The Ubiquitin-Conjugating System: Multiple Roles in Viral Replication and Infection



Abstract
:1. Introduction
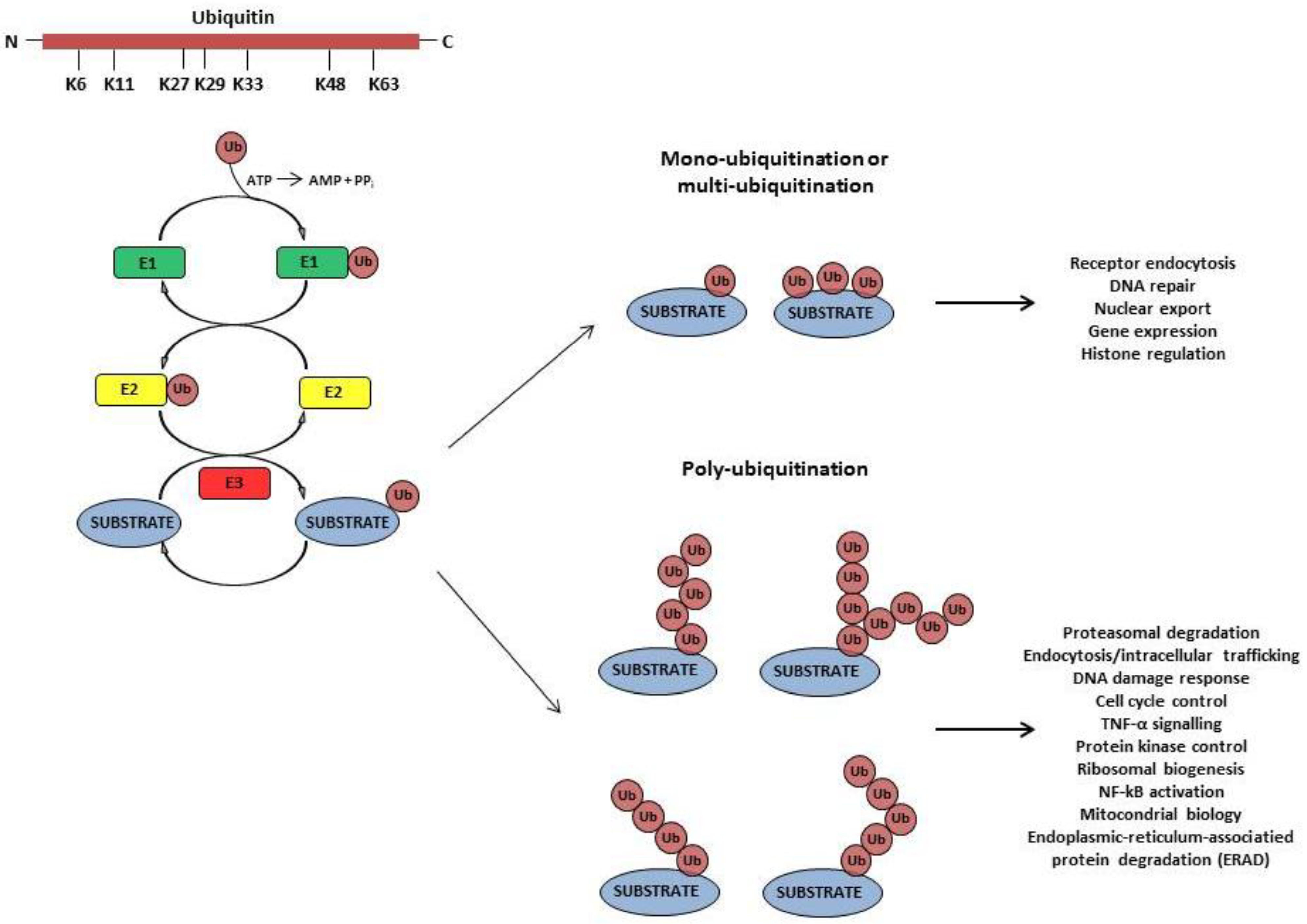
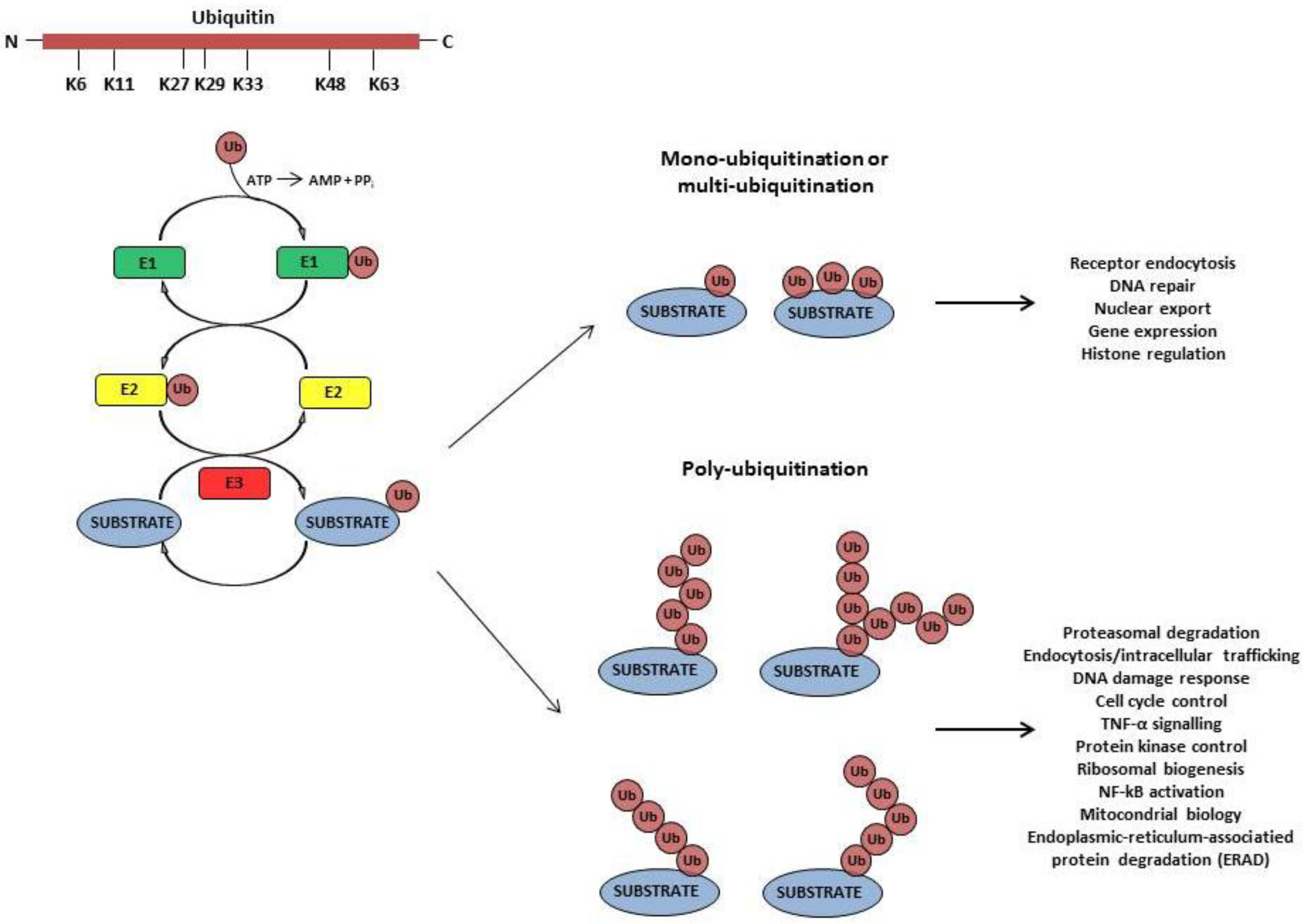
2. Viruses Employ the Ub-Conjugating System to Accomplish Different Steps of Their Replication and to Establish a Successful Infection
3. Viruses Usurp the Ub-Conjugating System to Evade Host Immune Responses
4. Viruses have Evolved Different Strategies to Exploit the Ub-Conjugating System
4.1. Viruses Subvert the Cellular Ub-Conjugating System
4.2. Viruses have Evolved Their Own Ub-Conjugating System

{kind=link}
{kind=link}
{kind=link}
| Virus | Viral protein | Target protein | Reference |
|---|---|---|---|
| Herpes Simplex Virus type 1 | ICP0 | pUL46 (viral) | Lin et al. 2013 [75] |
| p65, p50 | Zhang et al. 2013 [76] | ||
| CENPs | Gross et al. 2012 [77] | ||
| microtubule | Liu et al. 2010 [78] | ||
| RNF8,RNF168 | Lilley et al. 2010 [79] | ||
| PML | Isaacson et al. 2009 [80] | ||
| Sp100 | |||
| Cyclin D3 | |||
| p53 | |||
| USP7 | |||
| ICP0 (viral) | |||
| Kaposi Sarcoma-associated herpesvirus | K3 | MHC-I | Timms et al. 2013 [81] Isaacson et al. 2009 [80] |
| CD1d | Boname et al. 2011 [68] | ||
| PECAM | |||
| IFN-γ R1 | |||
| K5 | MHC-I | Boname et al. 2011 [68] | |
| Tetherin/BST-2 | Boname et al. 2011 [68] Pardieu et al. 2010 [82] | ||
| ICAM-1 | Timms et al. 2013 [81] Isaacson et al. 2009 [80] Boname et al. 2011 [68] | ||
| B7-2 | |||
| CD1d | |||
| HFE | |||
| PECAM | |||
| ALCAM | |||
| MIC-A/-B | |||
| AICL | |||
| DC-SIGN DC-SIGNR | Lang et al. 2013 [83] | ||
| Kaposi Sarcoma-associated herpesvirus | AICL | Boname et al. 2011 [68] | |
| VE-Cadherin | |||
| IFN-γ R1 | |||
| Syntaxin-4 | |||
| BMPRII | |||
| RTKs | Karki et al. 2011 [84] | ||
| Varicella Zoster Virus | ORF61p | ORF61p (viral) | Walters et al. 2010 [85] |
| IRF3 | Zhu et al. 2011 [46] | ||
| Adenovirus | E1B-55k,E4orf6 | p53 | Woo et al. 2007 [86] |
| MRN complex | |||
| Murine gamma herpesvirus 68 | ORF75c | PML | Sewatanon et al. 2013 [87] |
| Rodent herpesvirus Peru | pK3 | pK3 (viral)/MHC-1 | Herr et al. 2012 [88] |
| MHC-I membrane bound chaperons | |||
| Poxvirus | p28 | unknown | Huang et al. 2004 [89] |
| White Spot Syndrome Virus | WSSV222 | TSL | He et al. 2009 [90] |
| Nairovirus | Polymerase | RIG-I | van Kasteren et al. 2012 [91] |
| Murine Hepatitis Virus A59 | nsp3 | TBK1 | Wang et al. 2011 [92] |
| IRF3 | Zheng et al. 2008 [93] | ||
| Foot-and-mouth Disease Virus | L(pro) | RIG-I | Wang et al. 2011 [94] |
| TBK1 | |||
| TRAF6 | |||
| TRAF3 | |||
| Hepatitis B Virus | HBx | RIG-I | Jiang et al. 2010 [95] |
| TRAF3 |
| Virus | Viral protein | Target protein | Reference |
|---|---|---|---|
| Herpes simplex virus type 1 | UL36 | TRAF3 | Wang et al. 2013 [100] |
| UL36 (viral) | Bolstad et al. 2011 [101] | ||
| Human cytomegalovirus | UL48 | unknown | Kim et al. 2009 [102] |
| PseudoRabies Virus | UL36 | unknown | Bottcher et al. 2008 [103] |
| Kaposi Sarcoma-associated herpesvirus | ORF64 | RIG-I | Inn et al. 2011 [104] |
| RTA | IRF-7 | Isaacson et al. 2009 [80] | |
| Epstein-Barr Virus | BPLF1 | EBV ribonucleotide reductase (viral) | Whitehurst et al. 2009 [97] |
| PCNA | Kumar et al. 2014 [99] | ||
| Rad18 | |||
| Crimean-Congo Hemorrhagic Fever Virus | vOTU | Unknown | Akutsu et al. 2011 [105] |
| Marek's Disease Virus | UL36 | Unknown | Isaacson et al. 2009 [80] |
| Human coronavirus | PLpro | Unknown | Mielech et al. 2014 [106] |
| Turnip Yellow Mosaic Virus | PRO | Unknown | Lombardi et al. 2013 [107] |
| 98K | RdRp (viral) | Chenon et al. 2012 [108] | |
| Porcine Epidemic Diarrhea Virus | PLP2 | RIG-I | Xing et al. 2013 [109] |
| STING | |||
| Porcine Reproductive and Respiratory Syndrome Virus | nsp2 | IkBα | Sun et al. 2010 [110] |
| Adenovirus | Avp | Adenoviral and cellular proteins unknown | Balakirev et al. 2002 [111] |
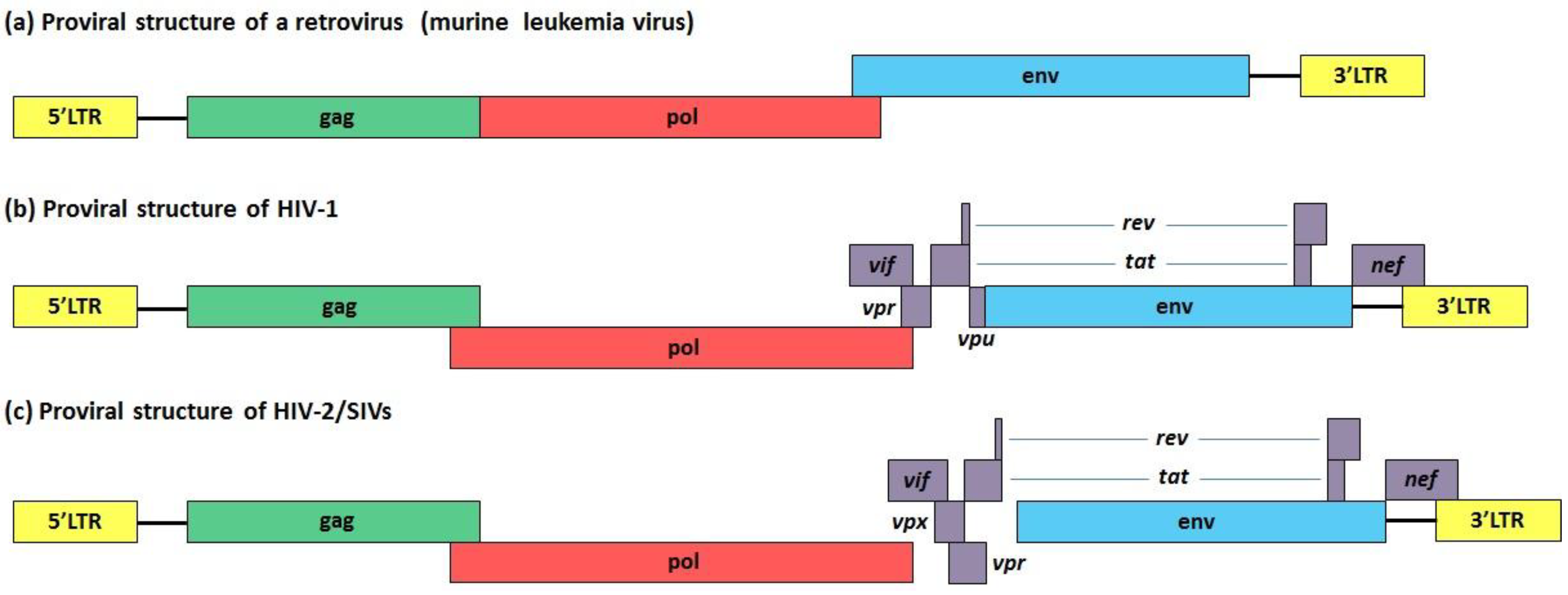
5. Vif, Vpu and Vpr: Three HIV-1 Accessory Proteins that Exploit Cullin-RING Finger Ub Ligase Complexes to Overcome Different Restriction Factors
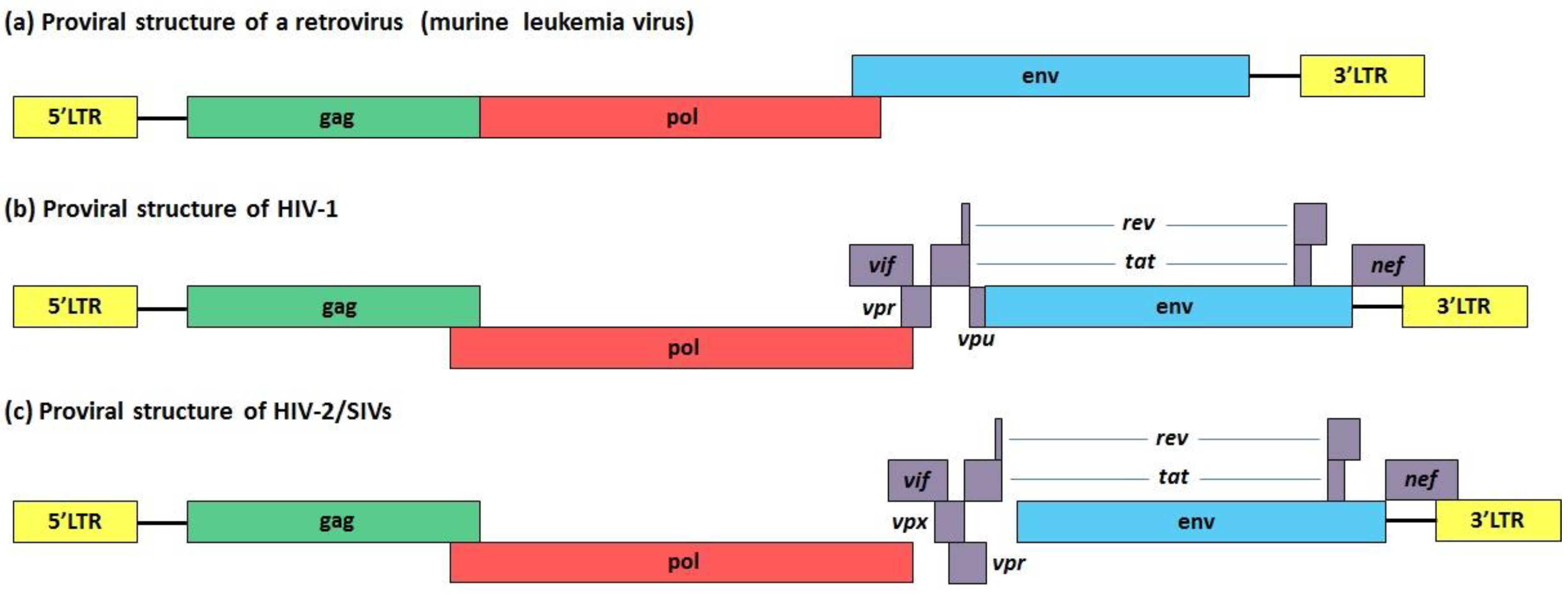
| Viral protein | Cullin-RING finger Ub ligase complex | Target protein | Biological effects | References |
|---|---|---|---|---|
| Vif (HIV-1) | CBF-β-ElonginB-ElonginC-Cullin5-Rbx | APOBEC3 (A3) | Prevention of A3s incorporation into the budding virions Prevention of proviral DNA hypermutation | Guo et al. 2014 [123] |
| Vpu (HIV-1) | Skp1-Cullin1-F box | CD4 | Retention in the ER and delivery to the ER-associated degradation (ERAD) pathway Prevention of superinfection | Nomaguchi et al. 2008 [124] |
| BST2/Tetherin | Promotion of viral egress | Goffinet et al. 2009 [125] Mangeat et al. 2009 [126] Douglas et al. 2009 [127] | ||
| p53 | Stabilization of p53 and enhancement of apoptosis | Verma et al. 2011 [128] | ||
| Ubiquitination of Vpu | Unknown (stabilization or proteasomal degradation) | Belaïdouni et al. 2007 [129] | ||
| Vpr (HIV-1) | Cullin4A-DDB1-DCAF1 Cullin4B also involved (Sharifi et al. 2014) | Unknown cellular substrate(s) | G2 cell cycle arrest | Le Rouzic et al. 2007 [130] |
| UNG2 and SMUG1 | Unknown | Eldin et al. 2014 [131] | ||
| Dicer | Suppression of RNA silencing pathway | Casey Klockow et al. 2013 [132] | ||
| Vpx (HIV-2, SIV) | Cullin4A-DDB1-DCAF1 Cullin 4B also involved (Sharifi et al. 2014) | SAMHD1 | Increase of the intacellular pool of dNTPs Efficient synthesis of viral DNA | Sze et al. 2013 [133] |
5.1. Vif and APOBEC Proteins
5.2. Vpu and Tetherin
5.3. Vpr and Vpx
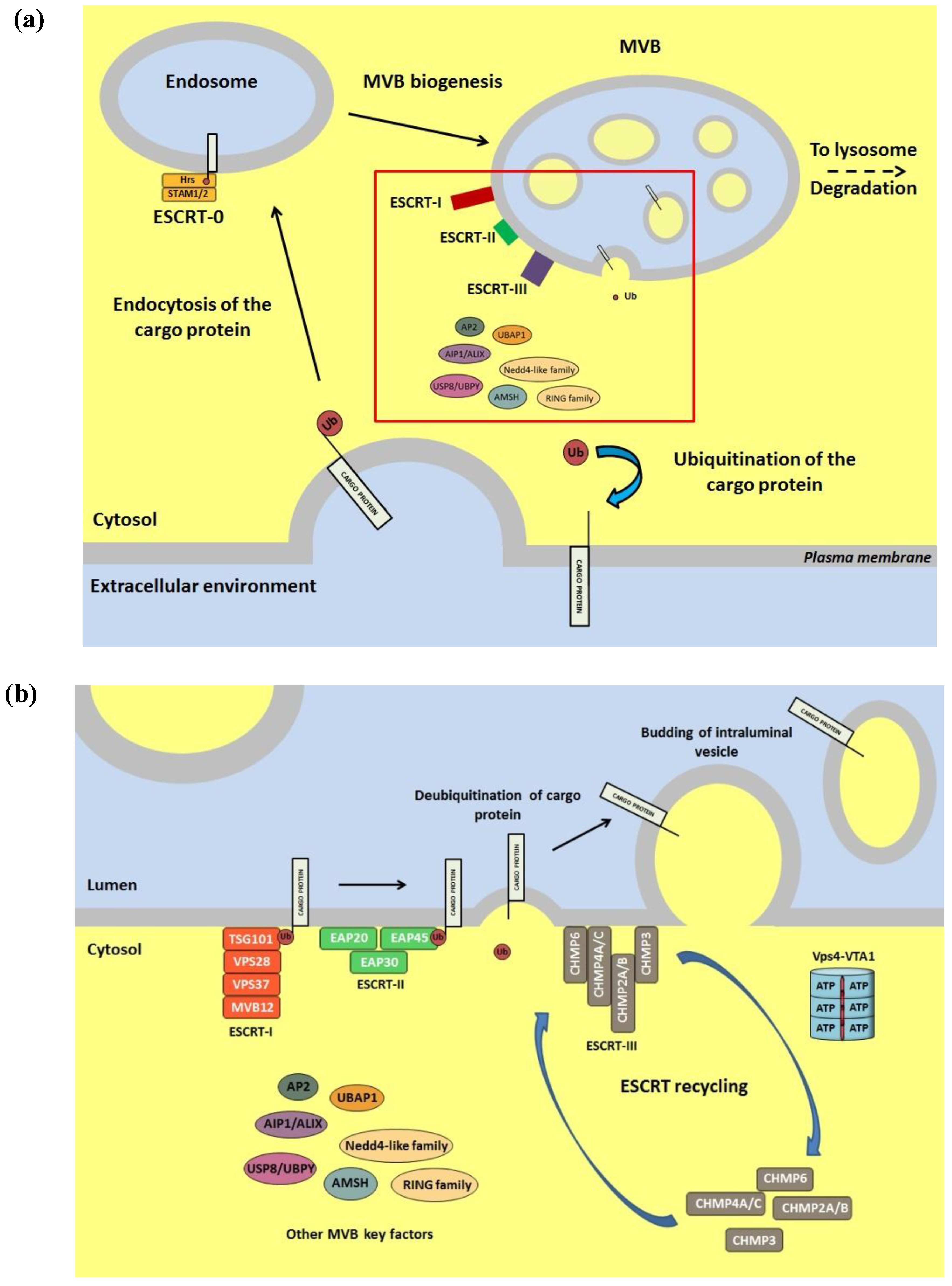
6. Role of the Ub Conjugation System in Viral Egress from Infected Cells
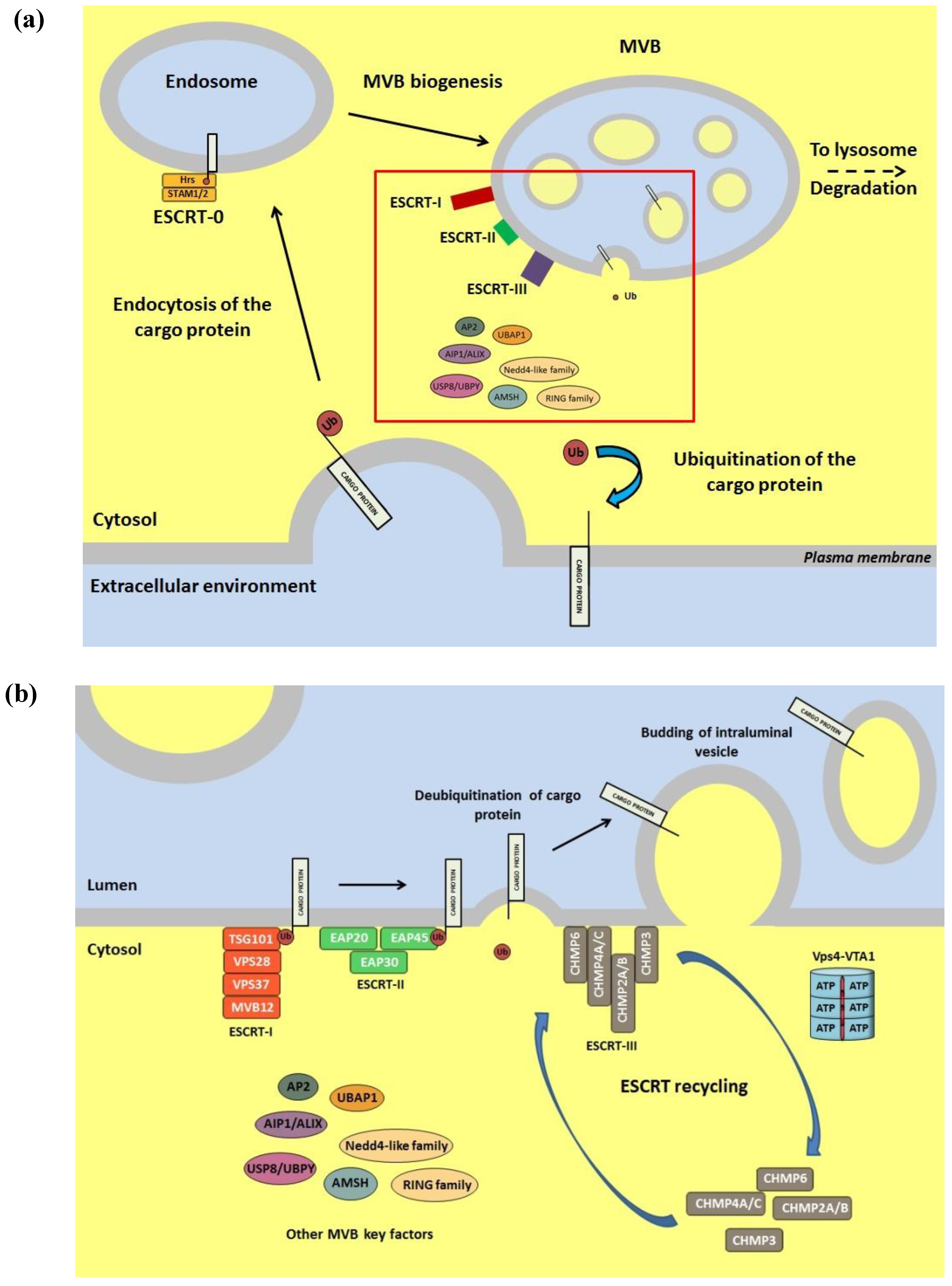
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Dikic, I.; Dötsch, V. Ubiquitin linkages make a difference. Nat. Struct. Mol. Biol. 2009, 16, 1209–1210. [Google Scholar] [CrossRef]
- Pickart, C.M. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef]
- Bernassola, F.; Karin, M.; Ciechanover, A.; Melino, G. The HECT family of E3 ubiquitin ligases: Multiple players in cancer development. Cancer Cell 2008, 14, 10–21. [Google Scholar] [CrossRef]
- Bosu, D.R.; Kipreos, E.T. Cullin-RING ubiquitin ligases: Global regulation and activation cycles. Cell Div. 2008, 3, 7. [Google Scholar] [CrossRef]
