Role of Scaffold Protein Proline-, Glutamic Acid-, and Leucine-Rich Protein 1 (PELP1) in the Modulation of Adrenocortical Cancer Cell Growth

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
3. Results
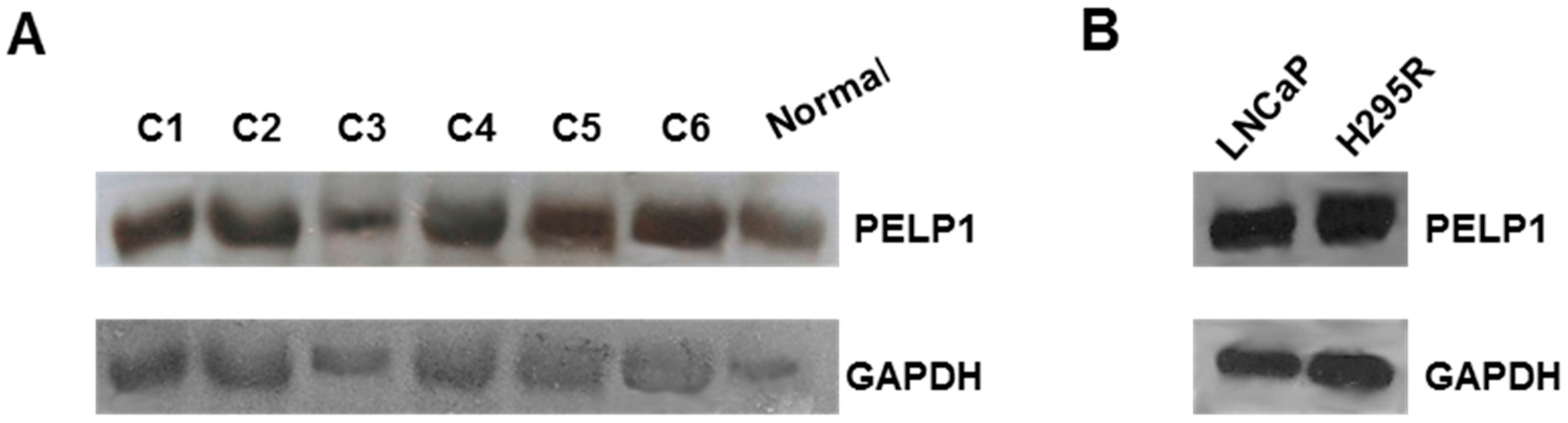
3.1. PELP1 Is Expressed in Human ACC Samples and in H295R Cells
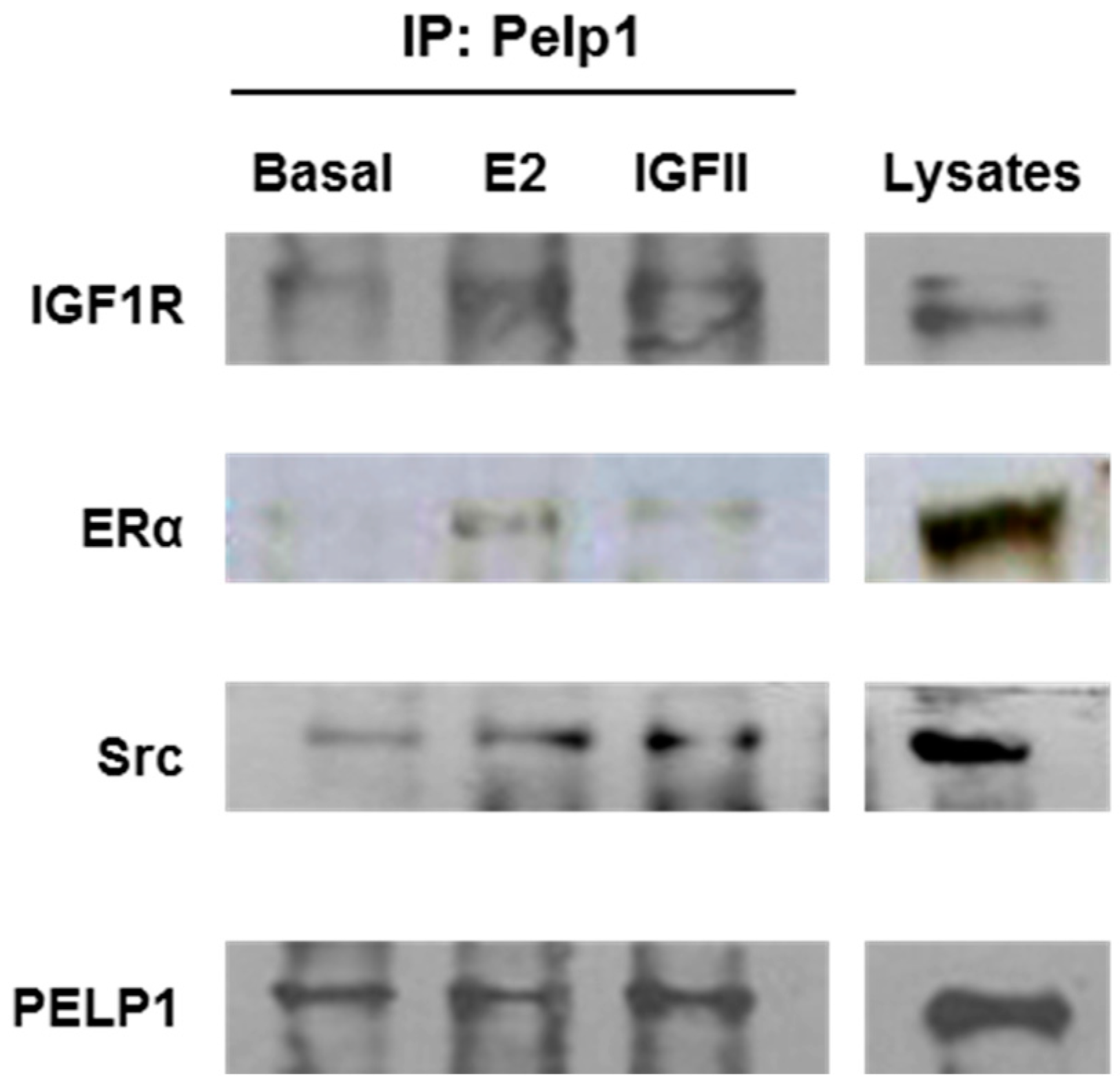
3.2. PELP1 Is Recruited to Form a Multiprotein Complex in H295R Cells after E2 and IGF-II Treatment
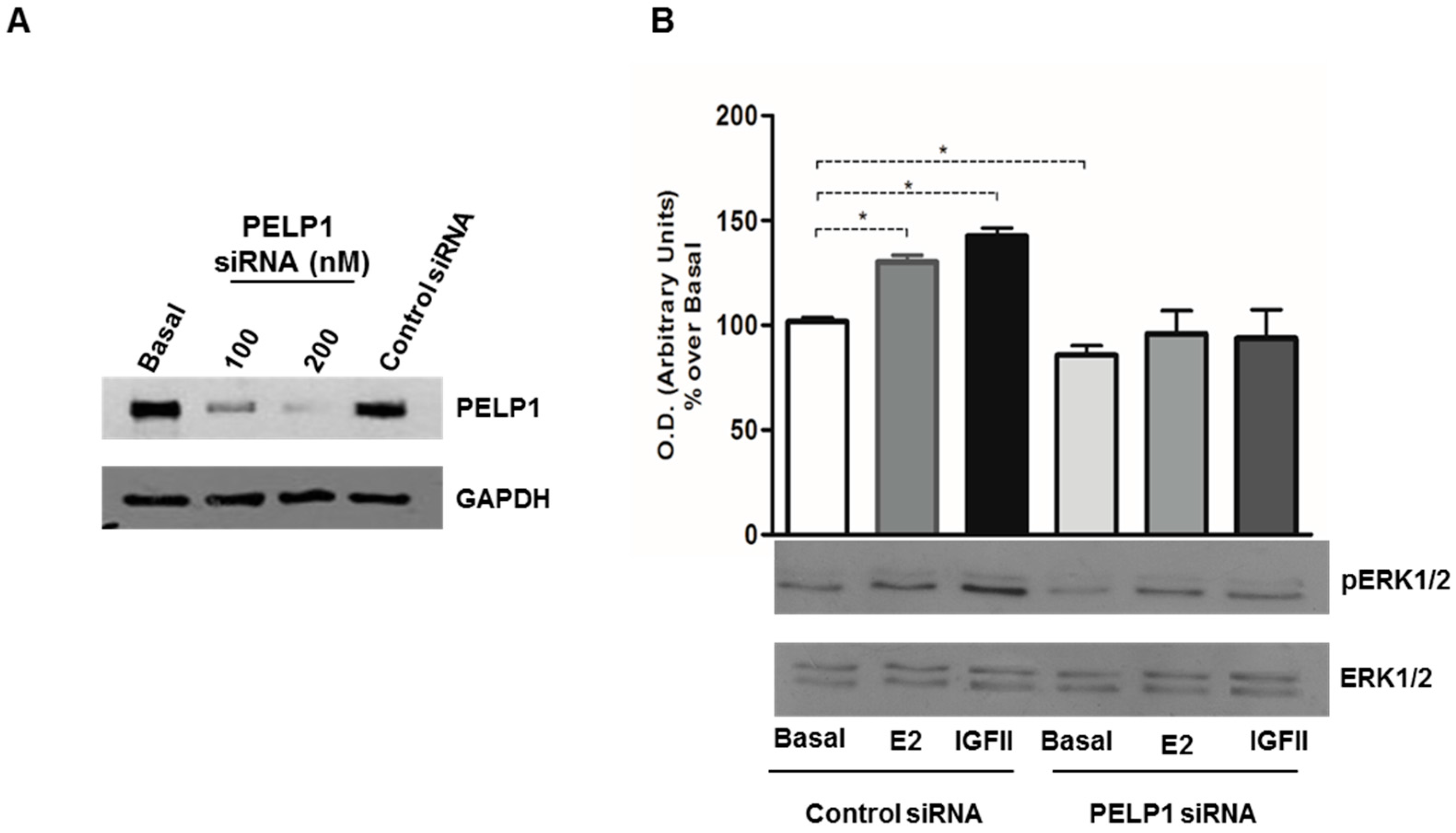
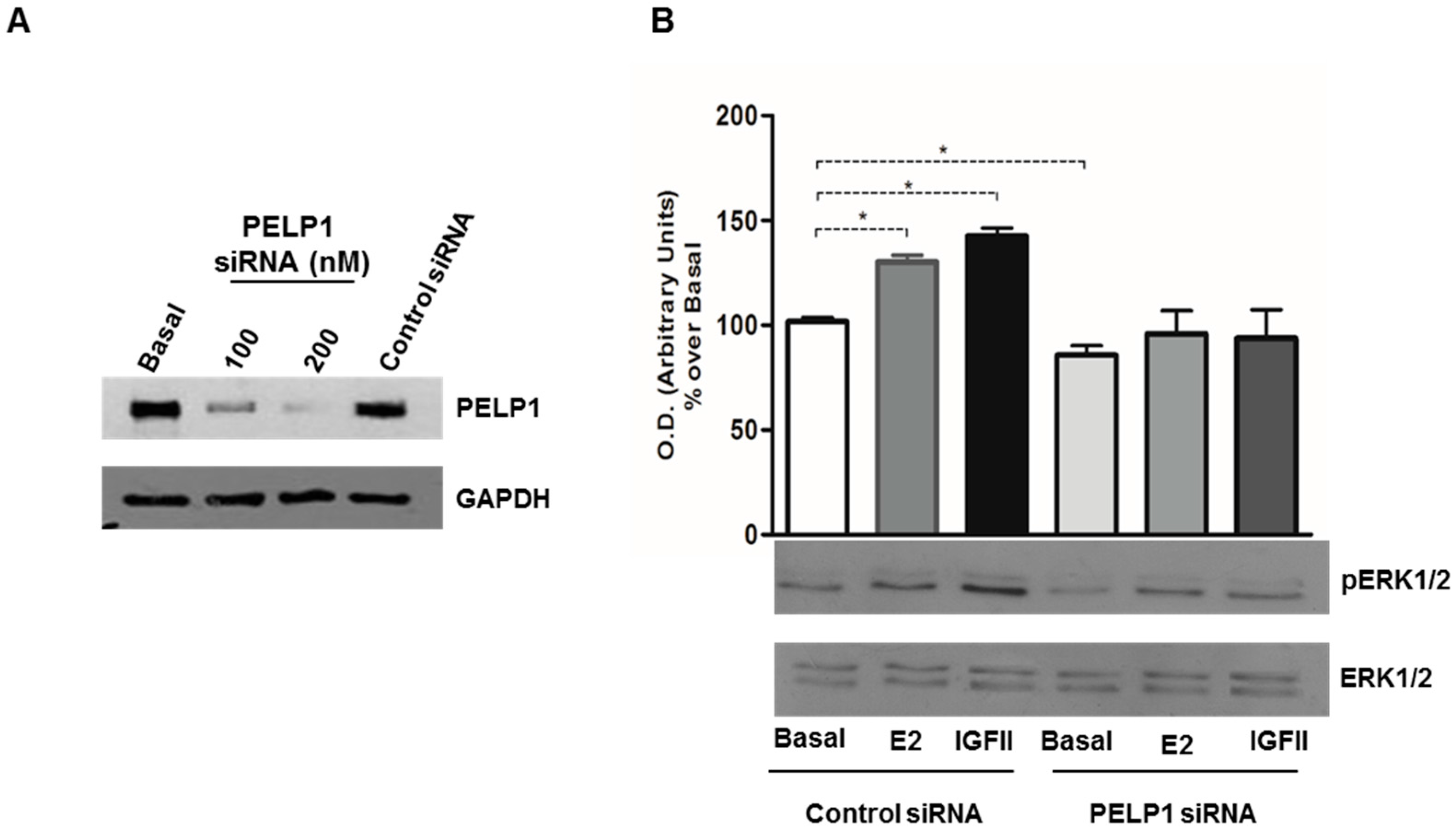
3.3. PELP1 Knockdown Decreases ERK1/2 Phosphorylation in H295R Cells
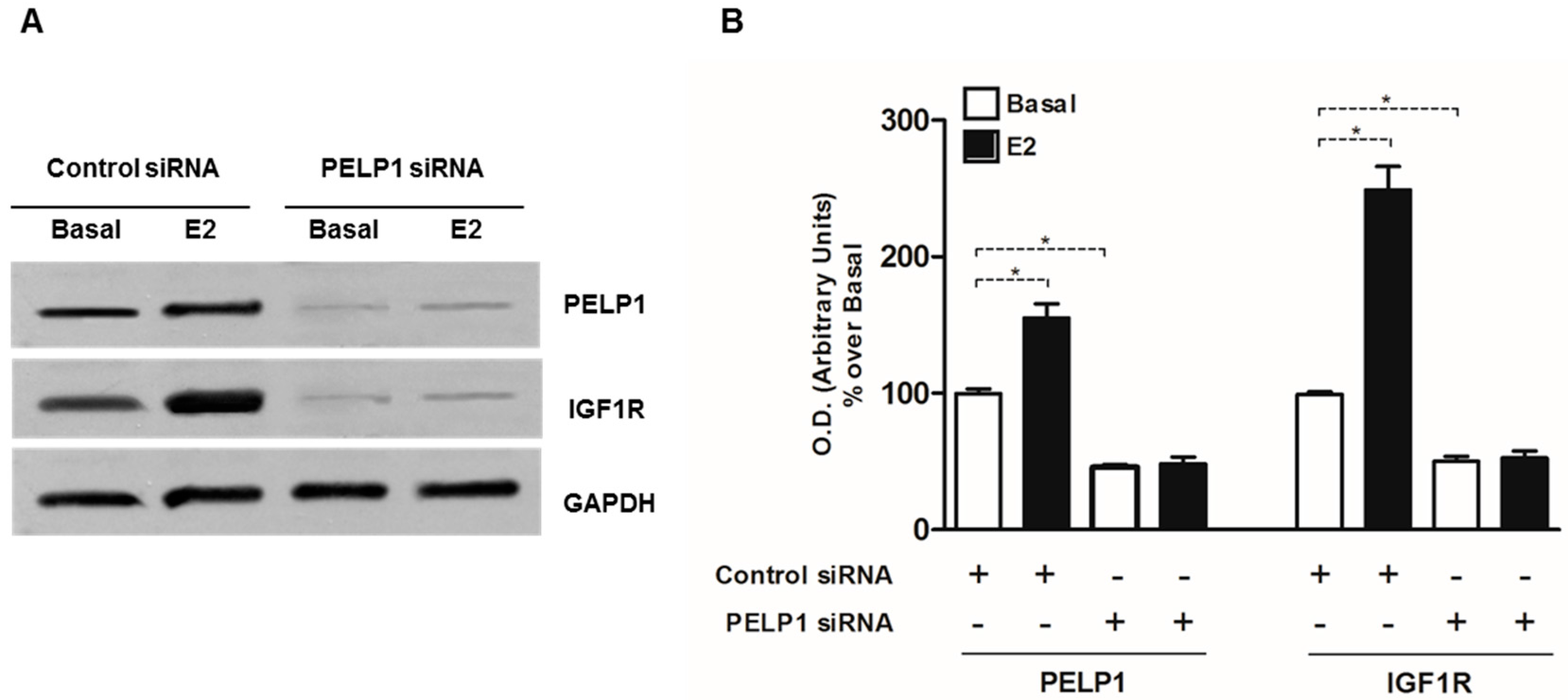
3.4. PELP1 Knockdown Decreases IGFIR Expression in H295R Cells
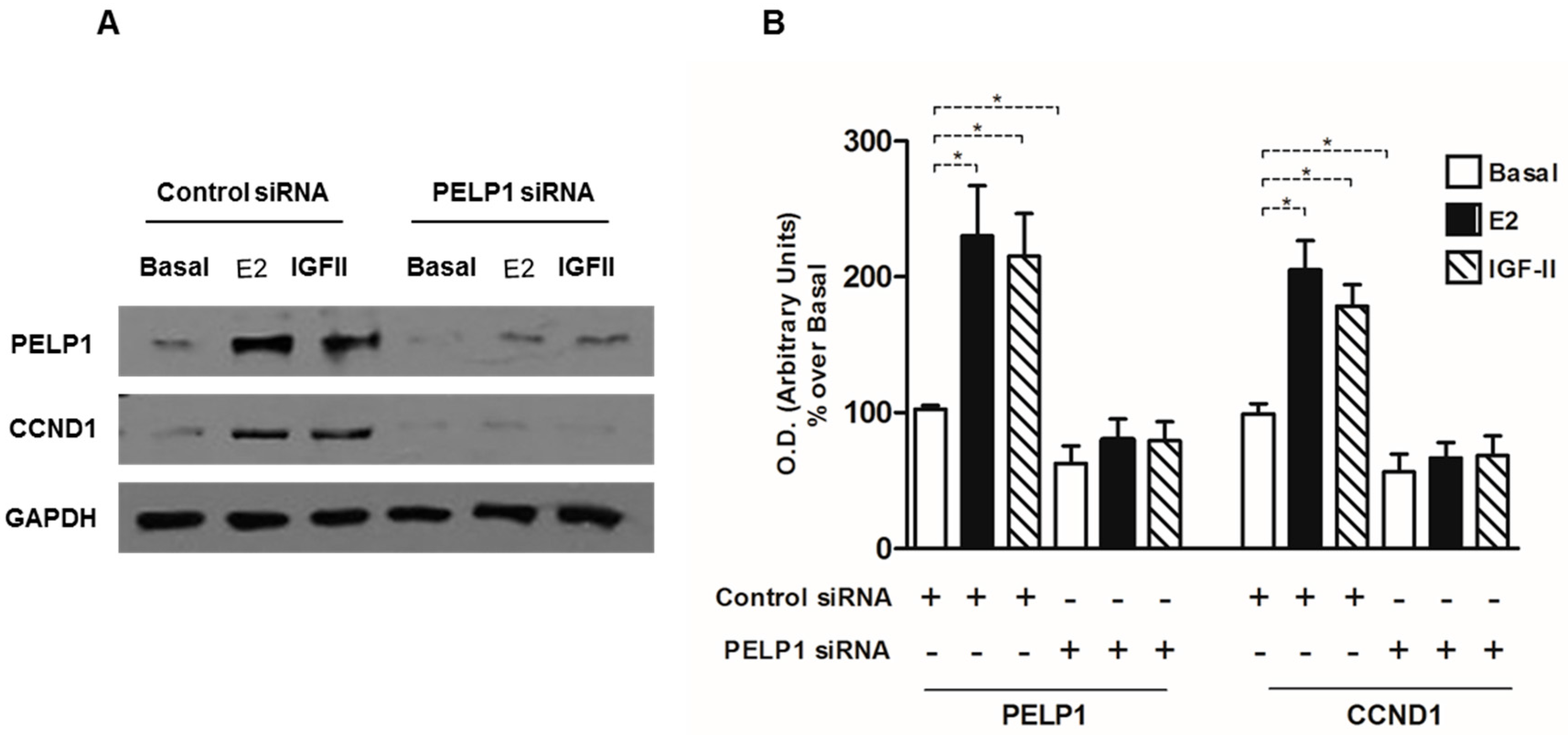
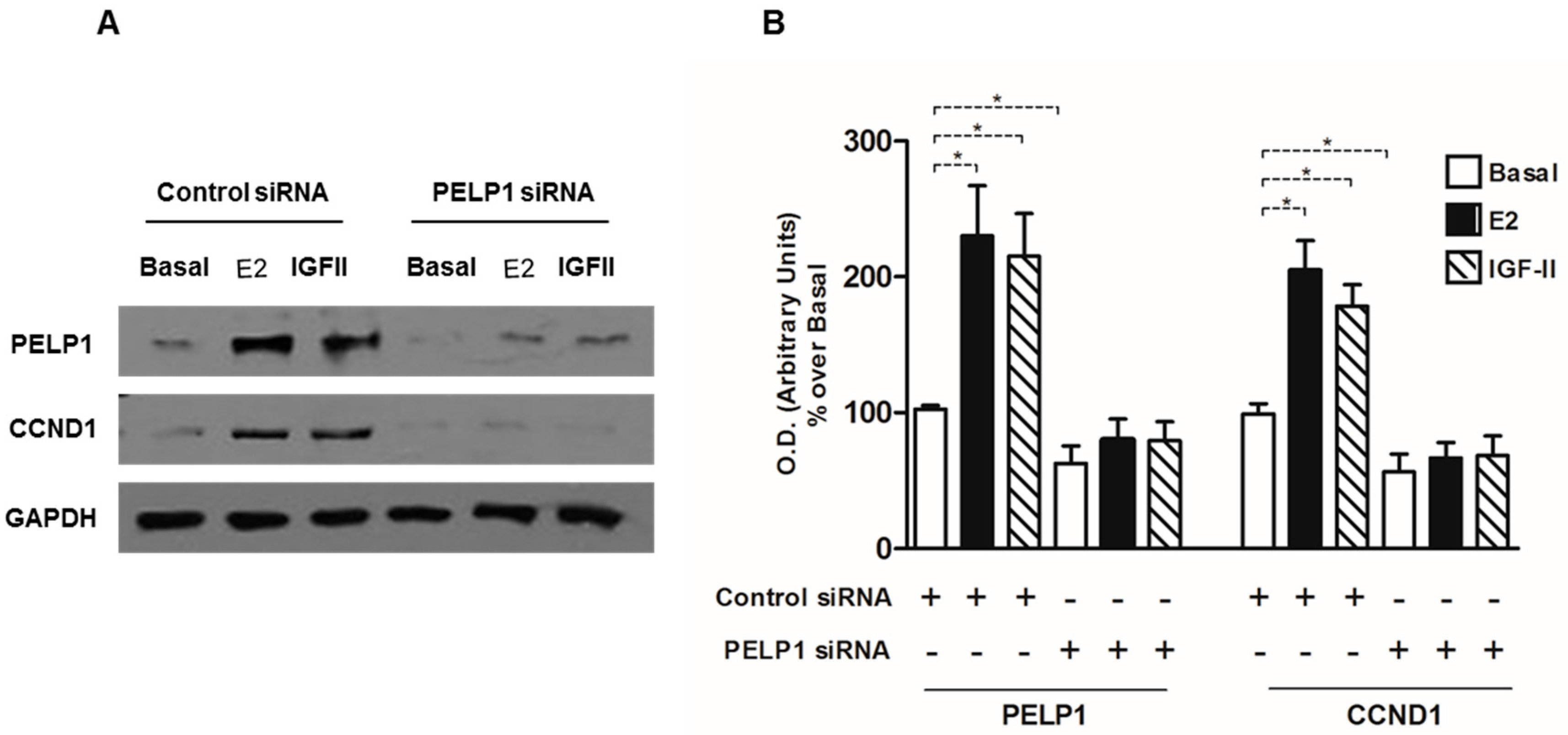
3.5. PELP1 Knockdown Decreases Cyclin D1 Expression in H295R Cells
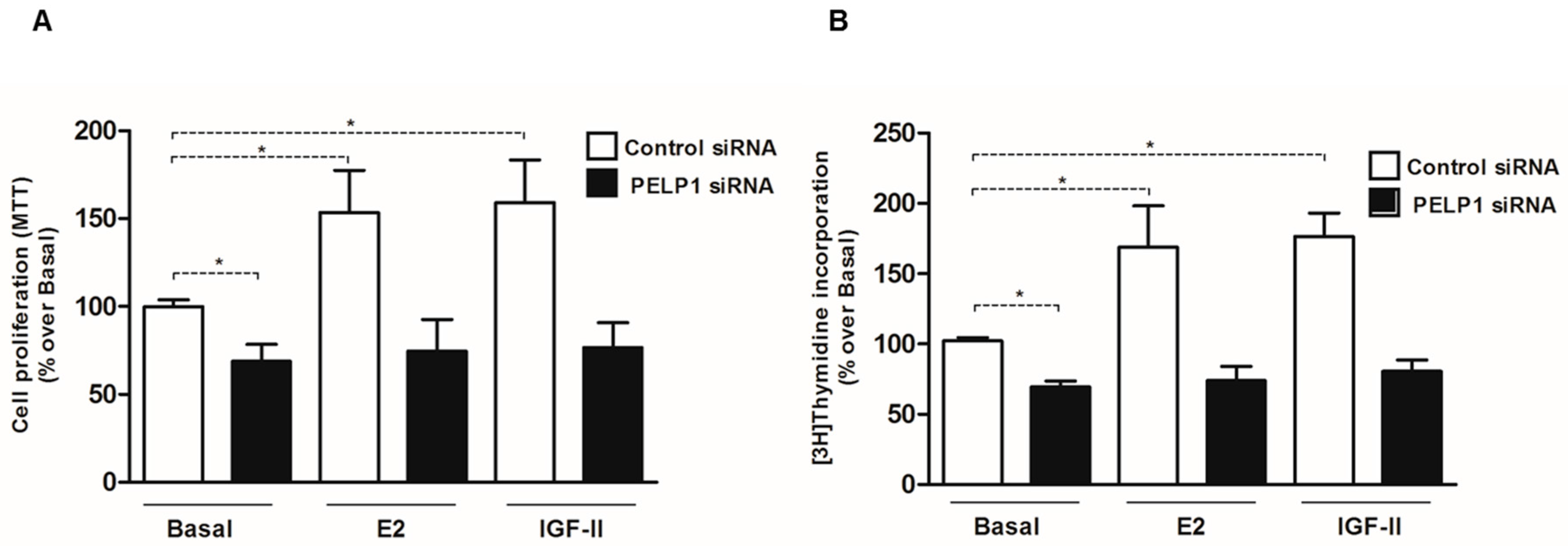
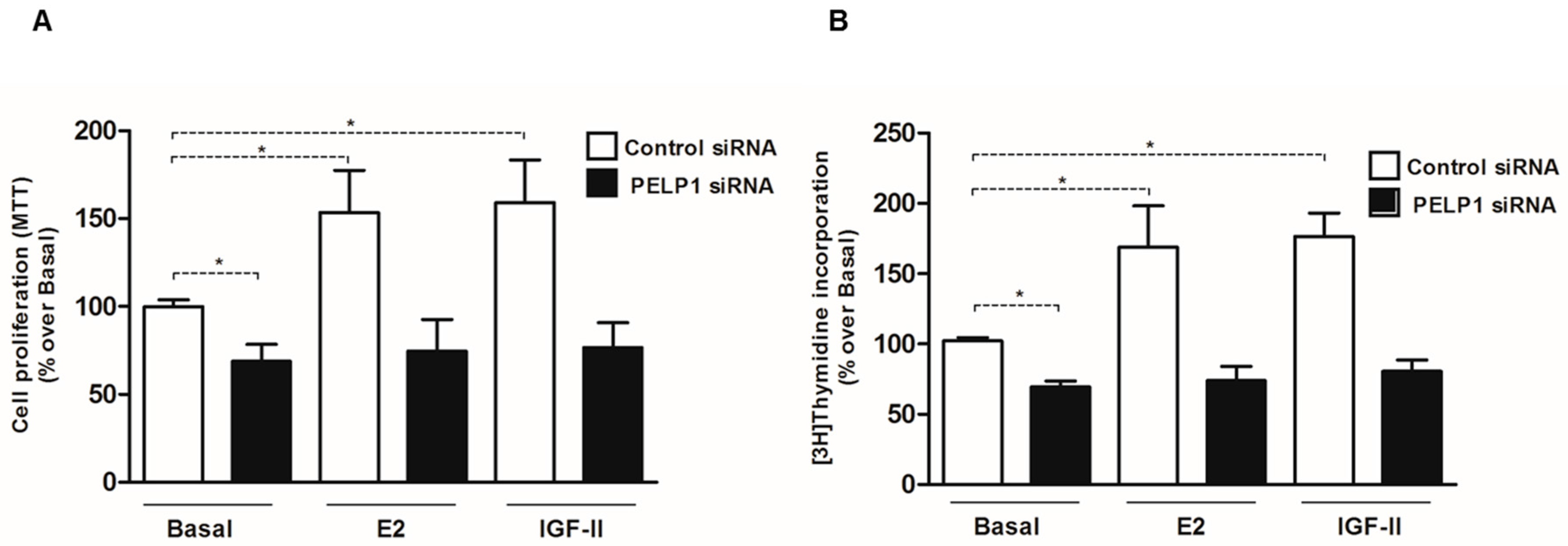
3.6. PELP1 Knockdown Reduces Proliferation of H295R Cells
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stojadinovic, A.; Ghossein, R.A.; Hoos, A.; Nissan, A.; Marshall, D.; Dudas, M.; Cordon-Cardo, C.; Jaques, D.P.; Brennan, M.F. Adrenocortical carcinoma: Clinical, morphologic, and molecular characterization. J. Clin. Oncol. 2002, 20, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Glover, A.R.; Ip, J.C.; Zhao, J.T.; Soon, P.S.; Robinson, B.G.; Sidhu, S.B. Current management options for recurrent adrenocortical carcinoma. Onco Targets Ther. 2013, 6, 635–643. [Google Scholar] [PubMed]
