Lafora Disease: A Ubiquitination-Related Pathology


Abstract
:1. Introduction
2. Genetic Basis of the Disease
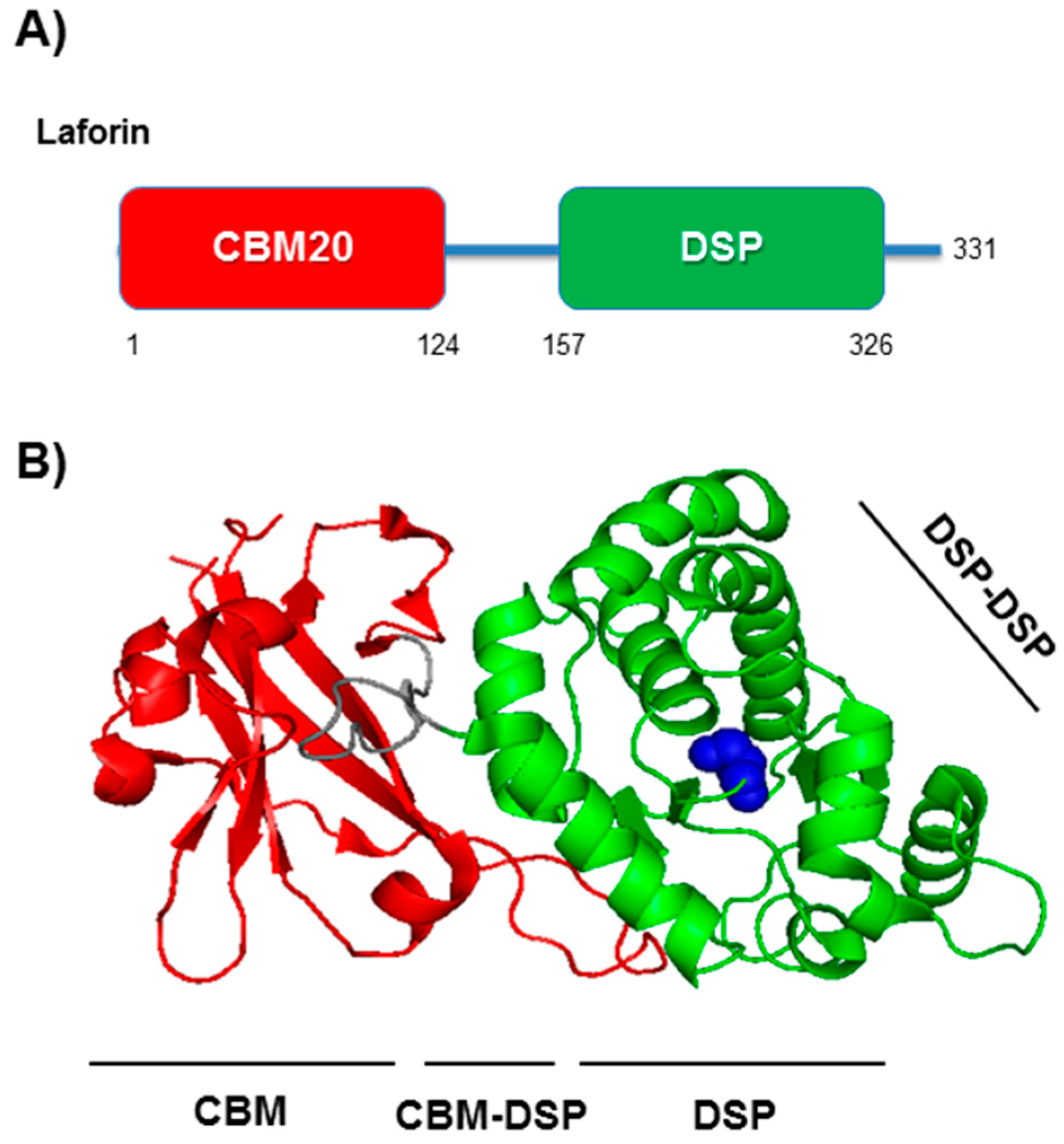
3. Laforin
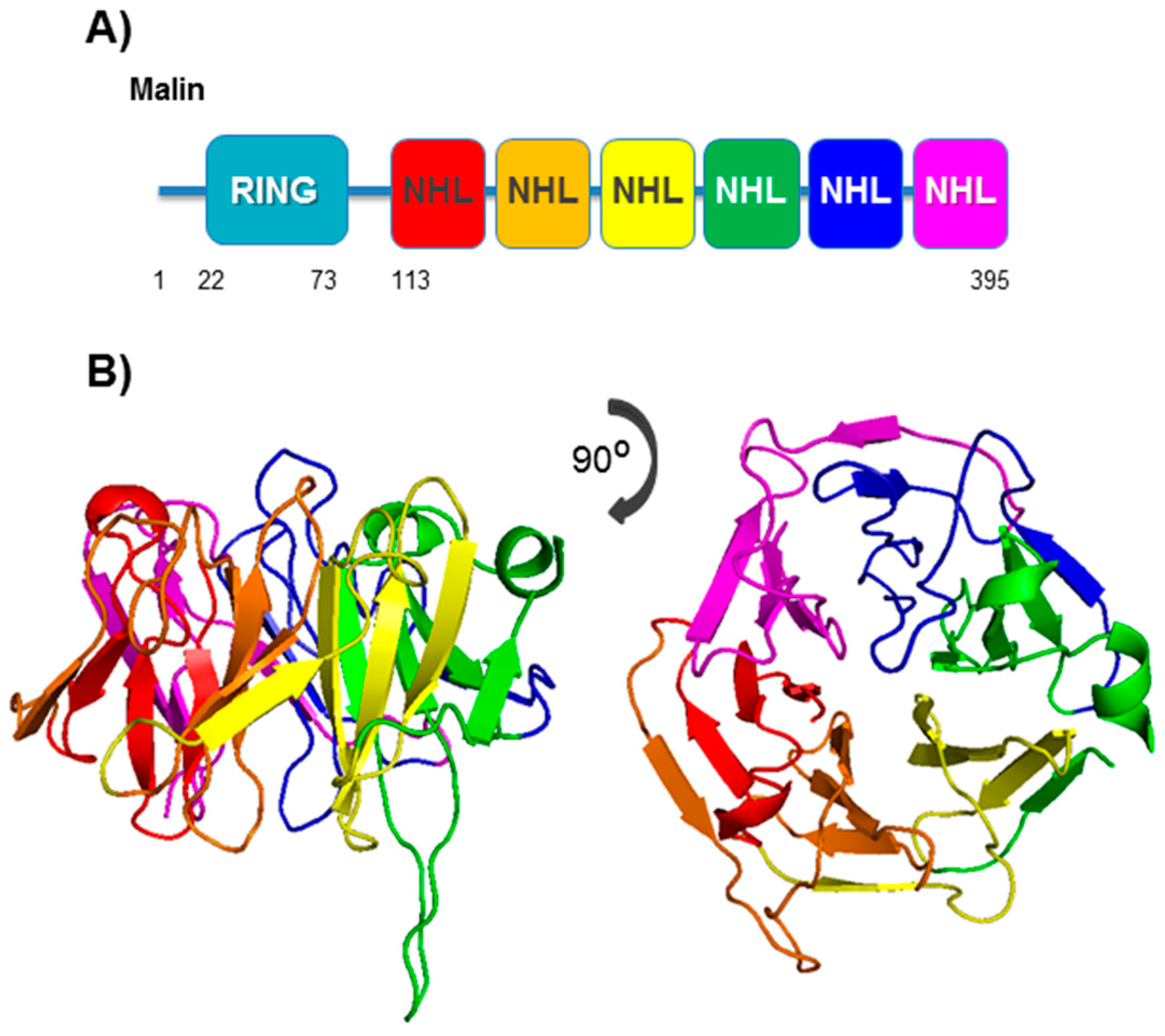
4. Malin
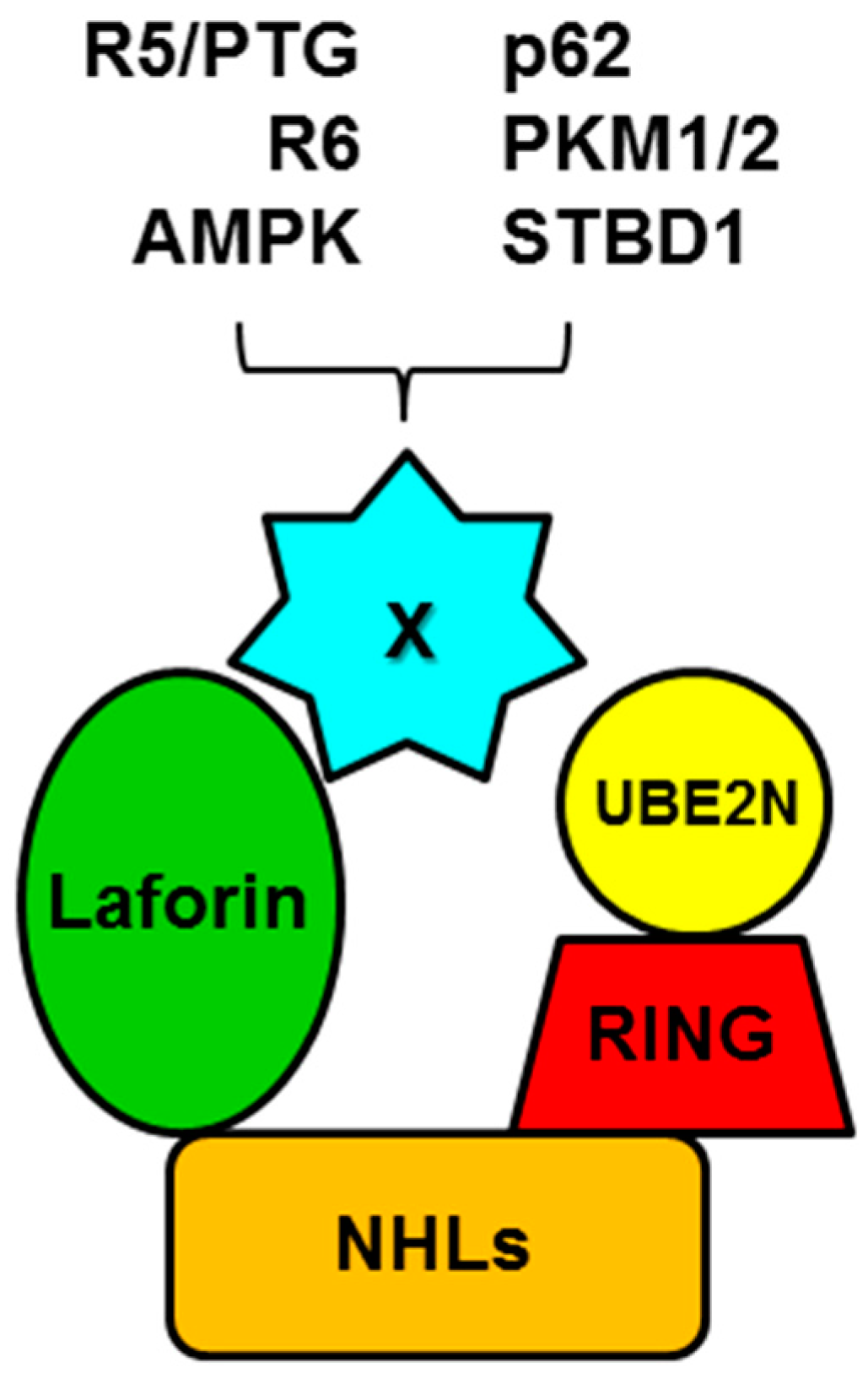
5. Laforin and Malin form a Functional Complex
6. Subcellular Localization of Laforin and Malin
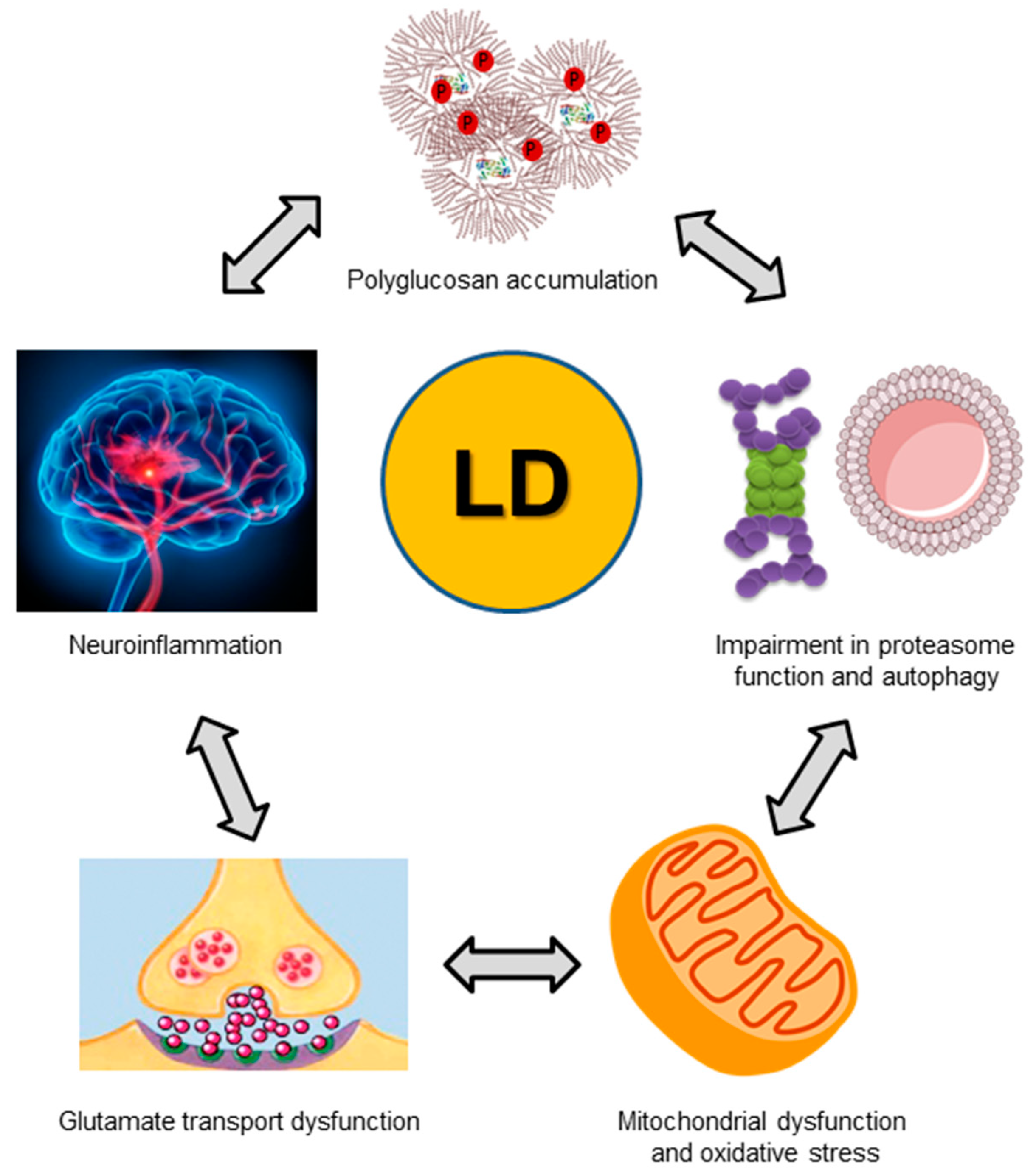
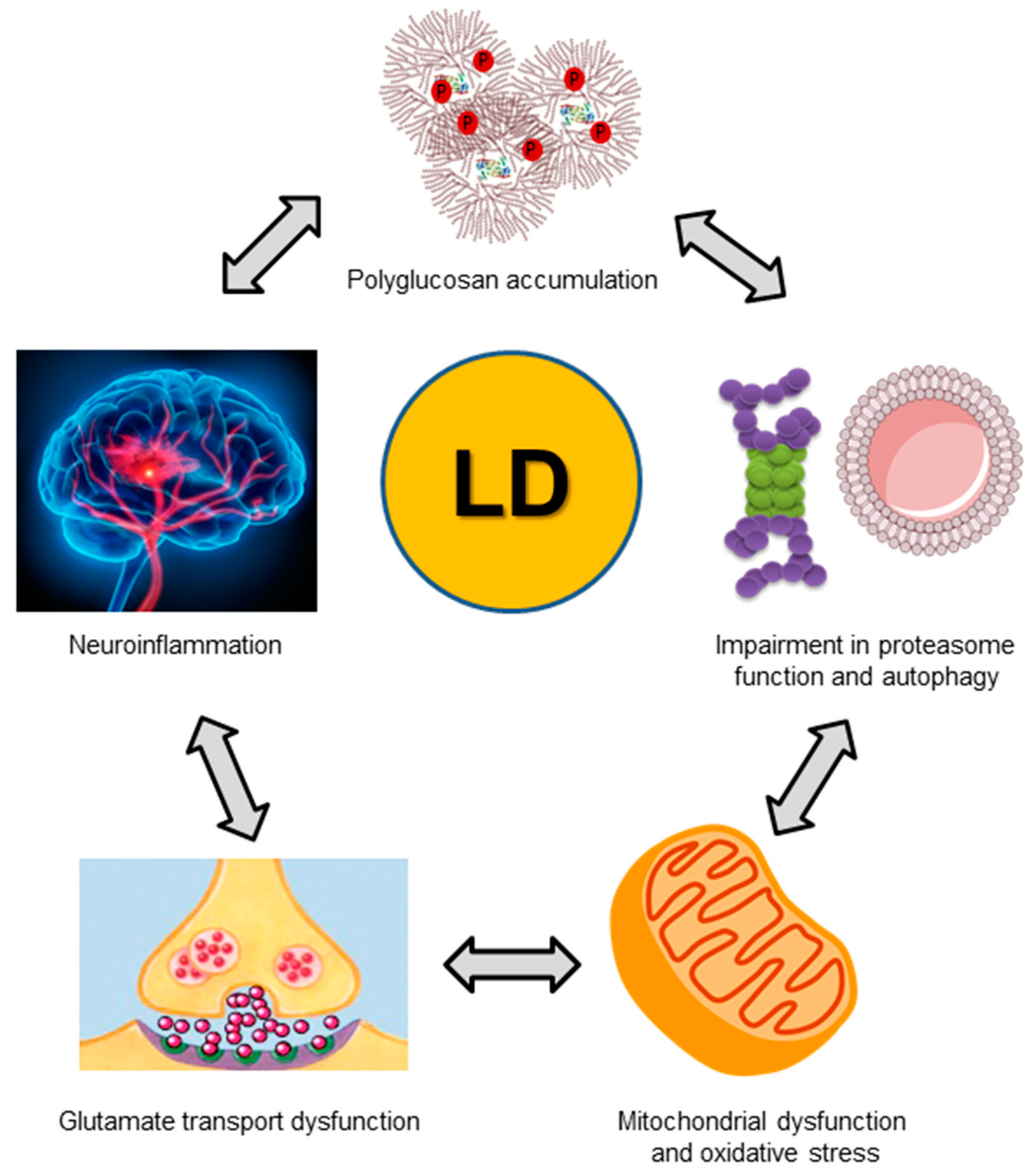
7. Pathophysiological Consequences of a Dysfunctional Laforin-Malin Complex
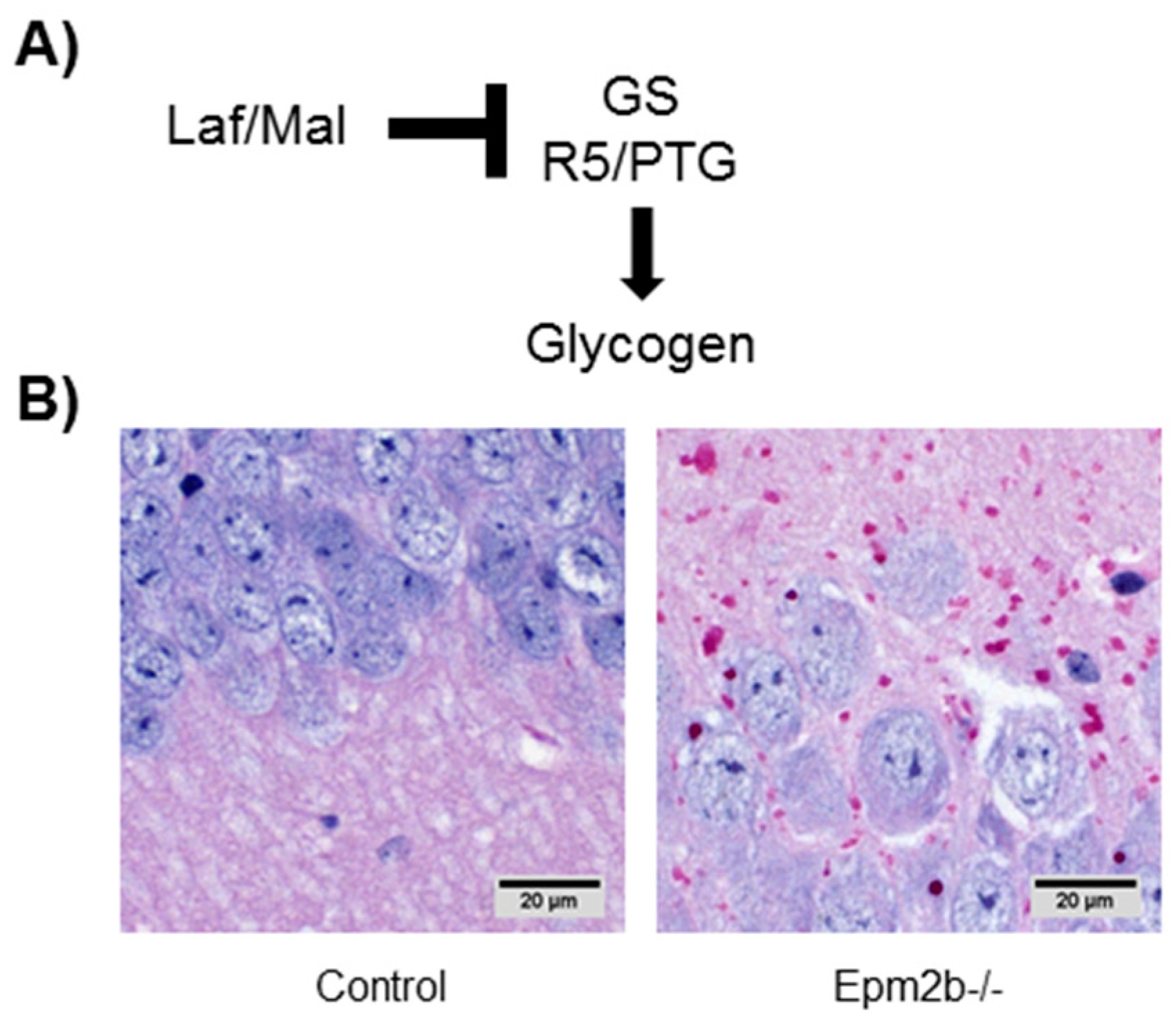
7.1. Altered Regulation of Glycogen Synthesis
7.2. Altered Homeostasis of Glucose Transporters
7.3. Altered Regulation of Proteostasis
7.4. Altered Homeostasis of the Astrocytic Glutamate Transporter
7.5. Enhanced Inflammatory Reaction
7.6. Altered Wnt Signaling Pathway
7.7. Altered Function of Processing Bodies
8. Treatment Strategies
8.1. Treatments Based on Decreasing Glycogen Synthesis
8.2. Treatments Based in Recovering Proteostasis and Decreasing Inflammation
8.3. Treatments Based in Decreasing Excitatory Neurotransmission
9. Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Zupanc, M.L.; Legros, B. Progressive myoclonic epilepsy. Cerebellum 2004, 3, 156–171. [Google Scholar] [CrossRef] [PubMed]
- Shahwan, A.; Farrell, M.; Delanty, N. Progressive myoclonic epilepsies: A review of genetic and therapeutic aspects. Lancet Neurol. 2005, 4, 239–248. [Google Scholar] [CrossRef]
- Minassian, B.A.; Striano, P.; Avanzini, G. Progressive myoclonus epilepsy: The gene-empowered era. Epileptic Disord. 2016, 18, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Lafora, G.R.; Glueck, B. Beitrag zur Histopathologie der myoklonischen Epilepsie. Z. Gesamte Neurol. Psychiatr. 1911, 6, 1–14. [Google Scholar] [CrossRef]
- Sakai, M.; Austin, J.; Witmer, F.; Trueb, L. Studies in myoclonus epilepsy (Lafora body form). Polyglucosans in the systemic deposits of myoclonus epilepsy and in corpora amylacea. Neurology 1970, 20, 160–176. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, T.S.; Delanty, N. Lafora disease: Epidemiology, pathophysiology and management. CNS Drugs 2010, 24, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, J.; Tiberia, E.; Striano, P.; Genton, P.; Carpenter, S.; Ackerley, C.A.; Minassian, B.A. Lafora disease. Epileptic Disord. 2016, 18, 38–62. [Google Scholar] [PubMed]
- Minassian, B.A.; Lee, J.R.; Herbrick, J.A.; Huizenga, J.; Soder, S.; Mungall, A.J.; Dunham, I.; Gardner, R.; Fong, C.Y.; Carpenter, S.; et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat. Genet. 1998, 20, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Serratosa, J.M.; Gomez-Garre, P.; Gallardo, M.E.; Anta, B.; de Bernabe, D.B.; Lindhout, D.; Augustijn, P.B.; Tassinari, C.A.; Malafosse, R.M.; Topcu, M.; et al. A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2A). Hum. Mol. Genet. 1999, 8, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.M.; Young, E.J.; Ianzano, L.; Munteanu, I.; Zhao, X.; Christopoulos, C.C.; Avanzini, G.; Elia, M.; Ackerley, C.A.; Jovic, N.J.; et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat. Genet. 2003, 35, 125–127. [Google Scholar] [CrossRef] [PubMed]
