Chemokine Receptors and Exercise to Tackle the Inadequacy of T Cell Homing to the Tumor Site


Abstract
:1. Introduction
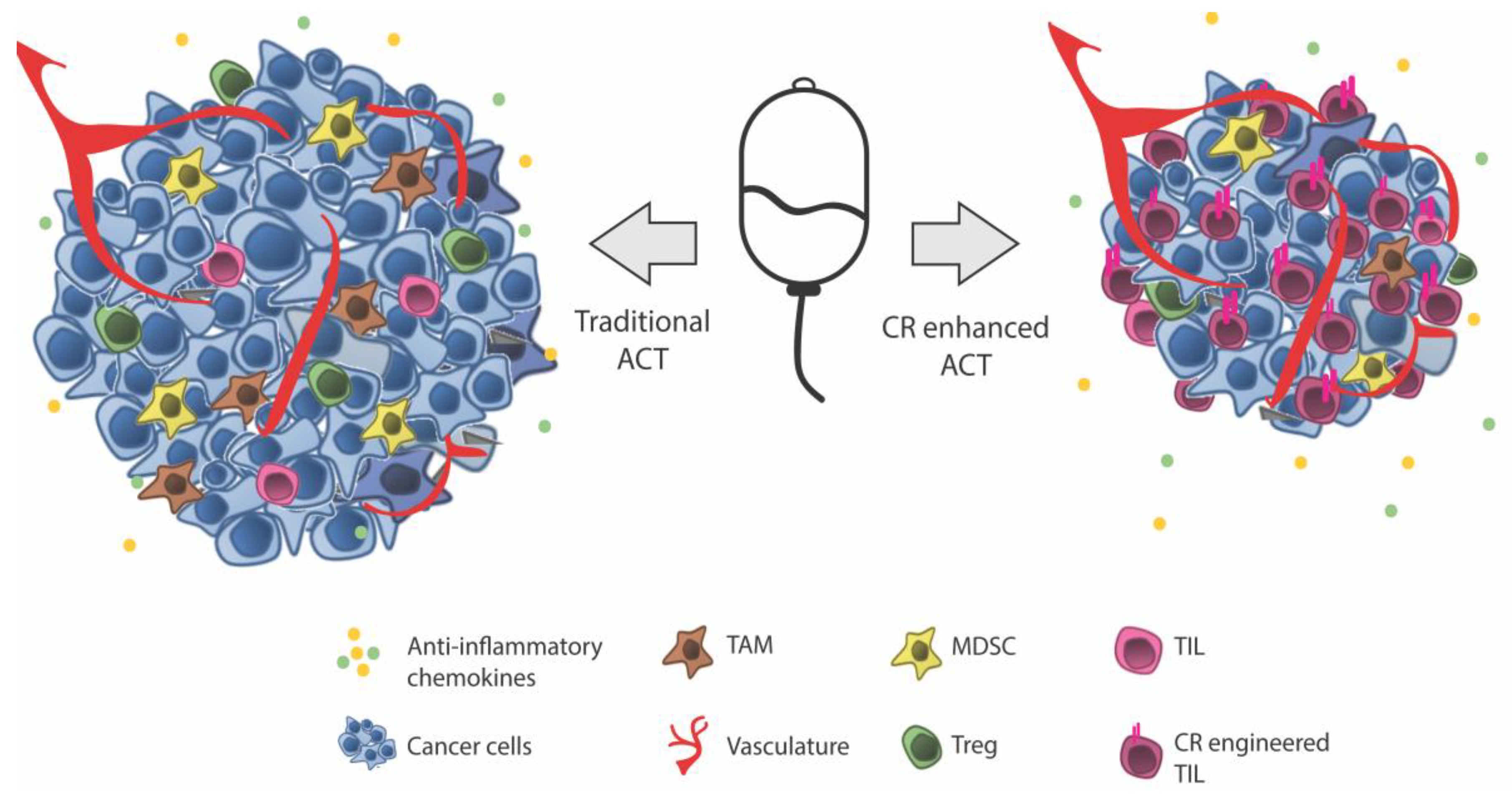
Adoptive Cell Therapy (ACT)
2. Chemokines in Cellular Homing and Immune Evasion
Exploiting Tumor-Specific Chemokine Axes
3. Disruption of Molecular Mediators of Homing
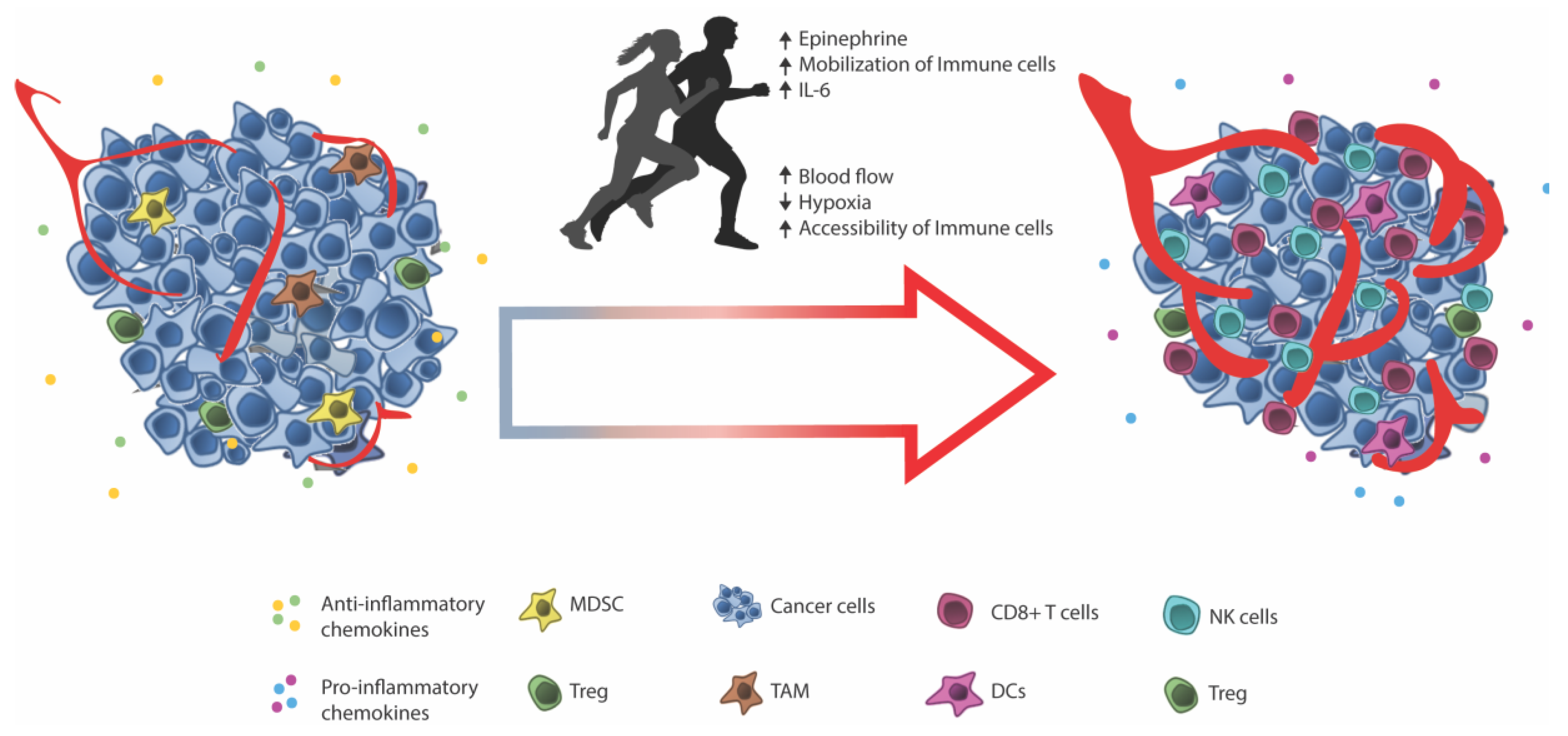
4. Facilitating Immune Infiltration through Exercise
5. Perspectives and Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sackstein, R.; Schatton, T.; Barthel, S.R. T-lymphocyte homing: An underappreciated yet critical hurdle for successful cancer immunotherapy. Lab. Investig. 2017, 97, 669–697. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, N.E.; Beniata, O.V.; Vitsos, P.; Tsitsilonis, O.; Samara, P. Harnessing the immune system to improve cancer therapy. Ann. Transl. Med. 2016, 4, 261. [Google Scholar] [CrossRef] [PubMed]
- Idorn, M.; Skadborg, S.K.; Kellermann, L.; Halldórsdóttir, H.R.; Holmen Olofsson, G.; Met, Ö.; thor Straten, P. Chemokine receptor engineering of T cells with CXCR2 improves homing towards subcutaneous human melanomas in xenograft mouse model. Oncoimmunology 2018, e1450715. [Google Scholar] [CrossRef]
- Weiss, S.A.; Han, S.W.; Lui, K.; Tchack, J.; Shapiro, R.; Berman, R.; Zhong, J.; Krogsgaard, M.; Osman, I.; Darvishian, F. Immunologic heterogeneity of tumor-infiltrating lymphocyte composition in primary melanoma. Hum. Pathol. 2016, 57, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; Wienert, S.; Poterie, A.; Loibl, S.; Budczies, J.; Badve, S.; Bago-Horvath, Z.; Bane, A.; Bedri, S.; Brock, J.; et al. Standardized evaluation of tumor-infiltrating lymphocytes in breast cancer: Results of the ring studies of the international immuno-oncology biomarker working group. Mod. Pathol. 2016, 29, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamid, O.; Schmidt, H.; Nissan, A.; Ridolfi, L.; Aamdal, S.; Hansson, J.; Guida, M.; Hyams, D.M.; Gómez, H.; Bastholt, L.; et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J. Transl. Med. 2011, 9, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-CeII recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef] [PubMed]
- Zsiros, E.; Duttagupta, P.; Dangaj, D.; Li, H.; Frank, R.; Garrabrant, T.; Hagemann, I.S.; Levine, B.L.; June, C.H.; Zhang, L.; et al. The ovarian cancer Chemokine landscape is conducive to homing of vaccine-primed and CD3/CD28-Costimulated T cells prepared for adoptive therapy. Clin. Cancer Res. 2015, 21, 2840–2850. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.; Sarukhan, A.; Bronte, V.; Molon, B. The pros and cons of chemokines in tumor immunology. Trends Immunol. 2012, 33, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Mikucki, M.E.; Fisher, D.T.; Matsuzaki, J.; Skitzki, J.J.; Gaulin, N.B.; Muhitch, J.B.; Ku, A.W.; Frelinger, J.G.; Odunsi, K.; Gajewski, T.F.; et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat. Commun. 2015, 6, 7458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudley, M.E.; Wunderlich, J.R.; Shelton, T.E.; Even, J.; Rosenberg, S.A. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J. Immunother. 2008, 26, 332–342. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.; Junker, N.; Ellebaek, E.; Andersen, M.H.; Straten, P.T.; Svane, I.M. Characterization and comparison of “Standard” and “Young” tumor infiltrating lymphocytes for adoptive cell therapy at a Danish Translational Research Institution. Scand. J. Immunol. 2012, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.; Donia, M.; Ellebaek, E.; Borch, T.H.; Kongsted, P.; Iversen, T.Z.; Hölmich, L.R.; Hendel, H.W.; Met, Ö.; Andersen, M.H.; et al. Long-Lasting complete responses in patients with metastatic melanoma after adoptive cell therapy with tumor-infiltrating lymphocytes and an attenuated il2 regimen. Clin. Cancer Res. 2016, 22, 3734–3745. [Google Scholar] [CrossRef] [PubMed]
- Radvanyi, L.G.; Bernatchez, C.; Zhang, M.; Fox, P.S.; Miller, P.; Chacon, J.; Wu, R.; Lizee, G.; Mahoney, S.; Alvarado, G.; et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin. Cancer Res. 2012, 18, 6758–6770. [Google Scholar] [CrossRef] [PubMed]