- Libe, R.; Fratticci, A.; Bertherat, J. Adrenocortical cancer: Pathophysiology and clinical management. Endocr.-Relat. Cancer 2007, 14, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Soon, P.S.; Libe, R.; Benn, D.E.; Gill, A.; Shaw, J.; Sywak, M.S.; Groussin, L.; Bertagna, X.; Gicquel, C.; Bertherat, J.; et al. Loss of heterozygosity of 17p13, with possible involvement of ACADVL and ALOX15B, in the pathogenesis of adrenocortical tumors. Ann. Surg. 2008, 247, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Giordano, T.J.; Kuick, R.; Else, T.; Gauger, P.G.; Vinco, M.; Bauersfeld, J.; Sanders, D.; Thomas, D.G.; Doherty, G.; Hammer, G. Molecular classification and prognostication of adrenocortical tumors by transcriptome profiling. Clin. Cancer Res. 2009, 15, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Lafemina, J.; Brennan, M.F. Adrenocortical carcinoma: Past, present, and future. J. Surg. Oncol. 2012, 106, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Giordano, T.J.; Thomas, D.G.; Kuick, R.; Lizyness, M.; Misek, D.E.; Smith, A.L.; Sanders, D.; Aljundi, R.T.; Gauger, P.G.; Thompson, N.W. Distinct transcriptional profiles of adrenocortical tumors uncovered by DNA microarray analysis. Am. J. Pathol. 2003, 162, 521–531. [Google Scholar] [CrossRef]
- Ribeiro, T.C.; Latronico, A.C. Insulin-like growth factor system on adrenocortical tumorigenesis. Mol. Cell. Endocrinol. 2012, 351, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Samani, A.A.; Yakar, S.; LeRoith, D.; Brodt, P. The role of the IGF system in cancer growth and metastasis: Overview and recent insights. Endocr. Rev. 2007, 28, 20–47. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Barzon, L.; Masi, G.; Pacenti, M.; Trevisan, M.; Fallo, F.; Remo, A.; Martignoni, G.; Montanaro, D.; Pezzi, V.; Palù, G. Expression of aromatase and estrogen receptors in human adrenocortical tumors. Virchows Arch. 2008, 452, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Montanaro, D.; Maggiolini, M.; Recchia, A.G.; Sirianni, R.; Aquila, S.; Barzon, L.; Fallo, F.; Ando, S.; Pezzi, V. Antiestrogens upregulate estrogen receptor beta expression and inhibit adrenocortical H295R cell proliferation. J. Mol. Endocrinol. 2005, 35, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Chimento, A.; Sirianni, R.; Casaburi, I.; Zolea, F.; Rizza, P.; Avena, P.; Malivindi, R.; De Luca, A.; Campana, C.; Martire, E. GPER agonist G-1 decreases adrenocortical carcinoma (ACC) cell growth in vitro and in vivo. Oncotarget 2015, 6, 19190–19203. [Google Scholar] [CrossRef] [PubMed]
- Casaburi, I.; Avena, P.; De Luca, A.; Chimento, A.; Sirianni, R.; Malivindi, R.; Rago, V.; Fiorillo, M.; Domanico, F.; Campana, C. Estrogen related receptor alpha (ERRalpha) a promising target for the therapy of adrenocortical carcinoma (ACC). Oncotarget 2015, 6, 25135–25148. [Google Scholar] [CrossRef] [PubMed]
