
Telomere Biology—Insights into an Intriguing Phenomenon

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Telomeres–Historical Perspective
2. Telomere Sequence
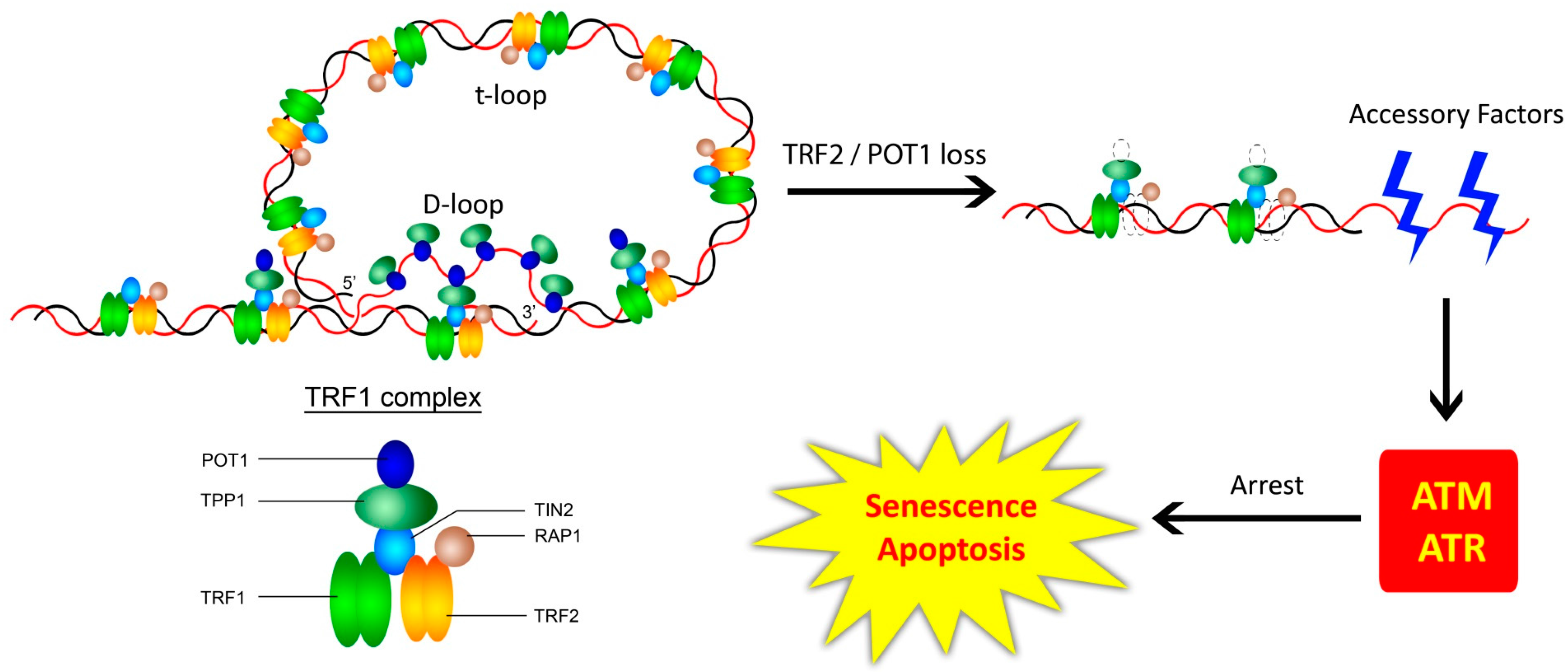
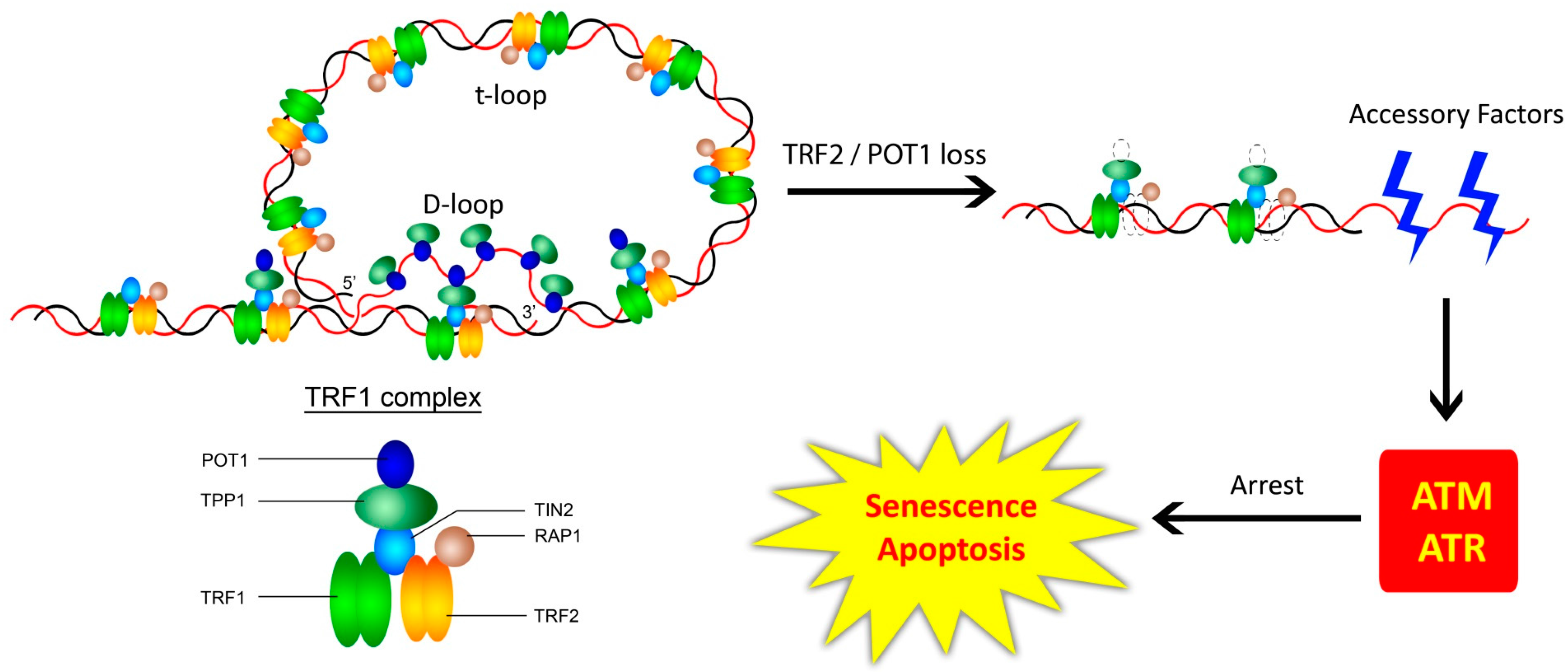
3. Telomere-Associated Protein Complexes and Other Accessory Factors
3.1. The Shelterin Complex, Telomeric Structure, and the Telomeric DNA Damage Response
3.2. DNA Repair Factors—Friend or Foe to the Telomeres?
3.3. New Kids on the Telomere Block
3.3.1. CST (CTC-STN1-TEN1)
3.3.2. HOT1 (Homeobox Telomere-Binding Protein 1)
3.4. Telomeric RNA
3.4.1. TERRA (Telomere Repeat-Containing RNA)
3.4.2. tDDRNA (Telomeric DNA Damage Response RNA)
4. Telomere Length Maintenance
4.1. Telomerase: What Is True of the Ciliates Is True of the Elephant Too!
4.2. Alternative Lengthening of Telomeres (ALT)
5. Telomeres and Cell Physiology
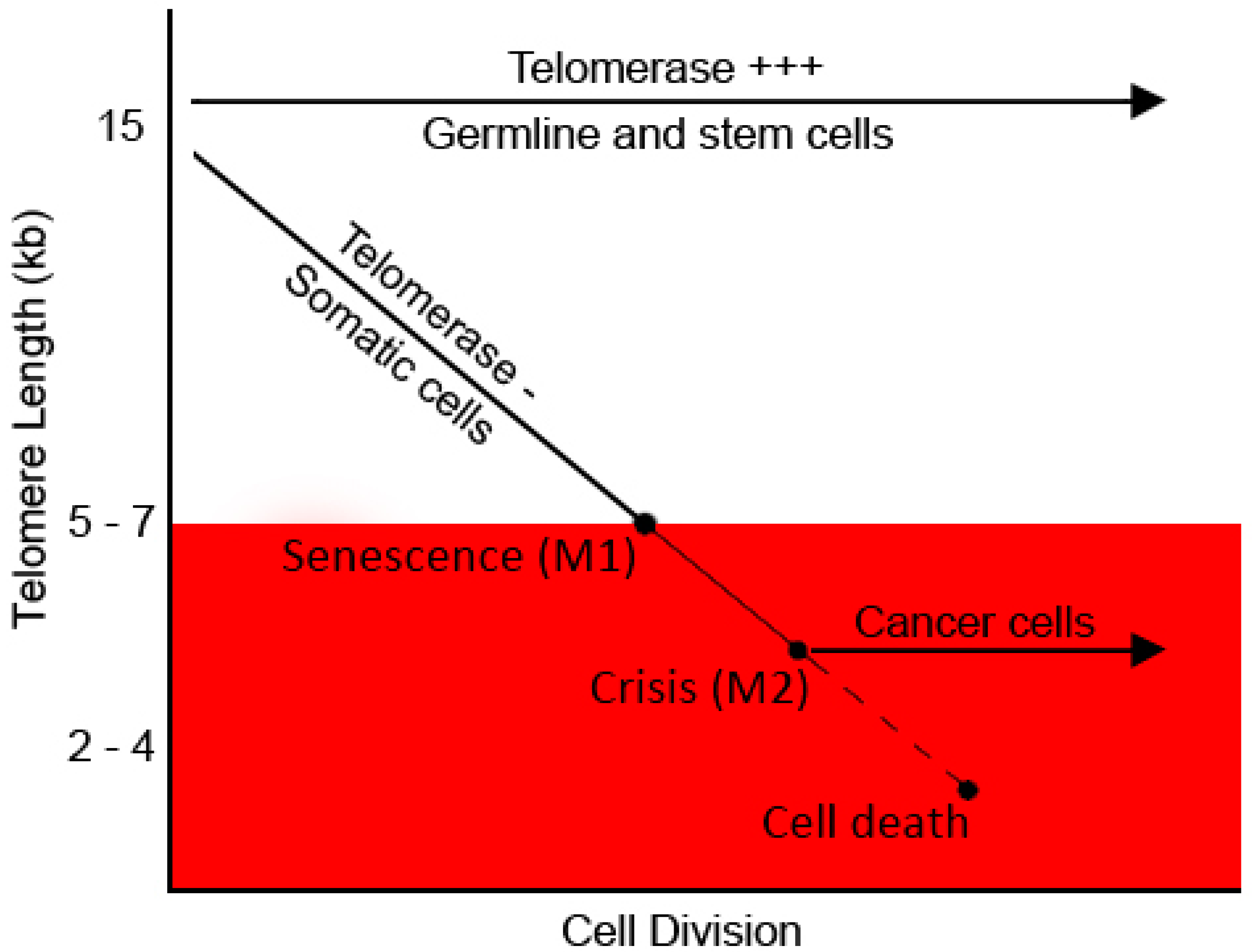
5.1. Telomeres and Cell Turnover
5.2. Telomere Shortening in Stress and Ageing
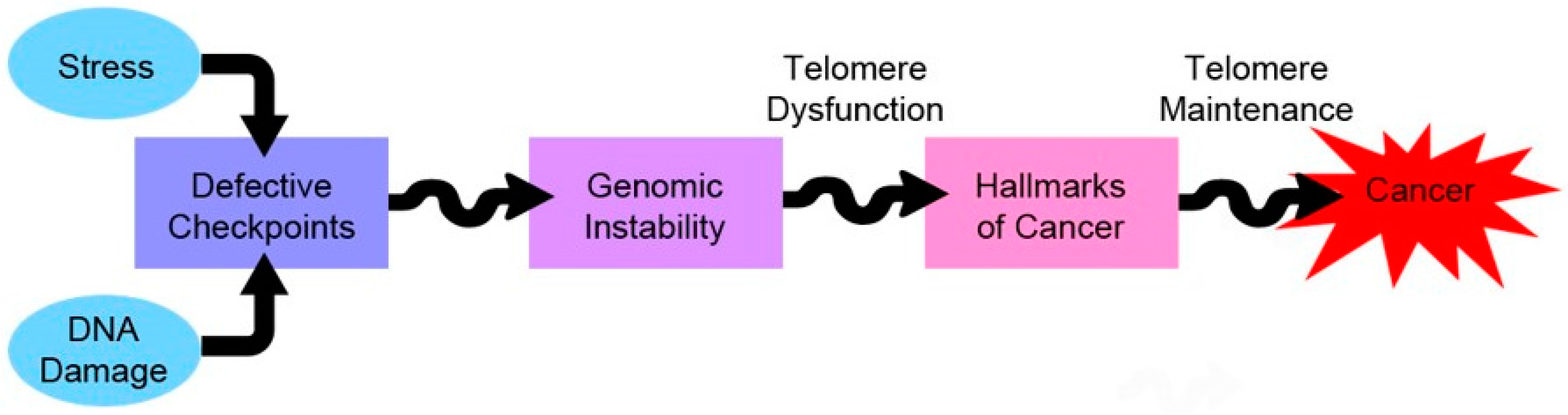
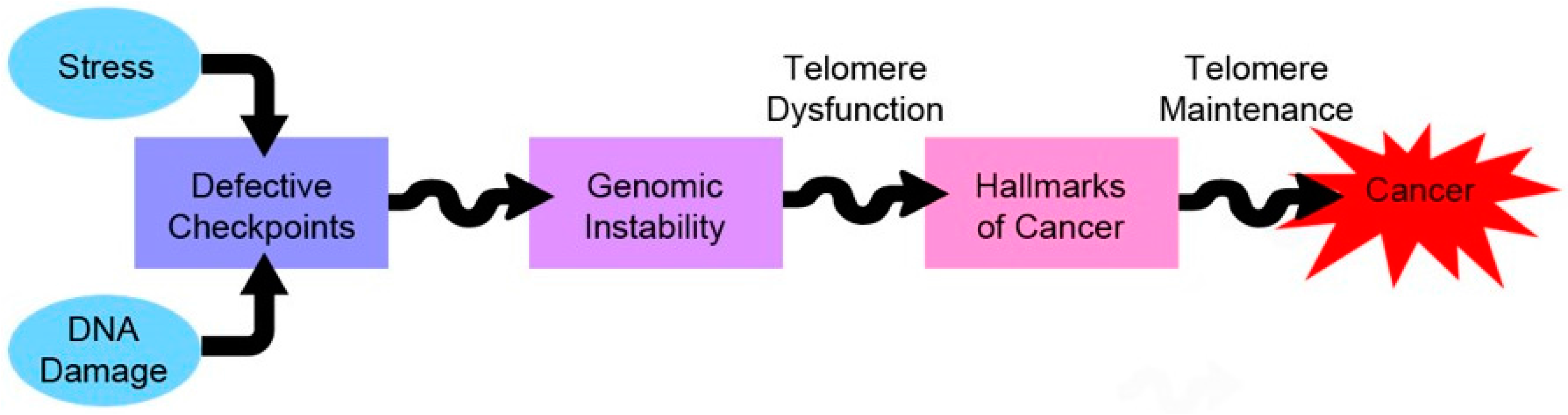
5.3. Telomeres and Cancer–Therapeutic Approaches
6. Concluding Remarks
Conflicts of Interest
References
- Muller, H.J. The remaking of chromosomes. Collect. Net 1938, 13, 181–195. [Google Scholar]
- McClintock, B. The Association of Mutants with Homozygous Deficiencies in Zea Mays. Genetics 1941, 26, 542–571. [Google Scholar] [PubMed]
- Shay, J.W.; Wright, W.E. Hayflick, his limit, and cellular ageing. Nat. Rev. Mol. Cell. Biol. 2000, 1, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.D. Origin of concatemeric T7 DNA. Nat. New Biol. 1972, 239, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Olovnikov, A.M. [Principle of marginotomy in template synthesis of polynucleotides]. Doklady Akademii Nauk SSSR 1971, 201, 1496–1499. [Google Scholar] [PubMed]
- Olovnikov, A.M. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J. Theor. Biol. 1973, 41, 181–190. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Gall, J.G. A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena. J. Mol. Biol. 1978, 120, 33–53. [Google Scholar] [CrossRef]
- Szostak, J.W.; Blackburn, E.H. Cloning yeast telomeres on linear plasmid vectors. Cell 1982, 29, 245–255. [Google Scholar] [CrossRef]
- Moyzis, R.K.; Buckingham, J.M.; Cram, L.S.; Dani, M.; Deaven, L.L.; Jones, M.D.; Meyne, J.; Ratliff, R.L.; Wu, J.R. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc. Natl. Acad. Sci. USA 1988, 85, 6622–6626. [Google Scholar] [CrossRef] [PubMed]
- Shampay, J.; Szostak, J.W.; Blackburn, E.H. DNA sequences of telomeres maintained in yeast. Nature 1984, 310, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Greider, C.W.; Blackburn, E.H. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 1985, 43, 405–413. [Google Scholar] [CrossRef]
- Cooke, H.J.; Smith, B.A. Variability at the telomeres of the human X/Y pseudoautosomal region. Cold Spring Harb. Symp. Quant. Biol. 1986, 51, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, J.; McGill, N.I.; Lindsey, L.A.; Green, D.K.; Cooke, H.J. In vivo loss of telomeric repeats with age in humans. Mutat. Res. 1991, 256, 45–48. [Google Scholar] [CrossRef]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Morin, G.B. The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell 1989, 59, 521–529. [Google Scholar] [CrossRef]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Bryan, T.M.; Englezou, A.; Gupta, J.; Bacchetti, S.; Reddel, R.R. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 1995, 14, 4240–4248. [Google Scholar] [PubMed]