- Hatakeyama, S.; Nakayama, K.I. U-Box proteins as a new family of ubiquitin ligases. Biochem. Biophys. Res. Commun. 2003, 302, 635–645. [Google Scholar] [CrossRef]
- Pickart, C.M. Ubiquitin enters the new millennium. Mol. Cell 2001, 8, 499–504. [Google Scholar] [CrossRef]
- Hanson, P.I.; Cashikar, A. Multivesicular body morphogenesis. Annu. Rev. Cell Dev. Biol. 2012, 28, 337–362. [Google Scholar] [CrossRef]
- Welchman, R.L.; Gordon, C.; Mayer, R.J. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat. Rev. Mol. Cell Biol. 2005, 6, 599–609. [Google Scholar] [CrossRef]
- Pickart, C.M. Back to the future with ubiquitin. Cell 2004, 116, 181–190. [Google Scholar] [CrossRef]
- Pickart, C.M. Ubiquitin in chains. Trends Biochem. Sci. 2000, 25, 544–548. [Google Scholar] [CrossRef]
- Pickart, C.M.; Fushman, D. Polyubiquitin chains: Polymeric protein signals. Curr. Opin. Chem. Biol. 2004, 8, 610–616. [Google Scholar] [CrossRef]
- Bedford, L.; Lowe, J.; Dick, L.R.; Mayer, R.J.; Brownell, J.E. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat. Rev. Drug Discov. 2011, 10, 29–46. [Google Scholar] [CrossRef]
- Randow, F.; Lehner, P.J. Viral avoidance and exploitation of the ubiquitin system. Nat. Cell Biol. 2009, 11, 527–534. [Google Scholar] [CrossRef]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Delboy, M.G.; Roller, D.G.; Nicola, A.V. Cellular proteasome activity facilitates Herpes simplex virus entry at a postpenetration step. J. Virol. 2008, 82, 3381–3390. [Google Scholar] [CrossRef]
- Tran, K.; Mahr, J.A.; Spector, D.H. Proteasome subunits relocalize during Human Cytomegalovirus infection, and proteasome activity is necessary for efficient viral gene transcription. J. Virol. 2010, 84, 3079–3093. [Google Scholar] [CrossRef]
- Satheshkumar, P.S.; Anton, L.C.; Sanz, P.; Moss, B. Inhibition of the ubiquitin-proteasome system prevents vaccinia virus DNA replication and expression of intermediate and late genes. J. Virol. 2009, 83, 2469–2479. [Google Scholar] [CrossRef]
- Teale, A.; Campbell, S.; Van Buuren, N.; Magee, W.C.; Watmough, K.; Couturier, B.; Shipclark, R.; Barry, M. Orthopoxviruses require a functional ubiquitin-proteasome system for productive replication. J. Virol. 2009, 83, 2099–2108. [Google Scholar] [CrossRef]
- Bandi, P.; Garcia, M.L.; Booth, C.J.; Chisari, F.V.; Robek, M.D. Bortezomib inhibits hepatitis B virus replication in transgenic mice. Antimicrob. Agents Chemother. 2010, 54, 749–756. [Google Scholar] [CrossRef]
- Gupta, A.; Jha, S.; Engel, D.A.; Ornelles, D.A.; Dutta, A. Tip60 degradation by adenovirus relieves transcriptional repression of viral transcriptional activator EIA. Oncogene 2013, 32, 5017–5025. [Google Scholar] [CrossRef]
- Widjaja, I.; de Vries, E.; Tscherne, D.M.; García-Sastre, A.; Rottier, P.J.; de Haan, C.A. Inhibition of the ubiquitin-proteasome system affects influenza A virus infection at a postfusion step. J. Virol. 2010, 84, 9625–9631. [Google Scholar]
- Schubert, U.; Ott, D.E.; Chertova, E.N.; Welker, R.; Tessmer, U.; Princiotta, M.F.; Bennink, J.R.; Krausslich, H.G.; Yewdell, J.W. Proteasome inhibition interferes with Gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc. Natl. Acad. Sci. USA 2000, 97, 13057–13062. [Google Scholar] [CrossRef]
- Strack, B.; Calistri, A.; Accola, M.A.; Palu, G.; Gottlinger, H.G. A role for ubiquitin ligase recruitment in retrovirus release. Proc. Natl. Acad. Sci. USA 2000, 97, 13063–13068. [Google Scholar] [CrossRef]
- Ott, D.E.; Coren, L.V.; Sowder, R.C.; Adams, J.; Schubert, U. Retroviruses have differing requirements for proteasome function in the budding process. J. Virol. 2003, 77, 3384–3393. [Google Scholar] [CrossRef]
- Yu, G.Y.; Lai, M.M. The ubiquitin-proteasome system facilitates the transfer of murine coronavirus from endosome to cytoplasm during virus entry. J. Virol. 2005, 79, 644–648. [Google Scholar] [CrossRef]
- Lupfer, C.; Pastey, M.K. Decreased replication of human respiratory syncytial virus treated with the proteasome inhibitor MG-132. Virus Res. 2010, 149, 36–41. [Google Scholar] [CrossRef]
- Si, X.; Gao, G.; Wong, J.; Wang, Y.; Zhang, J.; Luo, H. Ubiquitination is required for effective replication of coxsackievirus B3. PLoS One 2008, 3, e2585. [Google Scholar]
- López, T.; Silva-Ayala, D.; López, S.; Arias, C.F. Replication of the rotavirus genome requires an active ubiquitin-proteasome system. J. Virol. 2011, 85, 11964–11971. [Google Scholar] [CrossRef]
- Delboy, M.G.; Nicola, A.V. A pre-immediate-early role for tegument ICP0 in the proteasome-dependent entry of Herpes simplex virus. J. Virol. 2011, 85, 5910–5918. [Google Scholar] [CrossRef]
- Greene, W.; Zhang, W.; He, M.; Witt, C.; Ye, F.; Gao, S.J. The ubiquitin/proteasome system mediates entry and endosomal trafficking of Kaposi’s Sarcoma-associated herpesvirus in endothelial cells. PLoS Pathog. 2012, 8, e1002703. [Google Scholar] [CrossRef]
- Chen, C.; Zhuang, X. Epsin 1 is a cargo-specific adaptor for the clathrin-mediated endocytosis of the influenza virus. Proc. Natl. Acad. Sci. USA 2008, 105, 11790–11795. [Google Scholar] [CrossRef]
- Wodrich, H.; Henaff, D.; Jammart, B.; Segura-Morales, C.; Seelmeir, S.; Coux, O.; Ruzsics, Z.; Wiethoff, C.M.; Kremer, E.J. A capsid-encoded PPXY-motif facilitates adenovirus entry. PLoS Pathog. 2010, 6, e1000808. [Google Scholar] [CrossRef]
- Sarkari, F.; Sanchez-Alcaraz, T.; Wang, S.; Holowaty, M.N.; Sheng, Y.; Frappier, L. EBNA1-mediated recruitment of a histone H2B deubiquitylating complex to the Epstein-Barr virus latent origin of DNA replication. PLoS Pathog. 2009, 5, e1000624. [Google Scholar] [CrossRef]
- Brès, V.; Kiernan, R.E.; Linares, L.K.; Chable-Bessia, C.; Plechakova, O.; Tréand, C.; Emiliani, S.; Peloponese, J.M.; Jeang, K.T.; Coux, O.; et al. A non-proteolytic role for ubiquitin in Tat-mediated transactivation of the HIV-1 promoter. Nat. Cell Biol. 2003, 5, 754–761. [Google Scholar] [CrossRef]
- Peloponese, J.M.; Iha, H.; Yedavalli, V.R.; Miyazato, A.; Li, Y.; Haller, K.; Benkirane, M.; Jeang, K.T. Ubiquitination of human T-cell leukemia virus type 1 tax modulates its activity. J. Virol. 2004, 78, 11686–11695. [Google Scholar] [CrossRef]
- Yang, Z.; Yan, Z.; Wood, C. Kaposi’s Sarcoma-associated herpesvirus transactivator RTA promotes degradation of the repressors to regulate viral lytic replication. J. Virol. 2008, 82, 3590–3603. [Google Scholar] [CrossRef]
