Liver Immune Cells Release Type 1 Interferon Due to DNA Sensing and Amplify Liver Injury from Acetaminophen Overdose

 ,
,  , ,
, , 
 add
Show full author list
add
Show full author list
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Drug-Induced Liver Injury Model
2.3. In Vivo Mouse Imaging
2.4. Primary Murine Hepatocytes
2.5. Liver Non-Parenchymal Cell (NPC) Isolation
2.6. RNA Extraction and Real-Time PCR
2.7. GSH Quantification Assay
2.8. Statistical Analysis
3. Results
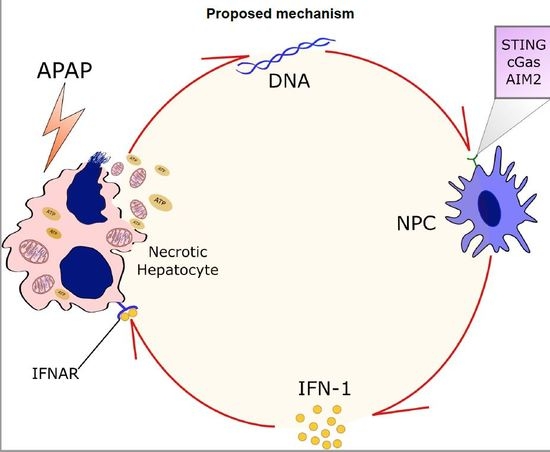
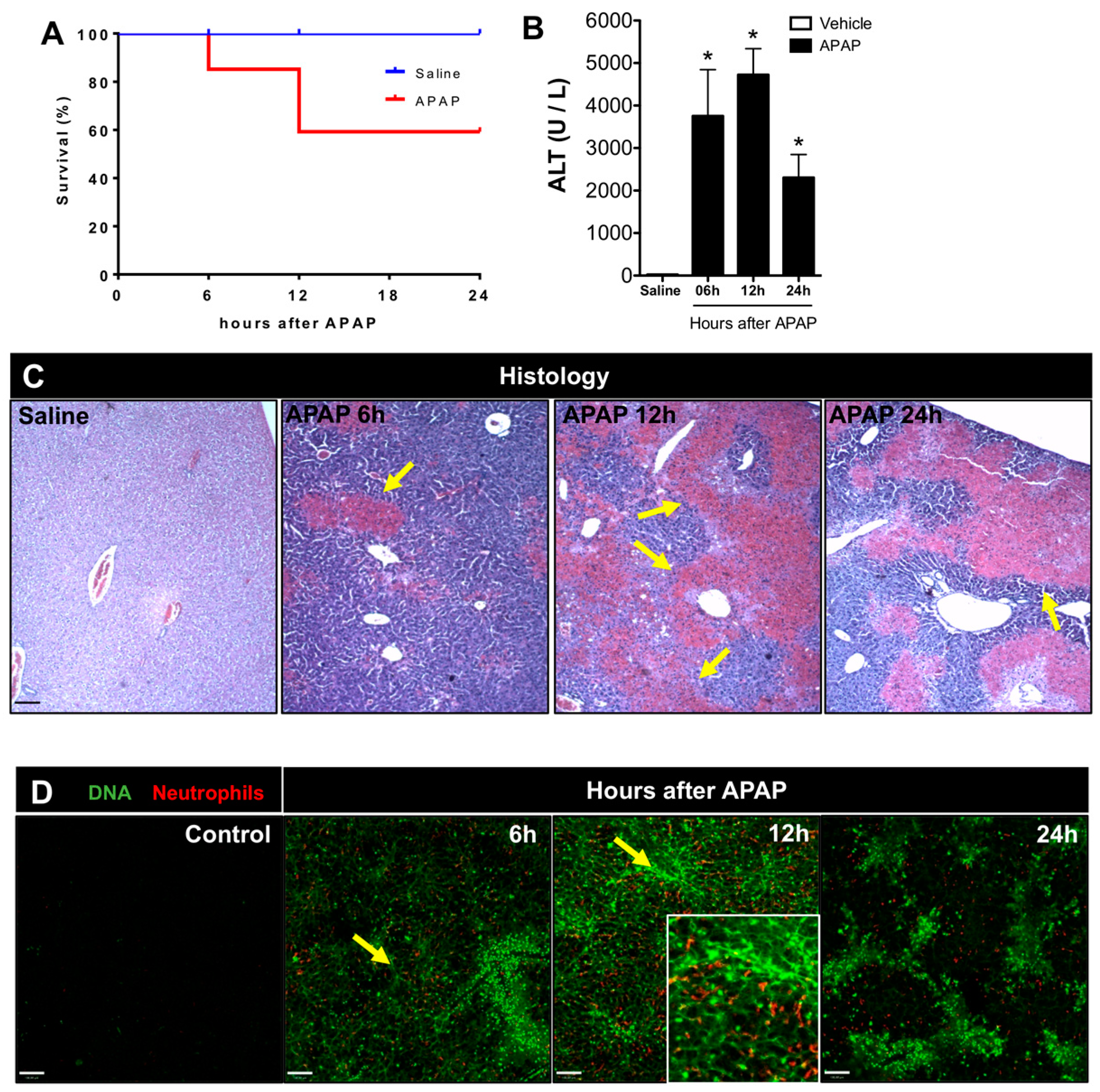
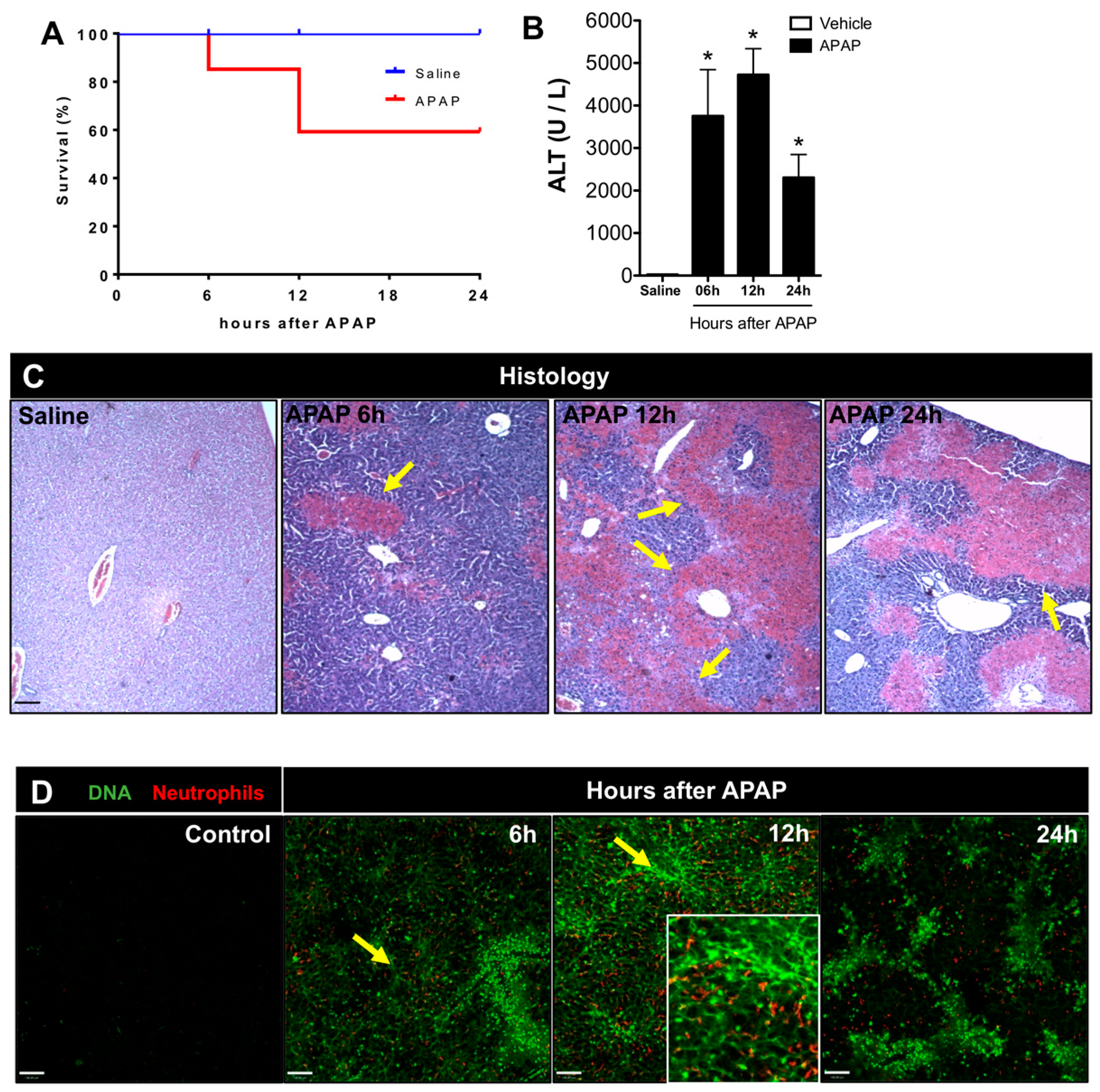
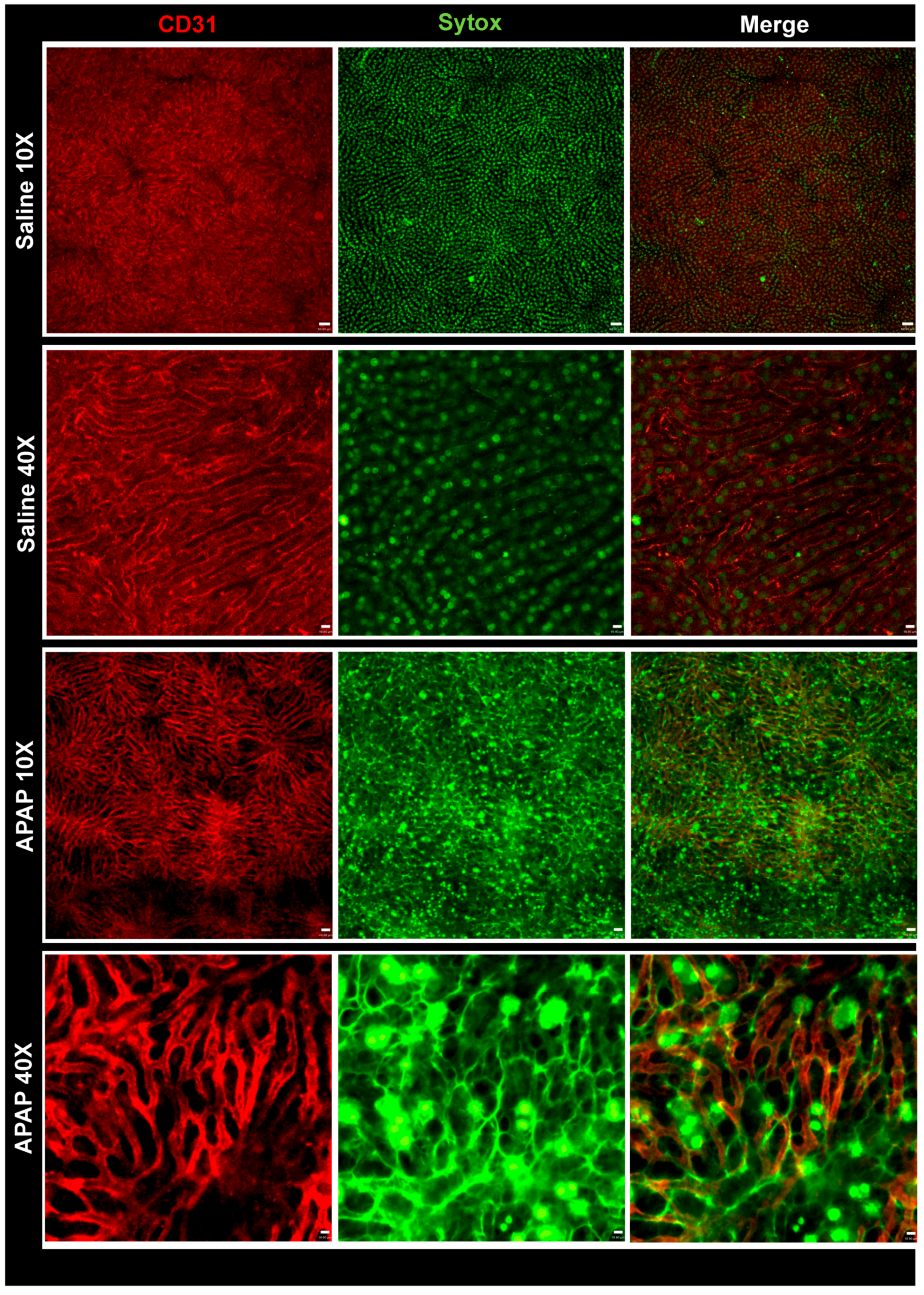
3.1. APAP Overdose Causes Massive DNA Accumulation within Necrotic Areas and Liver Sinusoids
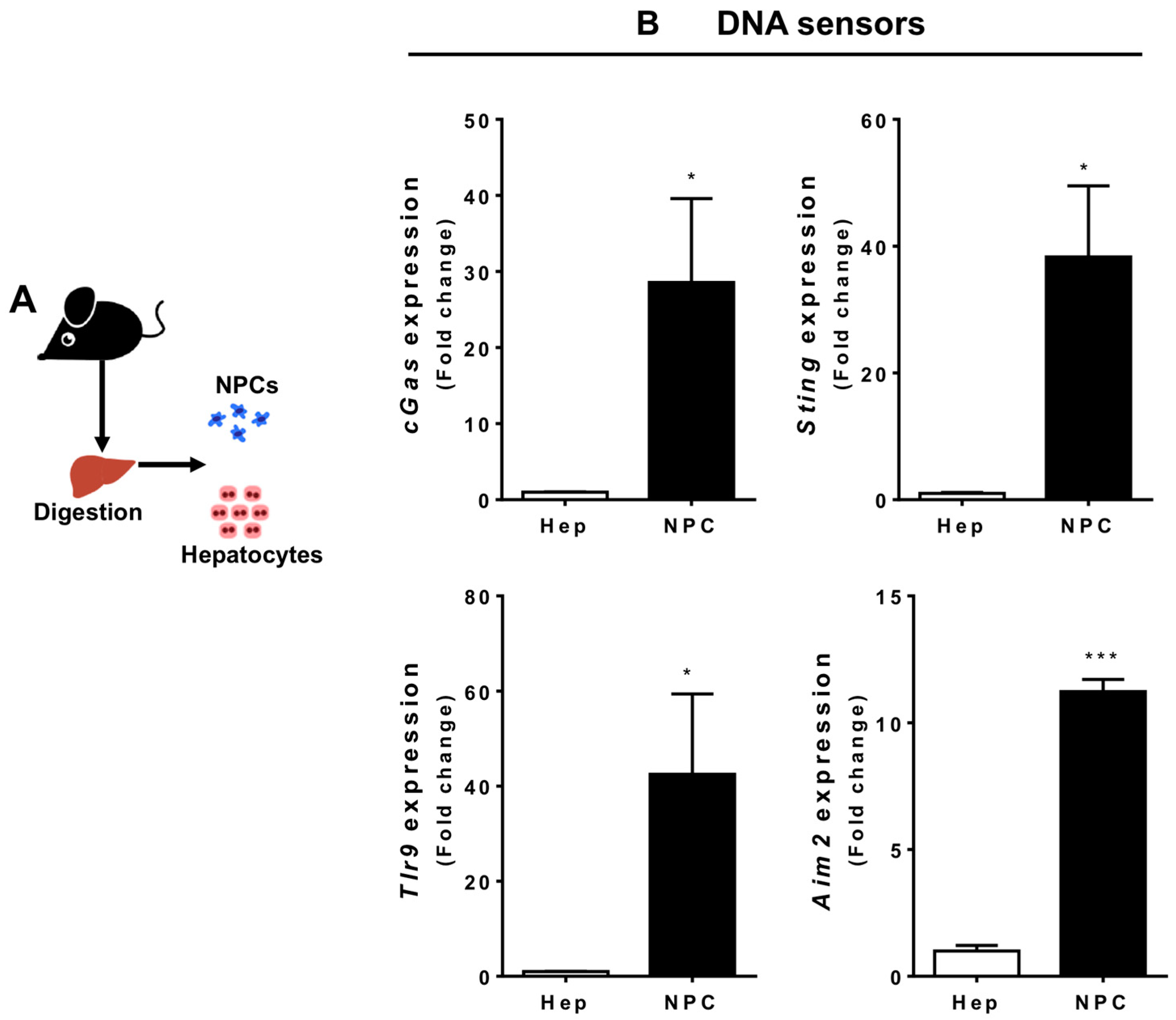
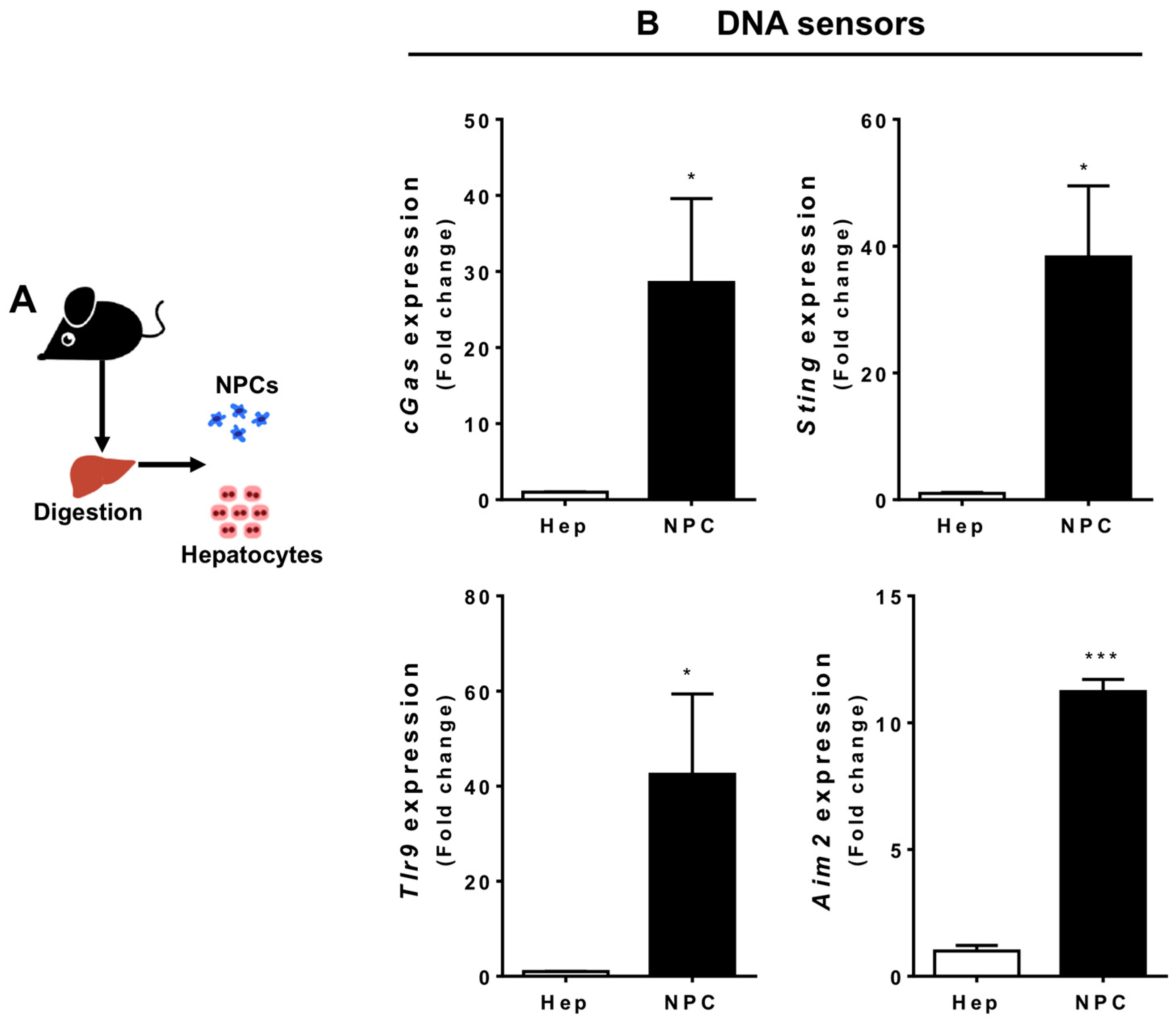
3.2. Liver Non-Parenchymal Cells Express Higher Levels of DNA Sensors than Hepatocytes during Homeostasis
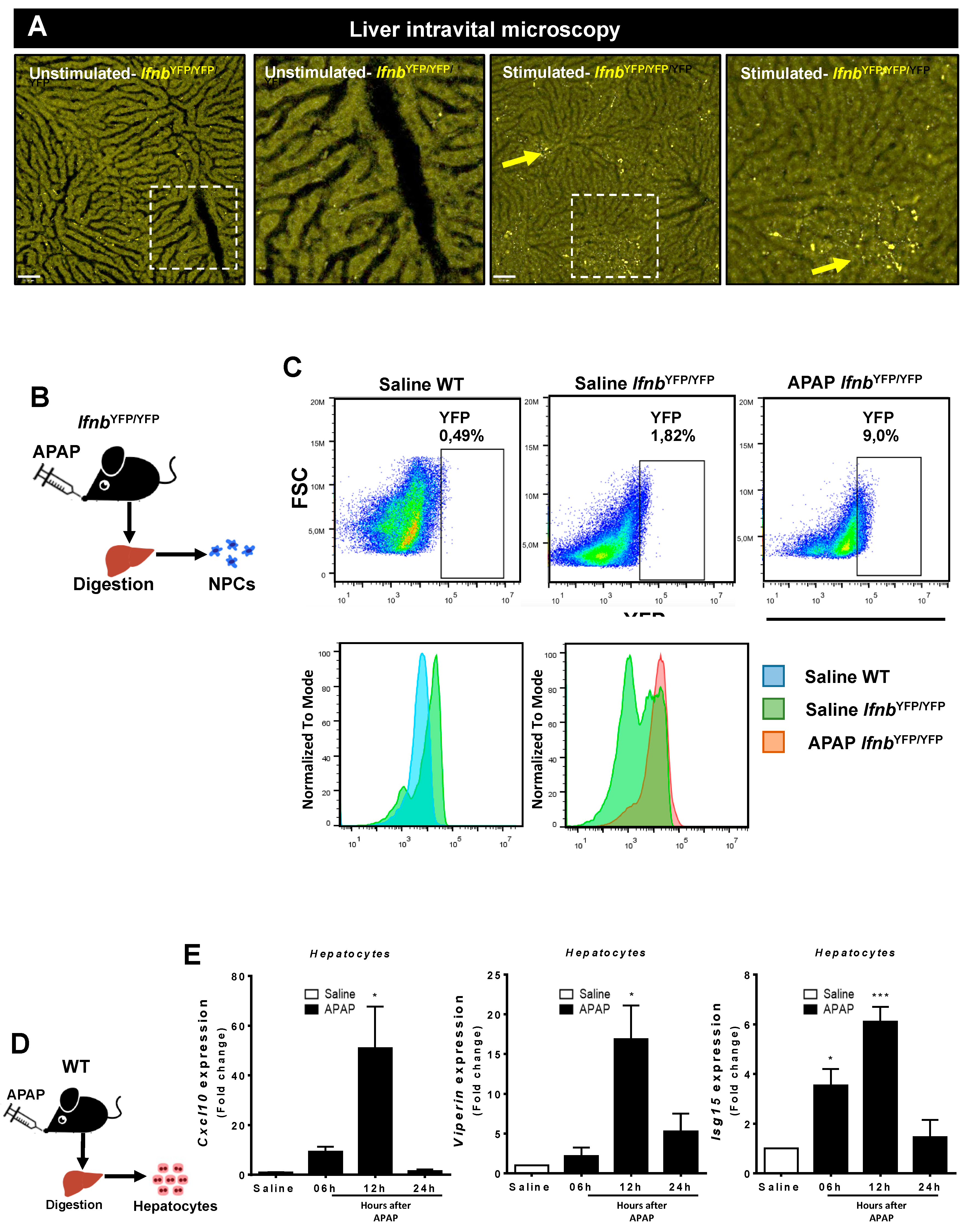