- Feng, J.; Funk, W.D.; Wang, S.S.; Weinrich, S.L.; Avilion, A.A.; Chiu, C.P.; Adams, R.R.; Chang, E.; Allsopp, R.C.; Yu, J.; et al. The RNA component of human telomerase. Science 1995, 269, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.M.; Morin, G.B.; Chapman, K.B.; Weinrich, S.L.; Andrews, W.H.; Lingner, J.; Harley, C.B.; Cech, T.R. Telomerase catalytic subunit homologs from fission yeast and human. Science 1997, 277, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.A.; Lee, H.W.; Hande, M.P.; Samper, E.; Lansdorp, P.M.; DePinho, R.A.; Greider, C.W. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 1997, 91, 25–34. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H. Telomere states and cell fates. Nature 2000, 408, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Collins, K. Mammalian telomeres and telomerase. Curr. Opin. Cell Biol. 2000, 12, 378–383. [Google Scholar] [CrossRef]
- Giraud-Panis, M.J.; Giraud-Panis, M.J.; Pisano, S.; Benarroch-Popivker, D.; Pei, B.; Le Du, M.H.; Gilson, E. One identity or more for telomeres? Front. Oncol. 2013, 3, 48. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Smith, D.L.; Blackburn, E.H. Mutant telomere sequences lead to impaired chromosome separation and a unique checkpoint response. Mol. Biol. Cell 2004, 15, 1623–1634. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Shiue, L.; Kaplan, S.; de Lange, T. A mammalian factor that binds telomeric TTAGGG repeats in vitro. Mol. Cell. Biol. 1992, 12, 4834–4843. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P.; Cech, T.R. Pot1, the putative telomere end-binding protein in fission yeast and humans. Science 2001, 292, 1171–1175. [Google Scholar] [CrossRef] [PubMed]
- Bilaud, T.; Brun, C.; Ancelin, K.; Koering, C.E.; Laroche, T.; Gilson, E. Telomeric localization of TRF2, a novel human telobox protein. Nat. Genet. 1997, 17, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Broccoli, D.; Smogorzewska, A.; Chong, L.; de Lange, T. Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat. Genet. 1997, 17, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Chong, L.; van Steensel, B.; Broccoli, D.; Erdjument-Bromage, H.; Hanish, J.; Tempst, P.; de Lange, T. A human telomeric protein. Science 1995, 270, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Houghtaling, B.R.; Cuttonaro, L.; Chang, W.; Smith, S. A dynamic molecular link between the telomere length regulator TRF1 and the chromosome end protector TRF2. Curr. Biol. 2004, 14, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kaminker, P.; Campisi, J. TIN2, a new regulator of telomere length in human cells. Nat. Genet. 1999, 23, 405–512. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Oestreich, S.; de Lange, T. Identification of human Rap1: Implications for telomere evolution. Cell 2000, 101, 471–483. [Google Scholar] [CrossRef]
- Bianchi, A.; Stansel, R.M.; Fairall, L.; Griffith, J.D.; Rhodes, D.; de Lange, T. TRF1 binds a bipartite telomeric site with extreme spatial flexibility. EMBO J. 1999, 18, 5735–5744. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Palm, W.; de Lange, T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008, 42, 301–334. [Google Scholar] [CrossRef] [PubMed]
- Court, R.; Chapman, L.; Fairall, L.; Rhodes, D. How the human telomeric proteins TRF1 and TRF2 recognize telomeric DNA: A view from high-resolution crystal structures. EMBO Rep. 2005, 6, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Podell, E.R.; Cech, T.R. Structure of human POT1 bound to telomeric single-stranded DNA provides a model for chromosome end-protection. Nat. Struct. Mol. Biol. 2004, 11, 1223–1239. [Google Scholar] [CrossRef] [PubMed]
- Loayza, D.; De Lange, T. POT1 as a terminal transducer of TRF1 telomere length control. Nature 2003, 423, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Giraud-Panis, M.J.; Pisano, S.; Poulet, A.; Le Du, M.H.; Gilson, E. Structural identity of telomeric complexes. FEBS Lett. 2010, 584, 3785–3799. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; de Lange, T. Mammalian telomeres end in a large duplex loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef]