- Forget, M.-A.; Haymaker, C.; Hess, K.R.; Meng, Y.J.; Creasy, C.; Karpinets, T.V.; Fulbright, O.J.; Roszik, J.; Woodman, S.E.; Kim, Y.U.; et al. Prospective analysis of adoptive TIL therapy in patients with metastatic melanoma: Response, impact of anti-CTLA4, and biomarkers to predict clinical outcome. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Dudley, M.E. Adoptive Cell Therapy for the Treatment of Patients with Metastatic MelanomaPublic Access. Curr. Opin. Immunol. 2009, 21, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Gross, C.A.; Somerville, R.P.T.; Hong, Y.; Schaub, N.P.; Rosati, S.F.; White, D.E.; Nathan, D.; Restifo, N.P.; Steinberg, S.M.; et al. Randomized selection design trial evaluating CD8+-enriched versus unselected tumor-infiltrating lymphocytes for adoptive cell therapy for patients with melanoma. J. Clin. Oncol. 2013, 31, 2152–2159. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, H.; Lindberg, M.F.; Donia, M.; Söderberg, E.M.V.; Andersen, R.; Keller, U.; Ny, L.; Svane, I.M.; Nilsson, L.M.; Nilsson, J.A. Clinical responses to adoptive T-cell transfer can be modeled in an autologous immune-humanized mouse model. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; June, C.H. Going viral: Chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol. Rev. 2015, 263, 68–89. [Google Scholar] [CrossRef] [PubMed]
- Liddy, N.; Bossi, G.; Adams, K.J.; Lissina, A.; Mahon, T.M.; Hassan, N.J.; Gavarret, J.; Bianchi, F.C.; Pumphrey, N.J.; Ladell, K.; et al. Monoclonal TCR-redirected tumor cell killing. Nat. Med. 2012, 18, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Kakarla, S.; Gottschalk, S. CAR T cells for solid tumors: Armed and ready to go? Cancer J. 2014, 20, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, B.J.; Gerry, A.B.; Dukes, J.; Harper, J.V.; Kannan, V.; Bianchi, F.C.; Grand, F.; Brewer, J.E.; Gupta, M.; Plesa, G.; et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med. 2013, 5, 197ra103. [Google Scholar] [CrossRef] [PubMed]
- Debets, R.; Donnadieu, E.; Chouaib, S.; Coukos, G. TCR-engineered T cells to treat tumors: Seeing but not touching? Semin. Immunol. 2016, 28, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Jindal, V.; Arora, E.; Gupta, S. Challenges and prospects of chimeric antigen receptor T cell therapy in solid tumors. Med. Oncol. 2018, 35, 87. [Google Scholar] [CrossRef] [PubMed]
- Bobisse, S.; Rondina, M.; Merlo, A.; Tisato, V.; Mandruzzato, S.; Amendola, M.; Naldini, L.; Willemsen, R.A.; Debets, R.; Zanovello, P.; et al. Reprogramming T lymphocytes for melanoma adoptive immunotherapy by T-cell receptor gene transfer with lentiviral vectors. Cancer Res. 2009, 69, 9385–9394. [Google Scholar] [CrossRef] [PubMed]
- Pockaj, B.A.; Sherry, R.M.; Wei, J.P.; Yannelli, J.R.; Carter, C.S.; Leitman, S.F.; Carasquillo, J.A.; Steinberg, S.M.; Rosenberg, S.A.; Yang, J.C. Localization of 111indium-labeled tumor infiltrating lymphocytes to tumor in patients receiving adoptive immunotherapy. Augmentation with cyclophosphamide and correlation with response. Cancer 1994, 73, 1731–1737. [Google Scholar] [CrossRef]
- Fisher, B.; Packard, B.S.; Read, E.J.; Carrasquillo, J.A.; Carter, C.S.; Topalian, S.L.; Yang, J.C.; Yolles, P.; Larson, S.M.; Rosenberg, S.A. Tumor localization of adoptively transferred indium-111 labeled tumor infiltrating lymphocytes in patients with metastatic melanoma. J. Clin. Oncol. 1989, 7, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Idorn, M.; Hojman, P. Exercise-Dependent Regulation of NK Cells in Cancer Protection. Trends Mol. Med. 2016, 22, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Idorn, M.; thor Straten, P. Exercise and cancer: From “healthy” to “therapeutic”? Cancer Immunol. Immunother. 2017, 66, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [PubMed]
