
The Ubiquitination of NF-κB Subunits in the Control of Transcription


Abstract
:1. Introduction
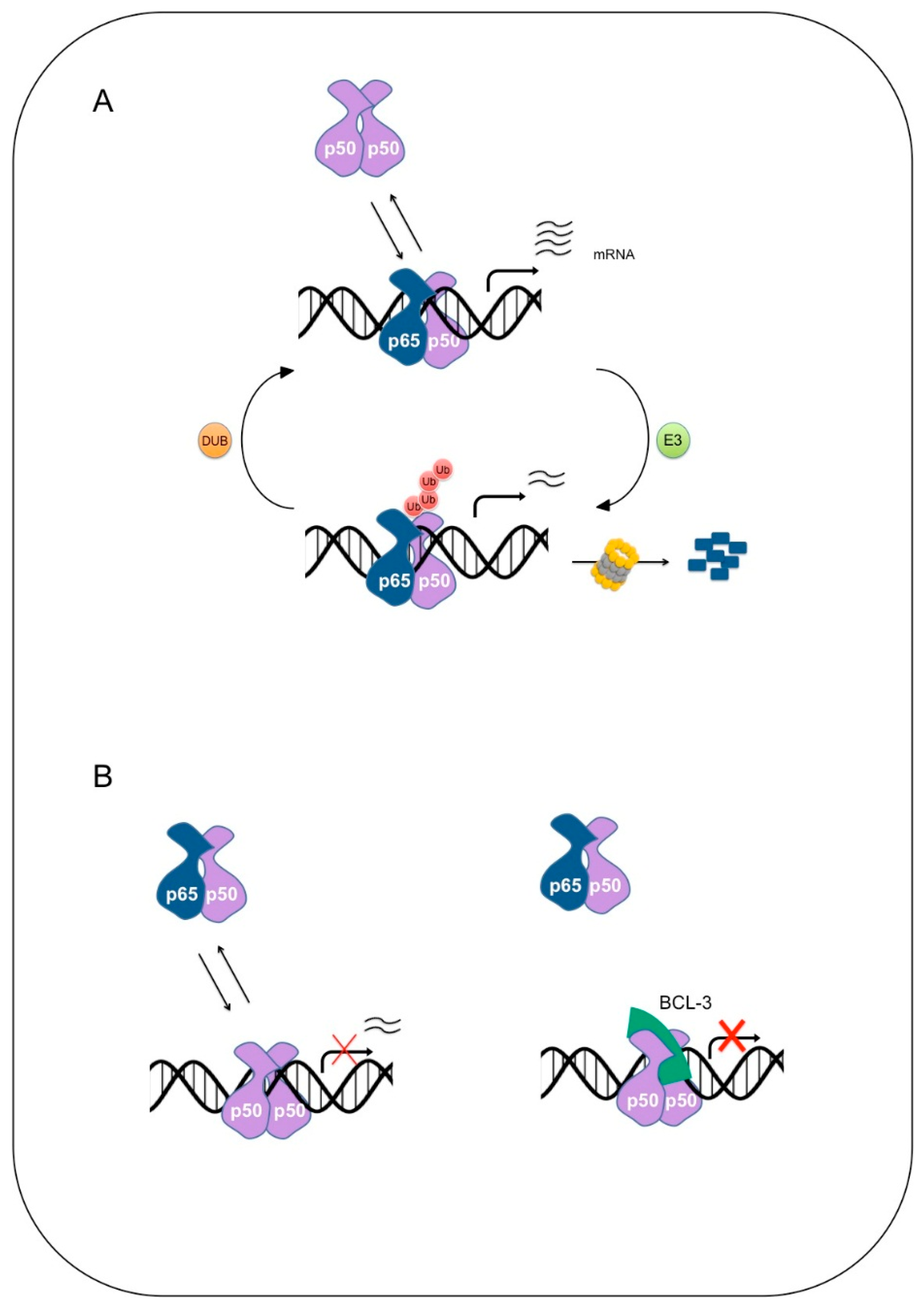
2. NF-κB
Degradation of NF-κB
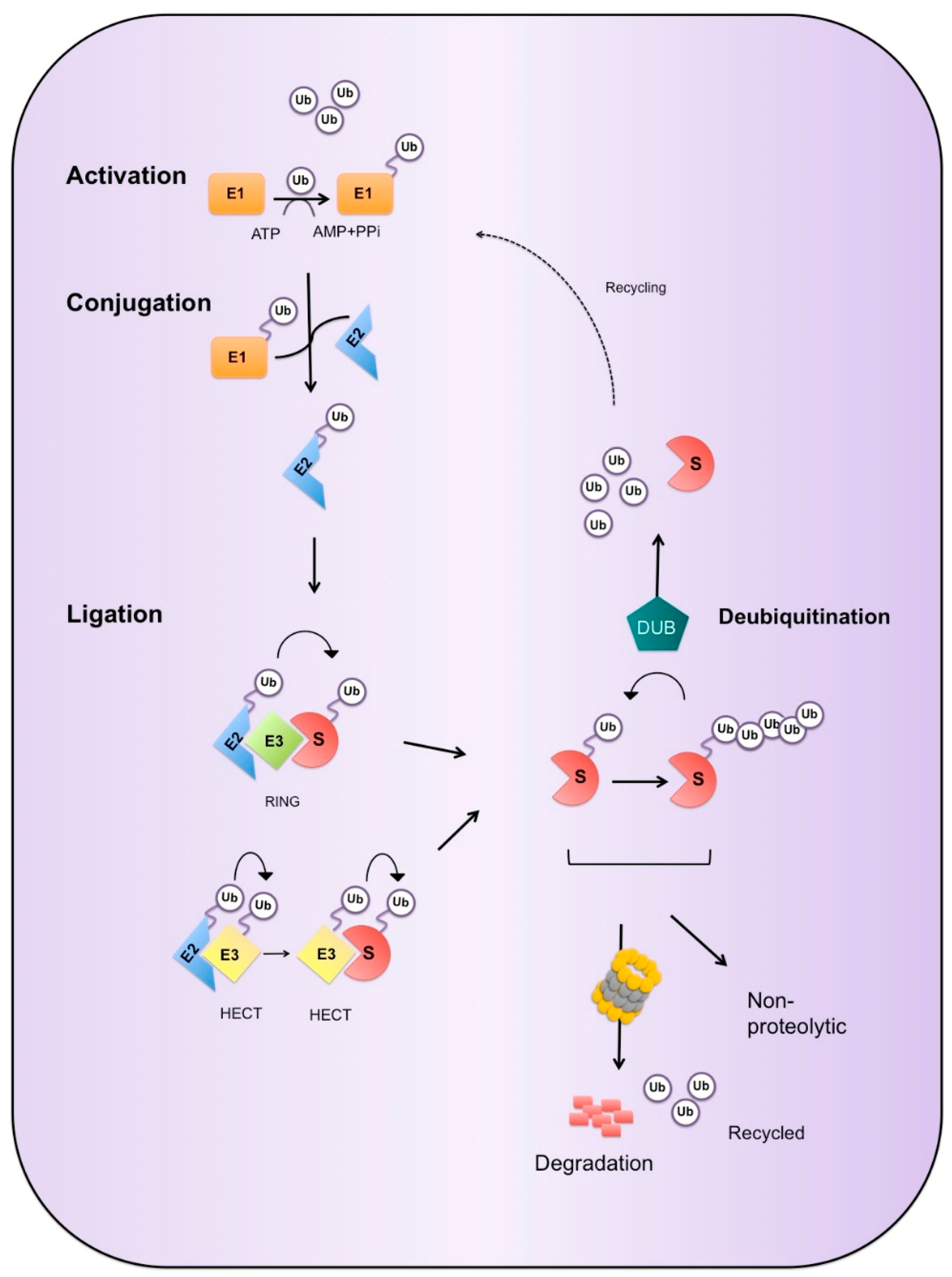
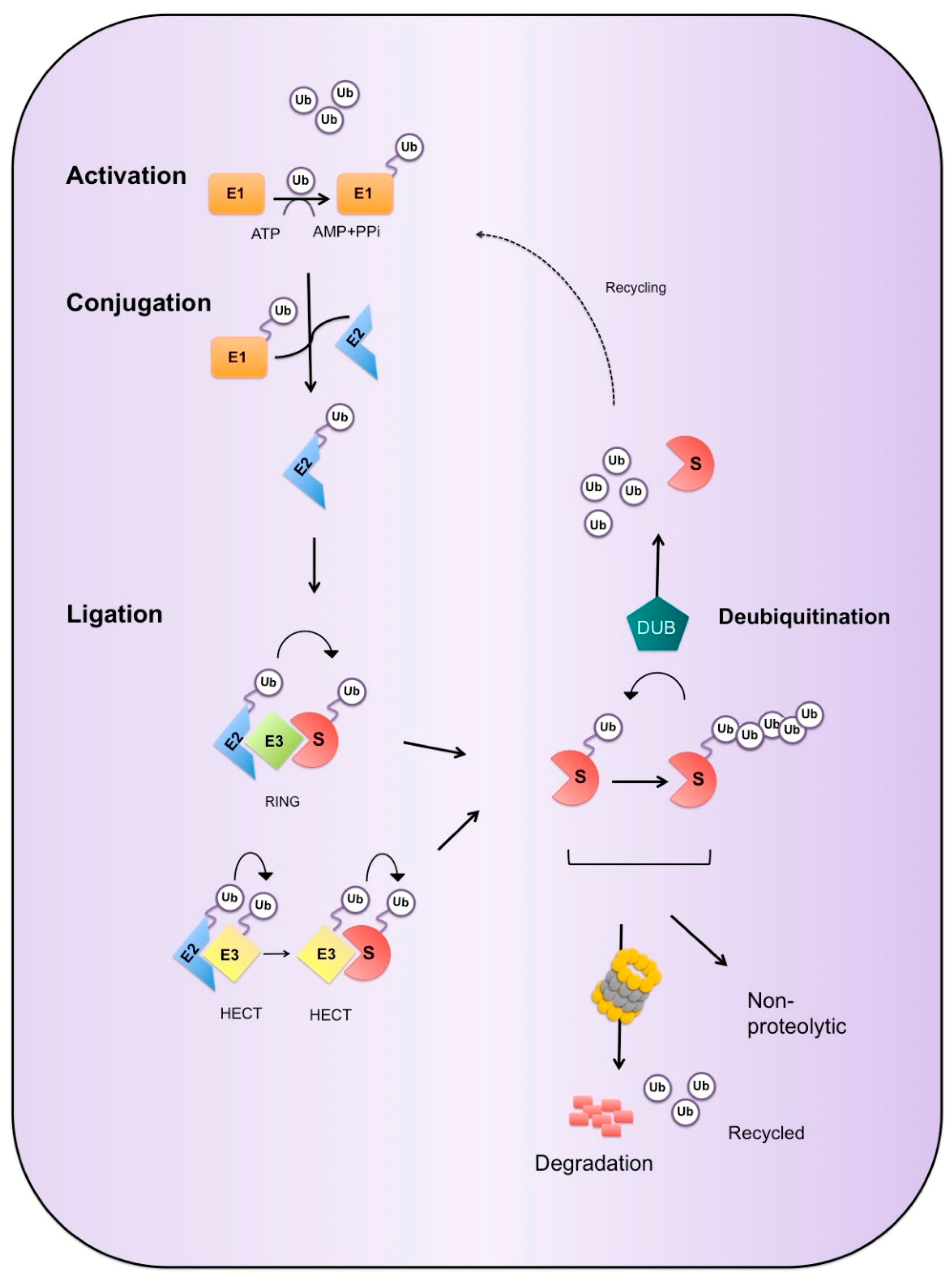
3. Ubiquitin Proteasome System
3.1. Ubiquitin Cascade
3.2. Enzymes of the UPS
3.2.1. E3 Ligases
3.2.2. Deubiquitinases
3.3. Ubiquitin Mediated Degradation
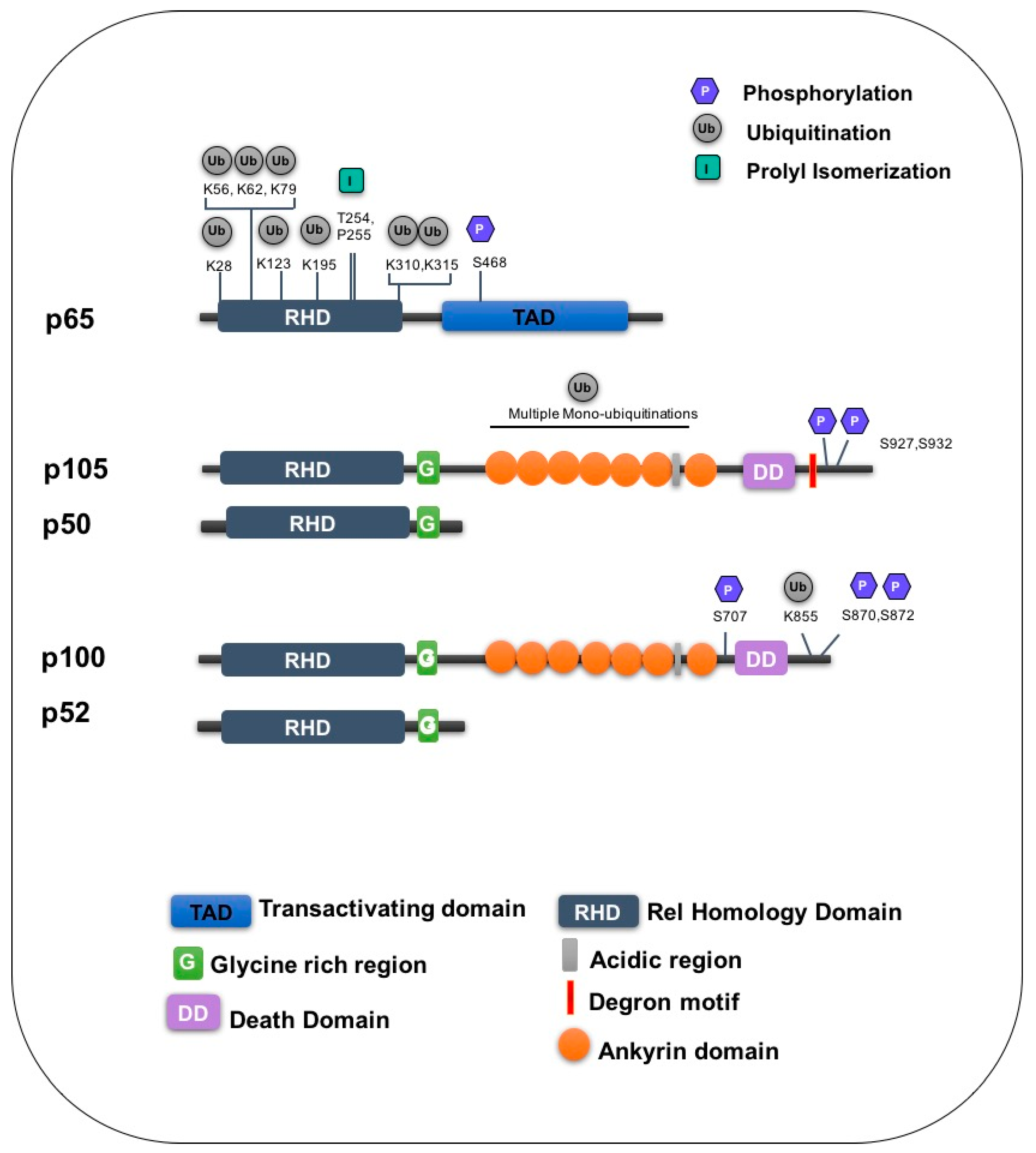
4. Ubiquitination of NF-κB
5. E3 Ligases of NF-κB
5.1. SOCS1
5.2. ORF73
5.3. PDLIM2
5.4. PPARγ
5.5. ING4
5.6. HSV-1 ICP0
5.7. Peli 1
5.8. cIAP
5.9. HERC3
6. Deubiquitinases of NF-κB
6.1. USP7
6.2. USP48
7. Regulation of NF-κB Precursors
7.1. p105
7.2. p100
8. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ARD | Ankyrin repeat domain |
| ATP | Adensoine triphosphate |
| BAG1 | BCL2-associated athanogene 1 |
| cIAP | Inhibitors of apoptosis proteins |
| COMMD1 | Copper metabolism Murr1 domain-containing 1 |
| CRL | Cullin-RING ligase |
| DUB | Deubiquitinase |
| E1 | Ubiquitin-activating enzymes |
| E2 | Ubiquitin-conjugating enzyme |
| E3 | Ubiquitin ligase |
| GRR | Glycine rich region |
| HECT | Homology to E6AP C terminus |
| HOIL-1L | Hame-oxidized IRP2 ligase-1 |
| HOIP | HOIL-1-interacting protein |
| IKK | IκB kinase |
| ING | Inhibitor of growth |
| IκB | Inhibitor of κB |
| LAP | Leukemia-associated protein |
| LPS | Lipopolysaccharide |
| LUBAC | Linear ubiquitination assembly complex |
| JAMM | JAB1/MPN/Mov34 |
| NEMO | NF-kb essential modulator |
| NF-κB | Nuclear factor kappa b |
| OUT | Ovarian tumor proteases |
| PDLIM2 | PDZ -LIM domain containing protein 2 |
| PHD | Plant homeodomain |
| PMA | Phorbol 12-myristate 13-acetate |
| PPAR | Peroxisome proliferator activated receptor |
| RBR | RING-between-RING |
| RHD | REL homology domain |
| RING | Really interesting new gene |
| SCF | Skp1–Cullin1–F-box protein |
| SHARPIN | Shank1, a postsynaptic density-enriched protein |
| SOC1 | Suppressor of cytokine signalling 1 |
| TLR | Toll like receptor |
| TNFR | TNF receptor |
| UCH | Ubiquitin carboxy-terminal hydrolases |
| USP | Ubiquitin specific proteases |
| UPS | Ubiquitin proteasome system |
References
- Schlesinger, D.H.; Goldstein, G.; Niall, H.D. Complete amino acid sequence of ubiquitin, an adenylate cyclase stimulating polypeptide probably universal in living cells. Biochemistry 1975, 14, 2214–2218. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, G.; Scheid, M.; Hammerling, U.; Schlesinger, D.H.; Niall, H.D.; Boyse, E.A. Isolation of a polypeptide that has lymphocyte-differentiating properties and is probably represented universally in living cells. Proc. Natl. Acad. Sci. USA 1975, 72, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Heller, H.; Elias, S.; Haas, A.L.; Hershko, A. Atp-dependent conjugation of reticulocyte proteins with the polypeptide required for protein degradation. Proc. Natl. Acad. Sci. USA 1980, 77, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A.; Heller, H.; Haas, A.L.; Rose, I.A. Proposed role of atp in protein breakdown: Conjugation of protein with multiple chains of the polypeptide of atp-dependent proteolysis. Proc. Natl. Acad. Sci. USA 1980, 77, 1783–1786. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, K.D.; Urban, M.K.; Haas, A.L. Ubiquitin is the atp-dependent proteolysis factor i of rabbit reticulocytes. J. Biol. Chem. 1980, 255, 7529–7532. [Google Scholar] [PubMed]
