develop IKKβ inhibitors with the combination of properties necessary for clinical success, including high selectivity, nanomolar potency, a transient/reversible mode-of-action (since long-term inhibition is unfavourable), desirable pharmacokinetics/pharmacodynamics and amenability to targeted delivery.
Intensive research within the NF-κB signalling field has uncovered the complexity governing cell-type and stimulus-specific NF-κB transcriptional programs, including IKK-isoform and NF-κB-subunit specific functions. This complexity acts both as a barrier and an opportunity to develop highly selective therapies that overcome the issues inherent with systemic targeting of IKKβ.
Funding
This research received no external funding. The APC was funded by MDPI.
Acknowledgments
We would like to thank members of the Cook lab and Paul Smith and Christine Chresta (AstraZeneca) for many useful discussions. Work in the Cook lab relevant to this review was supported by a BBSRC CASE PhD Studentship CW249523 with AstraZeneca (JP) and Institute Strategic Programme Grants BB/J004456/1 and BB/P013384/1 from BBSRC (S.J.C.).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript
| ABC DLBCL | Activated B cell-like Diffuse large B-cell lymphoma |
| BCR | B-cell receptor |
| BET | Bromodomain and extra-terminal |
| BTK | Burton tyrosine kinase |
| β-TrCP | Beta-transducing repeat-containing protein |
| CAC | Colitis-associated carcinoma |
| CAF | Cancer-associated fibroblast |
| CLL | Chronic lymphomatic leukemia |
| CNV | Choroid neovascularization |
| COPD | Chronic obstructive pulmonary disease |
| CSN5 | COP9 signalosome 5 |
| CTL | Cytotoxic T-lymphocyte |
| DC | Dendritic cell |
| DEN | Diethylnitrosoamine |
| DMF | Dimethyl fumarate |
| DSB | Double-strand break |
| EGFR | Epidermal growth factor receptor |
| HCC | Hepatocellular carcinoma |
| HNSCC | Head and neck squamous cell carcinoma |
| ICD | Immunogenic cell death |
| IKK | IκB kinase |
| IL-1 | Interleukin-1 |
| iNUB | Inhibitor of NEMO-Ubiquitin binding |
| IκB | Inhibitor of kappa B |
| KD | Kinase domain |
| KRAS | Kirsten rat sarcoma viral oncogene homolog |
| LPS | Lipopolysaccharide |
| LUBAC | Linear ubiquitin chain assembly complex |
| MALT | Mucosa-associated lymphoid tissue |
| MCL | Mantle cell lymphoma |
| MM | Multiple myeloma |
| mTORC | Mammalian target of rapamycin complex |
| NBD | NEMO-binding domain |
| NF-κB | nuclear factor- “kappa-light-chain-enhancer” of activated B-cells |
| NSCLC | Non-small-cell lung carcinoma |
| OV | Oncolytic virus |
| PD/PDL | Programmed death/PD-ligand 1 |
| POC | Proof-of-concept |
| RA | Rheumatoid arthritis |
| RHD | Rel homology domain |
| SCF | S phase kinase-associated protein 1 (SKP1)-cullin 1-F-box protein |
| SCID | Severe combined immunodeficient |
| SDD | Scaffold/dimerization domain |
| SH2 | Src Homology 2 |
| TAM | Tumour-associated macrophage |
| TCR | T-cell receptor |
| TLR | Toll-like receptor |
| TME | Tumour microenvironment |
| TNFα | Tumor necrosis factor-alpha |
| Treg | Regulatory T cells |
| UBC | Ubiquitin-conjugating enzyme |
| ULD | Ubiquitin-like domain |
References
- Napetschnig, J.; Wu, H. Molecular basis of NK-kappaB signaling. Annu. Rev. Biophys. 2013, 42, 443–468. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. Non-canonical NK-kappaB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Integrating cell-signalling pathways with NK-kappaB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Xia, Y.; Parker, A.S.; Verma, I.M. IKK biology. Immunol. Rev. 2012, 246, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Misquitta, Y.R.; Olland, A.; Johnson, M.A.; Kelleher, K.S.; Kriz, R.; Lin, L.L.; Stahl, M.; Mosyak, L. Crystal structure of a human IkappaB kinase beta asymmetric dimer. J. Biol. Chem. 2013, 288, 22758–22767. [Google Scholar] [CrossRef] [PubMed]
- Polley, S.; Huang, D.B.; Hauenstein, A.V.; Fusco, A.J.; Zhong, X.; Vu, D.; Schrofelbauer, B.; Kim, Y.; Hoffmann, A.; Verma, I.M.; et al. A structural basis for IkappaB kinase 2 activation via oligomerization-dependent trans auto-phosphorylation. PLoS Biol. 2013, 11, e1001581. [Google Scholar] [CrossRef] [PubMed]
- Polley, S.; Passos, D.O.; Huang, D.B.; Mulero, M.C.; Mazumder, A.; Biswas, T.; Verma, I.M.; Lyumkis, D.; Ghosh, G. Structural basis for the activation of IKK1/alpha. Cell Rep. 2016, 17, 1907–1914. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, F.; Zhu, H.; Murray, B.W.; Shevchenko, A.; Bennett, B.L.; Li, J.; Young, D.B.; Barbosa, M.; Mann, M.; Manning, A.; et al. IKK-1 and IKK-2: Cytokine-activated IkappaB kinases essential for NK-kappaB activation. Science 1997, 278, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Delhase, M.; Hayakawa, M.; Chen, Y.; Karin, M. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science 1999, 284, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Scholefield, J.; Henriques, R.; Savulescu, A.F.; Fontan, E.; Boucharlat, A.; Laplantine, E.; Smahi, A.; Israël, A.; Agou, F.; Mhlanga, M.M. Super-resolution microscopy reveals a preformed nemo lattice structure that is collapsed in incontinentia pigmenti. Nat. Commun. 2016, 7, 12629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Clark, K.; Lawrence, T.; Peggie, M.W.; Cohen, P. An unexpected twist to the activation of IKKβ: TAK1 primes IKKβ for activation by autophosphorylation. Biochem. J. 2014, 461, 531–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henkel, T.; Machleidt, T.; Alkalay, I.; Kronke, M.; Ben-Neriah, Y.; Baeuerle, P.A. Rapid proteolysis of I kappa B-alpha is necessary for activation of transcription factor NK-kappa B. Nature 1993, 365, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Gerstberger, S.; Carlson, L.; Franzoso, G.; Siebenlist, U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science 1995, 267, 1485–1488. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hagler, J.; Palombella, V.J.; Melandri, F.; Scherer, D.; Ballard, D.; Maniatis, T. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 1995, 9, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Baldi, L.; Brown, K.; Franzoso, G.; Siebenlist, U. Critical role for lysines 21 and 22 in signal-induced, ubiquitin-mediated proteolysis of ib. J. Biol. Chem. 1996, 271, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Winston, J.T.; Strack, P.; Beer-Romero, P.; Chu, C.Y.; Elledge, S.J.; Harper, J.W. The SCFβ-TRCP–ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IκBα and β-catenin and stimulates IκBα ubiquitination in vitro. Genes Dev. 1999, 13, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-kappaB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene 2006, 25, 6717–6730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christian, F.; Smith, E.L.; Carmody, R.J. The regulation of NK-kappaB subunits by phosphorylation. Cells 2016, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NK-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, M.M.; Sung, B.; Yadav, V.R.; Kannappan, R.; Aggarwal, B.B. NF-kappaB addiction and its role in cancer: ‘One size does not fit all’. Oncogene 2011, 30, 1615–1630. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-kappaB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-kappaB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Chariot, A. The NK-kappaB-independent functions of IKK subunits in immunity and cancer. Trends Cell Biol. 2009, 19, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Lee, D.F.; Xia, W.; Golfman, L.S.; Ou-Yang, F.; Yang, J.Y.; Zou, Y.; Bao, S.; Hanada, N.; Saso, H.; et al. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell 2004, 117, 225–237. [Google Scholar] [CrossRef]
- Lee, D.F.; Kuo, H.P.; Chen, C.T.; Hsu, J.M.; Chou, C.K.; Wei, Y.; Sun, H.L.; Li, L.Y.; Ping, B.; Huang, W.C.; et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mtor pathway. Cell 2007, 130, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.W.; Chu, W.; Hu, Y.; Delhase, M.; Deerinck, T.; Ellisman, M.; Johnson, R.; Karin, M. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J. Exp. Med. 1999, 189, 1839–1845. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Van Antwerp, D.; Mercurio, F.; Lee, K.F.; Verma, I.M. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science 1999, 284, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y. Abnormal morphogenesis but intact IKK activation in mice lacking the IKK subunit of ib kinase. Science 1999, 284, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Senftleben, U.; Li, Z.-W.; Baud, V.; Karin, M. IKKβ is essential for protecting T cells from TNFα-induced apoptosis. Immunity 2001, 14, 217–230. [Google Scholar] [CrossRef]
- Kaisho, T.; Takeda, K.; Tsujimura, T.; Kawai, T.; Nomura, F.; Terada, N.; Akira, S. IkappaB kinase alpha is essential for mature B cell development and function. J. Exp. Med. 2001, 193, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Dejardin, E.; Droin, N.M.; Delhase, M.; Haas, E.; Cao, Y.; Makris, C.; Li, Z.W.; Karin, M.; Ware, C.F.; Green, D.R. The lymphotoxin-beta receptor induces different patterns of gene expression via two NK-kappaB pathways. Immunity 2002, 17, 525–535. [Google Scholar] [CrossRef]
- Bonizzi, G.; Bebien, M.; Otero, D.C.; Johnson-Vroom, K.E.; Cao, Y.; Vu, D.; Jegga, A.G.; Aronow, B.J.; Ghosh, G.; Rickert, R.C.; et al. Activation of IKKalpha target genes depends on recognition of specific kappaB binding sites by RelB:P52 dimers. EMBO J. 2004, 23, 4202–4210. [Google Scholar] [CrossRef] [PubMed]
- Anthony, N.G.; Baiget, J.; Berretta, G.; Boyd, M.; Breen, D.; Edwards, J.; Gamble, C.; Gray, A.I.; Harvey, A.L.; Hatziieremia, S.; et al. Inhibitory kappa B kinase alpha (IKKalpha) inhibitors that recapitulate their selectivity in cells against isoform-related biomarkers. J. Med. Chem. 2017, 60, 7043–7066. [Google Scholar] [CrossRef] [PubMed]
- Kobori, M.; Yang, Z.; Gong, D.; Heissmeyer, V.; Zhu, H.; Jung, Y.K.; Gakidis, M.A.; Rao, A.; Sekine, T.; Ikegami, F.; et al. Wedelolactone suppresses LPS-induced caspase-11 expression by directly inhibiting the IKK complex. Cell Death Differ. 2004, 11, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Nam, N.H. Naturally occurring NK-kappaB inhibitors. Mini Rev. Med. Chem. 2006, 6, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Llona-Minguez, S.; Baiget, J.; Mackay, S.P. Small-molecule inhibitors of IkappaB kinase (IKK) and IKK-related kinases. Pharm. Pat. Anal. 2013, 2, 481–498. [Google Scholar] [CrossRef] [PubMed]
- Arepalli, S.K.; Choi, M.; Jung, J.K.; Lee, H. Novel NK-kappaB inhibitors: A patent review (2011–2014). Expert Opin. Ther. Pat. 2015, 25, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Peggie, M.; Plater, L.; Sorcek, R.J.; Young, E.R.; Madwed, J.B.; Hough, J.; McIver, E.G.; Cohen, P. Novel cross-talk within the IKK family controls innate immunity. Biochem. J. 2011, 434, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagashima, K.; Sasseville, V.G.; Wen, D.; Bielecki, A.; Yang, H.; Simpson, C.; Grant, E.; Hepperle, M.; Harriman, G.; Jaffee, B.; et al. Rapid TNFR1-dependent lymphocyte depletion in vivo with a selective chemical inhibitor of IKKbeta. Blood 2006, 107, 4266–4273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hideshima, T.; Neri, P.; Tassone, P.; Yasui, H.; Ishitsuka, K.; Raje, N.; Chauhan, D.; Podar, K.; Mitsiades, C.; Dang, L.; et al. MLN120B, a novel IkappaB kinase beta inhibitor, blocks multiple myeloma cell growth in vitro and in vivo. Clin. Cancer Res. 2006, 12, 5887–5894. [Google Scholar] [CrossRef] [PubMed]
- Schopf, L.; Savinainen, A.; Anderson, K.; Kujawa, J.; DuPont, M.; Silva, M.; Siebert, E.; Chandra, S.; Morgan, J.; Gangurde, P.; et al. IKKbeta inhibition protects against bone and cartilage destruction in a rat model of rheumatoid arthritis. Arthritis Rheum. 2006, 54, 3163–3173. [Google Scholar] [CrossRef] [PubMed]
- Mbalaviele, G.; Sommers, C.D.; Bonar, S.L.; Mathialagan, S.; Schindler, J.F.; Guzova, J.A.; Shaffer, A.F.; Melton, M.A.; Christine, L.J.; Tripp, C.S.; et al. A novel, highly selective, tight binding IkappaB kinase-2 (IKK-2) inhibitor: A tool to correlate IKK-2 activity to the fate and functions of the components of the nuclear factor-kappaB pathway in arthritis-relevant cells and animal models. J. Pharmacol. Exp. Ther. 2009, 329, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Chiang, P.C.; Kishore, N.N.; Thompson, D.C. Combined use of pharmacokinetic modeling and a steady-state delivery approach allows early assessment of IkappaB kinase-2 (IKK-2) target safety and efficacy. J. Pharm. Sci. 2010, 99, 1278–1287. [Google Scholar] [CrossRef] [PubMed]
