Molecular Perspectives for mu/delta Opioid Receptor Heteromers as Distinct, Functional Receptors

Abstract
:1. Introduction and Historical Perspective
Nomenclature
2. Formation
2.1. Molecular Nature of Mu-Delta Interaction
2.1.1. Computational
2.1.2. Experimental Disruption
Summary, Molecular Nature of Mu-Delta Interaction
2.2. Coexpression of MOR and DOR
Summary, Coexpression of MOR and DOR
2.3. Induction of M/DOR Formation
Summary, Induction of M/DOR formation
2.4. Subcellular Location of Formation
Summary, Subcellular Location of Formation
3. Outwards Trafficking
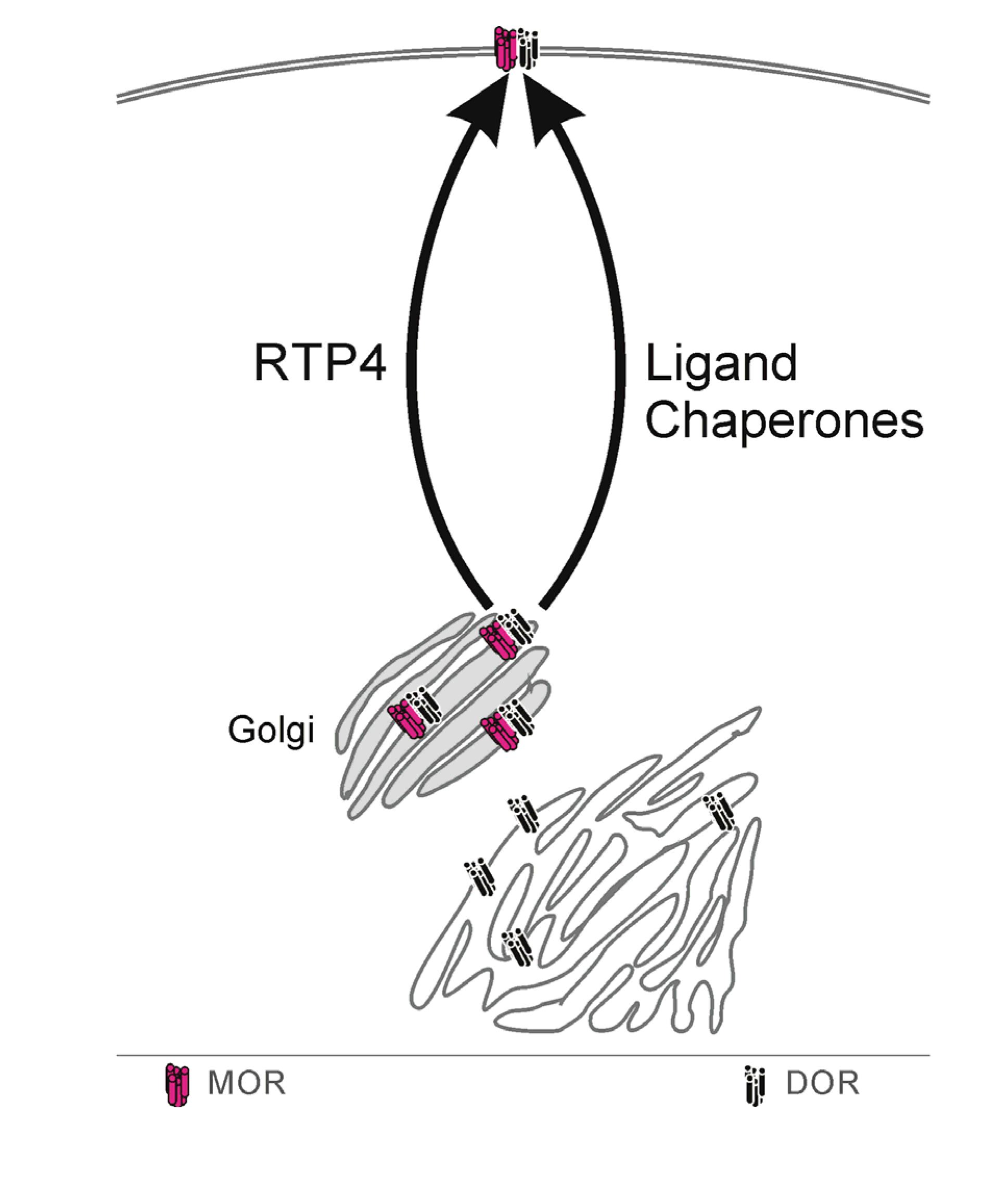
3.1. Protein Chaperone
Summary, Protein Chaperone

3.2. Perspective from DOR Results
Summary, Perspective from DOR Results
4. Function
4.1. Surface Expression
Summary, Surface Expression
4.2. Ligand Binding
Summary, Ligand Binding
4.2.1. M/DOR-Active Ligands