- Casciato, S.; Gambardella, S.; Mascia, A.; Quarato, P.P.; D’Aniello, A.; Ackurina, Y.; Albano, V.; Fornai, F.; Scala, S.; Di Gennaro, G. Severe and rapidly-progressive Lafora disease associated with NHLRC1 mutation: A case report. Int. J. Neurosci. 2017, 127, 1150–1153. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Abad, C.; Gómez-Garre, P.; Gutiérrez-Delicado, E.; Saygi, S.; Michelucci, R.; Tassinari, C.A.; Rodriguez de Cordoba, S.; Serratosa, J.M. Lafora disease due to EPM2B mutations. A clinical and genetic study. Neurology 2005, 64, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Franceschetti, S.; Gambardella, A.; Canafoglia, L.; Striano, P.; Lohi, H.; Gennaro, E.; Ianzano, L.; Veggiotti, P.; Sofia, V.; Biondi, R.; et al. Clinical and genetic findings in 26 italian patients with Lafora disease. Epilepsia 2006, 47, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Striano, P.; Zara, F.; Turnbull, J.; Girard, J.M.; Ackerley, C.A.; Cervasio, M.; De Rosa, G.; Del Basso-De Caro, M.L.; Striano, S.; Minassian, B.A. Typical progression of myoclonic epilepsy of the Lafora type: A case report. Nat. Clin. Pract. Neurol. 2008, 4, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, R.; Vernia, S.; Sanz, R.; Abreu-Rodriguez, I.; Almaraz, C.; Garcia-Hoyos, M.; Michelucci, R.; Tassinari, C.A.; Riguzzi, P.; Nobile, C.; et al. A PTG variant contributes to a milder phenotype in Lafora disease. PLoS ONE 2011, 6, e21294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kecmanovic, M.; Keckarevic-Markovic, M.; Keckarevic, D.; Stevanovic, G.; Jovic, N.; Romac, S. Genetics of Lafora progressive myoclonic epilepsy: Current perspectives. Appl. Clin. Genet. 2016, 9, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Gentry, M.S.; Roma-Mateo, C.; Sanz, P. Laforin, a protein with many faces: Glucan phosphatase, adapter protein, et alii. FEBS J. 2013, 280, 525–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The carbohydrate-active enzymes database (CAZY): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Stuckey, J.A.; Wishart, M.J.; Dixon, J.E. A unique carbohydrate binding domain targets the Lafora disease phosphatase to glycogen. J. Biol. Chem. 2002, 277, 2377–2380. [Google Scholar] [CrossRef] [PubMed]
- Boraston, A.B.; Bolam, D.N.; Gilbert, H.J.; Davies, G.J. Carbohydrate-binding modules: Fine-tuning polysaccharide recognition. Biochem. J. 2004, 382, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Janecek, S.; Svensson, B.; Macgregor, E.A. Structural and evolutionary aspects of two families of non-catalytic domains present in starch and glycogen binding proteins from microbes, plants and animals. Enzyme Microb. Technol. 2011, 49, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Kuchtova, A.; Gentry, M.S.; Janecek, S. The unique evolution of the carbohydrate-binding module CBM20 in laforin. FEBS Lett. 2018, 592, 586–598. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.; Tsurutani, N.; Suzuki, T.; Hoshii, Y.; Ishihara, T.; Delgado-Escueta, A.V.; Yamakawa, K. The carbohydrate-binding domain of Lafora disease protein targets Lafora polyglucosan bodies. Biochem. Biophys. Res. Commun. 2004, 313, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Gentry, M.S.; Dowen, R.H., 3rd; Worby, C.A.; Mattoo, S.; Ecker, J.R.; Dixon, J.E. The phosphatase laforin crosses evolutionary boundaries and links carbohydrate metabolism to neuronal disease. J. Cell Biol. 2007, 178, 477–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein tyrosine phosphatases in the human genome. Cell 2004, 117, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Moorhead, G.B.; De Wever, V.; Templeton, G.; Kerk, D. Evolution of protein phosphatases in plants and animals. Biochem. J. 2009, 417, 401–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worby, C.A.; Gentry, M.S.; Dixon, J.E. Laforin, a dual specificity phosphatase that dephosphorylates complex carbohydrates. J. Biol. Chem. 2006, 281, 30412–30418. [Google Scholar] [CrossRef] [PubMed]