- Bhoj, V.G.; Chen, Z.J. Ubiquitylation in innate and adaptive immunity. Nature 2009, 458, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Bergink, S.; Jentsch, S. Principles of ubiquitin and sumo modifications in DNA repair. Nature 2009, 458, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Muratani, M.; Tansey, W.P. How the ubiquitin-proteasome system controls transcription. Nat. Rev. Mol. Cell. Biol. 2003, 4, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.; Wenzel, S.; Tansey, W.P. Ubiquitin and proteasomes in transcription. Annu. Rev. Biochem. 2012, 81, 177–201. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, P.; Flick, K.; Wittenberg, C.; Reed, S.I. Regulation of transcription by ubiquitination without proteolysis: Cdc34/scf(met30)-mediated inactivation of the transcription factor met4. Cell 2000, 102, 303–314. [Google Scholar] [CrossRef]
- Hoppe, T.; Matuschewski, K.; Rape, M.; Schlenker, S.; Ulrich, H.D.; Jentsch, S. Activation of a membrane-bound transcription factor by regulated ubiquitin/proteasome-dependent processing. Cell 2000, 102, 577–586. [Google Scholar] [CrossRef]
- Kanarek, N.; London, N.; Schueler-Furman, O.; Ben-Neriah, Y. Ubiquitination and degradation of the inhibitors of nf-κb. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Archer, C.T.; Delahodde, A.; Gonzalez, F.; Johnston, S.A.; Kodadek, T. Activation domain-dependent monoubiquitylation of gal4 protein is essential for promoter binding in vivo. J. Biol. Chem. 2008, 283, 12614–12623. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.-C.; Feng, Q.; Lonard, D.M.; O'Malley, B.W. Src-3 coactivator functional lifetime is regulated by a phospho-dependent ubiquitin time clock. Cell 2007, 129, 1125–1140. [Google Scholar] [CrossRef] [PubMed]
- Saccani, S.; Marazzi, I.; Beg, A.A.; Natoli, G. Degradation of promoter-bound p65/rela is essential for the prompt termination of the nuclear factor kappab response. J. Exp. Med. 2004, 200, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. Transcriptional regulation by histone ubiquitination and deubiquitination. Genes Dev. 2003, 17, 2733–2740. [Google Scholar] [CrossRef] [PubMed]
- Weake, V.M.; Workman, J.L. Histone ubiquitination: Triggering gene activity. Mol. Cell 2008, 29, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Molinari, E.; Gilman, M.; Natesan, S. Proteasome-mediated degradation of transcriptional activators correlates with activation domain potency in vivo. EMBO J. 1999, 18, 6439–6447. [Google Scholar] [CrossRef] [PubMed]