- Delves, P.J.; Roitt, I.M. The immune system. First of two parts. N. Engl. J. Med. 2000, 343, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Historical insights into cytokines. Eur. J. Immunol. 2007, 37, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, A.; Tian, Y.; Wu, J.D.; Liu, Y.; Li, T.; Chen, Y.; Han, X.; Wu, K. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev. 2016, 31, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Yang, W.-M.; Chen, L.-P.; Yang, D.-H.; Zhou, Q.; Zhu, J.; Chen, J.-J.; Huang, R.-C.; Chen, Z.-S.; Huang, R.-P. Enhanced chemosensitization in multidrug-resistant human breast cancer cells by inhibition of IL-6 and IL-8 production. Breast Cancer Res. Treat. 2012, 135, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Wilson, T.; Johnston, P.G.; Longley, D.B.; Waugh, D.J.J. Interleukin-8 signaling attenuates TRAIL- and chemotherapy-induced apoptosis through transcriptional regulation of c-FLIP in prostate cancer cells. Mol. Cancer Ther. 2008, 7, 2649–2661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.; Purcell, C.; Seaton, A.; Oladipo, O.; Maxwell, P.J.; Sullivan, J.M.O.; Wilson, R.H.; Johnston, P.G.; Waugh, D.J.J. Chemotherapy-Induced CXC-Chemokine/CXC-Chemokine Receptor Signaling in Metastatic Prostate Cancer Cells Confers Resistance to Oxaliplatin through Potentiation of Nuclear Factor-B Transcription and Evasion of Apoptosis. Pharmacology 2008, 327, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Monteagudo, C.; Ramos, D.; Pellín-Carcelén, A.; Gil, R.; Callaghan, R.C.; Martín, J.M.; Alonso, V.; Murgui, A.; Navarro, L.; Calabuig, S.; et al. CCL27-CCR10 and CXCL12-CXCR4 chemokine ligand-receptor mRNA expression ratio: New predictive factors of tumor progression in cutaneous malignant melanoma. Clin. Exp. Metastasis 2012, 29, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, D.; Zhao, J. The Role of chemokine receptor CXCR4 in breast cancer metastasis. Am. J. Cancer Res. 2013, 3, 46–57. [Google Scholar] [PubMed]
- Darash-Yahana, M.; Pikarsky, E.; Abramovitch, R.; Zeira, E.; Pal, B.; Karplus, R.; Beider, K.; Avniel, S.; Kasem, S.; Galun, E.; et al. Role of high expression levels of CXCR4 in tumor growth, vascularization, and metastasis. FASEB J. 2004, 18, 1240–1242. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Burkhardt, A.M.; Homey, B. Homeostatic chemokine receptors and organ-specific metastasis. Nat. Rev. Immunol. 2011, 11, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Lesokhin, A.M.; Hohl, T.M.; Kitano, S.; Cortez, C.; Hirschhorn-Cymerman, D.; Avogadri, F.; Rizzuto, G.A.; Lazarus, J.J.; Pamer, E.G.; Houghton, A.N.; et al. Monocytic CCR2+ myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 2012, 72, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Obermajer, N.; Muthuswamy, R.; Odunsi, K.; Edwards, R.P.; Kalinski, P. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res. 2011, 71, 7463–7470. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Bromley, S.K.; Mempel, T.R.; Luster, A.D. Orchestrating the orchestrators: Chemokines in control of T cell traffic. Nat. Immunol. 2008, 9, 970–980. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.S.; Thrue, C.A.; Junker, N.; Lyngaa, R.; Donia, M.; Ellebæk, E.; Svane, I.M.; Schumacher, T.N.; Straten, P.T.; Hadrup, S.R. Dissection of T-cell antigen specificity in human melanoma. Cancer Res. 2012, 72, 1642–1650. [Google Scholar] [CrossRef] [PubMed]
- Bedognetti, D.; Spivey, T.L.; Zhao, Y.; Uccellini, L.; Tomei, S.; Dudley, M.E.; Ascierto, M.L.; De Giorgi, V.; Liu, Q.; Delogu, L.G.; et al. CXCR3/CCR5 pathways in metastatic melanoma patients treated with adoptive therapy and interleukin-2. Br. J. Cancer 2013, 109, 2412–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, Z.; Xiao, R.; Xiong, J.; Tembo, K.M.; Deng, X.; Xiong, M.; Liu, P.; Wang, M.; Zhang, Q. CCR9 in cancer: Oncogenic role and therapeutic targeting. J. Hematol. Oncol. 2016, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural Innate and Adaptive Immunity to Cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef] [PubMed]