- Tagliabracci, V.S.; Turnbull, J.; Wang, W.; Girard, J.M.; Zhao, X.; Skurat, A.V.; Delgado-Escueta, A.V.; Minassian, B.A.; Depaoli-Roach, A.A.; Roach, P.J. Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 19262–19266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raththagala, M.; Brewer, M.K.; Parker, M.W.; Sherwood, A.R.; Wong, B.K.; Hsu, S.; Bridges, T.M.; Paasch, B.C.; Hellman, L.M.; Husodo, S.; et al. Structural mechanism of laforin function in glycogen dephosphorylation and Lafora disease. Mol. Cell 2015, 57, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Roma-Mateo, C.; Sanz, P.; Gentry, M.S. Deciphering the role of malin in the Lafora progressive myoclonus epilepsy. IUBMB Life 2012, 64, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Gentry, M.S.; Worby, C.A.; Dixon, J.E. Insights into Lafora disease: Malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc. Natl. Acad. Sci. USA 2005, 102, 8501–8506. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martin, P.; Roma-Mateo, C.; Viana, R.; Sanz, P. Ubiquitin conjugating enzyme UBE2N and sequestosome-1 (p62) are components of the ubiquitination process mediated by the malin-laforin e3-ubiquitin ligase complex. Int. J. Biochem. Cell Biol. 2015, 69, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Solaz-Fuster, M.C.; Gimeno-Alcaniz, J.V.; Ros, S.; Fernandez-Sanchez, M.E.; Garcia-Fojeda, B.; Criado Garcia, O.; Vilchez, D.; Dominguez, J.; Garcia-Rocha, M.; Sanchez-Piris, M.; et al. Regulation of glycogen synthesis by the laforin-malin complex is modulated by the AMP-activated protein kinase pathway. Hum. Mol. Genet. 2008, 17, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio-Villena, C.; Garcia-Gimeno, M.A.; Sanz, P. Glycogenic activity of R6, a protein phosphatase 1 regulatory subunit, is modulated by the laforin-malin complex. Int. J. Biochem. Cell Biol. 2013, 45, 1479–1488. [Google Scholar] [CrossRef] [PubMed]
- Moreno, D.; Towler, M.C.; Hardie, D.G.; Knecht, E.; Sanz, P. The laforin-malin complex, involved in Lafora disease, promotes the incorporation of K63-linked ubiquitin chains into AMP-activated protein kinase beta subunits. Mol. Biol. Cell 2010, 21, 2578–2588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viana, R.; Lujan, P.; Sanz, P. The laforin/malin E3-ubiquitin ligase complex ubiquitinates pyruvate kinase M1/M2. BMC Biochem. 2015, 16, 24. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; Mulherkar, S.; Mukherjee, D.; Jana, N.R. Malin regulates WNT signaling pathway through degradation of dishevelled2. J. Biol. Chem. 2012, 287, 6830–6839. [Google Scholar] [CrossRef] [PubMed]