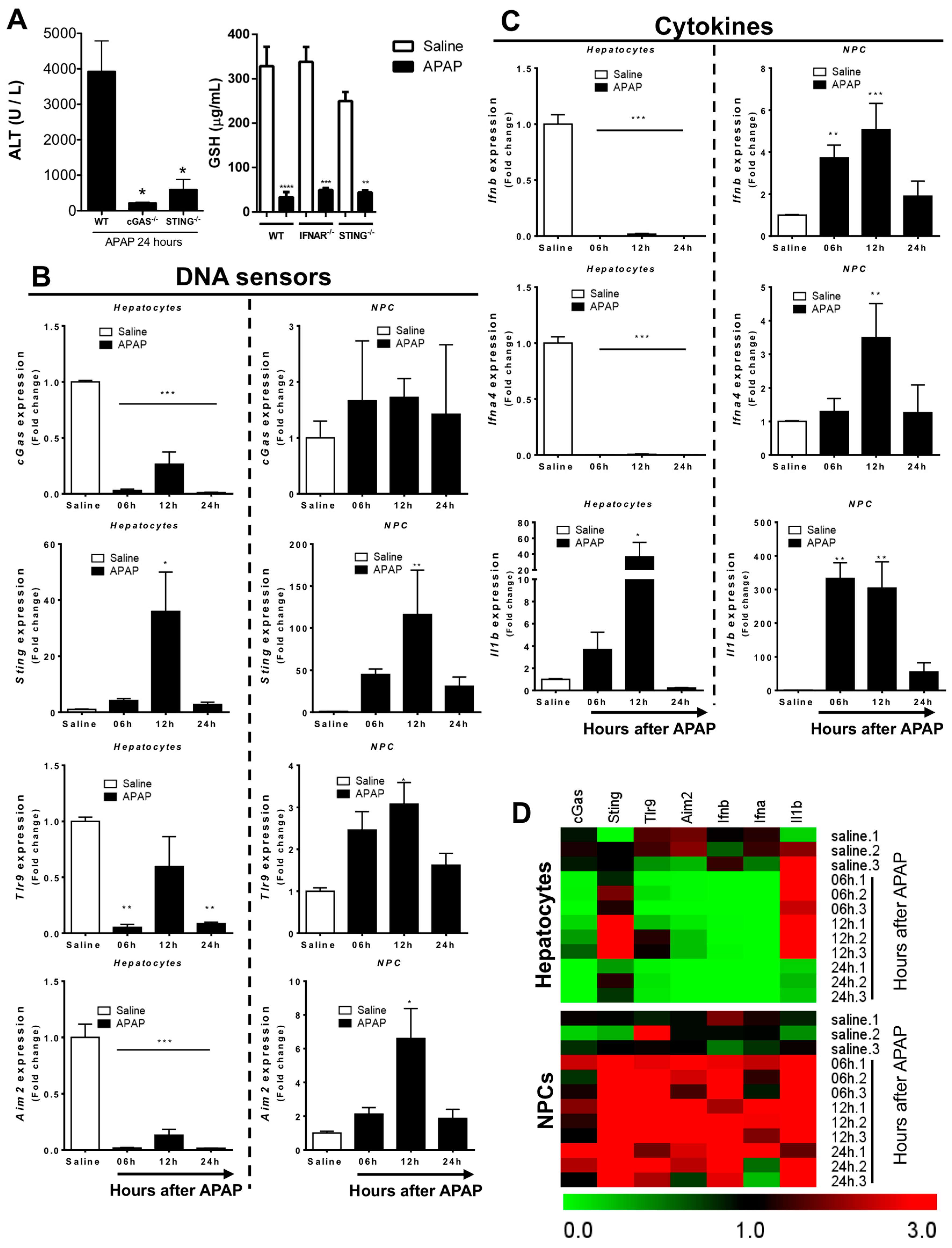
3.3. Liver Non-Parenchymal Cells Release Type 1 IFN during APAP Overdose, Which Is Concomitantly Sensed by Hepatocytes
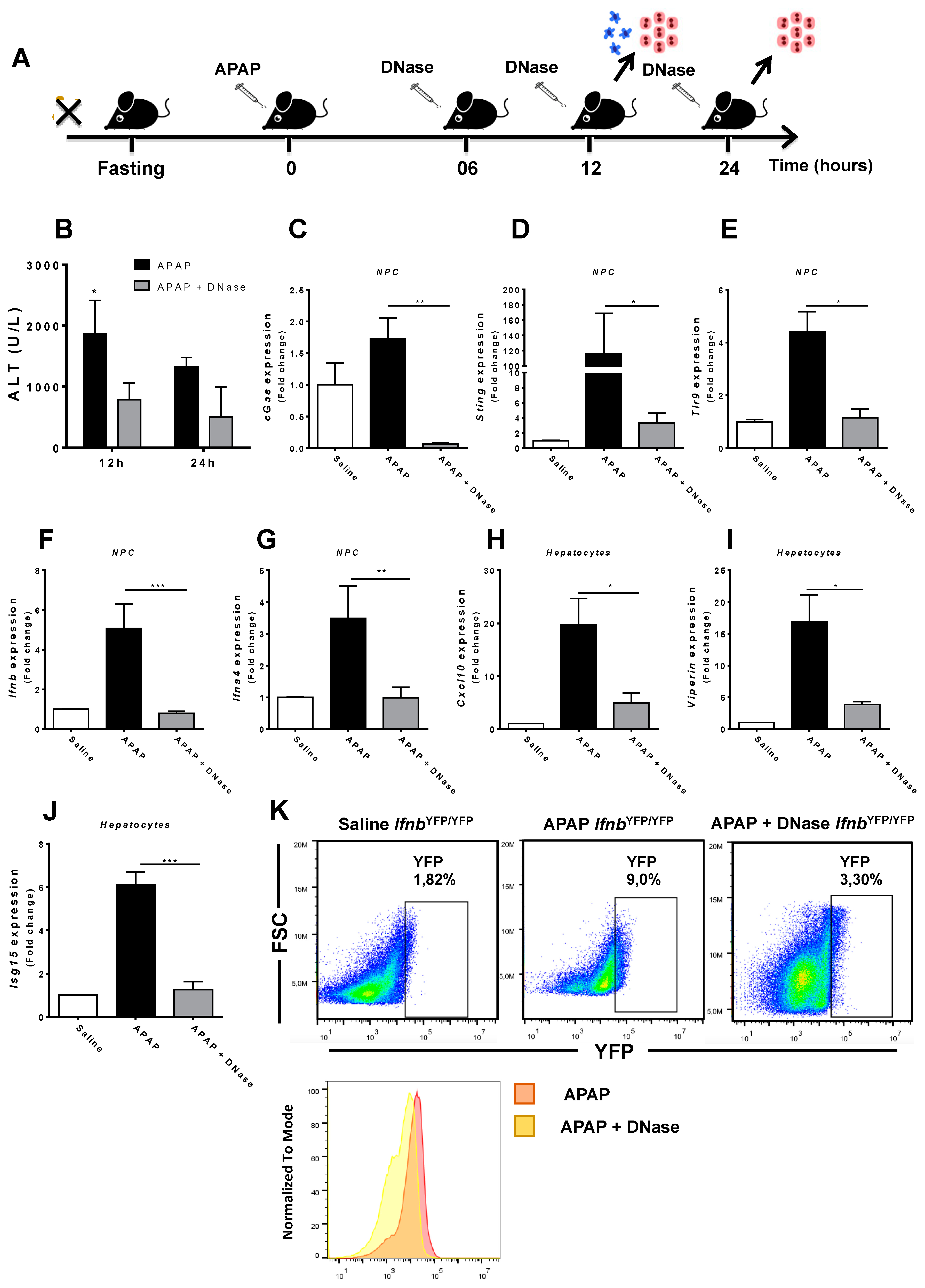
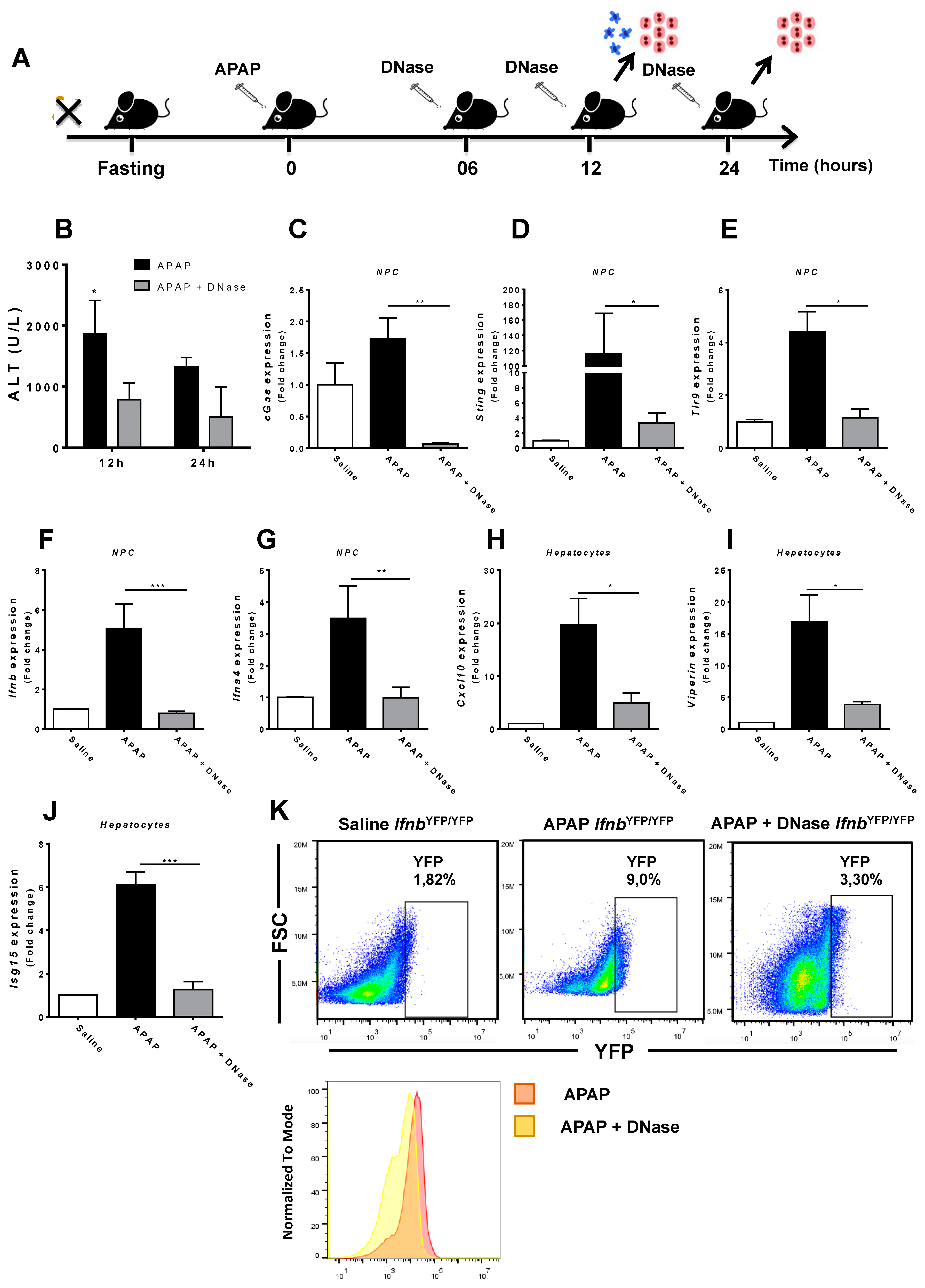
3.4. Absence of Extracellular DNA Abrogates IFN-Beta Production and Sensing within the Liver during APAP-Induced Injury
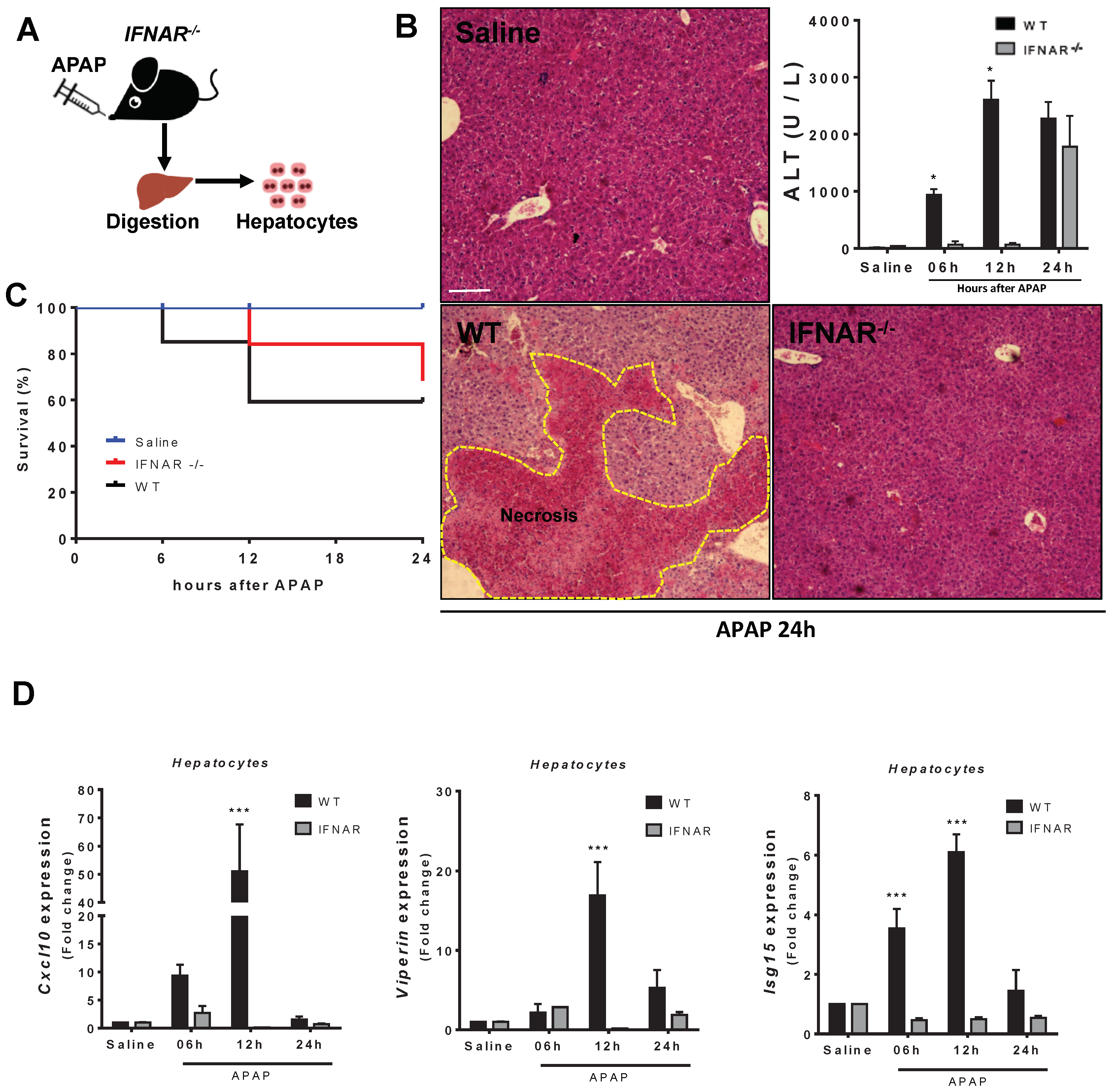
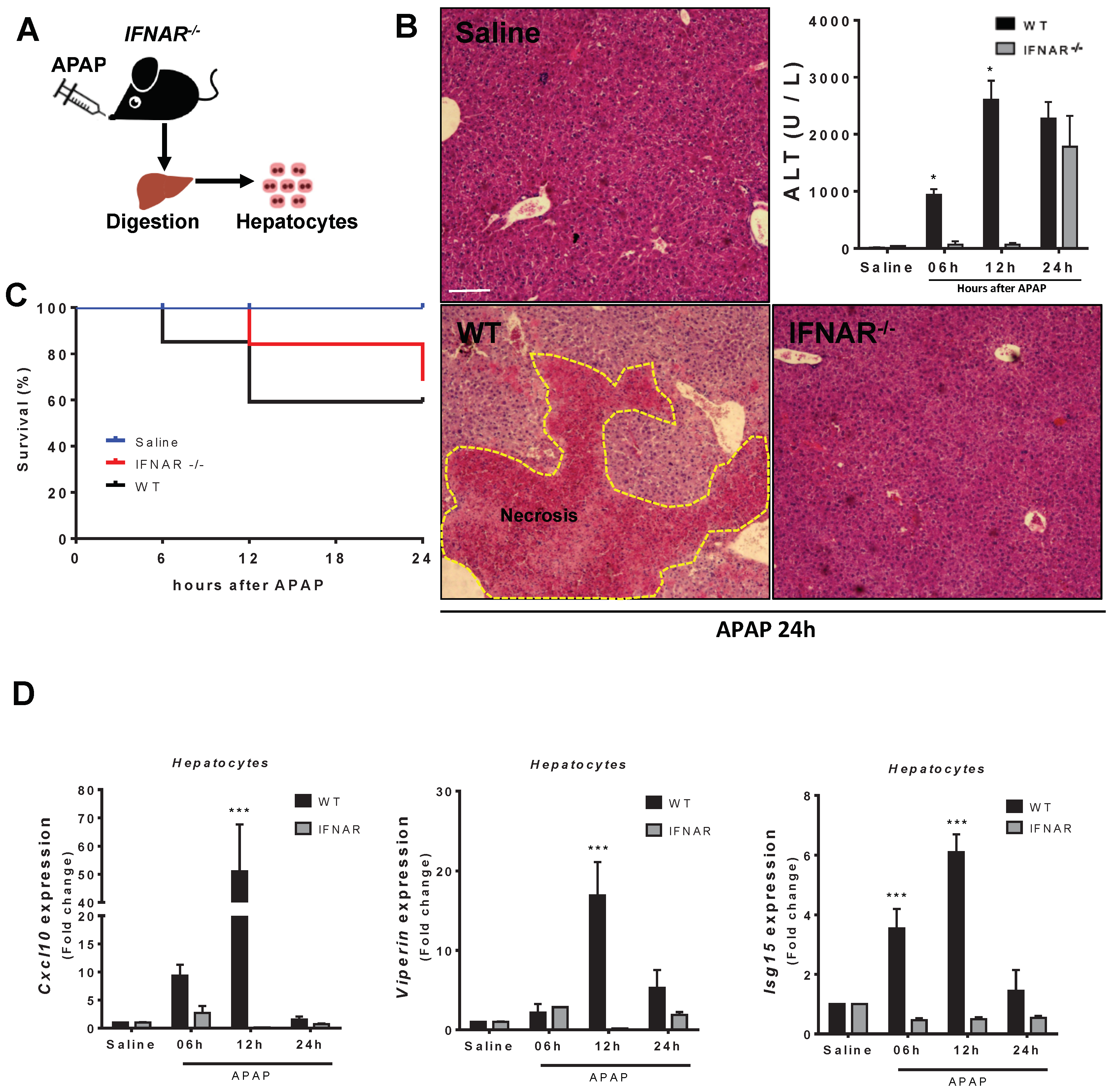
3.5. Lack of Type 1 IFN Signaling Protects Mice from APAP-Induced Liver Injury
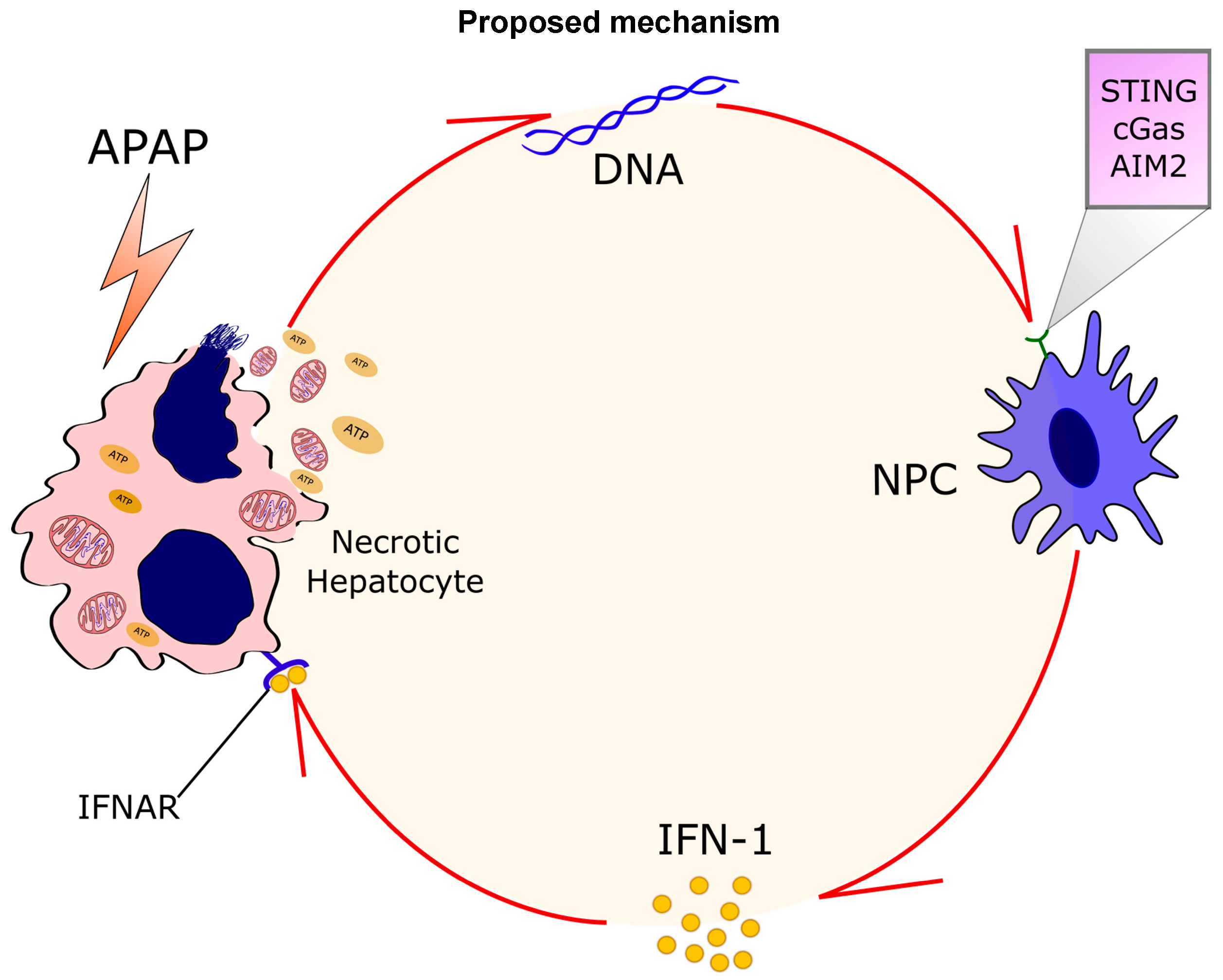
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lee, W.M. Acetaminophen toxicity: Changing perceptions on a social/medical issue. Hepatology 2007, 46, 966–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, P.E.; Amaral, S.S.; Pires, D.A.; Nogueira, L.L.; Soriani, F.M.; Lima, B.H.; Lopes, G.A.; Russo, R.C.; Avila, T.V.; Melgaco, J.G.; et al. Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology 2012, 56, 1971–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, P.E.; Antunes, M.M.; David, B.A.; Pereira, R.V.; Teixeira, M.M.; Menezes, G.B. Imaging liver biology in vivo using conventional confocal microscopy. Nat. Protoc. 2015, 10, 258–268. [Google Scholar] [CrossRef] [PubMed]
- James, L.P.; Mayeux, P.R.; Hinson, J.A. Acetaminophen-induced hepatotoxicity. Drug Metab. Dispos. 2003, 31, 1499–1506. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H.; Kono, H.; Patel, Z.; Kimura, Y.; Rock, K.L. Evaluation of the contribution of multiple DAMPs and DAMP receptors in cell death-induced sterile inflammatory responses. PLoS ONE 2014, 9, e104741. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.W.; Taylor, M.H.; Hickey, R.D.; Hanlon Newell, A.E.; Lenzi, M.L.; Olson, S.B.; Finegold, M.J.; Grompe, M. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature 2010, 467, 707–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guidotti, J.E.; Bregerie, O.; Robert, A.; Debey, P.; Brechot, C.; Desdouets, C. Liver cell polyploidization: A pivotal role for binuclear hepatocytes. J. Biol. Chem. 2003, 278, 19095–19101. [Google Scholar] [CrossRef] [PubMed]
- Miyaoka, Y.; Ebato, K.; Kato, H.; Arakawa, S.; Shimizu, S.; Miyajima, A. Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Curr. Biol. 2012, 22, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.J.; Chen, F.; Lau, J.T.Y.; Hu, Y.P. Hepatocyte polyploidization and its association with pathophysiological processes. Cell Death Dis. 2017, 8, e2805. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Latz, E. Intracellular DNA recognition. Nat. Rev. Immunol. 2010, 10, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Paludan, S.R.; Bowie, A.G. Immune sensing of DNA. Immunity 2013, 38, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Radoshevich, L.; Dussurget, O. Cytosolic Innate Immune Sensing and Signaling upon Infection. Front. Microbiol. 2016, 7, 313. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. STING-dependent cytosolic DNA sensing pathways. Trends Immunol. 2014, 35, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signaling. Nature 2008, 456, 274. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- David, B.A.; Rubino, S.; Moreira, T.G.; Freitas-Lopes, M.A.; Araujo, A.M.; Paul, N.E.; Rezende, R.M.; Menezes, G.B. Isolation and high-dimensional phenotyping of gastrointestinal immune cells. Immunology 2017, 151, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Amaral, S.S.; Oliveira, A.G.; Marques, P.E.; Quintao, J.L.; Pires, D.A.; Resende, R.R.; Sousa, B.R.; Melgaco, J.G.; Pinto, M.A.; Russo, R.C.; et al. Altered responsiveness to extracellular ATP enhances acetaminophen hepatotoxicity. Cell Commun. Signal. 2013, 11, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, C.L.; Jensen, J.L.; Orntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. TM4: A free, open-source system for microarray data management and analysis. Biotechniques 2003, 34, 374–378. [Google Scholar] [PubMed]
- Howe, E.A.; Sinha, R.; Schlauch, D.; Quackenbush, J. RNA-Seq analysis in MeV. Bioinformatics 2011, 27, 3209–3210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tietze, F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: Applications to mammalian blood and other tissues. Anal. Biochem. 1969, 27, 502–522. [Google Scholar] [CrossRef]
- Marques, P.E.; Oliveira, A.G.; Pereira, R.V.; David, B.A.; Gomides, L.F.; Saraiva, A.M.; Pires, D.A.; Novaes, J.T.; Patricio, D.O.; Cisalpino, D.; et al. Hepatic DNA deposition drives drug-induced liver injury and inflammation in mice. Hepatology 2015, 61, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Hegazy, A.N.; Deigendesch, N.; Kosack, L.; Cupovic, J.; Kandasamy, R.K.; Hildebrandt, A.; Merkler, D.; Kuhl, A.A.; Vilagos, B.; et al. Superoxide Dismutase 1 Protects Hepatocytes from Type I Interferon-Driven Oxidative Damage. Immunity 