- Sommers, C.D.; Thompson, J.M.; Guzova, J.A.; Bonar, S.L.; Rader, R.K.; Mathialagan, S.; Venkatraman, N.; Holway, V.W.; Kahn, L.E.; Hu, G.; et al. Novel tight-binding inhibitory factor-kappaB kinase (IKK-2) inhibitors demonstrate target-specific anti-inflammatory activities in cellular assays and following oral and local delivery in an in vivo model of airway inflammation. J. Pharmacol. Exp. Ther. 2009, 330, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Rajendrasozhan, S.; Hwang, J.W.; Yao, H.; Kishore, N.; Rahman, I. Anti-inflammatory effect of a selective IkappaB kinase-beta inhibitor in rat lung in response to LPS and cigarette smoke. Pulm. Pharmacol. Ther. 2010, 23, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Podolin, P.L.; Callahan, J.F.; Bolognese, B.J.; Li, Y.H.; Carlson, K.; Davis, T.G.; Mellor, G.W.; Evans, C.; Roshak, A.K. Attenuation of murine collagen-induced arthritis by a novel, potent, selective small molecule inhibitor of IkappaB kinase 2, TPCA-1 (2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide), occurs via reduction of proinflammatory cytokines and antigen-induced T cell proliferation. J. Pharmacol. Exp. Ther. 2005, 312, 373–381. [Google Scholar] [PubMed]
- Sachse, F.; Becker, K.; Basel, T.J.; Weiss, D.; Rudack, C. IKK-2 inhibitor TPCA-1 represses nasal epithelial inflammation in vitro. Rhinology 2011, 49, 168–173. [Google Scholar] [PubMed]
- Du, Z.; Whitt, M.A.; Baumann, J.; Garner, J.M.; Morton, C.L.; Davidoff, A.M.; Pfeffer, L.M. Inhibition of type I interferon-mediated antiviral action in human glioma cells by the IKK inhibitors BMS-345541 and TPCA-1. J. Interferon Cytokine Res. 2012, 32, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Nan, J.; Du, Y.; Chen, X.; Bai, Q.; Wang, Y.; Zhang, X.; Zhu, N.; Zhang, J.; Hou, J.; Wang, Q.; et al. TPCA-1 is a direct dual inhibitor of STAT3 and NK-kappaB and regresses mutant EGFR-associated human non-small cell lung cancers. Mol. Cancer Ther. 2014, 13, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Birrell, M.A.; Wong, S.; Hardaker, E.L.; Catley, M.C.; McCluskie, K.; Collins, M.; Haj-Yahia, S.; Belvisi, M.G. IkappaB kinase-2-independent and -dependent inflammation in airway disease models: Relevance of IKK-2 inhibition to the clinic. Mol. Pharmacol. 2006, 69, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Lu, Q.; Gaddipati, S.; Kasetti, R.B.; Wang, W.; Pasparakis, M.; Kaplan, H.J.; Li, Q. IKK2 inhibition attenuates laser-induced choroidal neovascularization. PLoS ONE 2014, 9, e87530. [Google Scholar] [CrossRef] [PubMed]
- Kishore, N.; Sommers, C.; Mathialagan, S.; Guzova, J.; Yao, M.; Hauser, S.; Huynh, K.; Bonar, S.; Mielke, C.; Albee, L.; et al. A selective IKK-2 inhibitor blocks NK-kappa B-dependent gene expression in interleukin-1 beta-stimulated synovial fibroblasts. J. Biol. Chem. 2003, 278, 32861–32871. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.; Shi, Z.; Liu, Y.; Stack, M.S. Inhibitors of NK-kappaB reverse cellular invasion and target gene upregulation in an experimental model of aggressive oral squamous cell carcinoma. Oral Oncol. 2014, 50, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wu, H.; Chim, S.M.; Zhou, L.; Zhao, J.; Feng, H.; Wei, Q.; Wang, Q.; Zheng, M.H.; Tan, R.X.; et al. SC-514, a selective inhibitor of IKKbeta attenuates rankl-induced osteoclastogenesis and NK-kappaB activation. Biochem. Pharmacol. 2013, 86, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Negi, G.; Sharma, S.S. Inhibition of IkappaB kinase (IKK) protects against peripheral nerve dysfunction of experimental diabetes. Mol. Neurobiol. 2015, 51, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Lipstein, M.; Rodriguez, R.; Serrano, X.O.; McIntosh, C.; Tsai, W.Y.; Wasmuth, A.S.; Jaken, S.; O’Connor, O.A. The novel IKK2 inhibitor ly2409881 potently synergizes with histone deacetylase inhibitors in preclinical models of lymphoma through the downregulation of NK-kappaB. Clin. Cancer Res. 2015, 21, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Castro, A.C.; Dang, L.C.; Soucy, F.; Grenier, L.; Mazdiyasni, H.; Hottelet, M.; Parent, L.; Pien, C.; Palombella, V.; Adams, J. Novel IKK inhibitors: Beta-carbolines. Bioorg. Med. Chem. Lett. 2003, 13, 2419–2422. [Google Scholar] [CrossRef]
- Vodanovic-Jankovic, S.; Hari, P.; Jacobs, P.; Komorowski, R.; Drobyski, W.R. NF-kappaB as a target for the prevention of graft-versus-host disease: Comparative efficacy of bortezomib and PS-1145. Blood 2006, 107, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Chauhan, D.; Richardson, P.; Mitsiades, C.; Mitsiades, N.; Hayashi, T.; Munshi, N.; Dang, L.; Castro, A.; Palombella, V.; et al. NK-kappa B as a therapeutic target in multiple myeloma. J. Biol. Chem. 2002, 277, 16639–16647. [Google Scholar] [CrossRef] [PubMed]
- Lam, L.T.; Davis, R.E.; Pierce, J.; Hepperle, M.; Xu, Y.; Hottelet, M.; Nong, Y.; Wen, D.; Adams, J.; Dang, L.; et al. Small molecule inhibitors of iκb kinase are selectively toxic for subgroups of diffuse large B-cell lymphoma defined by gene expression profiling. Clin. Cancer Res. 2005, 11, 28–40. [Google Scholar] [PubMed]
- Choi, S.I.; Lee, S.Y.; Jung, W.J.; Lee, S.H.; Lee, E.J.; Min, K.H.; Hur, G.Y.; Lee, S.H.; Lee, S.Y.; Kim, J.H.; et al. The effect of an IkappaB-kinase-beta (IKKbeta) inhibitor on tobacco smoke-induced pulmonary inflammation. Exp. Lung Res. 2016, 42, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Ziegelbauer, K.; Gantner, F.; Lukacs, N.W.; Berlin, A.; Fuchikami, K.; Niki, T.; Sakai, K.; Inbe, H.; Takeshita, K.; Ishimori, M.; et al. A selective novel low-molecular-weight inhibitor of IkappaB kinase-beta (IKK-beta) prevents pulmonary inflammation and shows broad anti-inflammatory activity. Br. J. Pharmacol. 2005, 145, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Bassères, D.S.; Ebbs, A.; Cogswell, P.C.; Baldwin, A.S. IKK is a therapeutic target in kras-induced lung cancer with disrupted p53 activity. Genes Cancer 2014, 5, 41–55. [Google Scholar] [PubMed]
- Moss, N.C.; Stansfield, W.E.; Willis, M.S.; Tang, R.H.; Selzman, C.H. IKKbeta inhibition attenuates myocardial injury and dysfunction following acute ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2248–H2253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Qian, L.; Flood, P.M.; Shi, J.S.; Hong, J.S.; Gao, H.M. Inhibition of IkappaB kinase-beta protects dopamine neurons against lipopolysaccharide-induced neurotoxicity. J. Pharmacol. Exp. Ther. 2010, 333, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, S.B.; Carlson, C.B.; Riddle, S.M.; Zhao, J.; Vogel, K.W.; Nichols, R.J.; Bi, K. Screening for novel LRRK2 inhibitors using a high-throughput tr-fret cellular assay for LRRK2 SER935 phosphorylation. PLoS ONE 2012, 7, e43580. [Google Scholar] [CrossRef] [PubMed]
- Waelchli, R.; Bollbuck, B.; Bruns, C.; Buhl, T.; Eder, J.; Feifel, R.; Hersperger, R.; Janser, P.; Revesz, L.; Zerwes, H.G.; et al. Design and preparation of 2-benzamido-pyrimidines as inhibitors of IKK. Bioorg. Med. Chem. Lett. 2006, 16, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Sordi, R.; Chiazza, F.; Johnson, F.L.; Patel, N.S.; Brohi, K.; Collino, M.; Thiemermann, C. Inhibition of IkappaB kinase attenuates the organ injury and dysfunction associated with hemorrhagic shock. Mol. Med. 2015, 21, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Coldewey, S.M.; Rogazzo, M.; Collino, M.; Patel, N.S.; Thiemermann, C. Inhibition of IkappaB kinase reduces the multiple organ dysfunction caused by sepsis in the mouse. Dis. Models Mech. 2013, 6, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.S.; Tao, W.; Miao, Q.B.; Zhu, Y.B.; Yang, Y.F. Improvement of ventilation-induced lung injury in a rodent model by inhibition of inhibitory kappaB kinase. J. Trauma Acute Care Surg. 2014, 76, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Johnson, F.L.; Patel, N.S.A.; Purvis, G.S.D.; Chiazza, F.; Chen, J.; Sordi, R.; Hache, G.; Merezhko, V.V.; Collino, M.; Yaqoob, M.M.; et al. Inhibition of IkappaB kinase at 24 hours after acute kidney injury improves recovery of renal function and attenuates fibrosis. J. Am. Heart Assoc. 2017, 6, e005092. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Konno, M.; Muto, S.; Kambe, N.; Morii, E.; Nakahata, T.; Itai, A.; Matsuda, H. A novel NK-kappaB inhibitor, IMD-0354, suppresses neoplastic proliferation of human mast cells with constitutively activated c-kit receptors. Blood 2005, 105, 2324–2331. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Muto, S.; Konno, M.; Itai, A.; Matsuda, H. A new IkappaB kinase beta inhibitor prevents human breast cancer progression through negative regulation of cell cycle transition. Cancer Res. 2006, 66, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Kanduri, M.; Tobin, G.; Aleskog, A.; Nilsson, K.; Rosenquist, R. The novel NK-kappaB inhibitor IMD-0354 induces apoptosis in chronic lymphocytic leukemia. Blood Cancer J. 2011, 1, e12. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, T.; Saito, Y.; Saitoh, T.; Dewan, M.Z.; Shioya, A.; Kobayashi, M.; Kawachi, H.; Muto, S.; Itai, A.; Uota, S.; et al. Inhibition of IkappaB kinase beta restrains oncogenic proliferation of pancreatic cancer cells. J. Med. Dent. Sci. 2008, 55, 49–59. [Google Scholar] [PubMed]
- Uota, S.; Zahidunnabi Dewan, M.; Saitoh, Y.; Muto, S.; Itai, A.; Utsunomiya, A.; Watanabe, T.; Yamamoto, N.; Yamaoka, S. An IkappaB kinase 2 inhibitor IMD-0354 suppresses the survival of adult T-cell leukemia cells. Cancer Sci. 2012, 103, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Shimada, M.; Sakakibara, S.; Yoshino, T.; Masuda, T.; Shintani, T.; Sato, H.; Koriyama, Y.; Fukushima, K.; Nunami, N.; et al. Synthesis and structure-activity relationships of novel IKK-beta inhibitors. Part 3: Orally active anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2004, 14, 4019–4022. [Google Scholar] [CrossRef] [PubMed]
- Sanda, T.; Iida, S.; Ogura, H.; Asamitsu, K.; Murata, T.; Bacon, K.B.; Ueda, R.; Okamoto, T. Growth inhibition of multiple myeloma cells by a novel IkappaB kinase inhibitor. Clin. Cancer Res. 2005, 11, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Sanda, T.; Asamitsu, K.; Ogura, H.; Iida, S.; Utsunomiya, A.; Ueda, R.; Okamoto, T. Induction of cell death in adult T-cell leukemia cells by a novel IkappaB kinase inhibitor. Leukemia 2006, 20, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Victoriano, A.F.; Asamitsu, K.; Hibi, Y.; Imai, K.; Barzaga, N.G.; Okamoto, T. Inhibition of human immunodeficiency virus type 1 replication in latently infected cells by a novel IkappaB kinase inhibitor. Antimicrob. Agents Chemother. 2006, 50, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.R.; Pattoli, M.A.; Gregor, K.R.; Brassil, P.J.; MacMaster, J.F.; McIntyre, K.W.; Yang, X.; Iotzova, V.S.; Clarke, W.; Strnad, J.; et al. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NK-kappa B-dependent transcription in mice. J. Biol. Chem. 2003, 278, 1450–1456. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, K.W.; Shuster, D.J.; Gillooly, K.M.; Dambach, D.M.; Pattoli, M.A.; Lu, P.; Zhou, X.D.; Qiu, Y.; Zusi, F.C.; Burke, J.R. A highly selective inhibitor of I kappa B kinase, BMS-345541, blocks both joint inflammation and destruction in collagen-induced arthritis in mice. Arthritis Rheum. 2003, 48, 2652–2659. [Google Scholar] [CrossRef] [PubMed]
- MacMaster, J.F.; Dambach, D.M.; Lee, D.B.; Berry, K.K.; Qiu, Y.; Zusi, F.C.; Burke, J.R. An inhibitor of IkappaB kinase, BMS-345541, blocks endothelial cell adhesion molecule expression and reduces the severity of dextran sulfate sodium-induced colitis in mice. Inflamm. Res. 2003, 52, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Townsend, R.M.; Postelnek, J.; Susulic, V.; McIntyre, K.W.; Shuster, D.J.; Qiu, Y.; Zusi, F.C.; Burke, J.R. A highly selective inhibitor of IkappaB kinase, BMS-345541, augments graft survival mediated by suboptimal immunosuppression in a murine model of cardiac graft rejection. Transplantation 2004, 77, 1090–1094. [Google Scholar] [CrossRef] [PubMed]
- Buontempo, F.; Chiarini, F.; Bressanin, D.; Tabellini, G.; Melchionda, F.; Pession, A.; Fini, M.; Neri, L.M.; McCubrey, J.A.; Martelli, A.M. Activity of the selective IkappaB kinase inhibitor BMS-345541 against T-cell acute lymphoblastic leukemia: Involvement of FOXO3a. Cell Cycle 2012, 11, 2467–2475. [Google Scholar] [CrossRef] [PubMed]
- Ping, H.; Yang, F.; Wang, M.; Niu, Y.; Xing, N. IKK inhibitor suppresses epithelial-mesenchymal transition and induces cell death in prostate cancer. Oncol. Rep. 2016, 36, 1658–1664. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.S.; Oberley, C.; Hooper, C.P.; Grindle, K.; Wuerzberger-Davis, S.; Wolff, J.; McCool, K.; Rui, L.; Miyamoto, S. Withaferin a disrupts ubiquitin-based nemo reorganization induced by canonical NK-kappaB signaling. Exp. Cell Res. 2015, 331, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.C.; Choi, B.Y. Withaferin-A—A natural anticancer agent with pleitropic mechanisms of action. Int. J. Mol. Sci. 2016, 17, 290. [Google Scholar] [CrossRef] [PubMed]