- Gould, F.; Harrison, S.M.; Hewitt, E.W.; Whitehouse, A. Kaposi’s sarcoma-associated herpesvirus RTA promotes degradation of the Hey1 repressor protein through the ubiquitin proteasome pathway. J. Virol. 2009, 83, 6727–6738. [Google Scholar] [CrossRef]
- Ikeda, M.; Ikeda, A.; Longan, L.C.; Longnecker, R. The Epstein-Barr virus latent membrane protein 2A PY motif recruits WW domain-containing ubiquitin-protein ligases. Virology 2000, 268, 178–191. [Google Scholar] [CrossRef]
- Ning, S.; Pagano, J.S. The A20 deubiquitinase activity negatively regulates LMP1 activation of IRF7. J. Virol. 2010, 84, 6130–6138. [Google Scholar]
- Meng, B.; Lever, A.M. Wrapping up the bad news: HIV assembly and release. Retrovirology 2013, 10, 5. [Google Scholar] [CrossRef]
- Oudshoorn, D.; Versteeg, G.A.; Kikkert, M. Regulation of the innate immune system by ubiquitin and ubiquitin-like modifiers. Cytokine Growth Factor Rev. 2012, 23, 273–282. [Google Scholar] [CrossRef]
- Bhoj, V.G.; Chen, Z.J. Ubiquitylation in innate and adaptive immunity. Nature 2009, 458, 430–437. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef]
- Rodrigues, L.; Filipe, J.; Seldon, M.P.; Fonseca, L.; Anrather, J.; Soares, M.P.; Simas, J.P. Termination of NF-kappaB activity through a gammaherpesvirus protein that assembles an EC5S ubiquitin-ligase. EMBO J. 2009, 28, 1283–1295. [Google Scholar] [CrossRef]
- Graff, J.W.; Ettayebi, K.; Hardy, M.E. Rotavirus NSP1 inhibits NFkappaB activation by inducing proteasome-dependent degradation of beta-TrCP: A novel mechanism of IFN antagonism. PLoS Pathog. 2009, 5, e1000280. [Google Scholar] [CrossRef]
- Zhu, H.; Zheng, C.; Xing, J.; Wang, S.; Li, S.; Lin, R.; Mossman, K.L. Varicella-zoster virus immediate-early protein ORF61 abrogates the IRF3-mediated innate immune response through degradation of activated IRF3. J. Virol. 2011, 85, 11079–11089. [Google Scholar] [CrossRef]
- Gale, M.; Sen, G.C. Viral evasion of the interferon system. J. Interferon Cytokine Res. 2009, 29, 475–476. [Google Scholar] [CrossRef]
- Ulane, C.M.; Horvath, C.M. Paramyxoviruses SV5 and HPIV2 assemble STAT protein ubiquitin ligase complexes from cellular components. Virology 2002, 304, 160–166. [Google Scholar] [CrossRef]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar]
- Amsler, L.; Verweij, M.C.; DeFilippis, V.R. The tiers and dimensions of evasion of the type I interferon response by Human Cytomegalovirus. J. Mol. Biol. 2013, 425, 4857–4871. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef]
- Hughes, R.; Towers, G.; Noursadeghi, M. Innate immune interferon responses to human immunodeficiency virus-1 infection. Rev. Med. Virol. 2012, 22, 257–266. [Google Scholar] [CrossRef]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef]
- Rajsbaum, R.; García-Sastre, A.; Versteeg, G.A. Trimmunity: The roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J. Mol. Biol. 2014, 426, 1265–1284. [Google Scholar] [CrossRef]
- Lenschow, D.J.; Lai, C.; Frias-Staheli, N.; Giannakopoulos, N.V.; Lutz, A.; Wolff, T.; Osiak, A.; Levine, B.; Schmidt, R.E.; García-Sastre, A.; et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against Influenza, Herpes, and Sindbis viruses. Proc. Natl. Acad. Sci. USA 2007, 104, 1371–1376. [Google Scholar] [CrossRef]
- Calistri, A.; Salata, C.; Parolin, C.; Palù, G. Role of multivesicular bodies and their components in the egress of enveloped RNA viruses. Rev. Med. Virol. 2009, 19, 31–45. [Google Scholar] [CrossRef]
- Clementz, M.A.; Chen, Z.; Banach, B.S.; Wang, Y.; Sun, L.; Ratia, K.; Baez-Santos, Y.M.; Wang, J.; Takayama, J.; Ghosh, A.K.; et al. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 2010, 84, 4619–4629. [Google Scholar] [CrossRef]
- Lou, Z.; Wang, S. E3 ubiquitin ligases and human papillomavirus-induced carcinogenesis. J. Int. Med. Res. 2014, 42, 247–260. [Google Scholar] [CrossRef]
- Asiaf, A.; Ahmad, S.T.; Mohammad, S.O.; Zargar, M.A. Review of the current knowledge on the epidemiology, pathogenesis, and prevention of human papillomavirus infection. Eur. J. Cancer Prev. 2014, 23, 206–224. [Google Scholar]
- DeCaprio, J.A. How the Rb tumor suppressor structure and function was revealed by the study of adenovirus and SV40. Virology 2009, 384, 274–284. [Google Scholar] [CrossRef]
- Fehr, A.R.; Yu, D. Control the host cell cycle: Viral regulation of the anaphase-promoting complex. J. Virol. 2013, 87, 8818–8825. [Google Scholar] [CrossRef]
- Fehr, A.R.; Gualberto, N.C.; Savaryn, J.P.; Terhune, S.S.; Yu, D. Proteasome-dependent disruption of the E3 ubiquitin ligase anaphase-promoting complex by HCMV protein pUL21a. PLoS Pathog. 2012, 8, e1002789. [Google Scholar] [CrossRef]
- Holowaty, M.N.; Sheng, Y.; Nguyen, T.; Arrowsmith, C.; Frappier, L. Protein interaction domains of the ubiquitin-specific protease, USP7/HUASP. J. Biol. Chem. 2003, 278, 47753–47761. [Google Scholar]
- Holowaty, M.N.; Zeghouf, M.; Wu, H.; Tellam, J.; Athanasopoulos, V.; Greenblatt, J.; Frappier, L. Protein profiling with Epstein-Barr nuclear antigen-1 reveals an interaction with the herpesvirus-associated ubiquitin-specific protease HUASP/USP7. J. Biol. Chem. 2003, 278, 29987–29994. [Google Scholar]
- Holowaty, M.N.; Frappier, L. HUASP/USP7 as an Epstein-Barr virus target. Biochem. Soc. Trans. 2004, 32, 731–732. [Google Scholar] [CrossRef]
- Frappier, L. Contributions of Epstein-Barr nuclear antigen 1 (EBNA1) to cell immortalization and survival. Viruses 2012, 4, 1537–1547. [Google Scholar] [CrossRef]
- Reya, T.; Clevers, H. Wnt signaling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef]
- Boname, J.M.; Lehner, P.J. What has the study of the K3 and K5 viral ubiquitin E3 ligases taught us about ubiquitin-mediated receptor regulation? Viruses 2011, 3, 118–131. [Google Scholar] [CrossRef]
- Coscoy, L.; Ganem, D. A viral protein that selectively downregulates ICAM-1 and B7-2 and modulates T cell costimulation. J. Clin. Invest. 2001, 107, 1599–1606. [Google Scholar] [CrossRef]
- Griffin, B.D.; Verweij, M.C.; Wiertz, E.J. Herpesviruses and immunity: The art of evasion. Vet. Microbiol. 2010, 143, 89–100. [Google Scholar] [CrossRef]
- Jackson, S.E.; Mason, G.M.; Wills, M.R. Human cytomegalovirus immunity and immune evasion. Virus Res. 2011, 157, 151–160. [Google Scholar] [CrossRef]