- Salghetti, S.E.; Young Kim, S.; Tansey, W.P. Destruction of myc by ubiquitin-mediated proteolysis: Cancer-associated and transforming mutations stabilize myc. EMBO J. 1999, 18, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Salghetti, S.E.; Muratani, M.; Wijnen, H.; Futcher, B.; Tansey, W.P. Functional overlap of sequences that activate transcription and signal ubiquitin-mediated proteolysis. Proc. Natl. Acad. Sci. USA 2000, 97, 3118–3123. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Tyers, M. Transcriptional regulation: Kamikaze activators. Curr. Biol. 2000, 10, R341–R343. [Google Scholar] [CrossRef]
- Conaway, R.C.; Brower, C.S.; Conaway, J.W. Emerging roles of ubiquitin in transcription regulation. Science 2002, 296, 1254–1258. [Google Scholar] [CrossRef] [PubMed]
- Salghetti, S.E.; Caudy, A.A.; Chenoweth, J.G.; Tansey, W.P. Regulation of transcriptional activation domain function by ubiquitin. Science 2001, 293, 1651–1653. [Google Scholar] [CrossRef] [PubMed]
- Lipford, J.R.; Deshaies, R.J. Diverse roles for ubiquitin-dependent proteolysis in transcriptional activation. Nat. Cell. Biol. 2003, 5, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Von der Lehr, N.; Johansson, S.; Wu, S.; Bahram, F.; Castell, A.; Cetinkaya, C.; Hydbring, P.; Weidung, I.; Nakayama, K.; Nakayama, K.I.; et al. The f-box protein skp2 participates in c-myc proteosomal degradation and acts as a cofactor for c-myc-regulated transcription. Mol. Cell 2003, 11, 1189–1200. [Google Scholar] [CrossRef]
- Kim, S.Y.; Herbst, A.; Tworkowski, K.A.; Salghetti, S.E.; Tansey, W.P. Skp2 regulates myc protein stability and activity. Mol. Cell 2003, 11, 1177–1188. [Google Scholar] [CrossRef]
- Perkins, N.D. Post-translational modifications regulating the activity and function of the nuclear factor kappa b pathway. Oncogene 2000, 25, 6717–6730. [Google Scholar] [CrossRef] [PubMed]
- Moreno, R.; Sobotzik, J.-M.; Schultz, C.; Schmitz, M.L. Specification of the nf-κb transcriptional response by p65 phosphorylation and tnf-induced nuclear translocation of ikkε. Nucleic Acids Res. 2010, 38, 6029–6044. [Google Scholar] [CrossRef] [PubMed]
- Smale, S.T. Hierarchies of nf-[kappa]b target-gene regulation. Nat. Immunol. 2011, 12, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; Dixit, V.M. Signaling to NF-κB: Regulation by ubiquitination. Cold Spring Harb. Perspect. Biol. 2010, 2, a003350. [Google Scholar] [CrossRef] [PubMed]
- Skaug, B.; Jiang, X.; Chen, Z.J. The role of ubiquitin in nf-kappab regulatory pathways. Annu. Rev. Biochem. 2009, 78, 769–796. [Google Scholar] [CrossRef] [PubMed]
- Iwai, K. Diverse ubiquitin signaling in nf-kappab activation. Trends Cell Biol. 2012, 22, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Kanarek, N.; Ben-Neriah, Y. Regulation of nf-kappab by ubiquitination and degradation of the ikappabs. Immunol. Rev. 2012, 246, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T. Target genes of nf-kb. Available online: http://www.bu.edu/nf-kb/gene-resources/target-genes/.