- Kaileh, M.; Vanden Berghe, W.; Heyerick, A.; Horion, J.; Piette, J.; Libert, C.; De Keukeleire, D.; Essawi, T.; Haegeman, G. Withaferin a strongly elicits IkappaB kinase beta hyperphosphorylation concomitant with potent inhibition of its kinase activity. J. Biol. Chem. 2007, 282, 4253–4264. [Google Scholar] [CrossRef] [PubMed]
- Heyninck, K.; Lahtela-Kakkonen, M.; Van der Veken, P.; Haegeman, G.; Vanden Berghe, W. Withaferin a inhibits NK-kappaB activation by targeting cysteine 179 in IKKbeta. Biochem. Pharmacol. 2014, 91, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, W.; Sabbe, L.; Kaileh, M.; Haegeman, G.; Heyninck, K. Molecular insight in the multifunctional activities of withaferin a. Biochem. Pharmacol. 2012, 84, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Roh, E.; Lee, H.Y.; Lee, I.J.; Ahn, B.; Jung, S.H.; Lee, H.; Han, S.B.; Kim, Y. Benzoxathiole derivative blocks lipopolysaccharide-induced nuclear factor-kappaB activation and nuclear factor-kappaB-regulated gene transcription through inactivating inhibitory kappaB kinase beta. Mol. Pharmacol. 2008, 73, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.; Li, C.; Wang, X.; Dian, L.; Zhang, X.; Li, L.; Chen, S.; Cao, R.; Li, L.; Huang, N.; et al. Ainsliadimer a selectively inhibits IKKα/β by covalently binding a conserved cysteine. Nat. Commun. 2015, 6, 6522. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.A.; Collins, I. Measuring and interpreting the selectivity of protein kinase inhibitors. J. Chem. Biol. 2009, 2, 131–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, J.W.; Schoenleber, R.; Jesmok, G.; Best, J.; Moore, S.A.; Collins, T.; Gerritsen, M.E. Novel inhibitors of cytokine-induced IκBα phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effectsin vivo. J.Biol. Chem. 1997, 272, 21096–21103. [Google Scholar] [CrossRef] [PubMed]
- Strickson, S.; Campbell, D.G.; Emmerich, C.H.; Knebel, A.; Plater, L.; Ritorto, M.S.; Shpiro, N.; Cohen, P. The anti-inflammatory drug bay 11-7082 suppresses the myd88-dependent signalling network by targeting the ubiquitin system. Biochem. J. 2013, 451, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gray, N.S. Snapshot: Kinase inhibitors II. Mol. Cell 2015, 58, 710. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K.; Sung, B.; Kunnumakkara, A.B.; Sethi, G.; Chaturvedi, M.M.; Aggarwal, B.B. Berberine modifies cysteine 179 of IkappaBalpha kinase, suppresses nuclear factor-kappaB-regulated antiapoptotic gene products, and potentiates apoptosis. Cancer Res. 2008, 68, 5370–5379. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Sundaram, C.; Reuter, S.; Aggarwal, B.B. Inhibiting NK-kappaB activation by small molecules as a therapeutic strategy. Biochim. Biophys. Acta 2010, 1799, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Byun, M.S.; Choi, J.; Jue, D.M. Cysteine-179 of IkappaB kinase beta plays a critical role in enzyme activation by promoting phosphorylation of activation loop serines. Exp. Mol. Med. 2006, 38, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Bernier, M.; Kwon, Y.K.; Pandey, S.K.; Zhu, T.N.; Zhao, R.J.; Maciuk, A.; He, H.J.; Decabo, R.; Kole, S. Binding of manumycin a inhibits IkappaB kinase beta activity. J. Biol. Chem. 2006, 281, 2551–2561. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.C.; Bardhan, S.; Li, C.; Pace, E.A.; Porco, J.A., Jr.; Gilmore, T.D. Jesterone dimer, a synthetic derivative of the fungal metabolite jesterone, blocks activation of transcription factor nuclear factor kappaB by inhibiting the inhibitor of kappaB kinase. Mol. Pharmacol. 2003, 64, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Reynaert, N.L.; van der Vliet, A.; Guala, A.S.; McGovern, T.; Hristova, M.; Pantano, C.; Heintz, N.H.; Heim, J.; Ho, Y.S.; Matthews, D.E.; et al. Dynamic redox control of NK-kappaB through glutaredoxin-regulated s-glutathionylation of inhibitory kappaB kinase beta. Proc. Natl. Acad. Sci. USA 2006, 103, 13086–13091. [Google Scholar] [CrossRef] [PubMed]
- May, M.J.; D’Acquisto, F.; Madge, L.A.; Glockner, J.; Pober, J.S.; Ghosh, S. Selective inhibition of NK-kappaB activation by a peptide that blocks the interaction of nemo with the IkappaB kinase complex. Science 2000, 289, 1550–1554. [Google Scholar] [CrossRef] [PubMed]
- Alfonso, L.; Ai, G.; Spitale, R.C.; Bhat, G.J. Molecular targets of aspirin and cancer prevention. Br. J. Cancer 2014, 111, 61. [Google Scholar] [CrossRef] [PubMed]
- Kopp, E.; Ghosh, S. Inhibition of NK-kappa B by sodium salicylate and aspirin. Science 1994, 265, 956–959. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 1998, 396, 77–80. [Google Scholar] [CrossRef] [PubMed]
- McDade, T.P.; Perugini, R.A.; Vittimberga, F.J., Jr.; Carrigan, R.C.; Callery, M.P. Salicylates inhibit NK-kappaB activation and enhance tnf-alpha-induced apoptosis in human pancreatic cancer cells. J. Surg. Res. 1999, 83, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Yin, M.J.; Lin, K.M.; Gaynor, R.B. Sulindac inhibits activation of the NK-kappaB pathway. J. Biol. Chem. 1999, 274, 27307–27314. [Google Scholar] [CrossRef] [PubMed]
- Kinase Profiling Inhibitor Database, MRC Protein Phosphorylation Unit at the University of Dundee. Available online: http://www.kinase-screen.mrc.ac.uk/screening-compounds/349388?order=field_results_inhibition&sort=asc (accessed on 1 August 2018).
- Schwenger, P.; Alpert, D.; Skolnik, E.Y.; Vilcek, J. Activation of p38 mitogen-activated protein kinase by sodium salicylate leads to inhibition of tumor necrosis factor-induced IkappaB alpha phosphorylation and degradation. Mol. Cell. Biol. 1998, 18, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Alpert, D.; Vilček, J. Inhibition of iκb kinase activity by sodium salicylate in vitro does not reflect its inhibitory mechanism in intact cells. J. Biol. Chem. 2000, 275, 10925–10929. [Google Scholar] [CrossRef] [PubMed]
- Stark, L.A.; Din, F.V.; Zwacka, R.M.; Dunlop, M.G. Aspirin-induced activation of the NK-kappaB signaling pathway: A novel mechanism for aspirin-mediated apoptosis in colon cancer cells. FASEB J. 2001, 15, 1273–1275. [Google Scholar] [CrossRef] [PubMed]
- Stark, L.A.; Reid, K.; Sansom, O.J.; Din, F.V.; Guichard, S.; Mayer, I.; Jodrell, D.I.; Clarke, A.R.; Dunlop, M.G. Aspirin activates the NK-kappaB signalling pathway and induces apoptosis in intestinal neoplasia in two in vivo models of human colorectal cancer. Carcinogenesis 2007, 28, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Loveridge, C.J.; MacDonald, A.D.; Thoms, H.C.; Dunlop, M.G.; Stark, L.A. The proapoptotic effects of sulindac, sulindac sulfone and indomethacin are mediated by nucleolar translocation of the RelA(p65) subunit of NK-kappaB. Oncogene 2008, 27, 2648–2655. [Google Scholar] [CrossRef] [PubMed]
- Mladenova, D.; Pangon, L.; Currey, N.; Ng, I.; Musgrove, E.A.; Grey, S.T.; Kohonen-Corish, M.R. Sulindac activates NK-kappaB signaling in colon cancer cells. Cell Commun. Signal. 2013, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.B.; Yang, X.; Clark, R.; Choi, J.; Baek, S.J.; Lee, S.H. A mechanistic study of the proapoptotic effect of tolfenamic acid: Involvement of NK-kappaB activation. Carcinogenesis 2013, 34, 2350–2360. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Stark, L.A. Aspirin prevention of colorectal cancer: Focus on NK-kappaB signalling and the nucleolus. Biomedicines 2017, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Verstrepen, L.; Beyaert, R. Receptor proximal kinases in NK-kappaB signaling as potential therapeutic targets in cancer and inflammation. Biochem. Pharmacol. 2014, 92, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, Y.; Sawada, T.; Nagatani, K.; Komagata, Y.; Inoue, T.; Muto, S.; Itai, A.; Yamamoto, K. Effect of nuclear factor-kappaB inhibition on rheumatoid fibroblast-like synoviocytes and collagen induced arthritis. J. Rheumatol. 2005, 32, 1440–1447. [Google Scholar] [PubMed]
- Onai, Y.; Suzuki, J.; Kakuta, T.; Maejima, Y.; Haraguchi, G.; Fukasawa, H.; Muto, S.; Itai, A.; Isobe, M. Inhibition of IkappaB phosphorylation in cardiomyocytes attenuates myocardial ischemia/reperfusion injury. Cardiovasc. Res. 2004, 63, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Pippione, A.C.; Federico, A.; Ducime, A.; Sainas, S.; Boschi, D.; Barge, A.; Lupino, E.; Piccinini, M.; Kubbutat, M.; Contreras, J.-M.; et al. 4-Hydroxy-n-[3,5-bis(trifluoromethyl)phenyl]-1,2,5-thiadiazole-3-carboxamide: A novel inhibitor of the canonical NF-κB cascade. MedChemComm 2017, 8, 1850–1855. [Google Scholar] [CrossRef] [PubMed]
- Tegeder, I.; Niederberger, E.; Schmidt, R.; Kunz, S.; Guhring, H.; Ritzeler, O.; Michaelis, M.; Geisslinger, G. Specific inhibition of IkappaB kinase reduces hyperalgesia in inflammatory and neuropathic pain models in rats. J. Neurosci. 2004, 24, 1637–1645. [Google Scholar] [CrossRef] [PubMed]
- Grothe, K.; Flechsenhar, K.; Paehler, T.; Ritzeler, O.; Beninga, J.; Saas, J.; Herrmann, M.; Rudolphi, K. IkappaB kinase inhibition as a potential treatment of osteoarthritis—Results of a clinical proof-of-concept study. Osteoarthr. Cartil. 2017, 25, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Palanki, M.S.S.; Gayo-Fung, L.M.; Shevlin, G.I.; Erdman, P.; Sato, M.; Goldman, M.; Ransone, L.J.; Spooner, C. Structure–activity relationship studies of ethyl 2-[(3-methyl-2,5-dioxo(3-pyrrolinyl))amino]-4-(trifluoromethyl)pyrimidine-5-carboxylate: An inhibitor of ap-1 and NF-κB mediated gene expression. Bioorg. Med. Chem. Lett. 2002, 12, 2573–2577. [Google Scholar] [CrossRef]
- Frelin, C.; Imbert, V.; Griessinger, E.; Loubat, A.; Dreano, M.; Peyron, J.F. AS602868, a pharmacological inhibitor of IKK2, reveals the apoptotic potential of tnf-alpha in jurkat leukemic cells. Oncogene 2003, 22, 8187–8194. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liang, H.; Meng, H.; Deng, X.; Zhang, X.; Lai, L. A novel allosteric inhibitor that prevents IKKβ activation. MedChemComm 2018, 9, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger, LLC. The Pymol Molecular Graphics System, Version 1.8; Schrodinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Schrodinger, LLC. The Jymol Molecular Graphics Development Component, Version 1.8; Schrodinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Adli, M.; Merkhofer, E.; Cogswell, P.; Baldwin, A.S. IKKalpha and IKKbeta each function to regulate NK-kappaB activation in the tnf-induced/canonical pathway. PLoS ONE 2010, 5, e9428. [Google Scholar] [CrossRef] [PubMed]
- Nottingham, L.K.; Yan, C.H.; Yang, X.; Si, H.; Coupar, J.; Bian, Y.; Cheng, T.F.; Allen, C.; Arun, P.; Gius, D.; et al. Aberrant IKKalpha and IKKbeta cooperatively activate NK-kappaB and induce EGFR/ap1 signaling to promote survival and migration of head and neck cancer. Oncogene 2014, 33, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- Slotta, C.; Storm, J.; Pfisterer, N.; Henkel, E.; Kleinwachter, S.; Pieper, M.; Ruiz-Perera, L.M.; Greiner, J.F.W.; Kaltschmidt, B.; Kaltschmidt, C. IKK1/2 protect human cells from tnf-mediated ripk1-dependent apoptosis in an NK-kappaB-independent manner. Biochim. Biophys. Acta 2018, 1865, 1025–1033. [Google Scholar] [PubMed]
- Lam, L.T.; Davis, R.E.; Ngo, V.N.; Lenz, G.; Wright, G.; Xu, W.; Zhao, H.; Yu, X.; Dang, L.; Staudt, L.M. Compensatory IKKalpha activation of classical NK-kappaB signaling during IKKbeta inhibition identified by an RNA interference sensitization screen. Proc. Natl. Acad. Sci. USA 2008, 105, 20798–20803. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Zhang, M.; Sun, S.C. Beta-TRCP binding and processing of NK-kappaB2/p100 involve its phosphorylation at serines 866 and 870. Cell. Signal. 2006, 18, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; Sha, W.C.; Bronson, R.T.; Ghosh, S.; Baltimore, D. Embryonic lethality and liver degeneration in mice lacking the rela component of NK-kappa B. Nature 1995, 376, 167–170. [Google Scholar] [PubMed]
- Tanaka, M.; Fuentes, M.E.; Yamaguchi, K.; Durnin, M.H.; Dalrymple, S.A.; Hardy, K.L.; Goeddel, D.V. Embryonic lethality, liver degeneration, and impaired NK-kappa B activation in IKK-beta-deficient mice. Immunity 1999, 10, 421–429. [Google Scholar] [CrossRef]
- Chen, L.W.; Egan, L.; Li, Z.W.; Greten, F.R.; Kagnoff, M.F.; Karin, M. The two faces of IKK and NK-kappaB inhibition: Prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat. Med. 2003, 9, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Pannicke, U.; Baumann, B.; Fuchs, S.; Henneke, P.; Rensing-Ehl, A.; Rizzi, M.; Janda, A.; Hese, K.; Schlesier, M.; Holzmann, K.; et al. Deficiency of innate and acquired immunity caused by an IKBKB mutation. N. Engl. J. Med. 2013, 369, 2504–2514. [Google Scholar] [CrossRef] [PubMed]