- Boutell, C.; Sadis, S.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein ICP0 and is isolated ring finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 2002, 76, 841–850. [Google Scholar] [CrossRef]
- Everett, R.D.; Chelbi-Alix, M.K. PML and PML nuclear bodies: Implications in antiviral defence. Biochimie 2007, 89, 819–830. [Google Scholar] [CrossRef]
- Gu, H.; Roizman, B. The two functions of herpes simplex virus 1 ICP0, inhibition of silencing by the CoREST/REST/HDAC complex and degradation of PML, are executed in tandem. J. Virol. 2009, 83, 181–187. [Google Scholar]
- Lin, A.E.; Greco, T.M.; Döhner, K.; Sodeik, B.; Cristea, I.M. A proteomic perspective of inbuilt viral protein regulation: pUL46 tegument protein is targeted for degradation by ICP0 during herpes simplex virus type 1 infection. Mol. Cell Proteomics 2013, 12, 3237–3252. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, K.; Wang, S.; Zheng, C. Herpes simplex virus 1 E3 ubiquitin ligase ICP0 protein inhibits tumor necrosis factor alpha-induced NF-κB activation by interacting with p65/RelA and p50/NF-κB1. J. Virol. 2013, 87, 12935–12948. [Google Scholar] [CrossRef]
- Gross, S.; Catez, F.; Masumoto, H.; Lomonte, P. Centromere architecture breakdown induced by the viral E3 ubiquitin ligase ICP0 protein of herpes simplex virus type 1. PLoS One 2012, 7, e44227. [Google Scholar]
- Liu, M.; Schmidt, E.E.; Halford, W.P. ICP0 dismantles microtubule networks in herpes simplex virus-infected cells. PLoS One 2010, 5, e10975. [Google Scholar] [CrossRef]
- Lilley, C.E.; Chaurushiya, M.S.; Boutell, C.; Landry, S.; Suh, J.; Panier, S.; Everett, R.D.; Stewart, G.S.; Durocher, D.; Weitzman, M.D. A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J. 2010, 29, 943–955. [Google Scholar] [CrossRef]
- Isaacson, M.K.; Ploegh, H.L. Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell Host Microbe 2009, 5, 559–570. [Google Scholar] [CrossRef]
- Timms, R.T.; Duncan, L.M.; Tchasovnikarova, I.A.; Antrobus, R.; Smith, D.L.; Dougan, G.; Weekes, M.P.; Lehner, P.J. Haploid genetic screens identify an essential role for PLP2 in the downregulation of novel plasma membrane targets by viral E3 ubiquitin ligases. PLoS Pathog. 2013, 9, e1003772. [Google Scholar] [CrossRef]
- Pardieu, C.; Vigan, R.; Wilson, S.J.; Calvi, A.; Zang, T.; Bieniasz, P.; Kellam, P.; Towers, G.J.; Neil, S.J. The RING-CH ligase K5 antagonizes restriction of KSHV and HIV-1 particle release by mediating ubiquitin-dependent endosomal degradation of tetherin. PLoS Pathog. 2010, 6, e1000843. [Google Scholar] [CrossRef]
- Lang, S.M.; Bynoe, M.O.; Karki, R.; Tartell, M.A.; Means, R.E. Kaposi's sarcoma-associated herpesvirus K3 and K5 proteins down regulate both DC-SIGN and DC-SIGNR. PLoS One 2013, 8, e58056. [Google Scholar]
- Karki, R.; Lang, S.M.; Means, R.E. The march family E3 ubiquitin ligase K5 alters monocyte metabolism and proliferation through receptor tyrosine kinase modulation. PLoS Pathog. 2011, 7, e1001331. [Google Scholar] [CrossRef]
- Walters, M.S.; Kyratsous, C.A.; Silverstein, S.J. The RING finger domain of varicella-zoster virus ORF61p has E3 ubiquitin ligase activity that is essential for efficient autoubiquitination and dispersion of Sp100-containing nuclear bodies. J. Virol. 2010, 84, 6861–6865. [Google Scholar] [CrossRef]
- Woo, J.L.; Berk, A.J. Adenovirus ubiquitin-protein ligase stimulates viral late mRNA nuclear export. J. Virol. 2007, 81, 575–587. [Google Scholar] [CrossRef]
- Sewatanon, J.; Ling, P.D. Murine gammaherpesvirus 68 ORF75c contains ubiquitin E3 ligase activity and requires PML sumoylation but not other known cellular pml regulators, CK2 and E6AP, to mediate PML degradation. Virology 2013, 440, 140–149. [Google Scholar] [CrossRef]
- Herr, R.A.; Wang, X.; Loh, J.; Virgin, H.W.; Hansen, T.H. Newly discovered viral E3 ligase PK3 induces endoplasmic reticulum-associated degradation of class I major histocompatibility proteins and their membrane-bound chaperones. J. Biol. Chem. 2012, 287, 14467–14479. [Google Scholar]
- Huang, J.; Huang, Q.; Zhou, X.; Shen, M.M.; Yen, A.; Yu, S.X.; Dong, G.; Qu, K.; Huang, P.; Anderson, E.M.; et al. The poxvirus p28 virulence factor is an E3 ubiquitin ligase. J. Biol. Chem. 2004, 279, 54110–54116. [Google Scholar] [CrossRef]
- He, F.; Syed, S.M.; Hameed, A.S.; Kwang, J. Viral ubiquitin ligase WSSV222 is required for efficient white spot syndrome virus replication in shrimp. J. Gen. Virol. 2009, 90, 1483–1490. [Google Scholar] [CrossRef]
- Van Kasteren, P.B.; Beugeling, C.; Ninaber, D.K.; Frias-Staheli, N.; van Boheemen, S.; García-Sastre, A.; Snijder, E.J.; Kikkert, M. Arterivirus and nairovirus ovarian tumor domain-containing deubiquitinases target activated RIG-I to control innate immune signaling. J. Virol. 2012, 86, 773–785. [Google Scholar] [CrossRef]
- Wang, G.; Chen, G.; Zheng, D.; Cheng, G.; Tang, H. PLP2 of mouse hepatitis virus A59 (MHV-A59) targets TBK1 to negatively regulate cellular type I interferon signaling pathway. PLoS One 2011, 6, e17192. [Google Scholar]
- Zheng, D.; Chen, G.; Guo, B.; Cheng, G.; Tang, H. PLP2, a potent deubiquitinase from murine hepatitis virus, strongly inhibits cellular type I interferon production. Cell Res. 2008, 18, 1105–1113. [Google Scholar]
- Wang, D.; Fang, L.; Li, P.; Sun, L.; Fan, J.; Zhang, Q.; Luo, R.; Liu, X.; Li, K.; Chen, H.; et al. The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. J. Virol. 2011, 85, 3758–3766. [Google Scholar] [CrossRef]
- Jiang, J.; Tang, H. Mechanism of inhibiting type I interferon induction by hepatitis B virus X protein. Protein Cell 2010, 1, 1106–1117. [Google Scholar] [CrossRef]
- Schlieker, C.; Weihofen, W.A.; Frijns, E.; Kattenhorn, L.M.; Gaudet, R.; Ploegh, H.L. Structure of a herpesvirus-encoded cysteine protease reveals a unique class of deubiquitinating enzymes. Mol. Cell 2007, 25, 677–687. [Google Scholar] [CrossRef]
- Whitehurst, C.B.; Ning, S.; Bentz, G.L.; Dufour, F.; Gershburg, E.; Shackelford, J.; Langelier, Y.; Pagano, J.S. The Epstein-Barr virus (EBV) deubiquitinating enzyme BPLF1 reduces EBV ribonucleotide reductase activity. J. Virol. 2009, 83, 4345–4353. [Google Scholar] [CrossRef]
- Whitehurst, C.B.; Vaziri, C.; Shackelford, J.; Pagano, J.S. Epstein-Barr virus BPLF1 deubiquitinates pcna and attenuates polymerase η recruitment to DNA damage sites. J. Virol. 2012, 86, 8097–8106. [Google Scholar] [CrossRef]