- Sirianni, R.; Zolea, F.; Chimento, A.; Ruggiero, C.; Cerquetti, L.; Fallo, F.; Pilon, C.; Arnaldi, G.; Carpinelli, G.; Stigliano, A.; et al. Targeting estrogen receptor-alpha reduces adrenocortical cancer (ACC) cell growth in vitro and in vivo: Potential therapeutic role of selective estrogen receptor modulators (SERMs) for ACC treatment. J. Clin. Endocrinol. Metab. 2012, 97, E2238–E2250. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Balasenthil, S.; Nguyen, D.; Vadlamudi, R.K. Cloning and functional characterization of PELP1/MNAR promoter. Gene 2004, 330, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Rajhans, R.; Vadlamudi, R.K. Comprehensive analysis of recent biochemical and biologic findings regarding a newly discovered protein-PELP1/MNAR. Clin. Exp. Metast. 2006, 23, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Manavathi, B.; Kumar, R. Steering estrogen signals from the plasma membrane to the nucleus: Two sides of the coin. J. Cell Physiol. 2006, 207, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Vadlamudi, R.K.; Manavathi, B.; Balasenthil, S.; Nair, S.S.; Yang, Z.; Sahin, A.A.; Kumar, R. Functional implications of altered subcellular localization of PELP1 in breast cancer cells. Cancer Res. 2005, 65, 7724–7732. [Google Scholar] [CrossRef] [PubMed]
- Vadlamudi, R.K.; Balasenthil, S.; Broaddus, R.R.; Gustafsson, J.A.; Kumar, R. Deregulation of estrogen receptor coactivator proline-, glutamic acid-, and leucine-rich protein-1/modulator of nongenomic activity of estrogen receptor in human endometrial tumors. J. Clin. Endocrinol. Metab. 2004, 89, 6130–6138. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, A.; Di Domenico, M.; Castoria, G.; Nanayakkara, M.; Lombardi, M.; de Falco, A.; Bilancio, A.; Varricchio, L.; Ciociola, A.; Auricchio, F. Steroid receptor regulation of epidermal growth factor signaling through Src in breast and prostate cancer cells: Steroid antagonist action. Cancer Res. 2005, 65, 10585–10593. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, D.; Tekmal, R.R.; Vadlamudi, R.K. PELP1: A novel therapeutic target for hormonal cancers. IUBMB Life 2010, 62, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, J.K.; Nair, S.; Chakravarty, D.; Rajhans, R.; Pothana, S.; Brann, D.W.; Tekmal, R.R.; Vadlamudi, R.K. Growth factor regulation of estrogen receptor coregulator PELP1 functions via Protein Kinase A pathway. Mol. Cancer Res. 2008, 6, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Rainey, W.E.; Bird, I.M.; Mason, J.I. The NCI-H295 cell line: A pluripotent model for human adrenocortical studies. Mol. Cell. Endocrinol. 1994, 100, 45–50. [Google Scholar] [CrossRef]
- Weiss, L.M.; Medeiros, L.J.; Vickery, A.L., Jr. Pathologic features of prognostic significance in adrenocortical carcinoma. Am. J. Surg. Pathol. 1989, 13, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Aubert, S.; Wacrenier, A.; Leroy, X.; Devos, P.; Carnaille, B.; Proye, C.; Wemeau, J.L.; Lecomte-Houcke, M.; Leteurtre, E. Weiss system revisited: A clinicopathologic and immunohistochemical study of 49 adrenocortical tumors. Am. J. Surg. Pathol. 2002, 26, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Fassnacht, M.; Terzolo, M.; Allolio, B.; Baudin, E.; Haak, H.; Berruti, A.; Welin, S.; Schade-Brittinger, C.; Lacroix, A.; Jarzab, B.; et al. Combination chemotherapy in advanced adrenocortical carcinoma. N. Engl. J. Med. 2012, 366, 2189–2197. [Google Scholar] [CrossRef] [PubMed]