- Umansky, V.; Blattner, C.; Gebhardt, C.; Utikal, J. CCR5 in recruitment and activation of myeloid-derived suppressor cells in melanoma. Cancer Immunol. Immunother. 2017, 66, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Ilkovitch, D.; Lopez, D.M. Immune modulation by melanoma-derived factors. Exp. Dermatol. 2008, 17, 977–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, A.S.; Cornelius, L.A. The role of chemokines in melanoma tumor growth and metastasis. J. Investig. Dermatol. 2002, 118, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Raman, D.; Baugher, P.J.; Thu, Y.M.; Richmond, A. Role of chemokines in tumor growth. Cancer Lett. 2007, 256, 137–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarvaiya, P.J.; Guo, D.; Ulasov, I.; Gabikian, P.; Lesniak, M.S. Chemokines in tumor progression and metastasis. Oncotarget 2013, 4, 2171–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, M.T.; Luster, A.D. Chemokines in Cancer. Cancer Immunol. Res. 2014, 2, 1125–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koizumi, K.; Hojo, S.; Akashi, T.; Yasumoto, K.; Saiki, I. Chemokine receptors in cancer metastasis and cancer cell-derived chemokines in host immune response. Cancer Sci. 2007, 98, 1652–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franciszkiewicz, K.; Boissonnas, A.; Boutet, M.; Combadière, C.; Mami-Chouaib, F. Role of chemokines and chemokine receptors in shaping the effector phase of the antitumor immune response. Cancer Res. 2012, 72, 6325–6332. [Google Scholar] [CrossRef] [PubMed]
- Waugh, D.J.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Wang, G.; Westwood, J.A.; Pachynski, R.K.; Tiffany, H.L.; Marincola, F.M.; Wang, E.; Young, H.A.; Murphy, P.M.; Hwu, P. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum. Gene Ther. 2002, 13, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Sapoznik, S.; Ortenberg, R.; Galore-Haskel, G.; Kozlovski, S.; Levy, D.; Avivi, C.; Barshack, I.; Cohen, C.J.; Besser, M.J.; Schachter, J.; et al. CXCR1 as a novel target for directing reactive T cells toward melanoma: Implications for adoptive cell transfer immunotherapy. Cancer Immunol. Immunother. 2012, 61, 1833–1847. [Google Scholar] [CrossRef] [PubMed]
- Mauldin, I.S.; Wages, N.A.; Stowman, A.M.; Wang, E.; Smolkin, M.E.; Olson, W.C.; Deacon, D.H.; Smith, K.T.; Galeassi, N.V.; Chianese-Bullock, K.A.; et al. Intratumoral interferon-gamma increases chemokine production but fails to increase T cell infiltration of human melanoma metastases. Cancer Immunol. Immunother. 2016, 65, 1189–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klarquist, J.; Tobin, K.; Oskuei, P.F.; Henning, S.W.; Fernandez, M.F.; Dellacecca, E.R.; Navarro, F.C.; Eby, J.M.; Chatterjee, S.; Mehrotra, S.; et al. Ccl22 diverts T regulatory cells and controls the growth of melanoma. Cancer Res. 2016, 76, 6230–6240. [Google Scholar] [CrossRef] [PubMed]
- Idorn, M.; Olsen, M.; Halldórsdóttir, H.R.; Skadborg, S.K.; Pedersen, M.; Høgdall, C.; Høgdall, E.; Met, Ö.; Thor Straten, P. Improved migration of tumor ascites lymphocytes to ovarian cancer microenvironment by CXCR2 transduction. Oncoimmunology 2018, 7, e1412029. [Google Scholar] [CrossRef] [PubMed]
- Idorn, M.; Thor Straten, P.; Svane, I.M.; Met, Ö. Transfection of Tumor-Infiltrating T Cells with mRNA Encoding CXCR2. Methods Mol. Biol. 2016, 1428, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.-C.C.S.; Kapoor, V.; Predina, J.; Powell, D.J.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Ye, Y.; Rabinovich, B.A.; Liu, C.; Lou, Y.; Zhang, M.; Whittington, M.; Yang, Y.; Overwijk, W.W.; Lizée, G.; et al. Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses. Clin. Cancer Res. 2010, 16, 5458–5468. [Google Scholar] [CrossRef] [PubMed]
- Craddock, J.A.; Lu, A.; Bear, A.; Pule, M.; Brenner, M.K.; Rooney, C.M.; Foster, A.E. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 2010, 33, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, A.; De Angelis, B.; Rooney, C.M.; Zhang, L.; Mahendravada, A.; Foster, A.E.; Heslop, H.E.; Brenner, M.K.; Dotti, G.; Savoldo, B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 2009, 113, 6392–6402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapp, M.; Grassmann, S.; Chaloupka, M.; Layritz, P.; Kruger, S.; Ormanns, S.; Rataj, F.; Janssen, K.-P.; Endres, S.; Anz, D.; et al. C-C chemokine receptor type-4 transduction of T cells enhances interaction with dendritic cells, tumor infiltration and therapeutic efficacy of adoptive T cell transfer. Oncoimmunology 2016, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Xie, K. Interleukin-8 and human cancer biology. Cytokine Growth Factor Rev. 2001, 12, 375–391. [Google Scholar] [CrossRef]