- Kumar, R.; Whitehurst, C.B.; Pagano, J.S. The Rad6/18 ubiquitin complex interacts with the Epstein-Barr virus deubiquitinating enzyme, BPLF1, and contributes to virus infectivity. J. Virol. 2014. [Google Scholar] [CrossRef]
- Wang, S.; Wang, K.; Li, J.; Zheng, C. Herpes simplex virus 1 ubiquitin-specific protease UL36 inhibits beta interferon production by deubiquitinating TRAF3. J. Virol. 2013, 87, 11851–11860. [Google Scholar] [CrossRef]
- Bolstad, M.; Abaitua, F.; Crump, C.M.; O’Hare, P. Autocatalytic activity of the ubiquitin-specific protease domain of herpes simplex virus 1 VP1-2. J. Virol. 2011, 85, 8738–8751. [Google Scholar] [CrossRef]
- Kim, E.T.; Oh, S.E.; Lee, Y.O.; Gibson, W.; Ahn, J.H. Cleavage specificity of the UL48 deubiquitinating protease activity of human cytomegalovirus and the growth of an active-site mutant virus in cultured cells. J. Virol. 2009, 83, 12046–12056. [Google Scholar] [CrossRef]
- Böttcher, S.; Maresch, C.; Granzow, H.; Klupp, B.G.; Teifke, J.P.; Mettenleiter, T.C. Mutagenesis of the active-site cysteine in the ubiquitin-specific protease contained in large tegument protein pUL36 of pseudorabies virus impairs viral replication in vitro and neuroinvasion in vivo. J. Virol. 2008, 82, 6009–6016. [Google Scholar] [CrossRef]
- Inn, K.S.; Lee, S.H.; Rathbun, J.Y.; Wong, L.Y.; Toth, Z.; Machida, K.; Ou, J.H.; Jung, J.U. Inhibition of RIG-I-mediated signaling by Kaposi’s Sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J. Virol. 2011, 85, 10899–10904. [Google Scholar]
- Akutsu, M.; Ye, Y.; Virdee, S.; Chin, J.W.; Komander, D. Molecular basis for ubiquitin and ISG15 cross-reactivity in viral ovarian tumor domains. Proc. Natl. Acad. Sci. USA 2011, 108, 2228–2233. [Google Scholar] [CrossRef]
- Mielech, A.M.; Kilianski, A.; Baez-Santos, Y.M.; Mesecar, A.D.; Baker, S.C. MERS-CoV papain-like protease has deISGylating and deubiquitinating activities. Virology 2014, 450–451, 64–70. [Google Scholar] [CrossRef]
- Lombardi, C.; Ayach, M.; Beaurepaire, L.; Chenon, M.; Andreani, J.; Guerois, R.; Jupin, I.; Bressanelli, S. A compact viral processing proteinase/ubiquitin hydrolase from the OTU family. PLoS Pathog. 2013, 9, e1003560. [Google Scholar] [CrossRef]
- Chenon, M.; Camborde, L.; Cheminant, S.; Jupin, I. A viral deubiquitylating enzyme targets viral RNA-dependent RNA polymerase and affects viral infectivity. EMBO J. 2012, 31, 741–753. [Google Scholar] [CrossRef]
- Xing, Y.; Chen, J.; Tu, J.; Zhang, B.; Chen, X.; Shi, H.; Baker, S.C.; Feng, L.; Chen, Z. The papain-like protease of porcine epidemic diarrhea virus negatively regulates type I interferon pathway by acting as a viral deubiquitinase. J. Gen. Virol. 2013, 94, 1554–1567. [Google Scholar] [CrossRef]
- Sun, Z.; Chen, Z.; Lawson, S.R.; Fang, Y. The cysteine protease domain of porcine reproductive and respiratory syndrome virus nonstructural protein 2 possesses deubiquitinating and interferon antagonism functions. J. Virol. 2010, 84, 7832–7846. [Google Scholar] [CrossRef]
- Balakirev, M.Y.; Jaquinod, M.; Haas, A.L.; Chroboczek, J. Deubiquitinating function of adenovirus proteinase. J. Virol. 2002, 76, 6323–6331. [Google Scholar] [CrossRef]
- Barré-Sinoussi, F.; Ross, A.L.; Delfraissy, J.F. Past, present and future: 30 years of HIV research. Nat. Rev. Microbiol. 2013, 11, 877–883. [Google Scholar] [CrossRef]
- Greene, W.C. Regulation of HIV-1 gene expression. Annu. Rev. Immunol. 1990, 8, 453–475. [Google Scholar] [CrossRef]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar] [CrossRef]
- Bushman, F.D.; Malani, N.; Fernandes, J.; D’Orso, I.; Cagney, G.; Diamond, T.L.; Zhou, H.; Hazuda, D.J.; Espeseth, A.S.; König, R.; et al. Host cell factors in hiv replication: Meta-analysis of genome-wide studies. PLoS Pathog. 2009, 5, e1000437. [Google Scholar] [CrossRef]
- Friedrich, B.M.; Dziuba, N.; Li, G.; Endsley, M.A.; Murray, J.L.; Ferguson, M.R. Host factors mediating HIV-1 replication. Virus Res. 2011, 161, 101–114. [Google Scholar]
- Zheng, Y.H.; Jeang, K.T.; Tokunaga, K. Host restriction factors in retroviral infection: Promises in virus-host interaction. Retrovirology 2012, 9, 112. [Google Scholar] [CrossRef]
- Strebel, K. HIV accessory proteins versus host restriction factors. Curr. Opin. Virol. 2013, 3, 692–699. [Google Scholar] [CrossRef]
- Barry, M.; Früh, K. Viral modulators of Cullin RING ubiquitin ligases: Culling the host defense. Sci. STKE 2006, 2006, pe21. [Google Scholar]
- Malim, M.H. HIV: Ringside views. Nature 2014, 505, 167–168. [Google Scholar] [CrossRef]
- Genschik, P.; Marrocco, K.; Bach, L.; Noir, S.; Criqui, M.C. Selective protein degradation: A rheostat to modulate cell-cycle phase transitions. J. Exp. Bot. 2013. [Google Scholar] [CrossRef]
- Genschik, P.; Sumara, I.; Lechner, E. The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s): cellular functions and disease implications. EMBO J. 2013, 32, 2307–2320. [Google Scholar] [CrossRef]
- Guo, Y.; Dong, L.; Qiu, X.; Wang, Y.; Zhang, B.; Liu, H.; Yu, Y.; Zang, Y.; Yang, M.; Huang, Z. Structural basis for hijacking CBF-β and CUL5 E3 ligase complex by HIV-1 Vif. Nature 2014, 505, 229–233. [Google Scholar] [CrossRef]
- Nomaguchi, M.; Fujita, M.; Adachi, A. Role of HIV-1 Vpu protein for virus spread and pathogenesis. Microbes Infect. 2008, 10, 960–967. [Google Scholar] [CrossRef]
- Goffinet, C.; Allespach, I.; Homann, S.; Tervo, H.M.; Habermann, A.; Rupp, D.; Oberbremer, L.; Kern, C.; Tibroni, N.; Welsch, S.; et al. HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe 2009, 5, 285–297. [Google Scholar] [CrossRef]
- Mangeat, B.; Gers-Huber, G.; Lehmann, M.; Zufferey, M.; Luban, J.; Piguet, V. HIV-1 Vpu neutralizes the antiviral factor Tetherin/BST-2 by binding it and directing its beta-TrCP2-dependent degradation. PLoS Pathog. 2009, 5, e1000574. [Google Scholar] [CrossRef]
- Douglas, J.L.; Viswanathan, K.; McCarroll, M.N.; Gustin, J.K.; Früh, K.; Moses, A.V. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a {beta}TrCP-dependent mechanism. J. Virol. 2009, 83, 7931–7947. [Google Scholar] [CrossRef]
- Verma, S.; Ali, A.; Arora, S.; Banerjea, A.C. Inhibition of β-TrCP-dependent ubiquitination of p53 by HIV-1 Vpu promotes p53-mediated apoptosis in human T cells. Blood 2011, 117, 6600–6607. [Google Scholar] [CrossRef]