- De Lange, T. How telomeres solve the end-protection problem. Science 2009, 326, 948–952. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [PubMed]
- Lingner, J.; Cooper, J.P.; Cech, T.R. Telomerase and DNA end replication: No longer a lagging strand problem? Science 1995, 269, 1533–1534. [Google Scholar] [CrossRef] [PubMed]
- Nezu, T.; Hosomi, N.; Takahashi, T.; Anno, K.; Aoki, S.; Shimamoto, A.; Maruyama, H.; Hayashi, T.; Matsumoto, M.; Tahara, H. Telomere G-tail Length is a Promising Biomarker Related to White Matter Lesions and Endothelial Dysfunction in Patients With Cardiovascular Risk: A Cross-sectional Study. EBioMedicine 2015, 2, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Seimiya, H. Predicting Risk at the End of the End: Telomere G-tail as a Biomarker. EBioMedicine 2015, 2, 804–805. [Google Scholar] [CrossRef] [PubMed]
- Van Steensel, B.; de Lange, T. Control of telomere length by the human telomeric protein TRF1. Nature 1997, 385, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Smogorzewska, A.; van Steensel, B.; Bianchi, A.; Oelmann, S.; Schaefer, M.R.; Schnapp, G.; de Lange, T. Control of human telomere length by TRF1 and TRF2. Mol. Cell. Biol. 2000, 20, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Bandaria, J.N.; Qin, P.; Berk, V.; Chu, S.; Yildiz, A. Shelterin Protects Chromosome Ends by Compacting Telomeric Chromatin. Cell 2016, 164, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Timashev, L.A.; Babcock, H.; Zhuang, X.; de Lange, T. The DDR at telomeres lacking intact shelterin does not require substantial chromatin decompaction. Genes Dev. 2017, 31, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Vancevska, A.; Douglass, K.M.; Pfeiffer, V.; Manley, S.; Lingner, J. The telomeric DNA damage response occurs in the absence of chromatin decompaction. Genes Dev. 2017, 31, 567–577. [Google Scholar] [CrossRef] [PubMed]
- D’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Jobling, W.A.; Chen, B.P.; Chen, D.J.; Sedivy, J.M. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef]
- Zschenker, O.; Kulkarni, A.; Miller, D.; Reynolds, G.E.; Granger-Locatelli, M.; Pottier, G.; Sabatier, L.; Murnane, J.P. Increased sensitivity of subtelomeric regions to DNA double-strand breaks in a human cancer cell line. DNA Repair (Amst.) 2009, 8, 886–900. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Zschenker, O.; Reynolds, G.; Miller, D.; Murnane, J.P. Effect of telomere proximity on telomere position effect, chromosome healing, and sensitivity to DNA double-strand breaks in a human tumor cell line. Mol. Cell. Biol. 2010, 30, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Muraki, K.; Nyhan, K.; Han, L.; Murnane, J.P. Mechanisms of telomere loss and their consequences for chromosome instability. Front. Oncol. 2012, 2, 135. [Google Scholar] [CrossRef] [PubMed]
- Doksani, Y.; de Lange, T. Telomere-Internal Double-Strand Breaks Are Repaired by Homologous Recombination and PARP1/Lig3-Dependent End-Joining. Cell. Rep. 2016, 17, 1646–1656. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, J.A.; Parkhill, J.; Campbell, L.; Stacey, M.; Biggs, P.; Byrd, P.J.; Taylor, A.M. Accelerated telomere shortening in ataxia telangiectasia. Nat. Genet. 1996, 13, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Hande, M.P.; Balajee, A.S.; Tchirkov, A.; Wynshaw-Boris, A.; Lansdorp, P.M. Extra-chromosomal telomeric DNA in cells from Atm(-/-) mice and patients with ataxia-telangiectasia. Hum. Mol. Genet. 2001, 10, 519–528. [Google Scholar] [CrossRef] [PubMed]
- D’Adda di Fagagna, F.; Hande, M.P.; Tong, W.M.; Lansdorp, P.M.; Wang, Z.Q.; Jackson, S.P. Functions of poly(ADP-ribose) polymerase in controlling telomere length and chromosomal stability. Nat. Genet. 1999, 23, 76–80. [Google Scholar] [PubMed]
- Hsu, H.L.; Gilley, D.; Galande, S.A.; Hande, M.P.; Allen, B.; Kim, S.H.; Li, G.C.; Campisi, J.; Kohwi-Shigematsu, T.; Chen, D.J. Ku acts in a unique way at the mammalian telomere to prevent end joining. Genes Dev. 2000, 14, 2807–2812. [Google Scholar] [CrossRef] [PubMed]