- Vilchez, D.; Ros, S.; Cifuentes, D.; Pujadas, L.; Valles, J.; Garcia-Fojeda, B.; Criado-Garcia, O.; Fernandez-Sanchez, E.; Medrano-Fernandez, I.; Dominguez, J.; et al. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat. Neurosci. 2007, 10, 1407–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worby, C.A.; Gentry, M.S.; Dixon, J.E. Malin decreases glycogen accumulation by promoting the degradation of protein targeting to glycogen (PTG). J. Biol. Chem. 2008, 283, 4069–4076. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Zhang, M.; Gentry, M.S.; Worby, C.A.; Dixon, J.E.; Saltiel, A.R. A role for AGL ubiquitination in the glycogen storage disorders of Lafora and Cori’s disease. Genes Dev. 2007, 21, 2399–2409. [Google Scholar] [CrossRef] [PubMed]
- Roma-Mateo, C.; Moreno, D.; Vernia, S.; Rubio, T.; Bridges, T.M.; Gentry, M.S.; Sanz, P. Lafora disease E3-ubiquitin ligase malin is related to TRIM32 at both the phylogenetic and functional level. BMC Evol. Biol. 2011, 11, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.N.; Sharma, J.; Maity, R.; Jana, N.R. Co-chaperone CHIP stabilizes aggregate-prone malin, a ubiquitin ligase mutated in Lafora disease. J. Biol. Chem. 2010, 285, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Badhwar, I.; Upadhyay, M.; Singh, S.; Ganesh, S. Malin and laforin are essential components of a protein complex that protects cells from thermal stress. J. Cell Sci. 2011, 124, 2277–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohi, H.; Ianzano, L.; Zhao, X.C.; Chan, E.M.; Turnbull, J.; Scherer, S.W.; Ackerley, C.A.; Minassian, B.A. Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy. Hum. Mol. Genet. 2005, 14, 2727–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roma-Mateo, C.; Solaz-Fuster, M.C.; Gimeno-Alcaniz, J.V.; Dukhande, V.; Donderis, J.; Worby, C.A.; Marina, A.; Criado, O.; Koller, A.; Rodriguez de Cordoba, S.; et al. Laforin, a dual specificity protein phosphatase involved in Lafora disease, is phosphorylated at Ser25 by AMP-activated protein kinase. Biochem. J. 2011, 439, 265–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.K.; Singh, S.; Ganesh, S. The laforin-malin complex negatively regulates glycogen synthesis by modulating cellular glucose uptake via glucose transporters. Mol. Cell. Biol. 2012, 32, 652–663. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.; Agarwala, K.L.; Ueda, K.; Akagi, T.; Shoda, K.; Usui, T.; Hashikawa, T.; Osada, H.; Delgado-Escueta, A.V.; Yamakawa, K. Laforin, defective in the progressive myoclonus epilepsy of Lafora type, is a dual-specificity phosphatase associated with polyribosomes. Hum. Mol. Genet. 2000, 9, 2251–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minassian, B.A.; Andrade, D.M.; Ianzano, L.; Young, E.J.; Chan, E.; Ackerley, C.A.; Scherer, S.W. Laforin is a cell membrane and endoplasmic reticulum-associated protein tyrosine phosphatase. Ann. Neurol. 2001, 49, 271–275. [Google Scholar] [CrossRef]
- Tagliabracci, V.S.; Girard, J.M.; Segvich, D.; Meyer, C.; Turnbull, J.; Zhao, X.; Minassian, B.A.; Depaoli-Roach, A.A.; Roach, P.J. Abnormal metabolism of glycogen phosphate as a cause for Lafora disease. J. Biol. Chem. 2008, 283, 33816–33825. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, J.; Wang, P.; Girard, J.M.; Ruggieri, A.; Wang, T.J.; Draginov, A.G.; Kameka, A.P.; Pencea, N.; Zhao, X.; Ackerley, C.A.; et al. Glycogen hyperphosphorylation underlies Lafora body formation. Ann. Neurol. 