- Antony, V.B.; Hott, J.W.; Godbey, S.W.; Holm, K. Angiogenesis in mesotheliomas. Role of mesothelial cell derived IL-8. Chest 1996, 109, 21S–22S. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, F.A.; Pantschenko, A.G.; Miller, L.J.; Anderson, K.; Grunnet, M.; McKenna, P.H.; Kreutzer, D. Angiogenesis and neuroblastomas: Interleukin-8 and interleukin-8 receptor expression in human neuroblastoma. J. Urol. 2000, 164, 1016–1020. [Google Scholar] [CrossRef]
- Miyamoto, M.; Shimizu, Y.; Okada, K.; Kashii, Y.; Higuchi, K.; Watanabe, A. Effect of interleukin-8 on production of tumor-associated substances and autocrine growth of human liver and pancreatic cancer cells. Cancer Immunol. Immunother. 1998, 47, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Kitadai, Y.; Haruma, K.; Sumii, K.; Yamamoto, S.; Ue, T.; Yokozaki, H.; Yasui, W.; Ohmoto, Y.; Kajiyama, G.; Fidler, I.J.; et al. Expression of interleukin-8 correlates with vascularity in human gastric carcinomas. Am. J. Pathol. 1998, 152, 93–100. [Google Scholar] [PubMed]
- Reiland, J.; Furcht, L.T.; McCarthy, J.B. CXC-chemokines stimulate invasion and chemotaxis in prostate carcinoma cells through the CXCR2 receptor. Prostate 1999, 41, 78–88. [Google Scholar] [CrossRef]
- Green, A.R.; Green, V.L.; White, M.C.; Speirs, V. Expression of cytokine messenger RNA in normal and neoplastic human breast tissue: Identification of interleukin-8 as a potential regulatory factor in breast tumours. Int. J. Cancer 1997, 72, 937–941. [Google Scholar] [CrossRef]
- Schadendorf, D.; Möller, A.; Algermissen, B.; Worm, M.; Sticherling, M.; Czarnetzki, B.M. IL-8 produced by human malignant melanoma cells in vitro is an essential autocrine growth factor. J. Immunol. 1993, 151, 2667–2675. [Google Scholar] [PubMed]
- Luciani, M.G.; Stoppacciaro, A.; Peri, G.; Mantovani, A.; Ruco, L.P. The monocyte chemotactic protein a (MCP-1) and interleukin 8 (IL-8) in Hodgkin’s disease and in solid tumours. Mol. Pathol. 1998, 51, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Celle, P.; Carbone, A.; Marchis, D.; Zhou, D.; Sozzani, S.; Zupo, S.; Pini, M.; Mantovani, A.; Foa, R. Cytokine Gene Expression in B-Cell Chronic Lymphocytic Leukemia: Evidence of Constitutive Interleukin-8 (IL-8) mRNA Expression and Secretion of Biologically Active IL-8 Protein. Blood 1994, 84, 220–228. [Google Scholar] [PubMed]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Langenkamp, E.; Georganaki, M.; Loskog, A.; Fuchs, P.F.; Dieterich, L.C.; Kreuger, J.; Dimberg, A. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-κB-induced endothelial activation. FASEB J. 2015, 29, 227–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peske, J.D.; Woods, A.B.; Engelhard, V.H. Control of CD8 T-Cell Infiltration into Tumors by Vasculature and Microenvironment, 1st ed.; Elsevier Inc.: New York, NY, USA, 2015; Volume 128, ISBN 9780128023167. [Google Scholar]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Church, S.E.; Galon, J. Regulation of CTL infiltration within the tumor microenvironment. Adv. Exp. Med. Biol. 2017, 1036, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Dirkx, A.E.M.; Egbrink, M.G.A.; Kuijpers, M.J.E.; Heijnen, V.V.T.; Steege, J.C.A.B.; Wagstaff, J.; Griffioen, A.W. Tumor Angiogenesis Modulates Leukocyte-Vessel Wall Interactions in Vivo by Reducing Endothelial Adhesion Molecule Expression Tumor Angiogenesis Modulates Leukocyte-Vessel Wall Interactions in Vivo by Reducing Endothelial Adhesion Molecule Expression. Cancer Res. 2003, 63, 2322–2329. [Google Scholar] [PubMed]