- Gilley, D.; Tanaka, H.; Hande, M.P.; Kurimasa, A.; Li, G.C.; Oshimura, M.; Chen, D.J. DNA-PKcs is critical for telomere capping. Proc. Natl. Acad. Sci. USA 2001, 98, 15084–15088. [Google Scholar] [CrossRef] [PubMed]
- Hande, M.P. DNA repair factors and telomere-chromosome integrity in mammalian cells. Cytogenet. Genome Res. 2004, 104, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Hande, P.; Slijepcevic, P.; Silver, A.; Bouffler, S.; van Buul, P.; Bryant, P.; Lansdorp, P. Elongated telomeres in scid mice. Genomics 1999, 56, 221–223. [Google Scholar] [CrossRef] [PubMed]
- d’Adda di Fagagna, F.; Hande, M.P.; Tong, W.M.; Roth, D.; Lansdorp, P.M.; Wang, Z.Q.; Jackson, S.P. Effects of DNA nonhomologous end-joining factors on telomere length and chromosomal stability in mammalian cells. Curr. Biol. 2001, 11, 1192–1196. [Google Scholar] [CrossRef]
- Zhu, X.D.; Kuster, B.; Mann, M.; Petrini, J.H.; de Lange, T. Cell-cycle-regulated association of RAD50/MRE11/NBS1 with TRF2 and human telomeres. Nat. Genet. 2000, 25, 347–352. [Google Scholar] [PubMed]
- Miller, D.; Reynolds, G.E.; Mejia, R.; Stark, J.M.; Murnane, J.P. Subtelomeric regions in mammalian cells are deficient in DNA double-strand break repair. DNA Repair (Amst.) 2011, 10, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Tong, A.S.; Stern, J.L.; Sfeir, A.; Kartawinata, M.; de Lange, T.; Zhu, X.D.; Bryan, T.M. ATM and ATR Signaling Regulate the Recruitment of Human Telomerase to Telomeres. Cell Rep. 2015, 13, 1633–1646. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Bohrson, C.; Pike, A.M.; Wheelan, S.J.; Greider, C.W. ATM Kinase Is Required for Telomere Elongation in Mouse and Human Cells. Cell Rep. 2015, 13, 1623–1632. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.M. A sense of the end. Science 2000, 288, 1377–1379. [Google Scholar] [CrossRef] [PubMed]
- Francia, S.; Weiss, R.S.; d’Adda di Fagagna, F. Need telomere maintenance? Call 911. Cell Div. 2007, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Slijepcevic, P. The role of DNA damage response proteins at telomeres—An “integrative” model. DNA Repair (Amst.) 2006, 5, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Nakamura, M.; Nabetani, A.; Shimamura, S.; Tamura, M.; Yonehara, S.; Saito, M.; Ishikawa, F. RPA-like mammalian Ctc1-Stn1-Ten1 complex binds to single-stranded DNA and protects telomeres independently of the Pot1 pathway. Mol. Cell 2009, 36, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Surovtseva, Y.V.; Churikov, D.; Boltz, K.A.; Song, X.; Lamb, J.C.; Warrington, R.; Leehy, K.; Heacock, M.; Price, C.M.; Shippen, D.E. Conserved telomere maintenance component 1 interacts with STN1 and maintains chromosome ends in higher eukaryotes. Mol. Cell 2009, 36, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.A.; Wang, F.; Chaiken, M.F.; Kasbek, C.; Chastain, P.D., 2nd; Wright, W.E.; Price, C.M. Human CST promotes telomere duplex replication and general replication restart after fork stalling. EMBO J. 2012, 31, 3537–3549. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Y.; Redon, S.; Lingner, J. The human CST complex is a terminator of telomerase activity. Nature 2012, 488, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Stewart, J.A.; Kasbek, C.; Zhao, Y.; Wright, W.E.; Price, C.M. Human CST has independent functions during telomere duplex replication and C-strand fill-in. Cell Rep. 2012, 2, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Giraud-Panis, M.J.; Teixeira, M.T.; Geli, V.; Gilson, E. CST meets shelterin to keep telomeres in check. Mol. Cell 2010, 39, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Jady, B.E.; Richard, P.; Bertrand, E.; Kiss, T. Cell cycle-dependent recruitment of telomerase RNA and Cajal bodies to human telomeres. Mol. Biol. Cell. 2006, 17, 944–954. [Google Scholar] [CrossRef] [PubMed]