- Casaburi, I.; Avena, P.; Lanzino, M.; Sisci, D.; Giordano, F.; Maris, P.; Catalano, S.; Morelli, C.; Andò, S. Chenodeoxycholic acid through a TGR5-dependent CREB signaling activation enhances cyclin D1 expression and promotes human endometrial cancer cell proliferation. Cell Cycle 2012, 11, 2699–2710. [Google Scholar] [CrossRef] [PubMed]
- Greger, J.G.; Guo, Y.; Henderson, R.; Ross, J.F.; Cheskis, B.J. Characterization of MNAR expression. Steroids 2006, 71, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Vadlamudi, R.K.; Kumar, R. Functional and biological properties of the nuclear receptor coregulator PELP1/MNAR. Nucl. Recept. Signal. 2007, 5, e004. [Google Scholar] [CrossRef] [PubMed]
- Cheskis, B.J.; Greger, J.; Cooch, N.; McNally, C.; Mclarney, S.; Lam, H.S.; Rutledge, S.; Mekonnen, B.; Hauze, D.; Nagpal, S. MNAR plays an important role in ERa activation of Src/MAPK and PI3K/Akt signaling pathways. Steroids 2008, 73, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Boonyaratanakornkit, V. Scaffolding proteins mediating membrane-initiated extra-nuclear actions of estrogen receptor. Steroids 2011, 76, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Barletta, F.; Wong, C.W.; McNally, C.; Komm, B.S.; Katzenellenbogen, B.; Cheskis, B.J. Characterization of the interactions of estrogen receptor and MNAR in the activation of cSrc. Mol. Endocrinol. 