- Belaïdouni, N.; Marchal, C.; Benarous, R.; Besnard-Guérin, C. Involvement of the betaTrCP in the ubiquitination and stability of the HIV-1 Vpu protein. Biochem. Biophys. Res. Commun. 2007, 357, 688–693. [Google Scholar] [CrossRef]
- Le Rouzic, E.; Belaïdouni, N.; Estrabaud, E.; Morel, M.; Rain, J.C.; Transy, C. HIV1 Vpr arrests the cell cycle by recruiting DCAF1/VprBP, a receptor of the Cul4-DDB1 ubiquitin ligase. Cell Cycle 2007, 6, 182–188. [Google Scholar] [CrossRef]
- Eldin, P.; Chazal, N.; Fenard, D.; Bernard, E.; Guichou, J.F.; Briant, L. Vpr expression abolishes the capacity of HIV-1 infected cells to repair uracilated DNA. Nucleic Acids Res. 2014, 42, 1698–1710. [Google Scholar] [CrossRef]
- Casey Klockow, L.; Sharifi, H.J.; Wen, X.; Flagg, M.; Furuya, A.K.; Nekorchuk, M.; de Noronha, C.M. The HIV-1 protein Vpr targets the endoribonuclease DICER for proteasomal degradation to boost macrophage infection. Virology 2013, 444, 191–202. [Google Scholar] [CrossRef]
- Sze, A.; Olagnier, D.; Lin, R.; van Grevenynghe, J.; Hiscott, J. SAMHD1 host restriction factor: A link with innate immune sensing of retrovirus infection. J. Mol. Biol. 2013, 425, 4981–4994. [Google Scholar] [CrossRef]
- Schröfelbauer, B.; Yu, Q.; Landau, N.R. New insights into the role of vif in HIV-1 replication. AIDS Rev. 2004, 6, 34–39. [Google Scholar]
- Simon, J.H.; Gaddis, N.C.; Fouchier, R.A.; Malim, M.H. Evidence for a newly discovered cellular anti-HIV-1 phenotype. Nat. Med. 1998, 4, 1397–1400. [Google Scholar] [CrossRef]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Evans, S.L.; Schön, A.; Gao, Q.; Han, X.; Zhou, X.; Freire, E.; Yu, X.F. HIV-1 Vif N-terminal motif is required for recruitment of Cul5 to suppress APOBEC3. Retrovirology 2014, 11, 4. [Google Scholar] [CrossRef]
- Goila-Gaur, R.; Strebel, K. Hiv-1 vif, apobec, and intrinsic immunity. Retrovirology 2008, 5, 51. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Lv, M.; Zuo, T.; Wu, H.; Wang, J.; Liu, D.; Wang, C.; Zhang, J.; Li, X.; et al. Interactions between HIV-1 Vif and human ElonginB-ElonginC are important for CBF-β binding to Vif. Retrovirology 2013, 10, 94. [Google Scholar] [CrossRef]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef]
- Perez-Caballero, D.; Zang, T.; Ebrahimi, A.; McNatt, M.W.; Gregory, D.A.; Johnson, M.C.; Bieniasz, P.D. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 2009, 139, 499–511. [Google Scholar] [CrossRef]
- Neil, S.J. The antiviral activities of tetherin. Curr. Top. Microbiol. Immunol. 2013, 371, 67–104. [Google Scholar]
- Sauter, D.; Schindler, M.; Specht, A.; Landford, W.N.; Münch, J.; Kim, K.A.; Votteler, J.; Schubert, U.; Bibollet-Ruche, F.; Keele, B.F.; et al. Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe 2009, 6, 409–421. [Google Scholar] [CrossRef]
- Sauter, D.; Kirchhoff, F. Tetherin antagonism by primate lentiviral Nef proteins. Curr. HIV Res. 2011, 9, 514–523. [Google Scholar] [CrossRef]
- Celestino, M.; Calistri, A.; Del Vecchio, C.; Salata, C.; Chiuppesi, F.; Pistello, M.; Borsetti, A.; Palù, G.; Parolin, C. Feline tetherin is characterized by a short N-terminal region and is counteracted by the Feline Immunodeficiency Virus Envelope glycoprotein. J. Virol. 2012, 86, 6688–6700. [Google Scholar] [CrossRef]
- Sauter, D.; Unterweger, D.; Vogl, M.; Usmani, S.M.; Heigele, A.; Kluge, S.F.; Hermkes, E.; Moll, M.; Barker, E.; Peeters, M.; et al. Human tetherin exerts strong selection pressure on the HIV-1 group N Vpu protein. PLoS Pathog. 2012, 8, e1003093. [Google Scholar]
- Sauter, D.; Vogl, M.; Kirchhoff, F. Ancient origin of a deletion in human BST2/tetherin that confers protection against viral zoonoses. Hum. Mutat. 2011, 32, 1243–1245. [Google Scholar] [CrossRef]
- Götz, N.; Sauter, D.; Usmani, S.M.; Fritz, J.V.; Goffinet, C.; Heigele, A.; Geyer, M.; Bibollet-Ruche, F.; Learn, G.H.; Fackler, O.T.; et al. Reacquisition of Nef-mediated tetherin antagonism in a single in vivo passage of HIV-1 through its original chimpanzee host. Cell Host Microbe 2012, 12, 373–380. [Google Scholar] [CrossRef]
- Mitchell, R.S.; Katsura, C.; Skasko, M.A.; Fitzpatrick, K.; Lau, D.; Ruiz, A.; Stephens, E.B.; Margottin-Goguet, F.; Benarous, R.; Guatelli, J.C. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endo-lysosomal trafficking. PLoS Pathog. 2009, 5, e1000450. [Google Scholar] [CrossRef]
- Caillet, M.; Janvier, K.; Pelchen-Matthews, A.; Delcroix-Genête, D.; Camus, G.; Marsh, M.; Berlioz-Torrent, C. Rab7A is required for efficient production of infectious HIV-1. PLoS Pathog. 2011, 7, e1002347. [Google Scholar] [CrossRef]
- Janvier, K.; Pelchen-Matthews, A.; Renaud, J.B.; Caillet, M.; Marsh, M.; Berlioz-Torrent, C. The ESCRt-0 component HRS is required for HIV-1 Vpu-mediated BST-2/tetherin down-regulation. PLoS Pathog. 2011, 7, e1001265. [Google Scholar] [CrossRef]
- Tokarev, A.A.; Munguia, J.; Guatelli, J.C. Serine-threonine ubiquitination mediates downregulation of BST-2/tetherin and relief of restricted virion release by HIV-1 Vpu. J. Virol. 2011, 85, 51–63. [Google Scholar] [CrossRef]
- Dubé, M.; Roy, B.B.; Guiot-Guillain, P.; Binette, J.; Mercier, J.; Chiasson, A.; Cohen, E.A. Antagonism of tetherin restriction of HIV-1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 2010, 6, e1000856. [Google Scholar] [CrossRef]
- Tervo, H.M.; Homann, S.; Ambiel, I.; Fritz, J.V.; Fackler, O.T.; Keppler, O.T. B-TrCP is dispensable for Vpu’s ability to overcome the CD317/tetherin-imposed restriction to HIV-1 release. Retrovirology 2011, 8, 9. [Google Scholar] [CrossRef]
- Hauser, H.; Lopez, L.A.; Yang, S.J.; Oldenburg, J.E.; Exline, C.M.; Guatelli, J.C.; Cannon, P.M. HIV-1 Vpu and HIV-2 Env counteract BST-2/tetherin by sequestration in a perinuclear compartment. Retrovirology 2010, 7, 51. [Google Scholar] [CrossRef]
- Belzile, J.P.; Duisit, G.; Rougeau, N.; Mercier, J.; Finzi, A.; Cohen, E.A. HIV-1 Vpr-mediated G2 arrest involves the DDB1-CUL4AVPRBP E3 ubiquitin ligase. PLoS Pathog. 2007, 3, e85. [Google Scholar] [CrossRef]
- DeHart, J.L.; Zimmerman, E.S.; Ardon, O.; Monteiro-Filho, C.M.; Argañaraz, E.R.; Planelles, V. HIV-1 Vpr activates the G2 checkpoint through manipulation of the ubiquitin proteasome system. Virol. J. 2007, 4, 57. [Google Scholar] [CrossRef]
- Hrecka, K.; Gierszewska, M.; Srivastava, S.; Kozaczkiewicz, L.; Swanson, S.K.; Florens, L.; Washburn, M.P.; Skowronski, J. Lentiviral Vpr usurps Cul4-DDB1[VprBP] E3 ubiquitin ligase to modulate cell cycle. Proc. Natl. Acad. Sci. USA 2007, 104, 11778–11783. [Google Scholar] [CrossRef]