- Weishaupt, C.; Munoz, K.N.; Buzney, E.; Kupper, T.S.; Fuhlbrigge, R.C. T-cell distribution and adhesion receptor expression in metastatic melanoma. Clin. Cancer Res. 2007, 13, 2549–2556. [Google Scholar] [CrossRef] [PubMed]
- Yoong, K.F.; McNab, G.; Hübscher, S.G.; Adams, D.H. Vascular adhesion protein-1 and ICAM-1 support the adhesion of tumor-infiltrating lymphocytes to tumor endothelium in human hepatocellular carcinoma. J. Immunol. 1998, 160, 3978–3988. [Google Scholar] [PubMed]
- Bigley, A.B.; Rezvani, K.; Pistillo, M.; Reed, J.; Agha, N.; Kunz, H.; O’Connor, D.P.; Sekine, T.; Bollard, C.M.; Simpson, R.J. Acute exercise preferentially redeploys NK-cells with a highly-differentiated phenotype and augments cytotoxicity against lymphoma and multiple myeloma target cells. Part II: Impact of latent cytomegalovirus infection and catecholamine sensitivity. Brain Behav. Immun. 2015, 49, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Barra, N.G.; Fan, I.Y.; Gillen, J.B.; Chew, M.; Marcinko, K.; Steinberg, G.R.; Gibala, M.J.; Ashkar, A.A. High Intensity Interval Training Increases Natural Killer Cell Number and Function in Obese Breast Cancer-challenged Mice and Obese Women. J. Cancer Prev. 2017, 22, 260–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedenreich, C.M.; Neilson, H.K.; Lynch, B.M. State of the epidemiological evidence on physical activity and cancer prevention. Eur. J. Cancer 2010, 46, 2593–2604. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.; Idorn, M.; Olofsson, G.H.; Lauenborg, B.; Nookaew, I.; Hansen, R.H.; Johannesen, H.H.; Becker, J.C.; Pedersen, K.S.; Dethlefsen, C.; et al. Voluntary Running Suppresses Tumor Growth through Epinephrine- and IL-6-Dependent NK Cell Mobilization and Redistribution. Cell Metab. 2016. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, H.; Urhausen, A.; Kindermann, W. Mobilization of circulating leucocyte and lymphocyte subpopulations during and after short, anaerobic exercise. Eur. J. Appl. Physiol. Occup. Physiol. 1992, 65, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Walsh, N.P.; Gleeson, M.; Shephard, R.J.; Gleeson, M.; Woods, J.A.; Bishop, N.C.; Fleshner, M.; Green, C.; Pedersen, B.K.; Hoffman-Goetz, L.; et al. Position statement. Part one: Immune function and exercise. Exerc. Immunol. Rev. 2011, 17, 6–63. [Google Scholar] [PubMed]
- Gustafson, M.P.; DiCostanzo, A.C.; Wheatley, C.M.; Kim, C.-H.; Bornschlegl, S.; Gastineau, D.A.; Johnson, B.D.; Dietz, A.B. A systems biology approach to investigating the influence of exercise and fitness on the composition of leukocytes in peripheral blood. J. Immunother. Cancer 2017, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, T.; Puntschart, A.; Kaijser, L.; Jansson, E.; Sundberg, C.J. Exercise-induced expression of angiogenesis-related transcription and growth factors in human skeletal muscle. Am. J. Physiol. 1999, 276, H679–H685. [Google Scholar] [CrossRef] [PubMed]
- Prior, B.M.; Yang, H.T.; Terjung, R.L. What makes vessels grow with exercise training? J. Appl. Physiol. 2004, 97, 1119–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Z.; Okutsu, M.; Akhtar, Y.N.; Lira, V.A. Regulation of exercise-induced fiber type transformation, mitochondrial biogenesis, and angiogenesis in skeletal muscle. J. Appl. Physiol. 2011, 110, 264–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, L.W.; Viglianti, B.L.; Tashjian, J.A.; Kothadia, S.M.; Keir, S.T.; Freedland, S.J.; Potter, M.Q.; Moon, E.J.; Schroeder, T.; Herndon, J.E.; et al. Effect of aerobic exercise on tumor physiology in an animal model of human breast cancer. J. Appl. Physiol. 2010, 108, 343–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betof, A.S.; Lascola, C.D.; Weitzel, D.; Landon, C.; Scarbrough, P.M.; Devi, G.R.; Palmer, G.; Jones, L.W.; Dewhirst, M.W. Modulation of murine breast tumor vascularity, hypoxia, and chemotherapeutic response by exercise. J. Natl. Cancer Inst. 2015, 107, 1–5. [Google Scholar] [CrossRef] [PubMed]