2004, 18, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, D.; Nair, S.S.; Santhamma, B.; Nair, B.C.; Wang, L.; Bandyopadhyay, A.; Agyin, J.K.; Brann, D.; Sun, L.Z.; Yeh, I.T.; et al. Extranuclear functions of ER impact invasive migration and metastasis by breast cancer cells. Cancer Res. 2010, 70, 4092–4101. [Google Scholar] [CrossRef] [PubMed]
- Vallabhaneni, S.; Nair, B.C.; Cortez, V.; Challa, R.; Chakravarty, D.; Tekmal, R.R.; Vadlamudi, R.K. Significance of ER-Src axis in hormonal therapy resistance. Breast Cancer Res. Treat. 2011, 130, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Girard, B.J.; Daniel, A.R.; Lange, C.A.; Ostrander, J.H. PELP1: A review of PELP1 interactions, signaling, and biology. Mol. Cell Endocrinol. 2014, 382, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Cortez, V.; Mann, M.; Tekmal, S.; Suzuki, T.; Miyata, N.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Sood, A.K.; Vadlamudi, R.K. Targeting the PELP1-KDM1 axis as a potential therapeutic strategy for breast cancer. Breast Cancer Res. 2012, 14, R108. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of all the compounds used in the present work are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Age (Years) | Gender | Stage at Surgery | Syndrome | Weiss Score | Size (cm) | Outcome |
|---|---|---|---|---|---|---|---|
| C1 | 41 | M | IV | Cushing | 9 | 16 | Died, 1 year |
| C2 | 17 | F | IV | Cushing | 9 | 14 | Died, 18 months |