- Kappei, D.; Butter, F.; Benda, C.; Scheibe, M.; Draskovic, I.; Stevense, M.; Novo, C.L.; Basquin, C.; Araki, M.; Araki, K.; et al. HOT1 is a mammalian direct telomere repeat-binding protein contributing to telomerase recruitment. EMBO J. 2013, 32, 1681–1701. [Google Scholar] [CrossRef] [PubMed]
- Tarsounas, M. It’s getting HOT at telomeres. EMBO J. 2013, 32, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Azzalin, C.M.; Reichenbach, P.; Khoriauli, L.; Giulotto, E.; Lingner, J. Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science 2007, 318, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Le, P.N.; Maranon, D.G.; Altina, N.H.; Battaglia, C.L.; Bailey, S.M. TERRA, hnRNP A1, and DNA-PKcs Interactions at Human Telomeres. Front. Oncol. 2013, 3, 91. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.L.; Centore, R.C.; O’Sullivan, R.J.; Rai, R.; Tse, A.; Songyang, Z.; Chang, S.; Karlseder, J.; Zou, L. TERRA and hnRNPA1 orchestrate an RPA-to-POT1 switch on telomeric single-stranded DNA. Nature 2011, 471, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.J.; Lopez de Silanes, I.; Grana, O.; Blasco, M.A. Telomeric RNAs are essential to maintain telomeres. Nat. Commun. 2016, 7, 12534. [Google Scholar] [CrossRef] [PubMed]
- Hirashima, K.; Seimiya, H. Telomeric repeat-containing RNA/G-quadruplex-forming sequences cause genome-wide alteration of gene expression in human cancer cells in vivo. Nucleic Acids Res. 2015, 43, 2022–2032. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Misteli, T. Non-coding RNAs in DNA damage and repair. FEBS Lett. 2013, 587, 1832–1839. [Google Scholar] [CrossRef] [PubMed]
- Rossiello, F.; Aguado, J.; Sepe, S.; Iannelli, F.; Nguyen, Q.; Pitchiaya, S.; Carninci, P.; d’Adda di Fagagna, F. DNA damage response inhibition at dysfunctional telomeres by modulation of telomeric DNA damage response RNAs. Nat. Commun. 2017, 8, 13980. [Google Scholar] [CrossRef] [PubMed]
- Rivera, T.; Haggblom, C.; Cosconati, S.; Karlseder, J. A balance between elongation and trimming regulates telomere stability in stem cells. Nat. Struct. Mol. Biol. 2017, 24, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Li, J.S.; Miralles Fuste, J.; Simavorian, T.; Bartocci, C.; Tsai, J.; Karlseder, J.; Lazzerini Denchi, E. TZAP: A telomere-associated protein involved in telomere length control. Science 2017, 355, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.; Blasco, M.A. Replicating through telomeres: A means to an end. Trends Biochem. Sci. 2015, 40, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.S.; Zhang, B.; Spector, D.L. Biogenesis and function of nuclear bodies. Trends Genet. 2011, 27, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Nizami, Z.; Deryusheva, S.; Gall, J.G. The Cajal body and histone locus body. Cold Spring Harb. Perspect. Biol. 2010, 2, a000653. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, R.L.; Abreu, E.B.; Ziegler, T.; Ly, H.; Counter, C.M.; Terns, R.M.; Terns, M.P. Telomerase reverse transcriptase is required for the localization of telomerase RNA to cajal bodies and telomeres in human cancer cells. Mol. Biol. Cell 2008, 19, 3793–3800. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Deng, Z.; Jiang, S.; Hu, Q.; Liu, H.; Songyang, Z.; Ma, W.; Chen, S.; Zhao, Y. Human cells lacking coilin and Cajal bodies are proficient in telomerase assembly, trafficking and telomere maintenance. Nucleic Acids Res. 2015, 43, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Vogan, J.M.; Zhang, X.; Youmans, D.T.; Regalado, S.G.; Johnson, J.Z.; Hockemeyer, D.; Collins, K. Minimized human telomerase maintains telomeres and resolves endogenous roles of H/ACA proteins, TCAB1, and Cajal bodies. Elife 2016, 5, e18221. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Rhodes, D. Telomerase structure. Curr. Opin. Struct. Biol. 2014, 25, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W. Molecular pathogenesis of aging and cancer: Are telomeres and telomerase the connection? J. Clin. Pathol. 1997, 50, 799–800. [Google Scholar] [CrossRef] [PubMed]