- McCullough, D.J.; Stabley, J.N.; Siemann, D.W.; Behnke, B.J. Modulation of blood flow, hypoxia, and vascular function in orthotopic prostate tumors during exercise. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- McCullough, D.J.; Nguyen, L.M.-D.; Siemann, D.W.; Behnke, B.J. Effects of exercise training on tumor hypoxia and vascular function in the rodent preclinical orthotopic prostate cancer model. J. Appl. Physiol. 2013, 115, 1846–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terry, S.; Buart, S.; Chouaib, S. Hypoxic stress-induced tumor and immune plasticity, suppression, and impact on tumor heterogeneity. Front. Immunol. 2017, 8, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Baginska, J.; Viry, E.; Berchem, G.; Poli, A.; Noman, M.Z.; van Moer, K.; Medves, S.; Zimmer, J.; Oudin, A.; Niclou, S.P.; et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl. Acad. Sci. USA 2013, 110, 17450–17455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messai, Y.; Noman, M.Z.; Hasmim, M.; Janji, B.; Tittarelli, A.; Boutet, M.; Baud, V.; Viry, E.; Billot, K.; Nanbakhsh, A.; et al. ITPR1 Protects Renal Cancer Cells against Natural Killer Cells by Inducing Autophagy. Cancer Res. 2014, 74, 6820–6832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noman, M.Z.; Janji, B.; Kaminska, B.; Van Moer, K.; Pierson, S.; Przanowski, P.; Buart, S.; Berchem, G.; Romero, P.; Mami-Chouaib, F.; et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. 2011, 71, 5976–5986. [Google Scholar] [CrossRef] [PubMed]
- Sethumadhavan, S.; Silva, M.; Philbrook, P.; Nguyen, T.; Hatfield, S.M.; Ohta, A.; Sitkovsky, M.V. Hypoxia and hypoxia-inducible factor (HIF) downregulate antigen-presenting MHC class I molecules limiting tumor cell recognition by T cells. PLoS ONE 2017, 12, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, Y.; Yang, M.; Zhang, Y.; Xie, Q.; Li, Z.; Dong, Z.; Yang, Y.; Deng, B.; Feng, A.; et al. Hypoxia induces T-cell apoptosis by inhibiting chemokine C receptor 7 expression: The role of adenosine receptor A2. Cell. Mol. Immunol. 2010, 7, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tejchman, A.; Lamerant-Fayel, N.; Jacquinet, J.-C.; Bielawska-Pohl, A.; Mleczko-Sanecka, K.; Grillon, C.; Chouaib, S.; Ugorski, M.; Kieda, C. Tumor hypoxia modulates podoplanin/CCL21 interactions in CCR7+ NK cell recruitment and CCR7+ tumor cell mobilization. Oncotarget 2017, 8, 31876–31887. [Google Scholar] [CrossRef] [PubMed]
- Vuillefroy de Silly, R.; Dietrich, P.Y.; Walker, P.R. Hypoxia and antitumor CD8+ T cells: An incompatible alliance? Oncoimmunology 2016, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Burd, R.; Dziedzic, T.S.; Xu, Y.; Caligiuri, M.A.; Subjeck, J.R.; Repasky, E.A. Tumor cell apoptosis, lymphocyte recruitment and tumor vascular changes are induced by low temperature, long duration (fever-like) whole body hyperthermia. J. Cell. Physiol. 1998, 177, 137–147. [Google Scholar] [CrossRef]
- Ostberg, J.R.; Dayanc, B.E.; Yuan, M.; Oflazoglu, E.; Repasky, E.A. Enhancement of natural killer (NK) cell cytotoxicity by fever-range thermal stress is dependent on NKG2D function and is associated with plasma membrane NKG2D clustering and increased expression of MICA on target cells. J. Leukoc. Biol. 2007, 82, 1322–1331. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.T.; Chen, Q.; Skitzki, J.J.; Muhitch, J.B.; Zhou, L.; Appenheimer, M.M.; Vardam, T.D.; Weis, E.L.; Passanese, J.; Wang, W.C.; et al. IL-6 trans-signaling licenses mouse and human tumor microvascular gateways for trafficking of cytotoxic T cells. J. Clin. Investig. 2011, 121, 3846–3859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Fisher, D.T.; Clancy, K.A.; Gauguet, J.-M.M.; Wang, W.-C.; Unger, E.; Rose-John, S.; von Andrian, U.H.; Baumann, H.; Evans, S.S. Fever-range thermal stress promotes lymphocyte trafficking across high endothelial venules via an interleukin 6 trans-signaling mechanism. Nat. Immunol. 2006, 7, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- MacNeil, B.; Hoffman-Goetz, L. Effect of exercise on natural cytotoxicity and pulmonary tumor metastases in mice. Med. Sci. Sports Exerc. 1993, 25, 922–928. [Google Scholar] [CrossRef] [PubMed]