- Burns, S.O.; Plagnol, V.; Gutierrez, B.M.; Al Zahrani, D.; Curtis, J.; Gaspar, M.; Hassan, A.; Jones, A.M.; Malone, M.; Rampling, D.; et al. Immunodeficiency and disseminated mycobacterial infection associated with homozygous nonsense mutation of IKKbeta. J. Allergy Clin. Immunol. 2014, 134, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Mousallem, T.; Yang, J.; Urban, T.J.; Wang, H.; Adeli, M.; Parrott, R.E.; Roberts, J.L.; Goldstein, D.B.; Buckley, R.H.; Zhong, X.P. A nonsense mutation in IKBKB causes combined immunodeficiency. Blood 2014, 124, 2046–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, C.; Jakobsen, M.A.; Larsen, M.J.; Muller, A.C.; Hansen, S.; Lillevang, S.T.; Fisker, N.; Barington, T. Immunodeficiency associated with a nonsense mutation of IKBKB. J. Clin. Immunol. 2014, 34, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Verma, I.M. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NK-kappaB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-kappaB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Firestein, G.S. NF-kappaB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M. Regulation of tissue homeostasis by NK-kappaB signalling: Implications for inflammatory diseases. Nat. Rev. Immunol. 2009, 9, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; Sha, W.C.; Bronson, R.T.; Baltimore, D. Constitutive NK-kappa B activation, enhanced granulopoiesis, and neonatal lethality in I kappa B alpha-deficient mice. Genes Dev. 1995, 9, 2736–2746. [Google Scholar] [CrossRef] [PubMed]
- Klement, J.F.; Rice, N.R.; Car, B.D.; Abbondanzo, S.J.; Powers, G.D.; Bhatt, P.H.; Chen, C.H.; Rosen, C.A.; Stewart, C.L. IkappaBalpha deficiency results in a sustained NK-kappaB response and severe widespread dermatitis in mice. Mol. Cell. Biol. 1996, 16, 2341–2349. [Google Scholar] [CrossRef] [PubMed]
- Courtois, G.; Gilmore, T.D. Mutations in the NK-kappaB signaling pathway: Implications for human disease. Oncogene 2006, 25, 6831–6843. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Splittgerber, R.; Yull, F.E.; Kantrow, S.; Ayers, G.D.; Karin, M.; Richmond, A. Conditional ablation of IKKb inhibits melanoma tumor development in mice. J. Clin. Investig. 2010, 120, 2563–2574. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; Kang, Y.; Zhao, R.; Xia, Q.; Lee, D.F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.B.; Wang, H.; et al. KrasG12D-induced IKK2/beta/NK-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. NF-kappaB: Tumor promoter or suppressor? Trends Cell Biol. 2004, 14, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.C.; Goode, J.; Miething, C.; Goktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S.; et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 2007, 130, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.-C.; Enzler, T.; Seita, J.; Timmer, A.M.; Lee, C.-Y.; Lai, T.-Y.; Yu, G.-Y.; Lai, L.-C.; Temkin, V.; Sinzig, U.; et al. IL-1β-driven neutrophilia preserves antibacterial defense in the absence of the kinase IKKβ. Nat. Immunol. 2010, 12, 144. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [PubMed]
- McLoed, A.G.; Sherrill, T.P.; Cheng, D.S.; Han, W.; Saxon, J.A.; Gleaves, L.A.; Wu, P.; Polosukhin, V.V.; Karin, M.; Yull, F.E.; et al. Neutrophil-derived IL-1beta impairs the efficacy of NK-kappaB inhibitors against lung cancer. Cell Rep. 2016, 16, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Pallangyo, C.K.; Ziegler, P.K.; Greten, F.R. IKKbeta acts as a tumor suppressor in cancer-associated fibroblasts during intestinal tumorigenesis. J. Exp. Med. 2015, 212, 2253–2266. [Google Scholar] [CrossRef] [PubMed]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NK-kappaB-dependent manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Koliaraki, V.; Pasparakis, M.; Kollias, G. IKKbeta in intestinal mesenchymal cells promotes initiation of colitis-associated cancer. J. Exp. Med. 2015, 212, 2235–2251. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hawkins, O.E.; Barham, W.; Gilchuk, P.; Boothby, M.; Ayers, G.D.; Joyce, S.; Karin, M.; Yull, F.E.; Richmond, A. Myeloid IKKbeta promotes antitumor immunity by modulating CCL11 and the innate immune response. Cancer Res. 2014, 74, 7274–7284. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Finkin, S.; Yuan, D.; Stein, I.; Taniguchi, K.; Weber, A.; Unger, K.; Browning, J.L.; Goossens, N.; Nakagawa, S.; Gunasekaran, G.; et al. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat. Immunol. 2015, 16, 1235–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, R.E.; Brown, K.D.; Siebenlist, U.; Staudt, L.M. Constitutive nuclear factor κb activity is required for survival of activated b cell–like diffuse large b cell lymphoma cells. J. Exp. Med. 2001, 194, 1861–1874. [Google Scholar] [CrossRef] [PubMed]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of multiple genes cause deregulation of NK-kappaB in diffuse large B-cell lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Staudt, L.M. Oncogenic activation of NK-kappaB. Cold Spring Harb. Perspect. Biol. 2010, 2, a000109. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Wright, G.W.; Emre, N.C.; Kohlhammer, H.; Dave, S.S.; Davis, R.E.; Carty, S.; Lam, L.T.; Shaffer, A.L.; Xiao, W.; et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 13520–13525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, X.; Wood, L.D.; Lindman, M.; Jones, S.; Buckhaults, P.; Polyak, K.; Sukumar, S.; Carter, H.; Kim, D.; Karchin, R.; et al. Somatic mutations in the notch, NF-KB, PIK3CA, and hedgehog pathways in human breast cancers. Genes Chromosomes Cancer 2012, 51, 480–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pflueger, D.; Terry, S.; Sboner, A.; Habegger, L.; Esgueva, R.; Lin, P.C.; Svensson, M.A.; Kitabayashi, N.; Moss, B.J.; MacDonald, T.Y.; et al. Discovery of non-ets gene fusions in human prostate cancer using next-generation RNA sequencing. Genome Res. 2011, 21, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Bredel, M.; Scholtens, D.M.; Yadav, A.K.; Alvarez, A.A.; Renfrow, J.J.; Chandler, J.P.; Yu, I.L.; Carro, M.S.; Dai, F.; Tagge, M.J.; et al. Nfkbia deletion in glioblastomas. N. Engl. J. Med. 2011, 364, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Dooley, A.L.; Feldser, D.M.; Shen, L.; Turk, E.; Ouyang, C.; Jacks, T. Requirement for NK-kappaB signalling in a mouse model of lung adenocarcinoma. Nature 2009, 462, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Basseres, D.S.; Ebbs, A.; Levantini, E.; Baldwin, A.S. Requirement of the NK-kappaB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res. 2010, 70, 3537–3546. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Yeddula, N.; Leblanc, M.; Ke, E.; Zhang, Y.; Oldfield, E.; Shaw, R.J.; Verma, I.M. Reduced cell proliferation by IKK2 depletion in a mouse lung-cancer model. Nat. Cell Biol. 2012, 14, 257–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniluk, J.; Liu, Y.; Deng, D.; Chu, J.; Huang, H.; Gaiser, S.; Cruz-Monserrate, Z.; Wang, H.; Ji, B.; Logsdon, C.D. An NK-kappaB pathway-mediated positive feedback loop amplifies ras activity to pathological levels in mice. J. Clin. Investig. 2012, 122, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Seguin, L.; Kato, S.; Franovic, A.; Camargo, M.F.; Lesperance, J.; Elliott, K.C.; Yebra, M.; Mielgo, A.; Lowy, A.M.; Husain, H.; et al. An integrin beta(3)-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell Biol. 2014, 16, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108. [Google Scholar] [CrossRef] [PubMed]
- Marazioti, A.; Lilis, I.; Vreka, M.; Apostolopoulou, H.; Kalogeropoulou, A.; Giopanou, I.; Giotopoulou, G.A.; Krontira, A.C.; Iliopoulou, M.; Kanellakis, N.I.; et al. Myeloid-derived interleukin-1beta drives oncogenic kras-NK-kappaBeta addiction in malignant pleural effusion. Nat. Commun. 2018, 9, 672. [Google Scholar] [CrossRef] [PubMed]
- Vreka, M.; Lilis, I.; Papageorgopoulou, M.; Giotopoulou, G.A.; Lianou, M.; Giopanou, I.; Kanellakis, N.I.; Spella, M.; Agalioti, T.; Armenis, V.; et al. IkappaB kinase alpha is required for development and progression of kras-mutant lung adenocarcinoma. Cancer Res. 2018, 78, 2939–2951. [Google Scholar] [CrossRef] [PubMed]
- Godwin, P.; Baird, A.M.; Heavey, S.; Barr, M.P.; O’Byrne, K.J.; Gately, K. Targeting nuclear factor-kappa B to overcome resistance to chemotherapy. Front. Oncol. 2013, 3, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Sethi, G. Targeting transcription factor NK-kappaB to overcome chemoresistance and radioresistance in cancer therapy. Biochim. Biophys. Acta 2010, 1805, 167–180. [Google Scholar] [PubMed]
- Voboril, R.; Weberova-Voborilova, J. Constitutive NK-kappaB activity in colorectal cancer cells: Impact on radiation-induced NK-kappaB activity, radiosensitivity, and apoptosis. Neoplasma 2006, 53, 518–523. [Google Scholar] [PubMed]
- Ishida, K.; Nishizuka, S.S.; Chiba, T.; Ikeda, M.; Kume, K.; Endo, F.; Katagiri, H.; Matsuo, T.; Noda, H.; Iwaya, T.; et al. Molecular marker identification for relapse prediction in 5-fu-based adjuvant chemotherapy in gastric and colorectal cancers. PLoS ONE 2012, 7, e43236. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NK-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.J.; Middleton, J.; Kim, T.; Lagana, A.; Piovan, C.; Secchiero, P.; Nuovo, G.J.; Cui, R.; Joshi, P.; Romano, G.; et al. A set of NK-kappaB-regulated micrornas induces acquired trail resistance in lung cancer. Proc. Natl. Acad. Sci. USA 2015, 112, E3355–E3364. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.; Lau, E.Y.; Ching, R.H.; Cheng, B.Y.; Ma, M.K.; Ng, I.O.; Lee, T.K. Nuclear factor kappa B-mediated CD47 up-regulation promotes sorafenib resistance and its blockade synergizes the effect of sorafenib in hepatocellular carcinoma in mice. Hepatology 2015, 62, 534–545. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Mani, A.M.; Wu, Z.H. DNA damage-induced nuclear factor-kappa B activation and its roles in cancer progression. J. Cancer Metastasis Treat. 2017, 3, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Al-Katib, A.; Arnold, A.A.; Aboukameel, A.; Sosin, A.; Smith, P.; Mohamed, A.N.; Beck, F.W.; Mohammad, R.M. I-kappa-kinase-2 (IKK-2) inhibition potentiates vincristine cytotoxicity in non-hodgkin’s lymphoma. Mol. Cancer 2010, 9, 228. [Google Scholar] [CrossRef] [PubMed]
- Bivona, T.G.; Hieronymus, H.; Parker, J.; Chang, K.; Taron, M.; Rosell, R.; Moonsamy, P.; Dahlman, K.; Miller, V.A.; Costa, C.; et al. Fas and NK-kappaB signalling modulate dependence of lung cancers on mutant EGFR. Nature 2011, 471, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Shostak, K.; Chariot, A. EGFR and NK-kappaB: Partners in cancer. Trends Mol. Med. 2015, 21, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Lam, L.T.; Wright, G.; Davis, R.E.; Lenz, G.; Farinha, P.; Dang, L.; Chan, J.W.; Rosenwald, A.; Gascoyne, R.D.; Staudt, L.M. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-{kappa}B pathways in subtypes of diffuse large B-cell lymphoma. Blood 2008, 111, 3701–3713. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Curr. Opin. Immunol. 2014, 27, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuser, C.; Gotot, J.; Piotrowski, E.C.; Philipp, M.S.; Courreges, C.J.F.; Otte, M.S.; Guo, L.; Schmid-Burgk, J.L.; Hornung, V.; Heine, A.; et al. Prolonged IKKbeta inhibition improves ongoing CTL antitumor responses by incapacitating regulatory T cells. Cell Rep. 2017, 21, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Barnes, S.E.; Wang, Y.; Chen, L.; Molinero, L.L.; Gajewski, T.F.; Evaristo, C.; Alegre, M.L. T cell-NK-kappaB activation is required for tumor control in vivo. J. Immunother. Cancer 2015, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Fife, B.T.; Pauken, K.E.; Eagar, T.N.; Obu, T.; Wu, J.; Tang, Q.; Azuma, M.; Krummel, M.F.; Bluestone, J.A. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat. Immunol. 2009, 10, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Balar, A.V.; Weber, J.S. PD-1 and PD-L1 antibodies in cancer: Current status and future directions. Cancer Immunol. Immunother. 2017, 66, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Wu, D.; Li, L.; Chai, Y.; Huang, J. PD-L1 and survival in solid tumors: A meta-analysis. PLoS ONE 2015, 10, e0131403. [Google Scholar] [CrossRef] [PubMed]