- DiDonato, J.A.; Mercurio, F.; Karin, M. Nf-κb and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Nf-κb, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.; Baltimore, D. I kappa b: A specific inhibitor of the nf-kappa b transcription factor. Science 1988, 242, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Baltimore, D. Activation of DNA-binding activity in an apparently cytoplasmic precursor of the nf-κb transcription factor. Cell 1988, 53, 211–217. [Google Scholar] [CrossRef]
- Beg, A.A.; Ruben, S.M.; Scheinman, R.I.; Haskill, S.; Rosen, C.A.; Baldwin, A.S. I kappa b interacts with the nuclear localization sequences of the subunits of nf-kappa b: A mechanism for cytoplasmic retention. Genes Dev. 1992, 6, 1899–1913. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.; Van Antwerp, D.; Hope, T.J. An n-terminal nuclear export signal is required for the nucleocytoplasmic shuttling of i[kappa]b[alpha]. EMBO J. 1999, 18, 6682–6693. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.S.; Thompson, J.; Hay, R.T.; Dargemont, C. Nuclear retention of iκbα protects it from signal-induced degradation and inhibits nuclear factor κb transcriptional activation. J. Biol. Chem. 1999, 274, 9108–9115. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.T.; Kudo, N.; Yoshida, M.; Miyamoto, S. A nuclear export signal in the n-terminal regulatory domain of iκbα controls cytoplasmic localization of inactive nf-κb/iκbα complexes. Proc. Natl. Acad. Sci. USA 2000, 97, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.F.; Lee, L.H.; Davis, L.; Sen, R. Cytoplasmic sequestration of rel proteins by iκbα requires crm1-dependent nuclear export. Mol. Cell. Biol. 2000, 20, 2269–2284. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Karin, M. Missing pieces in the nf-κb puzzle. Cell 2002, 109, S81–S96. [Google Scholar] [CrossRef]
- Mercurio, F.; Zhu, H.; Murray, B.W.; Shevchenko, A.; Bennett, B.L.; Li, J.W.; Young, D.B.; Barbosa, M.; Mann, M.; Manning, A.; et al. Ikk-1 and ikk-2: Cytokine-activated iκb kinases essential for nf-κb activation. Science 1997, 278, 860–866. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, J.A.; Hayakawa, M.; Rothwarf, D.M.; Zandi, E.; Karin, M. A cytokine-responsive i[kappa]b kinase that activates the transcription factor nf-[kappa]b. Nature 1997, 388, 548–554. [Google Scholar] [PubMed]
- Rothwarf, D.M.; Zandi, E.; Natoli, G.; Karin, M. Ikk-[gamma] is an essential regulatory subunit of the i[kappa]b kinase complex. Nature 1998, 395, 297–300. [Google Scholar] [PubMed]
- Fuchs, S.Y.; Spiegelman, V.S.; Suresh Kumar, K.G. The many faces of [beta]-trcp e3 ubiquitin ligases: Reflections in the magic mirror of cancer. Oncogene 0000, 23, 2028–2036. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Xu, G.; Schulman, B.A.; Jeffrey, P.D.; Harper, J.W.; Pavletich, N.P. Structure of a β-trcp1-skp1-β-catenin complex: Destruction motif binding and lysine specificity of the scfβ-trcp1 ubiquitin ligase. Mol. Cell 2003, 11, 1445–1456. [Google Scholar] [CrossRef]
- Alkalay, I.; Yaron, A.; Hatzubai, A.; Orian, A.; Ciechanover, A.; Ben-Neriah, Y. Stimulation-dependent i kappa b alpha phosphorylation marks the nf-kappa b inhibitor for degradation via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 1995, 92, 10599–10603. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hagler, J.; Palombella, V.J.; Melandri, F.; Scherer, D.; Ballard, D.; Maniatis, T. Signal-induced site-specific phosphorylation targets i kappa b alpha to the ubiquitin-proteasome pathway. Genes Dev. 1995, 9, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Yaron, A.; Hatzubai, A.; Davis, M.; Lavon, I.; Amit, S.; Manning, A.M.; Andersen, J.S.; Mann, M.; Mercurio, F.; Ben-Neriah, Y. Identification of the receptor component of the i[kappa]b[alpha]-ubiquitin ligase. Nature 1998, 396, 590–594. [Google Scholar] [PubMed]
- Winston, J.T.; Strack, P.; Beer-Romero, P.; Chu, C.Y.; Elledge, S.J.; Harper, J.W. The scfβ-trcp–ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in iκbα and β-catenin and stimulates iκbα ubiquitination in vitro. Genes Dev. 1999, 13, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Spencer, E.; Jiang, J.; Chen, Z.J. Signal-induced ubiquitination of iκbα by the f-box protein slimb/β-trcp. Genes Dev. 1999, 13, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Hake, S.B.; Allis, C.D. Histone h3 variants and their potential role in indexing mammalian genomes: The “h3 barcode hypothesis”. Proc. Natl. Acad. Sci. USA 2006, 103, 6428–6435. [Google Scholar] [CrossRef] [PubMed]
- Bosisio, D.; Marazzi, I.; Agresti, A.; Shimizu, N.; Bianchi, M.E.; Natoli, G. A hyper-dynamic equilibrium between promoter-bound and nucleoplasmic dimers controls nf-kappab-dependent gene activity. Embo J. 2006, 25, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Saccani, S.; Pantano, S.; Natoli, G. Modulation of nf-kappab activity by exchange of dimers. Mol. Cell 2003, 11, 1563–1574. [Google Scholar] [CrossRef]
- Hoffmann, A.; Leung, T.H.; Baltimore, D. Genetic analysis of NF-κB/Rel transcription factors defines functional specificities. EMBO J. 2003, 22, 5530–5539. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Gewert, D.; Hiscott, J. Differential transcriptional activation in vitro by nf-b/rel proteins. J. Biol. Chem. 1995, 270, 3123–3131. [Google Scholar] [PubMed]
- Leung, T.H.; Hoffmann, A.; Baltimore, D. One nucleotide in a κb site can determine cofactor specificity for nf-κb dimers. Cell 2004, 118, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Gerondakis, S.; Grumont, R.; Gugasyan, R.; Wong, L.; Isomura, I.; Ho, W.; Banerjee, A. Unravelling the complexities of the nf-[kappa]b signalling pathway using mouse knockout and transgenic models. Oncogene 2000, 25, 6781–6799. [Google Scholar] [CrossRef] [PubMed]
- Wiborg, O.; Pedersen, M.S.; Wind, A.; Berglund, L.E.; Marcker, K.A.; Vuust, J. The human ubiquitin multigene family: Some genes contain multiple directly repeated ubiquitin coding sequences. EMBO J. 1985, 4, 755–759. [Google Scholar] [PubMed]
- Redman, K.L.; Rechsteiner, M. Identification of the long ubiquitin extension as ribosomal protein s27a. Nature 1989, 338, 438–440. [Google Scholar] [CrossRef] [PubMed]
- Heride, C.; Urbé, S.; Clague, M.J. Ubiquitin code assembly and disassembly. Curr. Biol. 2014, 24, R215–R220. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Heller, H.; Elias, S.; Ciechanover, A. Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 1983, 258, 8206–8214. [Google Scholar] [PubMed]
- Kravtsova-Ivantsiv, Y.; Ciechanover, A. Non-canonical ubiquitin-based signals for proteasomal degradation. J. Cell Sci. 2012, 125, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Berndsen, C.E.; Wolberger, C. New insights into ubiquitin e3 ligase mechanism. Nat. Struct. Mol. Biol. 2014, 21, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. Hect and ring finger families of e3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Kumar, S. Mammalian hect ubiquitin-protein ligases: Biological and pathophysiological aspects. Biochim. Biophys. Acta (BBA) Mol. Cell Rese. 2014, 1843, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.B.; Pruneda, J.N.; Klevit, R.E.; Weissman, A.M. Ring-type e3 ligases: Master manipulators of e2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2014, 1843, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Hua, Z.; Vierstra, R.D. The cullin-ring ubiquitin-protein ligases. Annu. Rev. Plant Biol. 2011, 62, 299–334. [Google Scholar] [CrossRef] [PubMed]