- Schröfelbauer, B.; Hakata, Y.; Landau, N.R. HIV-1 Vpr function is mediated by interaction with the damage-specific DNA-binding protein DDB1. Proc. Natl. Acad. Sci. USA 2007, 104, 4130–4135. [Google Scholar] [CrossRef]
- Tan, L.; Ehrlich, E.; Yu, X.F. DDB1 and Cul4A are required for human immunodeficiency virus type 1 vpr-induced G2 arrest. J. Virol. 2007, 81, 10822–10830. [Google Scholar] [CrossRef]
- Wen, X.; Duus, K.M.; Friedrich, T.D.; de Noronha, C.M. The HIV1 protein Vpr acts to promote G2 cell cycle arrest by engaging a DDB1 and Cullin4A-containing ubiquitin ligase complex using VPRBP/DCAF1 as an adaptor. J. Biol. Chem. 2007, 282, 27046–27057. [Google Scholar] [CrossRef]
- Le Rouzic, E.; Morel, M.; Ayinde, D.; Belaïdouni, N.; Letienne, J.; Transy, C.; Margottin-Goguet, F. Assembly with the Cul4A-DDB1DCAF1 ubiquitin ligase protects HIV-1 Vpr from proteasomal degradation. J. Biol. Chem. 2008, 283, 21686–21692. [Google Scholar] [CrossRef]
- DeHart, J.L.; Bosque, A.; Harris, R.S.; Planelles, V. Human Immunodeficiency Virus type 1 Vif induces cell cycle delay via recruitment of the same E3 ubiquitin ligase complex that targets APOBEC3 proteins for degradation. J. Virol. 2008, 82, 9265–9272. [Google Scholar] [CrossRef]
- Gérard, F.C.; Yang, R.; Romani, B.; Poisson, A.; Belzile, J.P.; Rougeau, N.; Cohen, E.A. Defining the interactions and role of DCAF1/VPRBP in the DDB1-Cullin4A E3 ubiquitin ligase complex engaged by HIV-1 Vpr to induce a G2 cell cycle arrest. PLoS One 2014, 9, e89195. [Google Scholar]
- Mashiba, M.; Collins, K.L. Molecular mechanisms of HIV immune evasion of the innate immune response in myeloid cells. Viruses 2013, 5, 1–14. [Google Scholar] [CrossRef]
- Amie, S.M.; Noble, E.; Kim, B. Intracellular nucleotide levels and the control of retroviral infections. Virology 2013, 436, 247–254. [Google Scholar] [CrossRef]
- Schwefel, D.; Groom, H.C.; Boucherit, V.C.; Christodoulou, E.; Walker, P.A.; Stoye, J.P.; Bishop, K.N.; Taylor, I.A. Structural basis of lentiviral subversion of a cellular protein degradation pathway. Nature 2014, 505, 234–238. [Google Scholar]
- Sharifi, H.J.; Furuya, A.K.; Jellinger, R.M.; Nekorchuk, M.D.; de Noronha, C.M. Cullin 4A and Cullin4B are interchangeable for HIV Vpr and Vpx action through the CRL4 ubiquitin ligase complex. J. Virol. 2014. [Google Scholar] [CrossRef]
- Hurley, J.H.; Hanson, P.I. Membrane budding and scission by the ESCRT machinery: It’s all in the neck. Nat. Rev. Mol. Cell Biol. 2010, 11, 556–566. [Google Scholar]
- Rossman, J.S.; Lamb, R.A. Viral membrane scission. Annu. Rev. Cell Dev. Biol. 2013, 29, 551–569. [Google Scholar] [CrossRef]
- Göttlinger, H.G.; Dorfman, T.; Sodroski, J.G.; Haseltine, W.A. Effect of mutations affecting the p6 Gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. USA 1991, 88, 3195–3199. [Google Scholar] [CrossRef]
- Huang, M.; Orenstein, J.M.; Martin, M.A.; Freed, E.O. P6Gag is required for particle production from full-length human immunodeficiency virus type 1 molecular clones expressing protease. J. Virol. 1995, 69, 6810–6818. [Google Scholar]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef]
- Crump, C.M.; Yates, C.; Minson, T. Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. J. Virol. 2007, 81, 7380–7387. [Google Scholar] [CrossRef]
- Calistri, A.; Sette, P.; Salata, C.; Cancellotti, E.; Forghieri, C.; Comin, A.; Göttlinger, H.; Campadelli-Fiume, G.; Palù, G.; Parolin, C. Intracellular trafficking and maturation of herpes simplex virus type 1 gB and virus egress require functional biogenesis of multivesicular bodies. J. Virol. 2007, 81, 11468–11478. [Google Scholar] [CrossRef]
- Tandon, R.; AuCoin, D.P.; Mocarski, E.S. Human cytomegalovirus exploits ESCRT machinery in the process of virion maturation. J. Virol. 2009, 83, 10797–10807. [Google Scholar] [CrossRef]
- Li, Z.; Blissard, G.W. Cellular VPS4 is required for efficient entry and egress of budded virions of Autographa californica multiple nucleopolyhedrovirus. J. Virol. 2012, 86, 459–472. [Google Scholar] [CrossRef]
- Garcia, M.L.; Reynolds, T.D.; Mothes, W.; Robek, M.D. Functional characterization of the putative hepatitis B virus core protein late domain using retrovirus chimeras. PLoS One 2013, 8, e72845. [Google Scholar]
- Patnaik, A.; Chau, V.; Wills, J.W. Ubiquitin is part of the retrovirus budding machinery. Proc. Natl. Acad. Sci. USA 2000, 97, 13069–13074. [Google Scholar] [CrossRef]
- Joshi, A.; Munshi, U.; Ablan, S.D.; Nagashima, K.; Freed, E.O. Functional replacement of a retroviral late domain by ubiquitin fusion. Traffic 2008, 9, 1972–1983. [Google Scholar] [CrossRef]
- Putterman, D.; Pepinsky, R.B.; Vogt, V.M. Ubiquitin in avian leukosis virus particles. Virology 1990, 176, 633–637. [Google Scholar] [CrossRef]
- Ott, D.E.; Coren, L.V.; Copeland, T.D.; Kane, B.P.; Johnson, D.G.; Sowder, R.C.; Yoshinaka, Y.; Oroszlan, S.; Arthur, L.O.; Henderson, L.E. Ubiquitin is covalently attached to the p6Gag proteins of human immunodeficiency virus type 1 and simian immunodeficiency virus and to the p12Gag protein of Moloney murine leukemia virus. J. Virol. 1998, 72, 2962–2968. [Google Scholar]
- Martin-Serrano, J.; Eastman, S.W.; Chung, W.; Bieniasz, P.D. HECT ubiquitin ligases link viral and cellular PPXY motifs to the vacuolar protein-sorting pathway. J. Cell Biol. 2005, 168, 89–101. [Google Scholar]
- Usami, Y.; Popov, S.; Popova, E.; Göttlinger, H.G. Efficient and specific rescue of human immunodeficiency virus type 1 budding defects by a NEDD4-like ubiquitin ligase. J. Virol. 2008, 82, 4898–4907. [Google Scholar] [CrossRef]
- Chung, H.Y.; Morita, E.; von Schwedler, U.; Müller, B.; Kräusslich, H.G.; Sundquist, W.I. NEDD4L overexpression rescues the release and infectivity of human immunodeficiency virus type 1 constructs lacking PTAP and YPXL late domains. J. Virol. 2008, 82, 4884–4897. [Google Scholar] [CrossRef]
- Calistri, A.; Del Vecchio, C.; Salata, C.; Celestino, M.; Celegato, M.; Göttlinger, H.; Palù, G.; Parolin, C. Role of the feline immunodeficiency virus L-domain in the presence or absence of Gag processing: Involvement of ubiquitin and NEDD4-2s ligase in viral egress. J. Cell Physiol. 2009, 218, 175–182. [Google Scholar] [CrossRef]