- Gowrishankar, K.; Gunatilake, D.; Gallagher, S.J.; Tiffen, J.; Rizos, H.; Hersey, P. Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NK-kappaB. PLoS ONE 2015, 10, e0123410. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Hamanishi, J.; Matsumura, N.; Abiko, K.; Murat, K.; Baba, T.; Yamaguchi, K.; Horikawa, N.; Hosoe, Y.; Murphy, S.K.; et al. Chemotherapy induces programmed cell death-ligand 1 overexpression via the nuclear factor-kappaB to foster an immunosuppressive tumor microenvironment in ovarian cancer. Cancer Res. 2015, 75, 5034–5045. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.O.; Li, C.W.; Xia, W.; Cha, J.H.; Chan, L.C.; Wu, Y.; Chang, S.S.; Lin, W.C.; Hsu, J.M.; Hsu, Y.H.; et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell 2016, 30, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, F.; Montfort, A.; Marcheteau, E.; Imbert, C.; Gilhodes, J.; Filleron, T.; Rochaix, P.; Andrieu-Abadie, N.; Levade, T.; Meyer, N.; et al. Tnfalpha blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat. Commun. 2017, 8, 2256. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Filley, A.C.; Dey, M. Immune system, friend or foe of oncolytic virotherapy? Front. Oncol. 2017, 7, 106. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Ou, P.; Rousso, C.; Bergeron, A.; Krishnan, R.; Pikor, L.; Chen, A.; Keller, B.A.; Ilkow, C.; Bell, J.C.; et al. Dimethyl fumarate potentiates oncolytic virotherapy through NK-kappaB inhibition. Sci. Transl. Med. 2018, 10, eaao1613. [Google Scholar] [CrossRef] [PubMed]
- Cataldi, M.; Shah, N.R.; Felt, S.A.; Grdzelishvili, V.Z. Breaking resistance of pancreatic cancer cells to an attenuated vesicular stomatitis virus through a novel activity of IKK inhibitor TPCA-1. Virology 2015, 485, 340–354. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.; Wilson, J.; Burke, F.; Kulbe, H.; Li, N.F.; Pluddemann, A.; Charles, K.; Gordon, S.; Balkwill, F.R. Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. J. Immunol. 2006, 176, 5023–5032. [Google Scholar] [CrossRef] [PubMed]
- Sawa-Wejksza, K.; Kandefer-Szerszen, M. Tumor-associated macrophages as target for antitumor therapy. Arch. Immunol. Ther. Exp. 2018, 66, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.; Lawrence, T.; McNeish, I.; Charles, K.A.; Kulbe, H.; Thompson, R.G.; Robinson, S.C.; Balkwill, F.R. “Re-educating” tumor-associated macrophages by targeting NK-kappaB. J. Exp. Med. 2008, 205, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Saccani, A.; Schioppa, T.; Porta, C.; Biswas, S.K.; Nebuloni, M.; Vago, L.; Bottazzi, B.; Colombo, M.P.; Mantovani, A.; Sica, A. P50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 2006, 66, 11432–11440. [Google Scholar] [CrossRef] [PubMed]
- Torroella-Kouri, M.; Ma, X.; Perry, G.; Ivanova, M.; Cejas, P.J.; Owen, J.L.; Iragavarapu-Charyulu, V.; Lopez, D.M. Diminished expression of transcription factors nuclear factor kappaB and CCAAT/enhancer binding protein underlies a novel tumor evasion mechanism affecting macrophages of mammary tumor-bearing mice. Cancer Res. 2005, 65, 10578–10584. [Google Scholar] [CrossRef] [PubMed]
- Connelly, L.; Barham, W.; Onishko, H.M.; Chen, L.; Sherrill, T.P.; Zabuawala, T.; Ostrowski, M.C.; Blackwell, T.S.; Yull, F.E. NF-kappaB activation within macrophages leads to an anti-tumor phenotype in a mammary tumor lung metastasis model. Breast Cancer Res. 2011, 13, R83. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.P.; Mantovani, A. Targeting myelomonocytic cells to revert inflammation-dependent cancer promotion. Cancer Res. 2005, 65, 9113–9116. [Google Scholar] [CrossRef] [PubMed]
- Guiducci, C.; Vicari, A.P.; Sangaletti, S.; Trinchieri, G.; Colombo, M.P. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res. 2005, 65, 3437–3446. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.; Biswas, S.K.; Lawrence, T.; Sica, A.; Lewis, C.E. Regulation of macrophage function in tumors: The multifaceted role of NK-kappaB. Blood 2009, 113, 3139–3146. [Google Scholar] [CrossRef] [PubMed]
- Gaddipati, S.; Lu, Q.; Kasetti, R.B.; Miller, M.C.; Lu, Q.; Trent, J.O.; Kaplan, H.J.; Li, Q. IKK2 inhibition using TPCA-1-loaded PLGA microparticles attenuates laser-induced choroidal neovascularization and macrophage recruitment. PLoS ONE 2015, 10, e0121185. [Google Scholar] [CrossRef] [PubMed]
- Enzler, T.; Sano, Y.; Choo, M.K.; Cottam, H.B.; Karin, M.; Tsao, H.; Park, J.M. Cell-selective inhibition of NK-kappaB signaling improves therapeutic index in a melanoma chemotherapy model. Cancer Discov. 2011, 1, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Tan, Y.; Meng, T.; Liu, X.; Zhu, Y.; Hong, Y.; Yang, X.; Yuan, H.; Huang, X.; Hu, F. Simultaneous targeting therapy for lung metastasis and breast tumor by blocking the NK-kappaB signaling pathway using celastrol-loaded micelles. Drug Deliv. 2018, 25, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Begalli, F.; Bennett, J.; Capece, D.; Verzella, D.; D’Andrea, D.; Tornatore, L.; Franzoso, G. Unlocking the NK-kappaB conundrum: Embracing complexity to achieve specificity. Biomedicines 2017, 5, 50. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.; Capece, D.; Begalli, F.; Verzella, D.; D’Andrea, D.; Tornatore, L.; Franzoso, G. NF-kappaB in the crosshairs: Rethinking an old riddle. Int. J. Biochem. Cell Biol. 2018, 95, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.L.; Rule, S.; Martin, P.; Goy, A.; Auer, R.; Kahl, B.S.; Jurczak, W.; Advani, R.H.; Romaguera, J.E.; Williams, M.E.; et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2013, 369, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Noy, A.; de Vos, S.; Thieblemont, C.; Martin, P.; Flowers, C.R.; Morschhauser, F.; Collins, G.P.; Ma, S.; Coleman, M.; Peles, S.; et al. Targeting bruton tyrosine kinase with ibrutinib in relapsed/refractory marginal zone lymphoma. Blood 2017, 129, 2224–2232. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Ghia, P.; Polliack, A.; Tam, C.; Suri, D.; Clow, F.; Kraljevic, S.; James, D.F.; Kipps, T.J. Randomized, multicenter, open-label, phase III study of the btk inhibitor ibrutinib versus chlorambucil in patients 65 years or older with treatment-naive CLL/SLL (RESONATE-2, PCYC-1115-CA). J. Clin. Oncol. 2013, 31, TPS7130. [Google Scholar]
- Treon, S.P.; Tripsas, C.K.; Meid, K.; Warren, D.; Varma, G.; Green, R.; Argyropoulos, K.V.; Yang, G.; Cao, Y.; Xu, L.; et al. Ibrutinib in previously treated waldenstrom’s macroglobulinemia. N. Engl. J. Med. 2015, 372, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Miklos, D.; Cutler, C.S.; Arora, M.; Waller, E.K.; Jagasia, M.; Pusic, I.; Flowers, M.E.; Logan, A.C.; Nakamura, R.; Blazar, B.R.; et al. Ibrutinib for chronic graft-versus-host disease after failure of prior therapy. Blood 2017, 130, 2243–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demchenko, Y.N.; Glebov, O.K.; Zingone, A.; Keats, J.J.; Bergsagel, P.L.; Kuehl, W.M. Classical and/or alternative NK-kappaB pathway activation in multiple myeloma. Blood 2010, 115, 3541–3552. [Google Scholar] [CrossRef] [PubMed]
- Rothwarf, D.M.; Zandi, E.; Natoli, G.; Karin, M. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature 1998, 395, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Strickland, I.; Ghosh, S. Use of cell permeable NBD peptides for suppression of inflammation. Ann. Rheum. Dis. 2006, 65 (Suppl. 3), iii75–iii82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habineza Ndikuyeze, G.; Gaurnier-Hausser, A.; Patel, R.; Baldwin, A.S.; May, M.J.; Flood, P.; Krick, E.; Propert, K.J.; Mason, N.J. A phase i clinical trial of systemically delivered nemo binding domain peptide in dogs with spontaneous activated B-cell like diffuse large B-cell lymphoma. PLoS ONE 2014, 9, e95404. [Google Scholar] [CrossRef] [PubMed]
- Vincendeau, M.; Hadian, K.; Messias, A.C.; Brenke, J.K.; Halander, J.; Griesbach, R.; Greczmiel, U.; Bertossi, A.; Stehle, R.; Nagel, D.; et al. Inhibition of canonical NF-κB signaling by a small molecule targeting nemo-ubiquitin interaction. Sci. Rep. 2016, 6, 18934. [Google Scholar] [CrossRef] [PubMed]
- Colomer, C.; Marruecos, L.; Vert, A.; Bigas, A.; Espinosa, L. NF-kappaB members left home: NF-kappaB-independent roles in cancer. Biomedicines 2017, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- Dan, H.C.; Ebbs, A.; Pasparakis, M.; Van Dyke, T.; Basseres, D.S.; Baldwin, A.S. Akt-dependent activation of mTORC1 complex involves phosphorylation of mTOR (mammalian target of rapamycin) by IkappaB kinase alpha (IKKalpha). J. Biol. Chem. 2014, 289, 25227–25240. [Google Scholar] [CrossRef] [PubMed]
- Dan, H.C.; Antonia, R.J.; Baldwin, A.S. PI3K/Akt promotes feedforward mTORC2 activation through IKKalpha. Oncotarget 2016, 7, 21064–21075. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.W.; Irwin, D.; Stadtmauer, E.A.; Facon, T.; Harousseau, J.L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2005, 352, 2487–2498. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.I.; Bernstein, S.H.; Kahl, B.S.; Djulbegovic, B.; Robertson, M.J.; de Vos, S.; Epner, E.; Krishnan, A.; Leonard, J.P.; Lonial, S.; et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J. Clin. Oncol. 2006, 24, 4867–4874. [Google Scholar] [CrossRef] [PubMed]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Oliva, L.; Cascio, P.; Pengo, N.; Fontana, F.; Cerruti, F.; Orsi, A.; Pasqualetto, E.; Mezghrani, A.; Calbi, V.; et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood 2009, 113, 3040–3049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boothby, M. Specificity of SN50 for NK-kappa B? Nat. Immunol. 2001, 2, 471–472. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Grinberg-Bleyer, Y.; Liao, W.; Maloney, D.; Wang, P.; Wu, Z.; Wang, J.; Bhatt, D.M.; Heise, N.; Schmid, R.M.; et al. An NK-kappaB transcription-factor-dependent lineage-specific transcriptional program promotes regulatory T cell identity and function. Immunity 2017, 47, 450–465. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Tam, W.F.; Hughes, C.C.W.; Rath, S.; Sen, R. c-Rel is a target of pentoxifylline-mediated inhibition of T lymphocyte activation. Immunity 1997, 6, 165–174. [Google Scholar] [CrossRef]
- Grinberg-Bleyer, Y.; Oh, H.; Desrichard, A.; Bhatt, D.M.; Caron, R.; Chan, T.A.; Schmid, R.M.; Klein, U.; Hayden, M.S.; Ghosh, S. NF-kappaB c-Rel is crucial for the regulatory T cell immune checkpoint in cancer. Cell 2017, 170, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D.; Gerondakis, S. The c-Rel transcription factor in development and disease. Genes Cancer 2011, 2, 695–711. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, L.; Baldwin, A.S., Jr.; Friedman, A.D.; Paz-Priel, I. Loss of IKKbeta but not NK-kappaB p65 skews differentiation towards myeloid over erythroid commitment and increases myeloid progenitor self-renewal and functional long-term hematopoietic stem cells. PLoS ONE 2015, 10, e0130441. [Google Scholar]
- Brown, J.D.; Lin, C.Y.; Duan, Q.; Griffin, G.; Federation, A.; Paranal, R.M.; Bair, S.; Newton, G.; Lichtman, A.; Kung, A.; et al. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol. Cell 2014, 56, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Koche, R.; Zou, L.; van Galen, P.; Hill, C.E.; Dong, L.; De Groote, S.; Papalexi, E.; Hanasoge Somasundara, A.V.; Cordner, K.; et al. Dual targeting of oncogenic activation and inflammatory signaling increases therapeutic efficacy in myeloproliferative neoplasms. Cancer Cell 2018, 33, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Catrysse, L.; van Loo, G. Inflammation and the metabolic syndrome: The tissue-specific functions of NK-kappaB. Trends Cell Biol. 2017, 27, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.H.; Zheng, X.L.; Tang, C.K. Nuclear factor-kappaB activation as a pathological mechanism of lipid metabolism and atherosclerosis. Adv. Clin. Chem. 2015, 70, 1–30. [Google Scholar] [PubMed]
- Yan, J.; Greer, J.M. NK-kappa B, a potential therapeutic target for the treatment of multiple sclerosis. CNS Neurol. Disord. Drug Targets 2008, 7, 536–557. [Google Scholar] [CrossRef] [PubMed]
- Schuliga, M. NF-kappaB signaling in chronic inflammatory airway disease. Biomolecules 2015, 5, 1266–1283. [Google Scholar] [CrossRef] [PubMed]
- Acharyya, S.; Villalta, S.A.; Bakkar, N.; Bupha-Intr, T.; Janssen, P.M.; Carathers, M.; Li, Z.W.; Beg, A.A.; Ghosh, S.; Sahenk, Z.; et al. Interplay of IKK/NK-kappaB signaling in macrophages and myofibers promotes muscle degeneration in duchenne muscular dystrophy. J. Clin. Investig. 2007, 117, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Flood, P.M.; Qian, L.; Peterson, L.J.; Zhang, F.; Shi, J.S.; Gao, H.M.; Hong, J.S. Transcriptional factor NK-kappaB as a target for therapy in parkinson’s disease. Parkinsons Dis. 2011, 2011, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Atreya, I.; Atreya, R.; Neurath, M.F. NF-kappaB in inflammatory bowel disease. J. Intern. Med. 2008, 263, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Armaka, M.; Ospelt, C.; Pasparakis, M.; Kollias, G. The p55TNFR-IKK2-Ripk3 axis orchestrates arthritis by regulating death and inflammatory pathways in synovial fibroblasts. Nat. Commun. 2018, 9, 618. [Google Scholar] [CrossRef] [PubMed]