- Komada, M. Controlling receptor downregulation by ubiquitination and deubiquitination. Curr. Drug Discov. Technol. 2008, 5, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Huang, O.W.; Cochran, A.G. Regulation of deubiquitinase proteolytic activity. Curr. Opin. Struct. Biol. 2013, 23, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Clague, M.J.; Urbe, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Eletr, Z.M.; Wilkinson, K.D. Regulation of proteolysis by human deubiquitinating enzymes. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2014, 1843, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.B.; Luna-Vargas, M.P.A.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.G.; Sixma, T.K.; Bernards, R. A genomic and functional inventory of deubiquitinating enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Colleran, A.; Collins, P.E.; O'Carroll, C.; Ahmed, A.; Mao, X.; McManus, B.; Kiely, P.A.; Burstein, E.; Carmody, R.J. Deubiquitination of nf-kappab by ubiquitin-specific protease-7 promotes transcription. Proc. Natl. Acad. Sci. USA 2013, 110, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Tzimas, C.; Michailidou, G.; Arsenakis, M.; Kieff, E.; Mosialos, G.; Hatzivassiliou, E.G. Human ubiquitin specific protease 31 is a deubiquitinating enzyme implicated in activation of nuclear factor-kappab. Cell. Signal. 2006, 18, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, K.; Naumann, M. Csn-associated usp48 confers stability to nuclear nf-kappab/rela by trimming k48-linked ub-chains. Biochim. Biophys. Acta 2015, 1853, 453–469. [Google Scholar] [CrossRef] [PubMed]
- Kirisako, T.; Kamei, K.; Murata, S.; Kato, M.; Fukumoto, H.; Kanie, M.; Sano, S.; Tokunaga, F.; Tanaka, K.; Iwai, K. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006, 25, 4877–4887. [Google Scholar] [CrossRef] [PubMed]
- Spence, J.; Sadis, S.; Haas, A.L.; Finley, D. A ubiquitin mutant with specific defects in DNA repair and multiubiquitination. Mol. Cell. Biol. 1995, 15, 1265–1273. [Google Scholar] [CrossRef]
- Ikeda, F.; Dikic, I. Atypical ubiquitin chains: New molecular signals. Protein modifications: Beyond the usual suspects review series. EMBO Rep. 2008, 9, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Kulathu, Y.; Komander, D. Atypical ubiquitylation—The unexplored world of polyubiquitin beyond lys48 and lys63 linkages. Nat. Rev. Mol. Cell Biol. 2012, 13, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Thrower, J.S.; Hoffman, L.; Rechsteiner, M.; Pickart, C.M. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000, 19, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Boutet, S.C.; Biressi, S.; Iori, K.; Natu, V.; Rando, T.A. Taf1 regulates pax3 protein by monoubiquitination in skeletal muscle progenitors. Mol. Cell 2010, 40, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Carvallo, L.; Muñoz, R.; Bustos, F.; Escobedo, N.; Carrasco, H.; Olivares, G.; Larraín, J. Non-canonical wnt signaling induces ubiquitination and degradation of syndecan4. J. Biol. Chem. 2010, 285, 29546–29555. [Google Scholar] [CrossRef] [PubMed]
- Kravtsova-Ivantsiv, Y.; Cohen, S.; Ciechanover, A. Modification by single ubiquitin moieties rather than polyubiquitination is sufficient for proteasomal processing of the p105 nf-κb precursor. Mol. Cell 2009, 33, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.-J.; Rape, M. Enhanced protein degradation by branched ubiquitin chains. Cell 2014, 157, 910–921. [Google Scholar] [CrossRef] [PubMed]
- Grice, G.L.; Lobb, I.T.; Weekes, M.P.; Gygi, S.P.; Antrobus, R.; Nathan, J.A. The proteasome distinguishes between heterotypic and homotypic lysine-11-linked polyubiquitin chains. Cell Rep. 2015, 12, 545–553. [Google Scholar]
- Tanaka, T.; Grusby, M.J.; Kaisho, T. Pdlim2-mediated termination of transcription factor nf-kappab activation by intranuclear sequestration and degradation of the p65 subunit. Nat. Immunol. 2007, 8, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Bebien, M.; Liu, G.Y.; Nizet, V.; Karin, M. Ikk[alpha] limits macrophage nf-[kappa]b activation and contributes to the resolution of inflammation. Nature 2005, 434, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Burstein, E.; Hoberg, J.E.; Wilkinson, A.S.; Rumble, J.M.; Csomos, R.A.; Komarck, C.M.; Maine, G.N.; Wilkinson, J.C.; Mayo, M.W.; Duckett, C.S. Commd proteins, a novel family of structural and functional homologs of murr1. J. Biol. Chem. 2005, 280, 22222–22232. [Google Scholar] [CrossRef] [PubMed]
- Nozell, S.; Laver, T.; Moseley, D.; Nowoslawski, L.; DeVos, M.; Atkinson, G.P.; Harrison, K.; Nabors, L.B.; Benveniste, E.N. The ing4 tumor suppressor attenuates nf-κb activity at the promoters of target genes. Mol. Cell. Biol. 2008, 28, 6632–6645. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wittwer, T.; Weber, A.; Schneider, H.; Moreno, R.; Maine, G.N.; Kracht, M.; Schmitz, M.L.; Burstein, E. Regulation of nf-κb activity by competition between rela acetylation and ubiquitination. Oncogene 2012, 31, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Hochrainer, K.; Racchumi, G.; Zhang, S.; Iadecola, C.; Anrather, J. Monoubiquitination of nuclear rela negatively regulates nf-kappab activity independent of proteasomal degradation. Cell. Mol. Life Sci. CMLS 2012, 69, 2057–2073. [Google Scholar] [CrossRef] [PubMed]
- Ea, C.K.; Baltimore, D. Regulation of nf-kappab activity through lysine monomethylation of p65. Proc. Natl. Acad. Sci. USA 2009, 106, 18972–18977. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-D.; Tajkhorshid, E.; Chen, L.-F. Functional interplay between acetylation and methylation of the rela subunit of nf-κb. Mol. Cell. Biol. 2010, 30, 2170–2180. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-D.; Huang, B.; Li, M.; Lamb, A.; Kelleher, N.L.; Chen, L.-F. Negative regulation of nf-κb action by set9-mediated lysine methylation of the rela subunit. EMBO J. 2009, 28, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-F.; Williams, S.A.; Mu, Y.; Nakano, H.; Duerr, J.M.; Buckbinder, L.; Greene, W.C. Nf-κb rela phosphorylation regulates rela acetylation. Mol. Cell. Biol. 2005, 25, 7966–7975. [Google Scholar] [CrossRef] [PubMed]
- Buerki, C.; Rothgiesser, K.M.; Valovka, T.; Owen, H.R.; Rehrauer, H.; Fey, M.; Lane, W.S.; Hottiger, M.O. Functional relevance of novel p300-mediated lysine 314 and 315 acetylation of rela/p65. Nucleic Acids Res. 2008, 36, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.; Wittwer, T.; Dittrich-Breiholz, O.; Kracht, M.; Schmitz, M.L. Phosphorylation of nf-κb p65 at ser468 controls its commd1-dependent ubiquitination and target gene-specific proteasomal elimination. EMBO Rep. 2009, 10, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Gluck, N.; Li, D.; Maine, G.N.; Li, H.; Zaidi, I.W.; Repaka, A.; Mayo, M.W.; Burstein, E. Gcn5 is a required cofactor for a ubiquitin ligase that targets nf-κb/rela. Genes Dev. 2009, 23, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, C.Y.; Barberi, T.J.; Ghosh, P.; Longo, D.L. Phosphorylation of rela/p65 on serine 536 defines an i{kappa}b{alpha}-independent nf-{kappa}b pathway. J. Biol. Chem. 2005, 280, 34538–34547. [Google Scholar] [CrossRef] [PubMed]
- Nolan, G.P.; Fujita, T.; Bhatia, K.; Huppi, C.; Liou, H.C.; Scott, M.L.; Baltimore, D. The bcl-3 proto-oncogene encodes a nuclear i kappa b-like molecule that preferentially interacts with nf-kappa b p50 and p52 in a phosphorylation-dependent manner. Mol. Cell Biol. 1993, 13, 3557–3566. [Google Scholar] [CrossRef] [PubMed]
- Inoue, J.; Takahara, T.; Akizawa, T.; Hino, O. Bcl-3, a member of the i kappa b proteins, has distinct specificity towards the rel family of proteins. Oncogene 1993, 8, 2067–2073. [Google Scholar] [PubMed]
- Kerr, L.D.; Duckett, C.S.; Wamsley, P.; Zhang, Q.; Chiao, P.; Nabel, G.; McKeithan, T.W.; Baeuerle, P.A.; Verma, I.M. The proto-oncogene bcl-3 encodes an i kappa b protein. Genes Dev. 1992, 6, 2352–2363. [Google Scholar] [CrossRef] [PubMed]
- McKeithan, T.W.; Rowley, J.D.; Shows, T.B.; Diaz, M.O. Cloning of the chromosome translocation breakpoint junction of the t(14;19) in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 1987, 84, 9257–9260. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, V.; Melendez-Zajgla, J. Role of bcl-3 in solid tumors. Mol. Cancer 2011, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Carmody, R.J.; Ruan, Q.; Palmer, S.; Hilliard, B.; Chen, Y.H. Negative regulation of toll-like receptor signaling by nf-κb p50 ubiquitination blockade. Science 2007, 317, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Riemann, M.; Endres, R.; Liptay, S.; Pfeffer, K.; Schmid, R.M. The iκb protein bcl-3 negatively regulates transcription of the il-10 gene in macrophages. J. Immunol. 2005, 175, 3560–3568. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.E.; Kiely, P.A.; Carmody, R.J. Inhibition of transcription by b cell leukemia 3 (bcl-3) protein requires interaction with nuclear factor kappab (nf-kappab) p50. J. Biol. Chem. 2014, 289, 7059–7067. [Google Scholar] [CrossRef] [PubMed]