2010, 68, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Ballester, C.; Berthier, A.; Viana, R.; Sanz, P. Homeostasis of the astrocytic glutamate transporter Glt-1 is altered in mouse models of Lafora disease. Biochim. Biophys. Acta 2016, 1862, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Singh, S.; Ganesh, S. Activation of serum/glucocorticoid-induced kinase 1 (SGK1) underlies increased glycogen levels, mTOR activation, and autophagy defects in Lafora disease. Mol. Biol. Cell 2013, 24, 3776–3786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, J.; Gruart, A.; Garcia-Rocha, M.; Delgado-Garcia, J.M.; Guinovart, J.J. Glycogen accumulation underlies neurodegeneration and autophagy impairment in Lafora disease. Hum. Mol. Genet. 2014, 23, 3147–3156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthier, A.; Paya, M.; Garcia-Cabrero, A.M.; Ballester, M.I.; Heredia, M.; Serratosa, J.M.; Sanchez, M.P.; Sanz, P. Pharmacological interventions to ameliorate neuropathological symptoms in a mouse model of Lafora disease. Mol. Neurobiol. 2016, 53, 1296–1309. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gonzalez, I.; Viana, R.; Sanz, P.; Ferrer, I. Inflammation in Lafora disease: Evolution with disease progression in laforin and malin knock-out mouse models. Mol. Neurobiol. 2017, 54, 3119–3130. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.N.; Maity, R.; Sharma, J.; Dey, P.; Shankar, S.K.; Satishchandra, P.; Jana, N.R. Sequestration of chaperones and proteasome into Lafora bodies and proteasomal dysfunction induced by Lafora disease-associated mutations of malin. Hum. Mol. Genet. 2010, 19, 4726–4734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernia, S.; Rubio, T.; Heredia, M.; Rodriguez de Cordoba, S.; Sanz, P. Increased endoplasmic reticulum stress and decreased proteasomal function in Lafora disease models lacking the phosphatase laforin. PLoS ONE 2009, 4, e5907. [Google Scholar] [CrossRef] [PubMed]
- Roma-Mateo, C.; Aguado, C.; Garcia-Gimenez, J.L.; Ibanez-Cabellos, J.S.; Seco-Cervera, M.; Pallardo, F.V.; Knecht, E.; Sanz, P. Increased oxidative stress and impaired antioxidant response in Lafora disease. Mol. Neurobiol. 2015, 51, 932–946. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gimenez, J.L.; Seco-Cervera, M.; Aguado, C.; Roma-Mateo, C.; Dasi, F.; Priego, S.; Markovic, J.; Knecht, E.; Sanz, P.; Pallardo, F.V. Lafora disease fibroblasts exemplify the molecular interdependence between thioredoxin 1 and the proteasome in mammalian cells. Free Radic. Biol. Med. 2013, 65, 347–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criado, O.; Aguado, C.; Gayarre, J.; Duran-Trio, L.; Garcia-Cabrero, A.M.; Vernia, S.; San Millan, B.; Heredia, M.; Roma-Mateo, C.; Mouron, S.; et al. Lafora bodies and neurological defects in malin-deficient mice correlate with impaired autophagy. Hum. Mol. Genet. 2012, 21, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Aguado, C.; Sarkar, S.; Korolchuk, V.I.; Criado, O.; Vernia, S.; Boya, P.; Sanz, P.; de Cordoba, S.R.; Knecht, E.; Rubinsztein, D.C. Laforin, the most common protein mutated in Lafora disease, regulates autophagy. Hum. Mol. Genet. 2010, 19, 2867–2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, R.; Ganesh, S. Autophagy defects in Lafora disease: Cause or consequence? Autophagy 2012, 8, 289–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahuerta, M.; Aguado, C.; Sanchez-Martin, P.; Sanz, P.; Knecht, E. Degradation of altered mitochondria by autophagy is impaired in Lafora disease. FEBS J. 2018, 285, 2071–2090. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh, P.K.; Bhadauriya, P.; Ganesh, S. Lafora disease E3 ubiquitin ligase malin is recruited to the processing bodies and regulates the microRNA-mediated gene silencing process via the decapping enzyme Dcp1a. RNA Biol. 2012, 9, 1440–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DePaoli-Roach, A.A.; Tagliabracci, V.S.; Segvich, D.M.; Meyer, C.M.; Irimia, J.M.; Roach, P.J. Genetic depletion of the malin E3 ubiquitin ligase in mice leads to Lafora bodies and the accumulation of insoluble laforin. J. Biol. Chem. 2010, 285, 25372–25381. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Song, D.; Xue, Z.; Gu, L.; Hertz, L.; Peng, L. Requirement of glycogenolysis for uptake of increased extracellular K+ in astrocytes: Potential implications for K+ homeostasis and glycogen usage in brain. Neurochem. Res. 2013, 38, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A.; Cruz, N.F. Contributions of glycogen to astrocytic energetics during brain activation. Metab. Brain Dis. 2015, 30, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Fryer, K.L.; Brown, A.M. Pluralistic roles for glycogen in the central and peripheral nervous systems. Metab. Brain Dis. 2015, 30, 299–306. [Google Scholar] [CrossRef] [PubMed]
- DiNuzzo, M.; Mangia, S.; Maraviglia, B.; Giove, F. Does abnormal glycogen structure contribute to increased susceptibility to seizures in epilepsy? Metab. Brain Dis. 2015, 30, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Sinadinos, C.; Valles-Ortega, J.; Boulan, L.; Solsona, E.; Tevy, M.F.; Marquez, M.; Duran, J.; Lopez-Iglesias, C.; Calbo, J.; Blasco, E.; et al. Neuronal glycogen synthesis contributes to physiological aging. Aging Cell 2014, 13, 935–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garyali, P.; Siwach, P.; Singh, P.K.; Puri, R.; Mittal, S.; Sengupta, S.; Parihar, R.; Ganesh, S. The malin-laforin complex suppresses the cellular toxicity of misfolded proteins by promoting their degradation through the ubiquitin-proteasome system. Hum. Mol. Genet. 2009, 18, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Puri, R.; Suzuki, T.; Yamakawa, K.; Ganesh, S. Dysfunctions in endosomal-lysosomal and autophagy pathways underlie neuropathology in a mouse model for Lafora disease. Hum. Mol. Genet. 2012, 21, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Villena, C.; Viana, R.; Bonet, J.; Garcia-Gimeno, M.A.; Casado, M.; Heredia, M.; Sanz, P. Astrocytes: New players in progressive myoclonus epilepsy of Lafora type. Hum. Mol. Genet. 2018, 27, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Lee, S.G.; Kegelman, T.P.; Su, Z.Z.; Das, S.K.; Dash, R.; Dasgupta, S.; Barral, P.M.; Hedvat, M.; Diaz, P.; et al. Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration: Opportunities for developing novel therapeutics. J. Cell. Physiol. 2011, 226, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Kong, Q.; Cuny, G.D.; Glicksman, M.A. Glutamate transporter EAAT2: A new target for the treatment of neurodegenerative diseases. Future Med. Chem. 2012, 4, 1689–1700. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Danbolt, N.C. GABA and glutamate transporters in brain. Front. Endocrinol. 2013, 4, 165. [Google Scholar] [CrossRef] [PubMed]
- Grewer, C.; Gameiro, A.; Rauen, T. SLC1 glutamate transporters. Pflugers Arch. 2014, 466, 3–24. [Google Scholar] [CrossRef] [PubMed]