- Monaco, C.; Andreakos, E.; Kiriakidis, S.; Mauri, C.; Bicknell, C.; Foxwell, B.; Cheshire, N.; Paleolog, E.; Feldmann, M. Canonical pathway of nuclear factor kappa B activation selectively regulates proinflammatory and prothrombotic responses in human atherosclerosis. Proc. Natl. Acad. Sci. USA 2004, 101, 5634–5639. [Google Scholar] [CrossRef] [PubMed]
- Kempe, S.; Kestler, H.; Lasar, A.; Wirth, T. NF-kappaB controls the global pro-inflammatory response in endothelial cells: Evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 2005, 33, 5308–5319. [Google Scholar] [CrossRef] [PubMed]
- Gareus, R.; Kotsaki, E.; Xanthoulea, S.; van der Made, I.; Gijbels, M.J.; Kardakaris, R.; Polykratis, A.; Kollias, G.; de Winther, M.P.; Pasparakis, M. Endothelial cell-specific NK-kappaB inhibition protects mice from atherosclerosis. Cell Metab. 2008, 8, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Sui, Y.; Gizard, F.; Xu, J.; Rios-Pilier, J.; Helsley, R.N.; Han, S.S.; Zhou, C. Myeloid-specific IkappaB kinase beta deficiency decreases atherosclerosis in low-density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2869–2876. [Google Scholar] [CrossRef] [PubMed]
- Kanters, E.; Pasparakis, M.; Gijbels, M.J.; Vergouwe, M.N.; Partouns-Hendriks, I.; Fijneman, R.J.; Clausen, B.E.; Forster, I.; Kockx, M.M.; Rajewsky, K.; et al. Inhibition of NK-kappaB activation in macrophages increases atherosclerosis in LDL receptor-deficient mice. J. Clin. Investig. 2003, 112, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Meylan, E.; Oliver, T.G.; Feldser, D.M.; Winslow, M.M.; Bronson, R.; Jacks, T. Response and resistance to NK-kappaB inhibitors in mouse models of lung adenocarcinoma. Cancer Discov. 2011, 1, 236–247. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Overview of canonical and DNA damage-induced NF-κB signalling pathways. (A) Binding of IL-1/Toll-like receptor (TLR) ligands to the interleukin-1 receptor (IL-1R)/TLRs leads to the assembly of the so-called ‘Myddosome’, an oligomeric structure consisting of the adaptor protein MyD88, IL-1 Receptor (IL-1R)-Associated Kinase 4 (IRAK4), IRAK1 and IRAK2. IRAK4 activates IRAK1, allowing IRAK1 to autophosphorylate and subsequently phosphorylate the E3-ligase Pellino-1, which in turn causes K63-polyubiquitylation of IRAK1. This leads to the recruitment and activation of TRAF6, which along with the E2-conjugating complex Ubc13-Uev1a, generates K63-linked polyubiquitin chains that serve to recruit and activate the TAK1 complex or TAB1/2/3-TAK1. K63-linked chains also serve as a substrate for the LUBAC (linear ubiquitin assembly complex) complex, which conjugates M1-linked ubiquitin to these oligomers, to generate M1-K63-linked hybrid ubiquitin chains. The IKK complex is recruited to this complex through interaction of NEMO with M1-linked chains. The co-localisation of TAK1 and IKK to ubiquitin chains leads to activation of the IKK complex, which subsequently phosphorylates IκBα to activate the NF-κB pathway. (B) TNFα binding to the extracellular domain of the receptor leads to the recruitment of TRADD (Tumor necrosis factor receptor type 1-associated DEATH domain) to the cytoplasmic death domains of TNFR1. TRADD, in turn, recruits RIP kinase, and subsequently TRAF2 or TRAF5 adaptor proteins and cIAP1 or cIAP2 to assemble the TNFR1 complex I. cIAP1 and cIAP2 generate K63-linked polubiquitin chains on RIP1 and other components of the complex. This is necessary to recruit LUBAC, which stabilises complex I by catalysing the attachment of linear M1-linked polyubiquitin chains, typically to RIP1. K63-polyubiquitylated RIP1 also recruits the TAK1:TAB complex. LUBAC-mediated M1-linked linear polyubiquitylation of RIP1, meanwhile promotes the recruitment of NEMO, as part of the IKK complex. Membrane proximal recruitment of IKK kinases contributes to IKK activation through proximity to TAK1, which is thought to prime the activation of IKK via phosphorylation of S176/S177 of IKKα/IKKβ, and through oligomerisation of IKK complexes, which is thought to facilitate trans-autophosphorylation of the activation loop, leading to full activation. (C) Engagement of the TCR by a major histocompatibility complex (MHC)-antigen complex leads to recruitment of Src family kinases, including FYN and LCK, which phosphorylate the TCR to promote recruitment of the tyrosine kinase, ZAP-70. ZAP-70 phosphorylates the adapter proteins LAT and SLP-76, which along with VAV1 promote the recruitment and activation of PLCγ1. PLCγ1 generates the second messengers, inositol trisphosphate (IP3) and diacylglycerol (DAG), which, in turn, activate a specific PKC isoform, PKCθ. PKCθ-mediated phosphorylation of CARMA1 triggers a conformational change, enabling CARMA1 to bind to BCL10 and MALT1, to form the CBM complex. BCL10 and MALT1 become polyubiquitinated, possibly through TRAF6 activity, which promotes the recruitment of NEMO, as part of the IKK complex. (D) Antigen binding to BCRs leads to the recruitment and activation of SRC-family kinases, including BLK, LYN, FYN and SYK and adaptors, such as BLNK. This leads to activation of PLCγ2, which catalyses the generation of IP3 and DAG, which ultimately activate a specific PKC isoform, PKCβ. PKCβ phosphorylates CARMA1 to form the CBM complex and ultimately activate the IKK complex. (E) Genotoxic triggers the nuclear accumulation of ‘IKK-free’ NEMO. Within the nucleus NEMO forms a complex with PARP1, PIASy and ATM and undergoes a series of post-translational modification. PIASy promotes the sumoylation of NEMO, which promotes its nuclear localisation. ATM phosphorylates NEMO at Serine 85, which is necessary for the subsequent monoubiquitylation of NEMO. This is thought to trigger the nuclear export of the NEMO-ATM complex, which then, in an ill-defined mechanism, activates TAK1 and the IKK complex. Canonical and DNA damage-induced NF-κB signalling pathways converge at the activation of the IKK complex, which subsequently phosphorylates IκB proteins (at S32 and S36 IκBα). This promotes the recognition of the PEST motif degron within IκBα by β-TrCP, which is part of the E3 ubiquitin ligase SCFβ-TrCP (S phase kinase-associated protein 1 (SKP1)-cullin 1-F-box protein containing β-transducing repeat-containing protein), and its K48-linked ubiquitylation, which targets IκBα for proteasomal degradation. This enables NF-κB complexes (primarily p65-p50 and c-rel/p50 complexes in the case of canonical NF-κB pathways), to accumulate in the nucleus, where they regulate the expression of NF-κB-dependent genes.
Figure 1.
Overview of canonical and DNA damage-induced NF-κB signalling pathways. (A) Binding of IL-1/Toll-like receptor (TLR) ligands to the interleukin-1 receptor (IL-1R)/TLRs leads to the assembly of the so-called ‘Myddosome’, an oligomeric structure consisting of the adaptor protein MyD88, IL-1 Receptor (IL-1R)-Associated Kinase 4 (IRAK4), IRAK1 and IRAK2. IRAK4 activates IRAK1, allowing IRAK1 to autophosphorylate and subsequently phosphorylate the E3-ligase Pellino-1, which in turn causes K63-polyubiquitylation of IRAK1. This leads to the recruitment and activation of TRAF6, which along with the E2-conjugating complex Ubc13-Uev1a, generates K63-linked polyubiquitin chains that serve to recruit and activate the TAK1 complex or TAB1/2/3-TAK1. K63-linked chains also serve as a substrate for the LUBAC (linear ubiquitin assembly complex) complex, which conjugates M1-linked ubiquitin to these oligomers, to generate M1-K63-linked hybrid ubiquitin chains. The IKK complex is recruited to this complex through interaction of NEMO with M1-linked chains. The co-localisation of TAK1 and IKK to ubiquitin chains leads to activation of the IKK complex, which subsequently phosphorylates IκBα to activate the NF-κB pathway. (B) TNFα binding to the extracellular domain of the receptor leads to the recruitment of TRADD (Tumor necrosis factor receptor type 1-associated DEATH domain) to the cytoplasmic death domains of TNFR1. TRADD, in turn, recruits RIP kinase, and subsequently TRAF2 or TRAF5 adaptor proteins and cIAP1 or cIAP2 to assemble the TNFR1 complex I. cIAP1 and cIAP2 generate K63-linked polubiquitin chains on RIP1 and other components of the complex. This is necessary to recruit LUBAC, which stabilises complex I by catalysing the attachment of linear M1-linked polyubiquitin chains, typically to RIP1. K63-polyubiquitylated RIP1 also recruits the TAK1:TAB complex. LUBAC-mediated M1-linked linear polyubiquitylation of RIP1, meanwhile promotes the recruitment of NEMO, as part of the IKK complex. Membrane proximal recruitment of IKK kinases contributes to IKK activation through proximity to TAK1, which is thought to prime the activation of IKK via phosphorylation of S176/S177 of IKKα/IKKβ, and through oligomerisation of IKK complexes, which is thought to facilitate trans-autophosphorylation of the activation loop, leading to full activation. (C) Engagement of the TCR by a major histocompatibility complex (MHC)-antigen complex leads to recruitment of Src family kinases, including FYN and LCK, which phosphorylate the TCR to promote recruitment of the tyrosine kinase, ZAP-70. ZAP-70 phosphorylates the adapter proteins LAT and SLP-76, which along with VAV1 promote the recruitment and activation of PLCγ1. PLCγ1 generates the second messengers, inositol trisphosphate (IP3) and diacylglycerol (DAG), which, in turn, activate a specific PKC isoform, PKCθ. PKCθ-mediated phosphorylation of CARMA1 triggers a conformational change, enabling CARMA1 to bind to BCL10 and MALT1, to form the CBM complex. BCL10 and MALT1 become polyubiquitinated, possibly through TRAF6 activity, which promotes the recruitment of NEMO, as part of the IKK complex. (D) Antigen binding to BCRs leads to the recruitment and activation of SRC-family kinases, including BLK, LYN, FYN and SYK and adaptors, such as BLNK. This leads to activation of PLCγ2, which catalyses the generation of IP3 and DAG, which ultimately activate a specific PKC isoform, PKCβ. PKCβ phosphorylates CARMA1 to form the CBM complex and ultimately activate the IKK complex. (E) Genotoxic triggers the nuclear accumulation of ‘IKK-free’ NEMO. Within the nucleus NEMO forms a complex with PARP1, PIASy and ATM and undergoes a series of post-translational modification. PIASy promotes the sumoylation of NEMO, which promotes its nuclear localisation. ATM phosphorylates NEMO at Serine 85, which is necessary for the subsequent monoubiquitylation of NEMO. This is thought to trigger the nuclear export of the NEMO-ATM complex, which then, in an ill-defined mechanism, activates TAK1 and the IKK complex. Canonical and DNA damage-induced NF-κB signalling pathways converge at the activation of the IKK complex, which subsequently phosphorylates IκB proteins (at S32 and S36 IκBα). This promotes the recognition of the PEST motif degron within IκBα by β-TrCP, which is part of the E3 ubiquitin ligase SCFβ-TrCP (S phase kinase-associated protein 1 (SKP1)-cullin 1-F-box protein containing β-transducing repeat-containing protein), and its K48-linked ubiquitylation, which targets IκBα for proteasomal degradation. This enables NF-κB complexes (primarily p65-p50 and c-rel/p50 complexes in the case of canonical NF-κB pathways), to accumulate in the nucleus, where they regulate the expression of NF-κB-dependent genes.
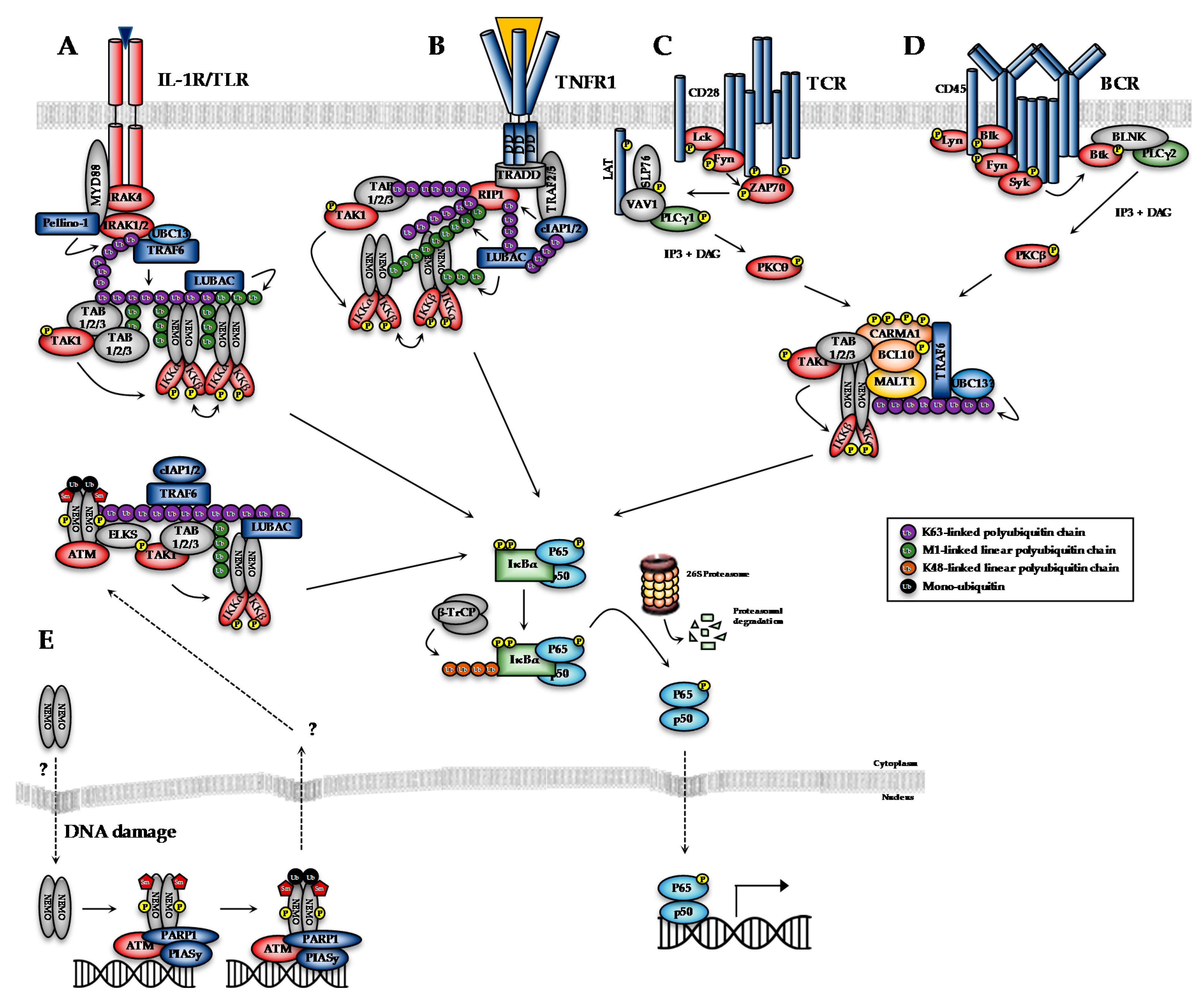
Figure 2.
X-ray crystal structures of human IKKβ and IKKα. (A) Ribbon diagram of the crystallographic structure of the human IKKβ dimer. The N-terminal lobe of the kinase domain (KD) (KD N; residues 1–109), C-terminal lobe of the KD (C; residues 110–307), ubiquitin-like domain (ULD; residues 308–404) and scaffold/dimerization domain (SDD; residues 410-664) are coloured in yellow, orange, blue and green, respectively. The NEMO-binding domain (NBD) at the extreme N-terminus was not resolved in the original structure. Figure adapted from [5]. PDB ID: 4KIK. (B) Ribbon diagram of the human IKKβ protomer showing the tri-modular architecture of KD, ULD, and the elongated, α-helical SDD. Figure adapted from [5]. PDB ID: 4KIK. (C) Ribbon diagram of a model of a human IKKα dimer derived from X-ray crystallographic data. Figure adapted from [7]. PDB ID: 5EBZ. (D) Ribbon diagram of the human IKKα protomer showing the same tri-modular architecture of domains as IKKβ. The NBD at the extreme N-terminus was also not resolved in the original structure. Figure adapted from [7]. PDB ID: 5EBZ. The pseudo two-fold axis of the IKKβ and IKKα dimers are indicated by a dashed line.
Figure 2.