- Hailfinger, S.; Nogai, H.; Pelzer, C.; Jaworski, M.; Cabalzar, K.; Charton, J.-E.; Guzzardi, M.; Décaillet, C.; Grau, M.; Dörken, B.; et al. Malt1-dependent relb cleavage promotes canonical nf-κb activation in lymphocytes and lymphoma cell lines. Proc. Natl. Acad. Sci. USA 2011, 108, 14596–14601. [Google Scholar] [CrossRef] [PubMed]
- Marienfeld, R.; Berberich-Siebelt, F.; Berberich, I.; Denk, A.; Serfling, E.; Neumann, M. Signal-specific and phosphorylation-dependent relb degradation: A potential mechanism of nf-kappab control. Oncogene 2001, 20, 8142–8147. [Google Scholar] [CrossRef] [PubMed]
- Leidner, J.; Palkowitsch, L.; Marienfeld, U.; Fischer, D.; Marienfeld, R. Identification of lysine residues critical for the transcriptional activity and polyubiquitination of the nf-kappab family member relb. Biochem. J. 2008, 416, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Moreau, F.; Chadee, K. Ppargamma is an e3 ligase that induces the degradation of nfkappab/p65. Nat. Commun. 2012, 3, 1300. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Zhang, Z.; Xu, Q.; Wang, H.; Xu, Y.; Chen, K. Inhibitor of growth 4 induces nfkappab/p65 ubiquitin-dependent degradation. Oncogene 2014, 33, 1997–2003. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Mao, R.; Zhao, Y.; Yu, Y.; Sun, W.; Song, P.; Shi, Z.; Zhang, D.; Yvon, E.; Zhang, H.; et al. Tumor necrosis factor-α induces rela degradation via ubiquitination at lysine 195 to prevent excessive nuclear factor-κb activation. J. Biol. Chem. 2009, 284, 29290–29297. [Google Scholar] [CrossRef] [PubMed]
- Ryo, A.; Suizu, F.; Yoshida, Y.; Perrem, K.; Liou, Y.-C.; Wulf, G.; Rottapel, R.; Yamaoka, S.; Lu, K.P. Regulation of nf-κb signaling by pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/rela. Mol. Cell 2003, 12, 1413–1426. [Google Scholar] [CrossRef]
- Salmeron, A.; Janzen, J.; Soneji, Y.; Bump, N.; Kamens, J.; Allen, H.; Ley, S.C. Direct phosphorylation of nf-kappab1 p105 by the ikappab kinase complex on serine 927 is essential for signal-induced p105 proteolysis. J. Biol. Chem. 2001, 276, 22215–22222. [Google Scholar] [CrossRef] [PubMed]
- Kravtsova-Ivantsiv, Y.; Shomer, I.; Cohen-Kaplan, V.; Snijder, B.; Superti-Furga, G.; Gonen, H.; Sommer, T.; Ziv, T.; Admon, A.; Naroditsky, I.; et al. Kpc1-mediated ubiquitination and proteasomal processing of nf-kappab1 p105 to p50 restricts tumor growth. Cell 2015, 161, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Haecker, H.; Karin, M.; Ciechanover, A. Mechanism of processing of the nf-kappa b2 p100 precursor: Identification of the specific polyubiquitin chain-anchoring lysine residue and analysis of the role of nedd8-modification on the scf(beta-trcp) ubiquitin ligase. Oncogene 2004, 23, 2540–2547. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Harhaj, E.W.; Sun, S.C. Nf-kappab-inducing kinase regulates the processing of nf-kappab2 p100. Mol. Cell 2001, 7, 401–409. [Google Scholar] [CrossRef]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krahn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.C.; et al. Activation by ikkalpha of a second, evolutionary conserved, nf-kappa b signaling pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef] [PubMed]
- Busino, L.; Millman, S.E.; Scotto, L.; Kyratsous, C.A.; Basrur, V.; O/'Connor, O.; Hoffmann, A.; Elenitoba-Johnson, K.S.; Pagano, M. Fbxw7[alpha]- and gsk3-mediated degradation of p100 is a pro-survival mechanism in multiple myeloma. Nat. Cell Biol. 2012, 14, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, H.; Matsumoto, A.; Inuzuka, H.; Zhai, B.; Lau, A.W.; Wan, L.; Gao, D.; Shaik, S.; Yuan, M.; Gygi, S.P.; et al. Scf(fbw7) modulates the nfkb signaling pathway by targeting nfkb2 for ubiquitination and destruction. Cell Rep. 2012, 1, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Alexander, W.S. Suppressors of cytokine signalling (socs) in the immune system. Nat. Rev. Immunol. 2002, 2, 410–416. [Google Scholar] [PubMed]
- Maine, G.N.; Mao, X.; Komarck, C.M.; Burstein, E. Commd1 promotes the ubiquitination of nf-κb subunits through a cullin-containing ubiquitin ligase. EMBO J. 2007, 26, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chan, L.; Bartuzi, P.; Melton, S.D.; Weber, A.; Ben–Shlomo, S.; Varol, C.; Raetz, M.; Mao, X.; Starokadomskyy, P.; et al. Copper metabolism domain-containing 1 represses genes that promote inflammation and protects mice from colitis and colitis-associated cancer. Gastroenterology 2014, 147, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Natoli, G.; Chiocca, S. Nuclear ubiquitin ligases, nf-kappab degradation, and the control of inflammation. Sci. Signal. 2008, 1, pe1. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; You, M.; Shi, H.; Hou, Y. Ubiquitin-mediated nfkappab degradation pathway. Cell. Mol. Immunol. 2015, 12, 653–655. [Google Scholar] [CrossRef] [PubMed]
- Baetz, A.; Koelsche, C.; Strebovsky, J.; Heeg, K.; Dalpke, A.H. Identification of a nuclear localization signal in suppressor of cytokine signaling 1. FASEB J. 2008, 22, 4296–4305. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; van de Sluis, B.; Groot, A.J.; Verbeek, D.; Vonk, W.I.; Maine, G.N.; Burstein, E.; Wijmenga, C.; Vooijs, M.; Reits, E.; et al. Nuclear-cytosolic transport of commd1 regulates nf-kappab and hif-1 activity. Traffic (Copenhagen, Denmark) 2009, 10, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Strebovsky, J.; Walker, P.; Lang, R.; Dalpke, A.H. Suppressor of cytokine signaling 1 (socs1) limits nfkappab signaling by decreasing p65 stability within the cell nucleus. FASEB J. 2011, 25, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.; Filipe, J.; Seldon, M.P.; Fonseca, L.; Anrather, J.; Soares, M.P.; Simas, J.P. Termination of nf-kappab activity through a gammaherpesvirus protein that assembles an ec5s ubiquitin-ligase. EMBO J. 2009, 28, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Soriano, M.A.; Grusby, M.J. Slim is a nuclear ubiquitin e3 ligase that negatively regulates stat signaling. Immunity 2005, 22, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Loughran, G.; Healy, N.C.; Kiely, P.A.; Huigsloot, M.; Kedersha, N.L.; O'Connor, R. Mystique is a new insulin-like growth factor-i-regulated pdz-lim domain protein that promotes cell attachment and migration and suppresses anchorage-independent growth. Mol. Biol. Cell 2005, 16, 1811–1822. [Google Scholar] [CrossRef] [PubMed]
- Torrado, M.; Senatorov, V.V.; Trivedi, R.; Fariss, R.N.; Tomarev, S.I. Pdlim2, a novel pdz-lim domain protein, interacts with alpha-actinins and filamin a. Investig. Ophthalmol. Visual Sci. 2004, 45, 3955–3963. [Google Scholar] [CrossRef] [PubMed]
- Healy, N.C.; O'Connor, R. Sequestration of pdlim2 in the cytoplasm of monocytic/macrophage cells is associated with adhesion and increased nuclear activity of nf-kappab. J. Leukoc. Biol. 2009, 85, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Fu, J.; Qu, Z.; Li, S.; Tanaka, T.; Grusby, M.J.; Xiao, G. Pdlim2 suppresses human t-cell leukemia virus type i tax-mediated tumorigenesis by targeting tax into the nuclear matrix for proteasomal degradation. Blood 2009, 113, 4370–4380. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Shibazaki, A.; Ono, R.; Kaisho, T. Hsp70 mediates degradation of the p65 subunit of nuclear factor kappab to inhibit inflammatory signaling. Sci. Signal. 2014, 7, ra119. [Google Scholar] [CrossRef] [PubMed]
- Stark, L.A.; Dunlop, M.G. Nucleolar sequestration of rela (p65) regulates nf-κb-driven transcription and apoptosis. Mol. Cell. Biol. 2005, 25, 5985–6004. [Google Scholar] [CrossRef] [PubMed]