- Hahn, W.C. Telomerase and cancer: Where and when? Clin. Cancer Res. 2001, 7, 2953–2954. [Google Scholar] [PubMed]
- Liu, R.; Xing, M. TERT promoter mutations in thyroid cancer. Endocr. Relat. Cancer 2016, 23, R143–R155. [Google Scholar] [PubMed]
- Matsuse, M.; Yabuta, T.; Saenko, V.; Hirokawa, M.; Nishihara, E.; Suzuki, K.; Yamashita, S.; Miyauchi, A.; Mitsutake, N. TERT promoter mutations and Ki-67 labeling index as a prognostic marker of papillary thyroid carcinomas: Combination of two independent factors. Sci. Rep. 2017, 7, 41752. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Keith, W.N. Targeting telomerase for cancer therapeutics. Br. J. Cancer 2008, 98, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Lundblad, V.; Blackburn, E.H. An alternative pathway for yeast telomere maintenance rescues est1-senescence. Cell 1993, 73, 347–360. [Google Scholar] [CrossRef]
- Hande, M.P.; Samper, E.; Lansdorp, P.; Blasco, M.A. Telomere length dynamics and chromosomal instability in cells derived from telomerase null mice. J. Cell Biol. 1999, 144, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Niida, H.; Shinkai, Y.; Hande, M.P.; Matsumoto, T.; Takehara, S.; Tachibana, M.; Oshimura, M.; Lansdorp, P.M.; Furuichi, Y. Telomere maintenance in telomerase-deficient mouse embryonic stem cells: Characterization of an amplified telomeric DNA. Mol. Cell Biol. 2000, 20, 4115–4127. [Google Scholar] [CrossRef] [PubMed]
- Rogan, E.M.; Bryan, T.M.; Hukku, B.; Maclean, K.; Chang, A.C.; Moy, E.L.; Englezou, A.; Warneford, S.G.; Dalla-Pozza, L.; Reddel, R.R. Alterations in p53 and p16INK4 expression and telomere length during spontaneous immortalization of Li-Fraumeni syndrome fibroblasts. Mol. Cell Biol. 1995, 15, 4745–4753. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Lafferty-Whyte, K.; Cairney, C.J.; Will, M.B.; Serakinci, N.; Daidone, M.G.; Zaffaroni, N.; Bilsland, A.; Keith, W.N. A gene expression signature classifying telomerase and ALT immortalization reveals an hTERT regulatory network and suggests a mesenchymal stem cell origin for ALT. Oncogene 2009, 28, 3765–3774. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Kaul, Z.; Cohen, S.B.; Napier, C.E.; Pickett, H.A.; Neumann, A.A.; Reddel, R.R. Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat. Struct. Mol. Biol. 2009, 16, 1244–1251. [Google Scholar] [CrossRef] [PubMed]
- Episkopou, H.; Draskovic, I.; Van Beneden, A.; Tilman, G.; Mattiussi, M.; Gobin, M.; Arnoult, N.; Londono-Vallejo, A.; Decottignies, A. Alternative Lengthening of Telomeres is characterized by reduced compaction of telomeric chromatin. Nucleic Acids Res. 2014, 42, 4391–4405. [Google Scholar] [CrossRef] [PubMed]
- Clynes, D.; Jelinska, C.; Xella, B.; Ayyub, H.; Scott, C.; Mitson, M.; Taylor, S.; Higgs, D.R.; Gibbons, R.J. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun. 2015, 6, 7538. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Shi, G.; Zhang, L.; Li, F.; Jiang, Y.; Jiang, S.; Ma, W.; Zhao, Y.; Songyang, Z.; Huang, J. Switch telomerase to ALT mechanism by inducing telomeric DNA damages and dysfunction of ATRX and DAXX. Sci. Rep. 2016, 6, 32280. [Google Scholar] [CrossRef] [PubMed]