X-ray crystal structures of human IKKβ and IKKα. (A) Ribbon diagram of the crystallographic structure of the human IKKβ dimer. The N-terminal lobe of the kinase domain (KD) (KD N; residues 1–109), C-terminal lobe of the KD (C; residues 110–307), ubiquitin-like domain (ULD; residues 308–404) and scaffold/dimerization domain (SDD; residues 410-664) are coloured in yellow, orange, blue and green, respectively. The NEMO-binding domain (NBD) at the extreme N-terminus was not resolved in the original structure. Figure adapted from [5]. PDB ID: 4KIK. (B) Ribbon diagram of the human IKKβ protomer showing the tri-modular architecture of KD, ULD, and the elongated, α-helical SDD. Figure adapted from [5]. PDB ID: 4KIK. (C) Ribbon diagram of a model of a human IKKα dimer derived from X-ray crystallographic data. Figure adapted from [7]. PDB ID: 5EBZ. (D) Ribbon diagram of the human IKKα protomer showing the same tri-modular architecture of domains as IKKβ. The NBD at the extreme N-terminus was also not resolved in the original structure. Figure adapted from [7]. PDB ID: 5EBZ. The pseudo two-fold axis of the IKKβ and IKKα dimers are indicated by a dashed line.
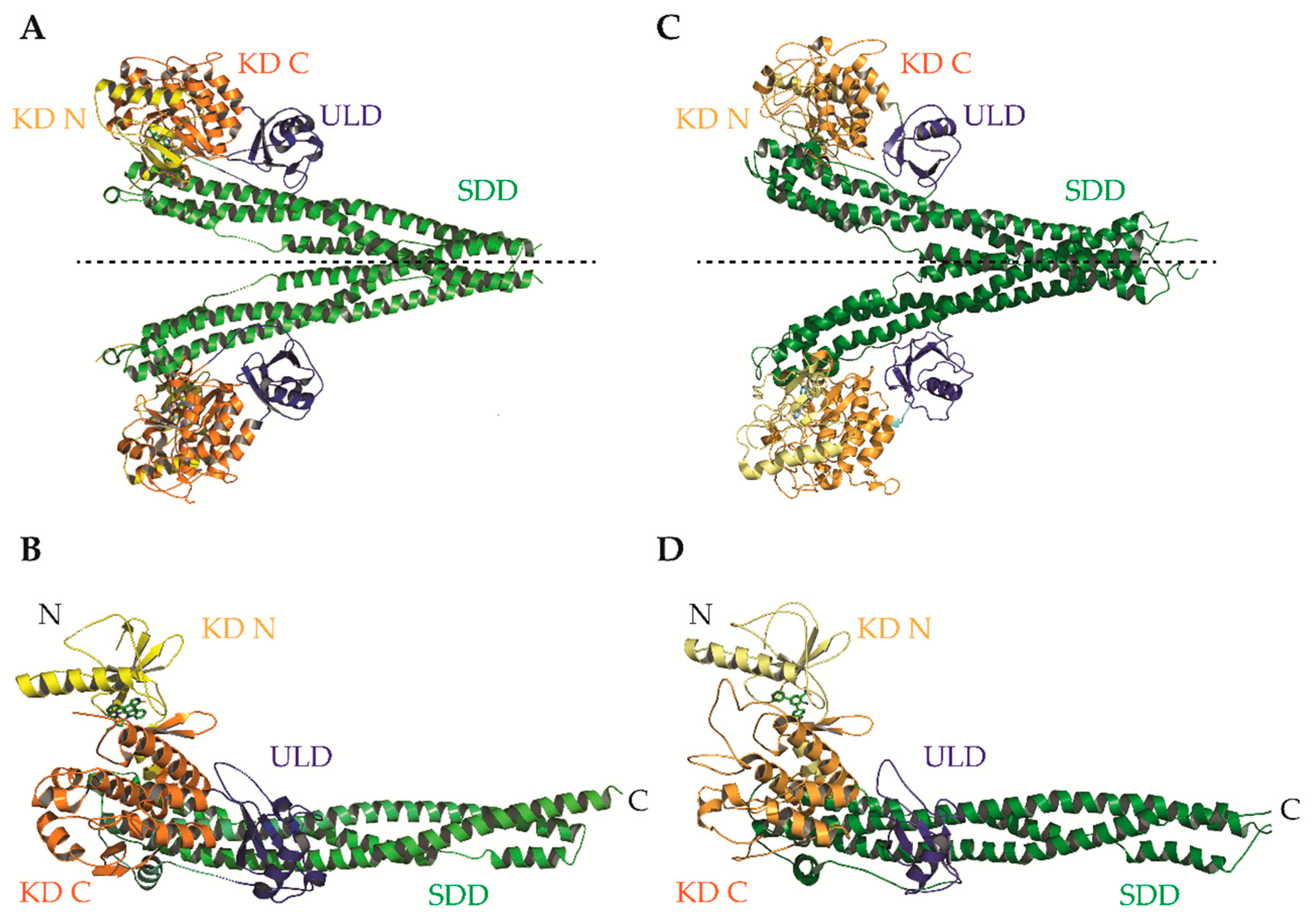
Figure 3.
Insights from X-ray crystallographic studies of IKKβ. (A) Predicted allosteric binding site between the KD (yellow) and ULD (blue) of the catalytically inactive conformation of the human IKKβ monomer. Surface representation of residues surrounding the binding pocket is shown in magenta. SDD, green. Liu et al. identified a compound that specifically binds to this allosteric pocket in the inactive conformation of IKKβ, but not the active conformation, and blocks IKKβ activation. Figure adapted from [130] PDB ID: 4KIK. (B) Ribbon diagram of a human IKKβ dimer (chains A and F) in a catalytically active conformation taken from the asymmetric unit of the crystallographic structure. The primary dimer interface is mediated by residues of the C-terminal portion of the SDD (dashed box). Figure adapted from [6]. PDB ID: 4E3C. (C) Close-up view of the boxed area from panel B. Displayed are residues mediating interactions at the dimer SDD interface that have been shown to be important for IKKβ catalytic activity in vitro via site-directed mutagenesis. Three pairs of residues were mutated (W655D/L658D, L654D/W655D and K482A/F485D) and in vitro kinase assays with human IKKβ performed [6]. Figure adapted from [6]. PDB ID: 4E3C. (D) Ribbon diagram showing the interaction of neighbouring, symmetry-related tetrameric assemblies of IKKβ protomers within the crystal. This oligomerisation positions two KDs (from chains A, green, and D’, magenta, in the representation provided) within close proximity to one another (dashed box). Figure adapted from [6]. PDB ID: 4E3C. (E) Close-up view of the boxed area from panel D. The arrangement of neighbouring KDs (from A and D’) positions the kinase activation loop (shown in yellow and blue) of one protomer directly over the active site of its neighbour, and potentially facilitates oligomerization-dependent trans auto-phosphorylation. Activation loop E177 and E181 (mutant forms of WT S177 and S181) are shown in cyan and red, respectively. The Cα positions of V229 and H232 are marked as orange spheres. Mutation of these, and other residues mediating interactions at this KD-KD oligomerisation interface inhibited IKKβ catalytic activity and activation loop phosphorylation in vitro [6]. Figure adapted from [6]. PDB ID: 4E3C. Small molecules designed to interfere with dimerization/oligomerization via the interfaces shown in panel C and E may function as specific inhibitors of IKKβ. Figures were prepared using program PyMOL [131,132].
Figure 3.
Insights from X-ray crystallographic studies of IKKβ. (A) Predicted allosteric binding site between the KD (yellow) and ULD (blue) of the catalytically inactive conformation of the human IKKβ monomer. Surface representation of residues surrounding the binding pocket is shown in magenta. SDD, green. Liu et al. identified a compound that specifically binds to this allosteric pocket in the inactive conformation of IKKβ, but not the active conformation, and blocks IKKβ activation. Figure adapted from [130] PDB ID: 4KIK. (B) Ribbon diagram of a human IKKβ dimer (chains A and F) in a catalytically active conformation taken from the asymmetric unit of the crystallographic structure. The primary dimer interface is mediated by residues of the C-terminal portion of the SDD (dashed box). Figure adapted from [6]. PDB ID: 4E3C. (C) Close-up view of the boxed area from panel B. Displayed are residues mediating interactions at the dimer SDD interface that have been shown to be important for IKKβ catalytic activity in vitro via site-directed mutagenesis. Three pairs of residues were mutated (W655D/L658D, L654D/W655D and K482A/F485D) and in vitro kinase assays with human IKKβ performed [6]. Figure adapted from [6]. PDB ID: 4E3C. (D) Ribbon diagram showing the interaction of neighbouring, symmetry-related tetrameric assemblies of IKKβ protomers within the crystal. This oligomerisation positions two KDs (from chains A, green, and D’, magenta, in the representation provided) within close proximity to one another (dashed box). Figure adapted from [6]. PDB ID: 4E3C. (E) Close-up view of the boxed area from panel D. The arrangement