- Thoms, H.C.; Loveridge, C.J.; Simpson, J.; Clipson, A.; Reinhardt, K.; Dunlop, M.G.; Stark, L.A. Nucleolar targeting of rela(p65) is regulated by commd1-dependent ubiquitination. Cancer Res. 2010, 70, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.B. The role of ppars in inflammation and immunity. J. Leukoc. Biol. 2002, 71, 388–400. [Google Scholar] [PubMed]
- Chung, S.W.; Kang, B.Y.; Kim, S.H.; Pak, Y.K.; Cho, D.; Trinchieri, G.; Kim, T.S. Oxidized low density lipoprotein inhibits interleukin-12 production in lipopolysaccharide-activated mouse macrophages via direct interactions between peroxisome proliferator-activated receptor-gamma and nuclear factor-kappa b. J. Biol. Chem. 2000, 275, 32681–32687. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Pownall, H.J.; Lodish, H.F. Troglitazone antagonizes tumor necrosis factor-α-induced reprogramming of adipocyte gene expression by inhibiting the transcriptional regulatory functions of nf-κb. J. Biol. Chem. 2003, 278, 28181–28192. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.; Campbell, J.I.; King, T.P.; Grant, G.; Jansson, E.A.; Coutts, A.G.P.; Pettersson, S.; Conway, S. Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of ppar-[gamma] and rela. Nat. Immunol. 2004, 5, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Garkavtsev, I.; Kozin, S.V.; Chernova, O.; Xu, L.; Winkler, F.; Brown, E.; Barnett, G.H.; Jain, R.K. The candidate tumour suppressor protein ing4 regulates brain tumour growth and angiogenesis. Nature 2004, 428, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Coles, A.H.; Gannon, H.; Cerny, A.; Kurt-Jones, E.; Jones, S.N. Inhibitor of growth-4 promotes ikappab promoter activation to suppress nf-kappab signaling and innate immunity. Proc. Natl. Acad. Sci. USA 2010, 107, 11423–11428. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D. Icp0, a regulator of herpes simplex virus during lytic and latent infection. BioEssays News Rev. Mol. Cell. Dev. Biol. 2000, 22, 761–770. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, K.; Wang, S.; Zheng, C. Herpes simplex virus 1 e3 ubiquitin ligase icp0 protein inhibits tumor necrosis factor alpha-induced nf-kappab activation by interacting with p65/rela and p50/nf-kappab1. J. Virol. 2013, 87, 12935–12948. [Google Scholar] [CrossRef] [PubMed]
- Boutell, C.; Sadis, S.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein icp0 and its isolated ring finger domain act as ubiquitin e3 ligases in vitro. J. Virol. 2002, 76, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Jin, W.; Chang, J.H.; Xiao, Y.; Brittain, G.C.; Yu, J.; Zhou, X.; Wang, Y.H.; Cheng, X.; Li, P.; et al. The ubiquitin ligase peli1 negatively regulates t cell activation and prevents autoimmunity. Nat. Immunol. 2011, 12, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Hrdlickova, R.; Nehyba, J.; Longo, D.L.; Bose, H.R., Jr.; Li, C.C. Degradation of proto-oncoprotein c-rel by the ubiquitin-proteasome pathway. J. Biol. Chem. 1998, 273, 35201–35207. [Google Scholar] [CrossRef] [PubMed]
- Zarnegar, B.J.; Wang, Y.; Mahoney, D.J.; Dempsey, P.W.; Cheung, H.H.; He, J.; Shiba, T.; Yang, X.; Yeh, W.-C.; Mak, T.W.; et al. Noncanonical nf-[kappa]b activation requires coordinated assembly of a regulatory complex of the adaptors ciap1, ciap2, traf2 and traf3 and the kinase nik. Nat. Immunol. 2008, 9, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Matsuzawa, A.; Zhang, W.; Tseng, P.H.; Keats, J.J.; Wang, H.; Vignali, D.A.; Bergsagel, P.L.; Karin, M. Nonredundant and complementary functions of traf2 and traf3 in a ubiquitination cascade that activates nik-dependent alternative nf-kappab signaling. Nat. Immunol. 2008, 9, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Xiao, Y.; Hu, H.; Zou, Q.; Li, Y.; Gao, Y.; Ge, W.; Cheng, X.; Sun, S.-C. Proinflammatory tlr signalling is regulated by a traf2-dependent proteolysis mechanism in macrophages. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.P.; Chen, Z.J. Traf2: A double-edged sword? Sci. STKE 2005, 2005, pe7. [Google Scholar] [CrossRef] [PubMed]
- Hacker, H.; Tseng, P.H.; Karin, M. Expanding traf function: Traf3 as a tri-faced immune regulator. Nat. Rev. Immunol. 2011, 11, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Hochrainer, K.; Pejanovic, N.; Olaseun, V.A.; Zhang, S.; Iadecola, C.; Anrather, J. The ubiquitin ligase herc3 attenuates nf-κb-dependent transcription independently of its enzymatic activity by delivering the rela subunit for degradation. Nucleic Acids Res. 2015, 43, 9889–9904. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, B.; Suresh Kumar, K.G. The multifaceted roles of usp7: New therapeutic opportunities. Cell Biochem. Biophys. 2011, 60, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Holowaty, M.N.; Sheng, Y.; Nguyen, T.; Arrowsmith, C.; Frappier, L. Protein interaction domains of the ubiquitin-specific protease, usp7/hausp. J. Biol. Chem. 2003, 278, 47753–47761. [Google Scholar] [CrossRef] [PubMed]
- Faesen, A.C.; Dirac, A.M.; Shanmugham, A.; Ovaa, H.; Perrakis, A.; Sixma, T.K. Mechanism of usp7/hausp activation by its c-terminal ubiquitin-like domain and allosteric regulation by gmp-synthetase. Mol. Cell 2011, 44, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Zapata, J.M.; Pawlowski, K.; Haas, E.; Ware, C.F.; Godzik, A.; Reed, J.C. A diverse family of proteins containing tumor necrosis factor receptor-associated factor domains. J. Biol. Chem. 2001, 276, 24242–24252. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, J.B.; Morgan, D.O. Protein-linked ubiquitin chain structure restricts activity of deubiquitinating enzymes. J. Biol. Chem. 2011, 286, 45186–45196. [Google Scholar] [CrossRef] [PubMed]
- Cheon, K.W.; Baek, K.H. Hausp as a therapeutic target for hematopoietic tumors (review). Int. J. Oncol. 2016, 28, 1209–1215. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, L.; Rouge, L.; Phillips, A.H.; Lam, C.; Liu, P.; Sandoval, W.; Helgason, E.; Murray, J.M.; Wertz, I.E.; Corn, J.E. Conformational stabilization of ubiquitin yields potent and selective inhibitors of usp7. Nat. Chem. Biol. 2013, 9, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Wrigley, J.D.; Eckersley, K.; Hardern, I.M.; Millard, L.; Walters, M.; Peters, S.W.; Mott, R.; Nowak, T.; Ward, R.A.; Simpson, P.B.; et al. Enzymatic characterisation of usp7 deubiquitinating activity and inhibition. Cell Biochem. Biophys. 2011, 60, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Kashiwaba, S.; Kanao, R.; Masuda, Y.; Kusumoto-Matsuo, R.; Hanaoka, F.; Masutani, C. Usp7 is a suppressor of pcna ubiquitination and oxidative-stress-induced mutagenesis in human cells. Cell Rep. 2015, 13, 2072–2080. [Google Scholar] [CrossRef] [PubMed]
- Zlatanou, A.; Sabbioneda, S.; Miller, E.S.; Greenwalt, A.; Aggathanggelou, A.; Maurice, M.M.; Lehmann, A.R.; Stankovic, T.; Reverdy, C.; Colland, F.; et al. Usp7 is essential for maintaining rad18 stability and DNA damage tolerance. Oncogene 2016, 35, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Rape, M.; Jentsch, S. Taking a bite: Proteasomal protein processing. Nat. Cell Biol. 2002, 4, E113–E116. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.M.; Maniatis, T. Generation of p50 subunit of nf-kappa b by processing of p105 through an atp-dependent pathway. Nature 1991, 354, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Palombella, V.J.; Rando, O.J.; Goldberg, A.L.; Maniatis, T. The ubiquitin-proteasome pathway is required for processing the nf-kappa b1 precursor protein and the activation of nf-kappa b. Cell 1994, 78, 773–785. [Google Scholar] [CrossRef]
- Betts, J.C.; Nabel, G.J. Differential regulation of nf-kappab2(p100) processing and control by amino-terminal sequences. Mol. Cell Biol. 1996, 16, 6363–6371. [Google Scholar] [CrossRef]
- Heusch, M.; Lin, L.; Geleziunas, R.; Greene, W.C. The generation of nfkb2 p52: Mechanism and efficiency. Oncogene 1999, 18, 6201–6208. [Google Scholar] [CrossRef] [PubMed]