- Draskovic, I.; Arnoult, N.; Steiner, V.; Bacchetti, S.; Lomonte, P.; Londono-Vallejo, A. Probing PML body function in ALT cells reveals spatiotemporal requirements for telomere recombination. Proc. Natl. Acad. Sci. USA 2009, 106, 15726–15731. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, N.; Chen, Y.C.; Spector, D.L.; de Lange, T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 2008, 456, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.W.; Dilley, R.L.; Lampson, M.A.; Greenberg, R.A. Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 2014, 159, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Villa, R.; Daidone, M.G.; Motta, R.; Venturini, L.; De Marco, C.; Vannelli, A.; Kusamura, S.; Baratti, D.; Deraco, M.; Costa, A.; et al. Multiple mechanisms of telomere maintenance exist and differentially affect clinical outcome in diffuse malignant peritoneal mesothelioma. Clin. Cancer Res. 2008, 14, 4134–4140. [Google Scholar] [CrossRef] [PubMed]
- Neumann, A.A.; Watson, C.M.; Noble, J.R.; Pickett, H.A.; Tam, P.P.; Reddel, R.R. Alternative lengthening of telomeres in normal mammalian somatic cells. Genes Dev. 2013, 27, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Plantinga, M.J.; Pascarelli, K.M.; Merkel, A.S.; Lazar, A.J.; von Mehren, M.; Lev, D.; Broccoli, D. Telomerase suppresses formation of ALT-associated single-stranded telomeric C-circles. Mol. Cancer Res. 2013, 11, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Conomos, D.; Stutz, M.D.; Hills, M.; Neumann, A.A.; Bryan, T.M.; Reddel, R.R.; Pickett, H.A. Variant repeats are interspersed throughout the telomeres and recruit nuclear receptors in ALT cells. J. Cell. Biol. 2012, 199, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Sieber, O.M.; Heinimann, K.; Tomlinson, I.P. Genomic instability—The engine of tumorigenesis? Nat. Rev. Cancer 2003, 3, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B.; Kim, N.W.; Prowse, K.R.; Weinrich, S.L.; Hirsch, K.S.; West, M.D.; Bacchetti, S.; Hirte, H.W.; Counter, C.M.; Greider, C.W.; et al. Telomerase, cell immortality, and cancer. Cold Spring Harb. Symp. Quant. Biol. 1994, 59, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.; Blasco, M.A. Telomere-driven diseases and telomere-targeting therapies. J. Cell Biol. 2017, 216, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Armanios, M.; Blackburn, E.H. The telomere syndromes. Nat. Rev. Genet. 2012, 13, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Holohan, B.; Wright, W.E.; Shay, J.W. Cell biology of disease: Telomeropathies: An emerging spectrum disorder. J. Cell Biol. 2014, 205, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Armanios, M. Telomeres and age-related disease: How telomere biology informs clinical paradigms. J. Clin. Investig. 2013, 123, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Shalev, I.; Moffitt, T.E.; Sugden, K.; Williams, B.; Houts, R.M.; Danese, A.; Mill, J.; Arseneault, L.; Caspi, A. Exposure to violence during childhood is associated with telomere erosion from 5 to 10 years of age: A longitudinal study. Mol. Psychiatry 2013, 18, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Vera, E.; Bernardes de Jesus, B.; Foronda, M.; Flores, J.M.; Blasco, M.A. The rate of increase of short telomeres predicts longevity in mammals. Cell Rep. 2012, 2, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Fyhrquist, F.; Saijonmaa, O. Telomere length and cardiovascular aging. Ann. Med. 2012, 44, S138–S142. [Google Scholar] [CrossRef] [PubMed]
- Robin, J.D.; Ludlow, A.T.; Batten, K.; Magdinier, F.; Stadler, G.; Wagner, K.R.; Shay, J.W.; Wright, W.E. Telomere position effect: Regulation of gene expression with progressive telomere shortening over long distances. Genes Dev. 2014, 28, 2464–2476. [Google Scholar] [CrossRef] [PubMed]
- Theodoris, C.V.; Mourkioti, F.; Huang, Y.; Ranade, S.S.; Liu, L.; Blau, H.M.; Srivastava, D. Long telomeres protect against age-dependent cardiac disease caused by NOTCH1 haploinsufficiency. J. Clin. Investig. 2017, 127, 1683–1688. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Wu, C.J.; Jaskelioff, M.; Ivanova, E.; Kost-Alimova, M.; Protopopov, A.; Chu, G.C.; Wang, G.; Lu, X.; Labrot, E.S.; et al. Telomerase reactivation following telomere dysfunction yields murine prostate tumors with bone metastases. Cell 2012, 148, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Hills, M.; Conomos, D.; Stutz, M.D.; Dagg, R.A.; Lau, L.M.; Reddel, R.R.; Pickett, H.A. Telomere extension by telomerase and ALT generates variant repeats by mechanistically distinct processes. Nucleic Acids Res. 2014, 42, 1733–1746. [Google Scholar] [CrossRef] [PubMed]