of neighbouring KDs (from A and D’) positions the kinase activation loop (shown in yellow and blue) of one protomer directly over the active site of its neighbour, and potentially facilitates oligomerization-dependent trans auto-phosphorylation. Activation loop E177 and E181 (mutant forms of WT S177 and S181) are shown in cyan and red, respectively. The Cα positions of V229 and H232 are marked as orange spheres. Mutation of these, and other residues mediating interactions at this KD-KD oligomerisation interface inhibited IKKβ catalytic activity and activation loop phosphorylation in vitro [6]. Figure adapted from [6]. PDB ID: 4E3C. Small molecules designed to interfere with dimerization/oligomerization via the interfaces shown in panel C and E may function as specific inhibitors of IKKβ. Figures were prepared using program PyMOL [131,132].


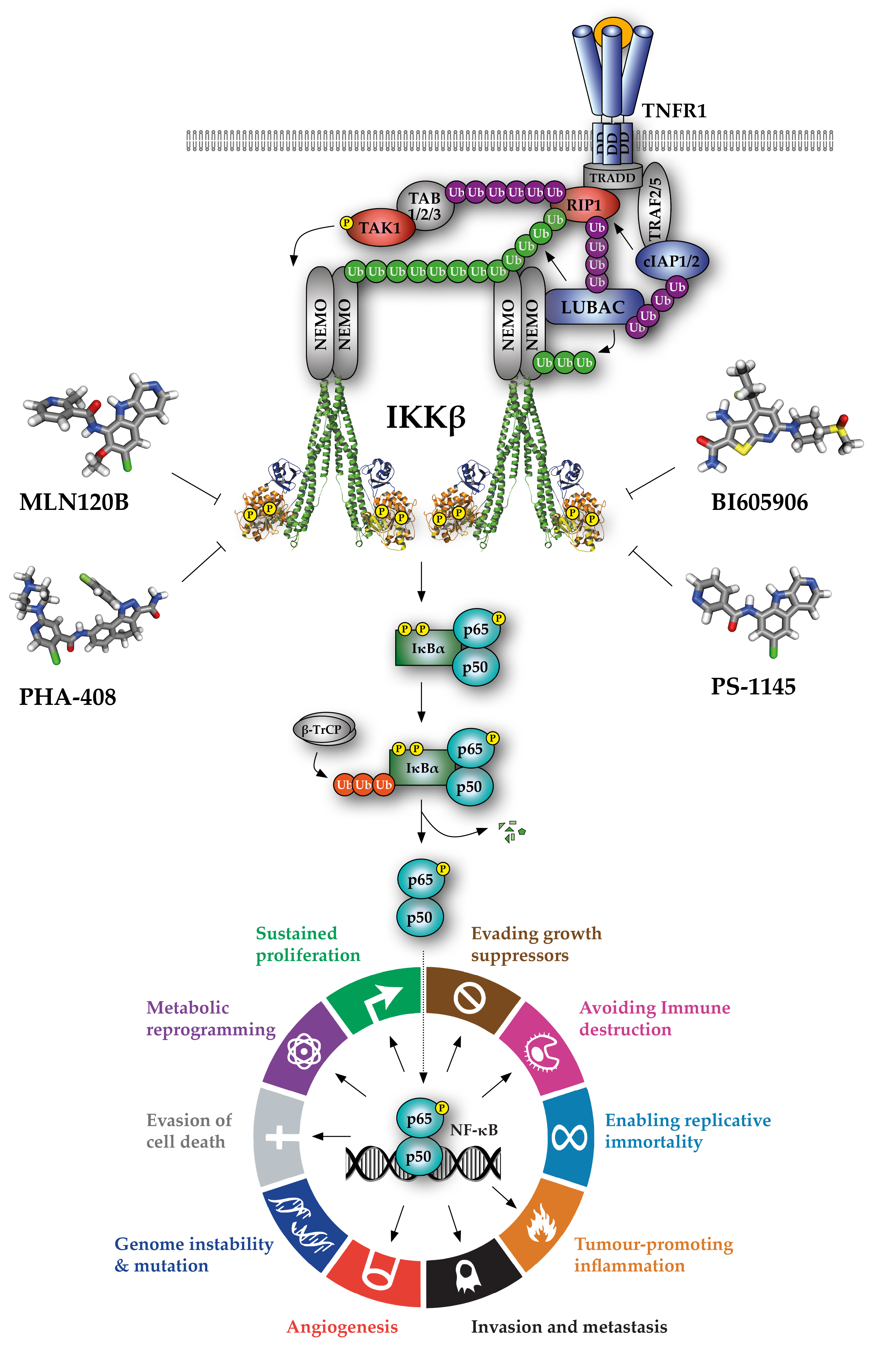{kind=link}
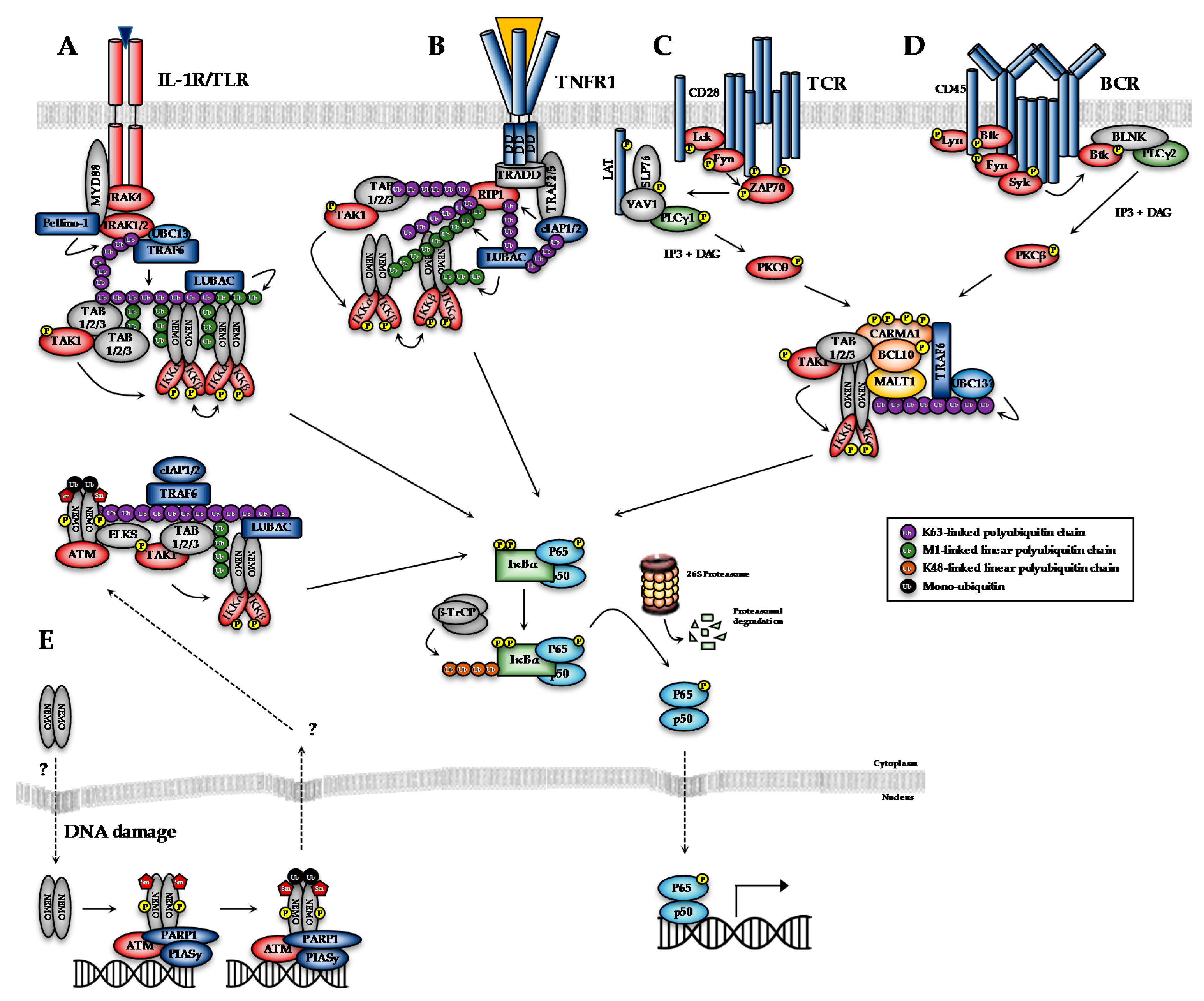{kind=link}
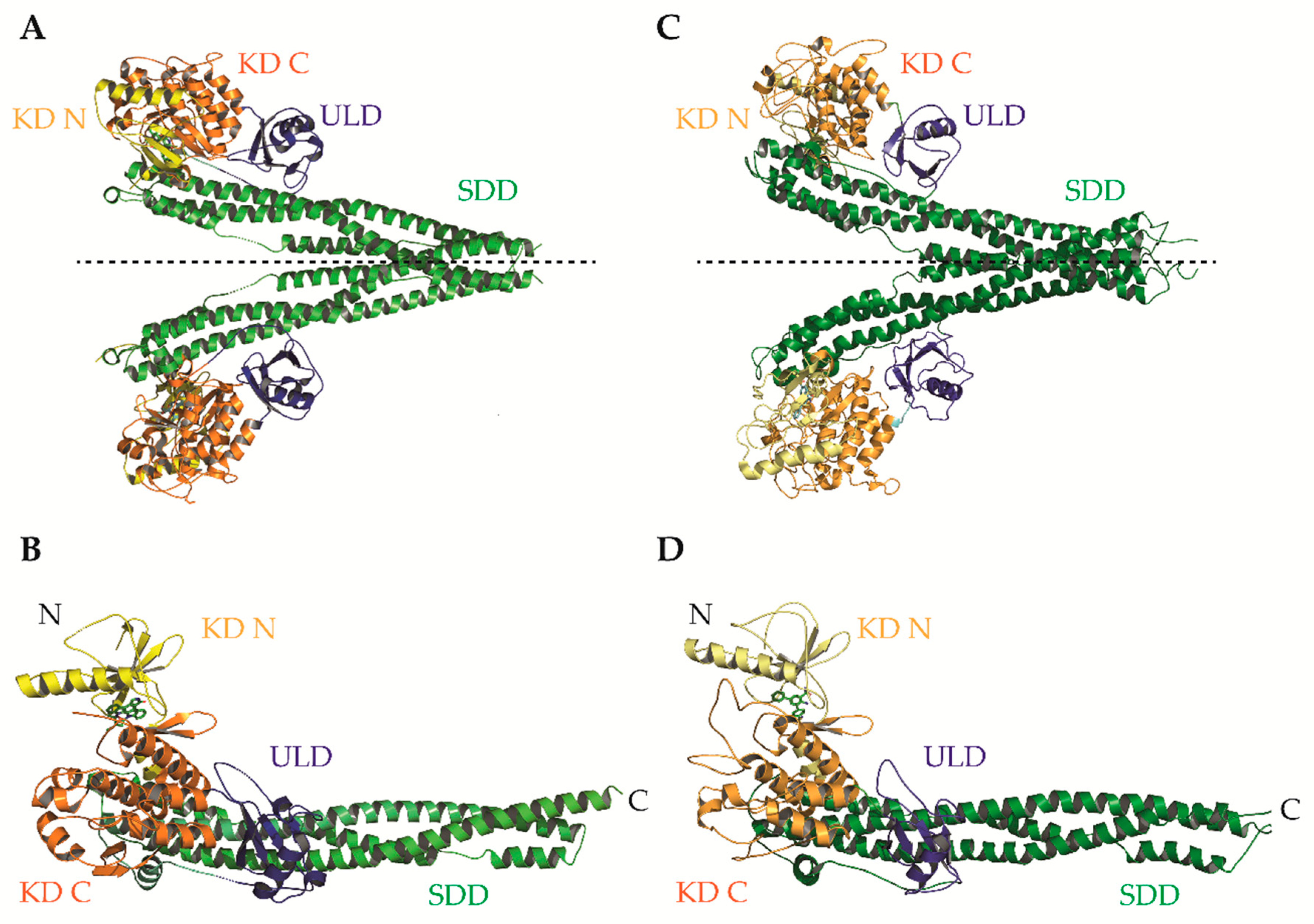{kind=link}
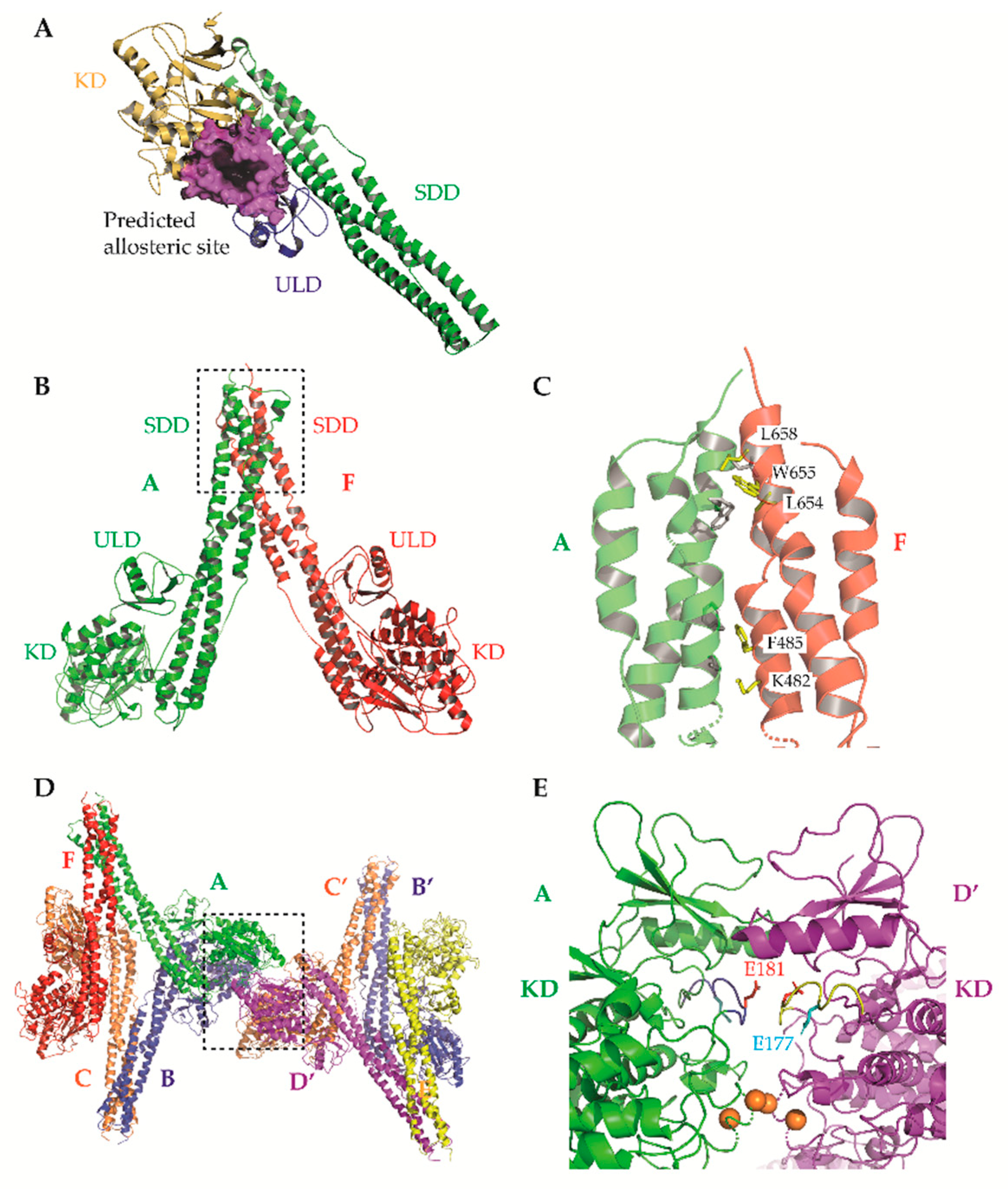{kind=link}
Table 1.
Commercially available IKKβ inhibitors.
| Inhibitor | Mechanism | Ki/IC50 for IKKβ (nM) * [Ref] | Selectivity Over IKKα | Known Off-Targets | Bio-Availability | Pre-Clinical Therapeutic Efficacy |
|---|---|---|---|---|---|---|
| BI605906 (BIX02514) | ATP-competitive | 380 [40] | >300 fold (>100 µM) | >300-fold selectivity over 100 representative tyr/ser-thr kinases IGF1 (7.6 µM) | N/A | N/A |
| MLN120B | ATP-competitive | 60 [41] | >1000 fold (>100 µM) | >1000-fold selectivity over 30 representative tyr/ser-thr kinases | Good oral bio-availability | Multiple myeloma [42] Arthritis [43] |
| PHA-408 | ATP-competitive | 10–40 [44,45] | >350 fold (14 µM) | >100-fold selectivity over 30 representative tyr/ser-thr kinases PIM-1 (0.6 µM) | Good oral bio-availability | Arthritis [44] COPD [46,47] |
| TPCA-1 (IKK inhibitor IV) | ATP-competitive | 18 [48] | ~22-fold (400 nM) | STAT3 | Poor oral bio-availabilityAdministered intra-peritoneally | Arthritis [48] Nasal epithelium inflammation [49] Glioma [50] NSCLC [51] COPD [52] Wet AMD [53] |
| SC-514 | ATP-competitive | 3000–12,000 [54] | >15-fold (>200 µM) | CDK2/CycA (61 µM) Aurora A (71 µM) PRAK (75 µM) MSK (123 µM) | Poor oral bio-availabilityAdministered intra-peritoneally | Rat model of inflammation [54] Oral squamous cell carcinoma [55] Osteoclast-related disorders [56] Diabetic neuropathy [57] |
| LY2409881 | ATP-competitive | 30 [58] | > 10-fold | >10-fold selectivity over panel of representative tyr/ser-thr kinases | Administered intra-peritoneally | DLBCL [58] |
| PS-1145 | ATP-competitive | 100 [59,60] | N/A | [61] | Administered intra-peritoneally | Multiple myeloma [61] DLBCL [62] Graft-versus-host disease [60] Tobacco smoke-induced pulmonary inflammation [63] |
| Compound A (Bay 65-1942) | ATP-competitive | Ki for GST-IκBα = 4 nM [64] | >30 fold (135 nM) | IKKε, MKK4, MKK7, ERK-1, Syk, Lck, Fyn, PI3Kγ, PKA and PKC (IC50 > 10 µM) | Good oral bio-availability | KRAS-induced lung cancer [65] Chronic pulmonary inflammation [64] Ischemia–reperfusion injury [66] LPS-induced neurotoxicity [67] |
| IKK-16 (IKK Inhibitor VII) | ATP-competitive | 40–70 [68,69] | 5-fold (200 nM) | LRKK2 (50 nM) | Good oral bio-availability | Multiple organ failure associated with hemorrhagic shock [70] Sepsis-associated multiple organ dysfunction [71] Ventilation-induced lung injury [72] Acute kidney injury [73] |
| IMD-0354 (and pro-drug IMD-1041) | ATP-competitive | ~1µM [74,75] | N/A | N/A | Administered intra-peritoneally | CLL [76] Pancreatic cancer [77] Adult T-cell leukemia [78] Breast cancer [75] |
| ACHP (IKK inhibitor VIII) | ATP-competitive | 8.5 [79] | 30-fold (250 nM) | IKKε, Syk, MKK4 (IC50 > 20 µM) | Good oral bio-availability | Multiple myeloma [80] Adult T-cell leukemia [81] HIV-1 replication [82] |
| BMS-345541 | Allosteric | 300 [83] | ~13-fold (4000 nM) | >300-fold selectivity over a small panel of representative tyr/ser-thr kinases | Good oral bio-availability | Arthritis [84] Colitis [85] Cardiac graft rejection [86] T-ALL [87] Glioma [50] Prostate cancer [88] |
| Withaferin A | Cys179-binding | [89,90,91,92] | N/A | Broad spectrum inhibitor [93]Vimentin, HSP90, β-tubulin, Desmin, Annexin-A2, Notch-1, STAT1/3 | Poor oral bioavailability | N/A |
| BOT-64 | Ser-177/181 binding | 1000–3000 [94] | N/A | N/A | Administered intra-peritoneally | N/A |
| Ainsliadimer A | Cysteine-46 binding | 30 [95] | N/A | No significant activity against 340 human kinases at 200 nM | Administered intravenously | N/A |
* Value as reported in the reference, from activity or binding assay, not corrected for ATP concentration.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Prescott, J.A.; Cook, S.J. Targeting IKKβ in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKβ Inhibitors. Cells 2018, 7, 115. https://doi.org/10.3390/cells7090115
AMA Style
Prescott JA, Cook SJ. Targeting IKKβ in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKβ Inhibitors. Cells. 2018; 7(9):115. https://doi.org/10.3390/cells7090115
Chicago/Turabian StylePrescott, Jack A., and Simon J. Cook. 2018. "Targeting IKKβ in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKβ Inhibitors" Cells 7, no. 9: 115. https://doi.org/10.3390/cells7090115
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.