2015, 43, 974–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomsen, M.K.; Nandakumar, R.; Stadler, D.; Malo, A.; Valls, R.M.; Wang, F.; Reinert, L.S.; Dagnaes-Hansen, F.; Hollensen, A.K.; Mikkelsen, J.G.; et al. Lack of immunological DNA sensing in hepatocytes facilitates hepatitis B virus infection. Hepatology 2016, 64, 746–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutz, P.; Metz, P.; Lempp, F.A.; Bender, S.; Qu, B.; Schoneweis, K.; Seitz, S.; Tu, T.; Restuccia, A.; Frankish, J.; et al. HBV Bypasses the Innate Immune Response and Does Not Protect HCV From Antiviral Activity of Interferon. Gastroenterology 2018, 154, 1791–1804. [Google Scholar] [CrossRef] [PubMed]
- Dusheiko, G. Side effects of alpha interferon in chronic hepatitis C. Hepatology 1997, 26, 112S–121S. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araujo, A.M.d.; Antunes, M.M.; Mattos, M.S.; Diniz, A.B.; Alvarenga, D.M.; Nakagaki, B.N.; Carvalho, É.D.; Lacerda, V.A.S.; Carvalho-Gontijo, R.; Goulart, J.; et al. Liver Immune Cells Release Type 1 Interferon Due to DNA Sensing and Amplify Liver Injury from Acetaminophen Overdose. Cells 2018, 7, 88. https://doi.org/10.3390/cells7080088
Araujo AMd, Antunes MM, Mattos MS, Diniz AB, Alvarenga DM, Nakagaki BN, Carvalho ÉD, Lacerda VAS, Carvalho-Gontijo R, Goulart J, et al. Liver Immune Cells Release Type 1 Interferon Due to DNA Sensing and Amplify Liver Injury from Acetaminophen Overdose. Cells. 2018; 7(8):88. https://doi.org/10.3390/cells7080088
Chicago/Turabian StyleAraujo, Alan Moreira de, Maísa Mota Antunes, Matheus Silvério Mattos, Ariane Barros Diniz, Débora Moreira Alvarenga, Brenda Naemi Nakagaki, Érika De Carvalho, Viviane Aparecida Souza Lacerda, Raquel Carvalho-Gontijo, Jorge Goulart, and et al. 2018. "Liver Immune Cells Release Type 1 Interferon Due to DNA Sensing and Amplify Liver Injury from Acetaminophen Overdose" Cells 7, no. 8: 88. https://doi.org/10.3390/cells7080088