- Orian, A.; Whiteside, S.; Israël, A.; Stancovski, I.; Schwartz, A.L.; Ciechanover, A. Ubiquitin-mediated processing of nf-κb transcriptional activator precursor p105: Reconstitution of a cell-free system and identification of the ubiquitin-carrier protein, e2, and a novel ubiquitin-protein ligase, e3, involved in conjugation. J. Biol. Chem. 1995, 270, 21707–21714. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; DeMartino, G.N.; Greene, W.C. Cotranslational biogenesis of nf-κb p50 by the 26s proteasome. Cell 1998, 92, 819–828. [Google Scholar] [CrossRef]
- Lin, L.; DeMartino, G.N.; Greene, W.C. Cotranslational dimerization of the Rel homology domain of NF-κB1 generates p50–p105 heterodimers and is required for effective p50 production. EMBO J. 2000, 19, 4712–4722. [Google Scholar] [CrossRef] [PubMed]
- Rice, N.R.; MacKichan, M.L.; Israel, A. The precursor of nf-kappa b p50 has i kappa b-like functions. Cell 1992, 71, 243–253. [Google Scholar] [CrossRef]
- Mercurio, F.; DiDonato, J.A.; Rosette, C.; Karin, M. P105 and p98 precursor proteins play an active role in nf-kappa b-mediated signal transduction. Genes Dev. 1993, 7, 705–718. [Google Scholar] [CrossRef]
- Basak, S.; Kim, H.; Kearns, J.D.; Tergaonkar, V.; O'Dea, E.; Werner, S.L.; Benedict, C.A.; Ware, C.F.; Ghosh, G.; Verma, I.M.; et al. A fourth iκb protein within the nf-κb signaling module. Cell 2007, 128, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Liou, H.C.; Nolan, G.P.; Ghosh, S.; Fujita, T.; Baltimore, D. The nf-kappa b p50 precursor, p105, contains an internal i kappa b-like inhibitor that preferentially inhibits p50. EMBO J. 1992, 11, 3003–3009. [Google Scholar] [PubMed]
- Savinova, O.V.; Hoffmann, A.; Ghosh, G. The nfkb1 and nfkb2 proteins p105 and p100 function as the core of high-molecular-weight heterogeneous complexes. Mol. Cell 2009, 34, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Coux, O.; Goldberg, A.L. Enzymes catalyzing ubiquitination and proteolytic processing of the p105 precursor of nuclear factor κb1. J. Biol. Chem. 1998, 273, 8820–8828. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.; Yasuda, H.; Sato, Y.; Yamamoto, K. A role for phosphorylation in the proteolytic processing of the human nf-kappa b1 precursor. Gene 1995, 165, 183–189. [Google Scholar] [CrossRef]
- MacKichan, M.L.; Logeat, F.; Israel, A. Phosphorylation of p105 pest sequence via a redox-insensitive pathway up-regulates processing of p50 nf-kappab. J. Biol. Chem. 1996, 271, 6084–6091. [Google Scholar] [PubMed]
- Orian, A.; Gonen, H.; Bercovich, B.; Fajerman, I.; Eytan, E.; Israel, A.; Mercurio, F.; Iwai, K.; Schwartz, A.L.; Ciechanover, A. Scf[beta]-trcp ubiquitin ligase-mediated processing of nf-[kappa]b p 105 requires phosphorylation of its c-terminus by i[kappa]b kinase. EMBO J. 2000, 19, 2580–2591. [Google Scholar] [CrossRef] [PubMed]
- Heissmeyer, V.; Krappmann, D.; Hatada, E.N.; Scheidereit, C. Shared pathways of iκb kinase-induced scfβtrcp-mediated ubiquitination and degradation for the nf-κb precursor p105 and iκbα. Mol. Cell. Biol. 2001, 21, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Lang, V.; Janzen, J.; Fischer, G.Z.; Soneji, Y.; Beinke, S.; Salmeron, A.; Allen, H.; Hay, R.T.; Ben-Neriah, Y.; Ley, S.C. Βtrcp-mediated proteolysis of nf-κb1 p105 requires phosphorylation of p105 serines 927 and 932. Mol. Cell. Biol. 2003, 23, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Achbert-Weiner, H.; Ciechanover, A. Dual effects of iκb kinase β-mediated phosphorylation on p105 fate: Scfβ-trcp-dependent degradation and scfβ-trcp-independent processing. Mol. Cell. Biol. 2004, 24, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Lahav-Baratz, S.; Ciechanover, A. Two distinct ubiquitin-dependent mechanisms are involved in nf-kappab p105 proteolysis. Biochem. Biophys. Res. Commun. 2006, 345, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Ghosh, S. A glycine-rich region in nf-kappab p105 functions as a processing signal for the generation of the p50 subunit. Molecular and Cellular Biology 1996, 16, 2248–2254. [Google Scholar] [CrossRef]
- Orian, A.; Schwartz, A.L.; Israël, A.; Whiteside, S.; Kahana, C.; Ciechanover, A. Structural motifs involved in ubiquitin-mediated processing of the nf-κb precursor p105: Roles of the glycine-rich region and a downstream ubiquitination domain. Mol. Cell. Biol. 1999, 19, 3664–3673. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Gonen, H.; Bercovich, B.; Cohen, S.; Fajerman, I.; Israel, A.; Mercurio, F.; Kahana, C.; Schwartz, A.L.; Iwai, K.; et al. Mechanisms of ubiquitin-mediated, limited processing of the nf-kappab1 precursor protein p105. Biochimie 2001, 83, 341–349. [Google Scholar] [CrossRef]
- Moorthy, A.K.; Savinova, O.V.; Ho, J.Q.; Wang, V.Y.-F.; Vu, D.; Ghosh, G. The 20s proteasome processes nf-κb1 p105 into p50 in a translation-independent manner. EMBO J. 2006, 25, 1945–1956. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. Non-canonical nf-kappab signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. The noncanonical nf-kappab pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef]
- Coope, H.J.; Atkinson, P.G.; Huhse, B.; Belich, M.; Janzen, J.; Holman, M.J.; Klaus, G.G.; Johnston, L.H.; Ley, S.C. Cd40 regulates the processing of nf-kappab2 p100 to p52. EMBO J. 2002, 21, 5375–5385. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Zhang, M.; Sun, S.C. Beta-trcp binding and processing of nf-kappab2/p100 involve its phosphorylation at serines 866 and 870. Cell. Signal. 2006, 18, 1309–1317. [Google Scholar] [CrossRef]
- Fong, A.; Sun, S.C. Genetic evidence for the essential role of beta-transducin repeat-containing protein in the inducible processing of nf-kappa b2/p100. J. Biol. Chem. 2002, 277, 22111–22114. [Google Scholar] [CrossRef]
- Fong, A.; Zhang, M.; Neely, J.; Sun, S.C. S9, a 19 s proteasome subunit interacting with ubiquitinated nf-kappab2/p100. J. Biol. Chem. 2002, 277, 40697–40702. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Tcherpakov, M. Will the ubiquitin system furnish as many drug targets as protein kinases? Cell 2010, 143, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-κB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Subunit | Amino Acid | Enzyme/Stimuli | Modification | Function | Ref |
|---|---|---|---|---|---|
| p65 | K28 | PPARY | Ubiquitination | Degradation | [116] |
| K62 | ING4 | Ubiquitination | Degradation | [95,96,117] | |
| K195 | TNF | Ubiquitination | Degradation | [95,118] | |
| K56, K79, K123, K310, K315 | Unknown | Ubiquitination | Unknown | [95,96] | |
| S468 | TNF | Phosphorylation | Promotes ubiquitination | [102,103] | |
| T254/P255 | Pin-1 | Proline isomerization | Inhibits ubiquitination | [119] | |
| p105 | S927,S932 | IKKβ | Phosphorylation | Recruits SCFβ−TrCP | [120] |
| Unknown | SCFβ−TrCP | Ubiquitination | Degradation | [49] | |
| Multiple lysines | KPC1 | Mono ubiquitination | Processing | [121] | |
| p100 | K855 | SCFβ−TrCP | Ubiquitination | Processing | [122] |
| S870/S872 | NIK/IKKα | Phosphorylation | Processing | [123,124] | |
| Unknown | SCFFbw7α | Ubiquitination | Degradation | [125,126] | |
| S707 | GSK3β | Phosphorylation | Promotes ubiquitination | [125,126] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Collins, P.E.; Mitxitorena, I.; Carmody, R.J. The Ubiquitination of NF-κB Subunits in the Control of Transcription. Cells 2016, 5, 23. https://doi.org/10.3390/cells5020023
Collins PE, Mitxitorena I, Carmody RJ. The Ubiquitination of NF-κB Subunits in the Control of Transcription. Cells. 2016; 5(2):23. https://doi.org/10.3390/cells5020023
Chicago/Turabian StyleCollins, Patricia E., Izaskun Mitxitorena, and Ruaidhrí J. Carmody. 2016. "The Ubiquitination of NF-κB Subunits in the Control of Transcription" Cells 5, no. 2: 23. https://doi.org/10.3390/cells5020023
APA StyleCollins, P. E., Mitxitorena, I., & Carmody, R. J. (2016). The Ubiquitination of NF-κB Subunits in the Control of Transcription. Cells, 5(2), 23. https://doi.org/10.3390/cells5020023