- MacNeil, B.; Hoffman-Goetz, L. Chronic exercise enhances in vivo and in vitro cytotoxic mechanisms of natural immunity in mice. J. Appl. Physiol. 1993, 74, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Millard, A.L.; Valli, P.V.; Stussi, G.; Mueller, N.J.; Yung, G.P.; Seebach, J.D. Brief exercise increases peripheral blood NK cell counts without immediate functional changes, but impairs their responses to ex vivo stimulation. Front. Immunol. 2013, 4, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdalla, D.R.; Murta, E.F.C.; Michelin, M.A. The influence of physical activity on the profile of immune response cells and cytokine synthesis in mice with experimental breast tumors induced by 7,12-dimethylbenzanthracene. Eur. J. Cancer Prev. 2013, 22, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Sweegers, M.G.; Altenburg, T.M.; Chinapaw, M.J.; Kalter, J.; Verdonck-De Leeuw, I.M.; Courneya, K.S.; Newton, R.U.; Aaronson, N.K.; Jacobsen, P.B.; Brug, J.; et al. Which exercise prescriptions improve quality of life and physical function in patients with cancer during and following treatment? A systematic review and meta-analysis of randomised controlled trials. Br. J. Sports Med. 2018, 52, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S. Tumor Heterogeneity and Tumor Immunity: A Chicken-and-Egg Problem. Trends Immunol. 2016, 37, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.; Hodi, F. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Chasalow, S.D.; Wang, L.; Hamid, O.; Schmidt, H.; Cogswell, J.; Alaparthy, S.; Berman, D.; Jure-Kunkel, M.; Siemers, N.O.; et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother. 2012, 61, 1019–1031. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Chemokine | Alternative Name | Receptor(s) | Function an Adaptive T Cell Response | Function in Tumor Immunology |
|---|---|---|---|---|
| CCL2 | MCP-1 | CCR2, CCR3 | Inflammatory monocyte trafficking | Recruitment of TAMs, MDSC, and neutrophils, Tumor infiltration of CD8+ T cells |
| CCL3 | MIP-1α | CCR5 | T cell-DC interactions | Recruitment and maturation of DCs, increased CD8+ T cell activation and tumor infiltration |
| CCL4 | MIP-1β | |||
| CCL5 | RANTES | |||
| CCL17 | TARC | CCR4 | Treg migration, Th2 response and migration | Treg recruitment and tumor infiltration |
| CCL19 | MIP-3β | CCR7 | T and DC homing to LNs | Formation of tumor associated TLS, recruitment and activation of Treg and MDSC, tolerogenic TLS and promotion of metastasis |
| CCL21 | SLC | |||
| CCL22 | MDC | CCR4 | Treg migration, Th2 response and migration | Treg recruitment and tumor infiltration |
| CCL25 | TECK | CCR9 | T cell precursor homing to thymus | Inhibit effector T cell function, chemotherapy resistance, and metastasis [56] |
| CXCL1 | Gro-α | CXCR1, CXCR2 | Neutrophil trafficking | Survival, proliferation, and metastasis (cancer cells), Neoangiogenesis, recruitment of MDSC and TANs |
| CXCL8 | IL-8 | CXCR1, CXCR2, | ||
| CXCL9 | MIG | CXCR3 | Th1 response, CD8, Th1 and NK trafficking to site of inflammation | Recruitment and infiltration of CD8+ T, NK, and NKT cells, inhibit tumor cell proliferation, vascular transmigration checkpoint [14] |
| CXCL10 | IP-10 | |||
| CXCL11 | I-TAC | |||
| CXCL12 | SDF-1 | CXCR4 | Bone marrow homing, LN homing | MDSC recruitment and promotion of metastasis |
| CXCL16 | LEC, etc. | CXCR6 | NKT and ILC migration and survival | Recruitment of activated T cells, NK cells, and monocytes |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Idorn, M.; Thor Straten, P. Chemokine Receptors and Exercise to Tackle the Inadequacy of T Cell Homing to the Tumor Site. Cells 2018, 7, 108. https://doi.org/10.3390/cells7080108
Idorn M, Thor Straten P. Chemokine Receptors and Exercise to Tackle the Inadequacy of T Cell Homing to the Tumor Site. Cells. 2018; 7(8):108. https://doi.org/10.3390/cells7080108
Chicago/Turabian StyleIdorn, Manja, and Per Thor Straten. 2018. "Chemokine Receptors and Exercise to Tackle the Inadequacy of T Cell Homing to the Tumor Site" Cells 7, no. 8: 108. https://doi.org/10.3390/cells7080108