- Weiss, E.R.; Popova, E.; Yamanaka, H.; Kim, H.C.; Huibregtse, J.M.; Göttlinger, H. Rescue of HIV-1 release by targeting widely divergent NEDD4-type ubiquitin ligases and isolated catalytic hect domains to Gag. PLoS Pathog. 2010, 6, e1001107. [Google Scholar] [CrossRef]
- Parent, L.J.; Bennett, R.P.; Craven, R.C.; Nelle, T.D.; Krishna, N.K.; Bowzard, J.B.; Wilson, C.B.; Puffer, B.A.; Montelaro, R.C.; Wills, J.W. Positionally independent and exchangeable late budding functions of the Rous sarcoma virus and human immunodeficiency virus Gag proteins. J. Virol. 1995, 69, 5455–5460. [Google Scholar]
- Yuan, B.; Campbell, S.; Bacharach, E.; Rein, A.; Goff, S.P. Infectivity of Moloney murine leukemia virus defective in late assembly events is restored by late assembly domains of other retroviruses. J. Virol. 2000, 74, 7250–7260. [Google Scholar] [CrossRef]
- Li, F.; Chen, C.; Puffer, B.A.; Montelaro, R.C. Functional replacement and positional dependence of homologous and heterologous L domains in equine infectious anemia virus replication. J. Virol. 2002, 76, 1569–1577. [Google Scholar] [CrossRef]
- Strack, B.; Calistri, A.; Craig, S.; Popova, E.; Göttlinger, H.G. AIP1/Alix is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 2003, 114, 689–699. [Google Scholar] [CrossRef]
- Zhadina, M.; Bieniasz, P.D. Functional interchangeability of late domains, late domain cofactors and ubiquitin in viral budding. PLoS Pathog. 2010, 6, e1001153. [Google Scholar] [CrossRef]
- Staub, O.; Dho, S.; Henry, P.; Correa, J.; Ishikawa, T.; McGlade, J.; Rotin, D. WW domains of NEDD4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. EMBO J. 1996, 15, 2371–2380. [Google Scholar]
- Staub, O.; Yeger, H.; Plant, P.J.; Kim, H.; Ernst, S.A.; Rotin, D. Immunolocalization of the ubiquitin-protein ligase Nedd4 in tissues expressing the epithelial Na+ channel (ENaC). Am. J. Physiol. 1997, 272, C1871–C1880. [Google Scholar]
- Staub, O.; Abriel, H.; Plant, P.; Ishikawa, T.; Kanelis, V.; Saleki, R.; Horisberger, J.D.; Schild, L.; Rotin, D. Regulation of the epithelial Na+ channel by NEDD4 and ubiquitination. Kidney Int. 2000, 57, 809–815. [Google Scholar] [CrossRef]
- Strack, B.; Calistri, A.; Göttlinger, H.G. Late assembly domain function can exhibit context dependence and involves ubiquitin residues implicated in endocytosis. J. Virol. 2002, 76, 5472–5479. [Google Scholar] [CrossRef]
- VerPlank, L.; Bouamr, F.; LaGrassa, T.J.; Agresta, B.; Kikonyogo, A.; Leis, J.; Carter, C.A. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag). Proc. Natl. Acad. Sci. USA 2001, 98, 7724–7729. [Google Scholar] [CrossRef]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Côté, M.; Rich, R.L.; et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef]
- Henry, A.G.; White, I.J.; Marsh, M.; von Zastrow, M.; Hislop, J.N. The role of ubiquitination in lysosomal trafficking of δ-opioid receptors. Traffic 2011, 12, 170–184. [Google Scholar] [CrossRef]
- Hislop, J.N.; Henry, A.G.; von Zastrow, M. Ubiquitination in the first cytoplasmic loop of μ-opioid receptors reveals a hierarchical mechanism of lysosomal down-regulation. J. Biol. Chem. 2011, 286, 40193–40204. [Google Scholar] [CrossRef]
- Jouvenet, N. Dynamics of ESCRT proteins. Cell Mol. Life Sci. 2012, 69, 4121–4133. [Google Scholar] [CrossRef]
- Clague, M.J.; Urbé, S. Ubiquitin: Same molecule, different degradation pathways. Cell 2010, 143, 682–685. [Google Scholar] [CrossRef]
- Shields, S.B.; Piper, R.C. How ubiquitin functions with ESCRTS. Traffic 2011, 12, 1306–1317. [Google Scholar] [CrossRef]
- Shields, S.B.; Oestreich, A.J.; Winistorfer, S.; Nguyen, D.; Payne, J.A.; Katzmann, D.J.; Piper, R. ESCRT ubiquitin-binding domains function cooperatively during MVB cargo sorting. J. Cell Biol. 2009, 185, 213–224. [Google Scholar] [CrossRef]
- Hurley, J.H.; Ren, X. The circuitry of cargo flux in the ESCRT pathway. J. Cell Biol. 2009, 185, 185–187. [Google Scholar] [CrossRef]
- Amit, I.; Yakir, L.; Katz, M.; Zwang, Y.; Marmor, M.D.; Citri, A.; Shtiegman, K.; Alroy, I.; Tuvia, S.; Reiss, Y.; et al. Tal, a TSG101-specific E3 ubiquitin ligase, regulates receptor endocytosis and retrovirus budding. Genes Dev. 2004, 18, 1737–1752. [Google Scholar] [CrossRef]
- Pashkova, N.; Gakhar, L.; Winistorfer, S.C.; Sunshine, A.B.; Rich, M.; Dunham, M.J.; Yu, L.; Piper, R.C. The yeast Alix homolog bro1 functions as a ubiquitin receptor for protein sorting into multivesicular endosomes. Dev. Cell 2013, 25, 520–533. [Google Scholar] [CrossRef]
- Pornillos, O.; Alam, S.L.; Rich, R.L.; Myszka, D.G.; Davis, D.R.; Sundquist, W.I. Structure and functional interactions of the TSG101 UEV domain. EMBO J. 2002, 21, 2397–2406. [Google Scholar] [CrossRef]
- Pornillos, O.; Alam, S.L.; Davis, D.R.; Sundquist, W.I. Structure of the Tsg101 UEV domain in complex with the PTAP motif of the HIV-1 p6 protein. Nat. Struct. Biol. 2002, 9, 812–817. [Google Scholar]
- Sette, P.; Nagashima, K.; Piper, R.C.; Bouamr, F. Ubiquitin conjugation to Gag is essential for ESCRT-mediated HIV-1 budding. Retrovirology 2013, 10, 79. [Google Scholar] [CrossRef]
- Gottwein, E.; Kräusslich, H.G. Analysis of human immunodeficiency virus type 1 Gag ubiquitination. J. Virol. 2005, 79, 9134–9144. [Google Scholar] [CrossRef]
- Zhadina, M.; McClure, M.O.; Johnson, M.C.; Bieniasz, P.D. Ubiquitin-dependent virus particle budding without viral protein ubiquitination. Proc. Natl. Acad. Sci. USA 2007, 104, 20031–20036. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Calistri, A.; Munegato, D.; Carli, I.; Parolin, C.; Palù, G. The Ubiquitin-Conjugating System: Multiple Roles in Viral Replication and Infection. Cells 2014, 3, 386-417. https://doi.org/10.3390/cells3020386
Calistri A, Munegato D, Carli I, Parolin C, Palù G. The Ubiquitin-Conjugating System: Multiple Roles in Viral Replication and Infection. Cells. 2014; 3(2):386-417. https://doi.org/10.3390/cells3020386
Chicago/Turabian StyleCalistri, Arianna, Denis Munegato, Ilaria Carli, Cristina Parolin, and Giorgio Palù. 2014. "The Ubiquitin-Conjugating System: Multiple Roles in Viral Replication and Infection" Cells 3, no. 2: 386-417. https://doi.org/10.3390/cells3020386
APA StyleCalistri, A., Munegato, D., Carli, I., Parolin, C., & Palù, G. (2014). The Ubiquitin-Conjugating System: Multiple Roles in Viral Replication and Infection. Cells, 3(2), 386-417. https://doi.org/10.3390/cells3020386