| C3 | 43 | F | III | None | 4 | 9 | Died, 8 years |
| C4 | 46 | M | III | None | 3 | 18 | Remission, 7 years |
| C5 | 47 | M | IV | Cushing | 9 | 14 | Died, 1 year |
| C6 | 57 | M | II | Subclinical Cushing | 5 | 14 | Remission, 4 years |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Luca, A.; Avena, P.; Sirianni, R.; Chimento, A.; Fallo, F.; Pilon, C.; Casaburi, I.; Pezzi, V. Role of Scaffold Protein Proline-, Glutamic Acid-, and Leucine-Rich Protein 1 (PELP1) in the Modulation of Adrenocortical Cancer Cell Growth. Cells 2017, 6, 42. https://doi.org/10.3390/cells6040042
De Luca A, Avena P, Sirianni R, Chimento A, Fallo F, Pilon C, Casaburi I, Pezzi V. Role of Scaffold Protein Proline-, Glutamic Acid-, and Leucine-Rich Protein 1 (PELP1) in the Modulation of Adrenocortical Cancer Cell Growth. Cells. 2017; 6(4):42. https://doi.org/10.3390/cells6040042
Chicago/Turabian StyleDe Luca, Arianna, Paola Avena, Rosa Sirianni, Adele Chimento, Francesco Fallo, Catia Pilon, Ivan Casaburi, and Vincenzo Pezzi. 2017. "Role of Scaffold Protein Proline-, Glutamic Acid-, and Leucine-Rich Protein 1 (PELP1) in the Modulation of Adrenocortical Cancer Cell Growth" Cells 6, no. 4: 42. https://doi.org/10.3390/cells6040042
APA StyleDe Luca, A., Avena, P., Sirianni, R., Chimento, A., Fallo, F., Pilon, C., Casaburi, I., & Pezzi, V. (2017). Role of Scaffold Protein Proline-, Glutamic Acid-, and Leucine-Rich Protein 1 (PELP1) in the Modulation of Adrenocortical Cancer Cell Growth. Cells, 6(4), 42. https://doi.org/10.3390/cells6040042