- Parpura, V.; Baker, B.J.; Jeras, M.; Zorec, R. Regulated exocytosis in astrocytic signal integration. Neurochem. Int. 2010, 57, 451–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Gonzalez, I.M.; Garcia-Tardon, N.; Gimenez, C.; Zafra, F. PKC-dependent endocytosis of the Glt1 glutamate transporter depends on ubiquitylation of lysines located in a C-terminal cluster. Glia 2008, 56, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Villarreal, J.; Garcia Tardon, N.; Ibanez, I.; Gimenez, C.; Zafra, F. Cell surface turnover of the glutamate transporter Glt-1 is mediated by ubiquitination/deubiquitination. Glia 2012, 60, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Tardon, N.; Gonzalez-Gonzalez, I.M.; Martinez-Villarreal, J.; Fernandez-Sanchez, E.; Gimenez, C.; Zafra, F. Protein kinase c (PKC)-promoted endocytosis of glutamate transporter Glt-1 requires ubiquitin ligase Nedd4-2-dependent ubiquitination but not phosphorylation. J. Biol. Chem. 2012, 287, 19177–19187. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W.; Maulucci-Gedde, M.; Kriegstein, A.R. Glutamate neurotoxicity in cortical cell culture. J. Neurosci. 1987, 7, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Bauer, S.; Bozzi, Y.; Caleo, M.; Dingledine, R.; Gorter, J.A.; Henshall, D.C.; Kaufer, D.; Koh, S.; Loscher, W.; et al. Neuroinflammatory targets and treatments for epilepsy validated in experimental models. Epilepsia 2017, 58, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Cerri, C.; Caleo, M.; Bozzi, Y. Chemokines as new inflammatory players in the pathogenesis of epilepsy. Epilepsy Res. 2017, 136, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Koh, S. Role of neuroinflammation in evolution of childhood epilepsy. J. Child Neurol. 2018, 33, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, J.; DePaoli-Roach, A.A.; Zhao, X.; Cortez, M.A.; Pencea, N.; Tiberia, E.; Piliguian, M.; Roach, P.J.; Wang, P.; Ackerley, C.A.; et al. PTG depletion removes Lafora bodies and rescues the fatal epilepsy of Lafora disease. PLoS Genet. 2011, 7, e1002037. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, J.; Epp, J.R.; Goldsmith, D.; Zhao, X.; Pencea, N.; Wang, P.; Frankland, P.W.; Ackerley, C.A.; Minassian, B.A. PTG protein depletion rescues malin-deficient Lafora disease in mouse. Ann. Neurol. 2014, 75, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Pederson, B.A.; Turnbull, J.; Epp, J.R.; Weaver, S.A.; Zhao, X.; Pencea, N.; Roach, P.J.; Frankland, P.; Ackerley, C.A.; Minassian, B.A. Inhibiting glycogen synthesis prevents Lafora disease in a mouse model. Ann. Neurol. 2013, 74, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Wang, P.; Goldsmith, D.; Zhao, X.; Xue, Y.; Christians, U.; Minassian, B.A. Everolimus does not prevent Lafora body formation in murine Lafora disease. Neurol. Genet. 2017, 3, e127. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Chern, Y. AMPK-mediated regulation of neuronal metabolism and function in brain diseases. J. Neurogenet. 2015, 29, 50–58. [Google Scholar] [CrossRef] [PubMed]