- Prescott, J.; Wentzensen, I.M.; Savage, S.A.; De Vivo, I. Epidemiologic evidence for a role of telomere dysfunction in cancer etiology. Mutat. Res. 2012, 730, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Bertorelle, R.; Rampazzo, E.; Pucciarelli, S.; Nitti, D.; De Rossi, A. Telomeres, telomerase and colorectal cancer. World J. Gastroenterol. 2014, 20, 1940–1950. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Espiridion, B.; Chen, M.; Chang, J.Y.; Lu, C.; Chang, D.W.; Roth, J.A.; Wu, X.; Gu, J. Telomere length in peripheral blood leukocytes and lung cancer risk: A large case-control study in Caucasians. Cancer Res. 2014, 74, 2476–2486. [Google Scholar] [CrossRef] [PubMed]
- Boscolo-Rizzo, P.; Da Mosto, M.C.; Rampazzo, E.; Giunco, S.; Del Mistro, A.; Menegaldo, A.; Baboci, L.; Mantovani, M.; Tirelli, G.; De Rossi, A. Telomeres and telomerase in head and neck squamous cell carcinoma: From pathogenesis to clinical implications. Cancer Metastasis Rev. 2016, 35, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Khaw, A.K.; Hande, M.P.; Kalthur, G.; Hande, M.P. Curcumin inhibits telomerase and induces telomere shortening and apoptosis in brain tumour cells. J. Cell. Biochem. 2013, 114, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Khaw, A.K.; Yong, J.W.; Kalthur, G.; Hande, M.P. Genistein induces growth arrest and suppresses telomerase activity in brain tumor cells. Genes Chromosomes Cancer 2012, 51, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Gurung, R.L.; Lim, S.N.; Khaw, A.K.; Soon, J.F.; Shenoy, K.; Mohamed Ali, S.; Jayapal, M.; Sethu, S.; Baskar, R.; Hande, M.P. Thymoquinone induces telomere shortening, DNA damage and apoptosis in human glioblastoma cells. PLoS ONE 2010, 5, e12124. [Google Scholar] [CrossRef] [PubMed]
- Gurung, R.L.; Lim, H.K.; Venkatesan, S.; Lee, P.S.; Hande, M.P. Targeting DNA-PKcs and telomerase in brain tumour cells. Mol. Cancer 2014, 13, 232. [Google Scholar] [CrossRef] [PubMed]
- Khaw, A.K.; Sameni, S.; Venkatesan, S.; Kalthur, G.; Hande, M.P. Plumbagin alters telomere dynamics, induces DNA damage and cell death in human brain tumour cells. Mutat Res. Genet. Toxicol. Environ. Mutagen. 2015, 793, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Buseman, C.M.; Wright, W.E.; Shay, J.W. Is telomerase a viable target in cancer? Mutat. Res. 2012, 730, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Heaphy, C.M.; Subhawong, A.P.; Hong, S.M.; Goggins, M.G.; Montgomery, E.A.; Gabrielson, E.; Netto, G.J.; Epstein, J.I.; Lotan, T.L.; Westra, W.H.; et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am. J. Pathol. 2011, 179, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Hwang, S.S.; Liesa, M.; Gan, B.; Sahin, E.; Jaskelioff, M.; Ding, Z.; Ying, H.; Boutin, A.T.; Zhang, H.; et al. Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell 2012, 148, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suva, M.L.; Benes, C.H.; et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273–277. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venkatesan, S.; Khaw, A.K.; Hande, M.P. Telomere Biology—Insights into an Intriguing Phenomenon. Cells 2017, 6, 15. https://doi.org/10.3390/cells6020015
Venkatesan S, Khaw AK, Hande MP. Telomere Biology—Insights into an Intriguing Phenomenon. Cells. 2017; 6(2):15. https://doi.org/10.3390/cells6020015
Chicago/Turabian StyleVenkatesan, Shriram, Aik Kia Khaw, and Manoor Prakash Hande. 2017. "Telomere Biology—Insights into an Intriguing Phenomenon" Cells 6, no. 2: 15. https://doi.org/10.3390/cells6020015