- DiTacchio, K.A.; Heinemann, S.F.; Dziewczapolski, G. Metformin treatment alters memory function in a mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 44, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Marinangeli, C.; Didier, S.; Vingtdeux, V. AMPK in neurodegenerative diseases: Implications and therapeutic perspectives. Curr. Drug Targets 2016, 17, 890–907. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, G.R. Cellular energy sensing and metabolism-implications for treating diabetes: The 2017 outstanding scientific achievement award lecture. Diabetes 2018, 67, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Elexpuru, G.; Serratosa, J.M.; Sanchez, M.P. Sodium selenate treatment improves symptoms and seizure susceptibility in a malin-deficient mouse model of Lafora disease. Epilepsia 2017, 58, 467–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirani, M.; Nasreddine, W.; Abdulla, F.; Beydoun, A. Seizure control and improvement of neurological dysfunction in Lafora disease with perampanel. Epilepsy Behav. Case Rep. 2014, 2, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, D.; Minassian, B.A. Efficacy and tolerability of perampanel in ten patients with Lafora disease. Epilepsy Behav. 2016, 62, 132–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Function | Topology of Ubiquitin Labeling | References |
|---|---|---|---|
| R5/PTG (PPP1R3C) | Glycogen metabolism | K63-Ub | [34,40,41] |
| R6 (PPP1R3D) | Glycogen metabolism | Mono-Ub and K63-Ub | [36] |
| GL (PPP1R3B) | Glycogen metabolism | Unknown | [41] |
| Glycogen synthase | Glycogen metabolism | Unknown | [40] |
| Debranching enzyme AGL | Glycogen metabolism | Unknown | [42] |
| Starch-binding domain containing protein 1 | Glycogen metabolism | K63-Ub | Unpublished results |
| Pyruvate kinase, isozymes M1 and M2 | Glycolysis | K63-Ub | [38] |
| AMPK, subunits α and β | Energy sensor | K63-Ub | [37] |
| Laforin | Unknown, ancillary protein of malin? | K63-Ub | [32] |
| p62 | Autophagy receptor | K63-Ub | [33] |
| Dishevelled2 | Wnt-β-catenin signaling | K48- and K63-Ub | [39] |
| Cellular Component/Process | General Defect in Lafora Disease | References |
|---|---|---|
| Glycogen synthesis | Accumulation of poorly branched glycogen-like inclusions (Lafora bodies) | [34,36,40,41,42] |
| Glutamate transport | Impaired astrocytic glutamate uptake | [53] |
| Glucose transporters | Increased glucose uptake due to altered homeostasis of GLUT1 and GLUT3 | [48,54] |
| Inflammation | Increased reactive astrocytes and microglia. Increased expression of pro-inflammatory markers, cytokines and chemokines | [55,56,57] |
| Wnt signaling | Increased Wnt signaling | [39] |
| ER-stress | Increased sensitivity to unfolded protein response | [58,59] |
| Oxidative stress | Increased oxidative stress | [60] |
| Proteasomes | Inhibition of proteasome activity | [59,61] |
| Macroautophagy | Impairment in the initial steps of macroautophagy | [62,63,64] |
| Mitochondria | Dysfunctional mitochondria | [60,65] |
| Processing bodies | Dysregulation of RNA metabolism | [66] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Gimeno, M.A.; Knecht, E.; Sanz, P. Lafora Disease: A Ubiquitination-Related Pathology. Cells 2018, 7, 87. https://doi.org/10.3390/cells7080087
García-Gimeno MA, Knecht E, Sanz P. Lafora Disease: A Ubiquitination-Related Pathology. Cells. 2018; 7(8):87. https://doi.org/10.3390/cells7080087
Chicago/Turabian StyleGarcía-Gimeno, Maria Adelaida, Erwin Knecht, and Pascual Sanz. 2018. "Lafora Disease: A Ubiquitination-Related Pathology" Cells 7, no. 8: 87. https://doi.org/10.3390/cells7080087