{kind=link}
{kind=link}
{kind=link}
| Ligand | Monomer action | Reported heteromer action | Methods | Source |
|---|---|---|---|---|
| ADL5859 | DOR agonist | M/DOR agonism (reduced potency) | G-protein activation screening assay | [51] |
| ARM1000390 | DOR agonist | M/DOR agonism (reduced potency) | G-protein activation screening assay | [51] |
| Deltorphin II | DOR agonist | M/DOR agonism | Competitive binding | [15] |
| Deltorphin II | DOR agonist | M/DOR agonism | Calcium mobilization | [52] |
| Deltorphin II | DOR agonist | M/DOR agonism | G-protein activation screening assay | [51] |
| DPDPE | DOR agonist | none | Competitive binding | [15] |
| DPDPE | DOR agonist | M/DOR agonism | Calcium mobilization | [53] |
| DPDPE | DOR agonist | M/DOR agonism | Calcium mobilization | [52] |
| SNC80 | DOR agonist | M/DOR agonism | Calcium mobilization | [54] |
| SNC80 | DOR agonist | M/DOR agonism (reduced potency) | G-protein activation screening assay | [51] |
| DAMGO | MOR agonist | none | Competitive binding | [15] |
| DAMGO | MOR agonist | M/DOR agonism | Calcium mobilization | [53] |
| DAMGO | MOR agonist | M/DOR agonism | Calcium mobilization, GTPɣS binding | [52] |
| Methadone | MOR agonist | M/DOR agonism | Biotin protection, calcium mobilization | [55] |
| Morphine | MOR agonist | M/DOR agonism | Calcium mobilization, GTPɣS binding | [52] |
| Endomorphin-2 | MOR agonist | M/DOR agonism | Inhibition of forskolin-evoked cAMP | [42] |
| Bremazocine | KOR agonist | M/DOR partial agonism | Calcium mobilization | [52] |
| U69593 | KOR agonist | none | Calcium mobilization | [52] |
| Naltriben | DOR antagonist | Blocks M/DOR endocytosis but not signaling | Biotin protection, calcium mobilization | [55] |
| DAMGO & Deltorphin II | MOR agonist & DOR agonist | M/DOR agonism | GTPɣS binding | [56] |
| DAMGO & Deltorphin II | MOR agonist & DOR agonist | none | Antinociception | [57] |
| DAMGO & DPDPE | MOR agonist & DOR agonist | none | Antinociception | [57] |
| DAMGO & TIPPψ | MOR agonist & DOR antagonist | M/DOR agonism | GTPɣS binding | [56] |
| DAMGO & TICPψ | MOR agonist & DOR antagonist | M/DOR agonism | Antinociception | [57] |
| DAMGO & TIPP | MOR agonist & DOR antagonist | M/DOR agonism | Antinociception | [57] |
| DAMGO & Naltriben | MOR agonist & DOR antagonist | M/DOR agonism | GTPɣS binding | [56] |
| DAMGO & ICI 174,864 | MOR agonist & DOR inverse agonist | M/DOR agonism | GTPɣS binding | [56] |
| Morphine & Deltorphin II | MOR agonist & DOR agonist | M/DOR agonism | GTPɣS binding | [56] |
| Morphine & TIPPψ | MOR agonist & DOR antagonist | M/DOR agonism | GTPɣS binding, cAMP inhibition, antinociception | [56] |
| Morphine & Naltriben | MOR agonist & DOR antagonist | M/DOR agonism | GTPɣS binding | [56] |
| Morphine & Naltrindole | MOR agonist & DOR antagonist | M/DOR antagonism | Calcium mobilization, antinociception | [58] |
| Morphine & ICI 174,864 | MOR agonist & DOR inverse agonist | M/DOR agonism | GTPɣS binding | [56] |
| Fentanyl & Deltorphin II | MOR agonist & DOR agonist | M/DOR agonism | GTPɣS binding | [56] |
| Fentanyl & TIPPψ | MOR agonist & DOR antagonist | M/DOR agonism | GTPɣS binding | [56] |
| Fentanyl & Naltrindole | MOR agonist & DOR antagonist | M/DOR antagonism | Calcium mobilization, antinociception | [58] |
| Methadone & Deltorphin II | MOR agonist & DOR agonist | M/DOR agonism | GTPɣS binding | [56] |
| Methadone & TIPPψ | MOR agonist & DOR antagonist | M/DOR agonism | GTPɣS binding | [56] |
| Methadone & Naltrindole | MOR agonist & DOR antagonist | M/DOR antagonism | Calcium mobilization, antinociception | [58] |
| MDAN 16 to 21 | Novel bivalent ligand | M/DOR agonism | Antinociception | [59] |
| CYM51010 | Novel ligand | M/DOR agonism | M/DOR activation screening assay | [60] |
4.2.2. M/DOR Specific/Selective Ligands
Summary, M/DOR Specific/Selective Ligands
4.2.3. Ligand Combinations
Summary, Ligand Combinations
4.2.4. Bivalent Ligands
Summary, Bivalent Ligands
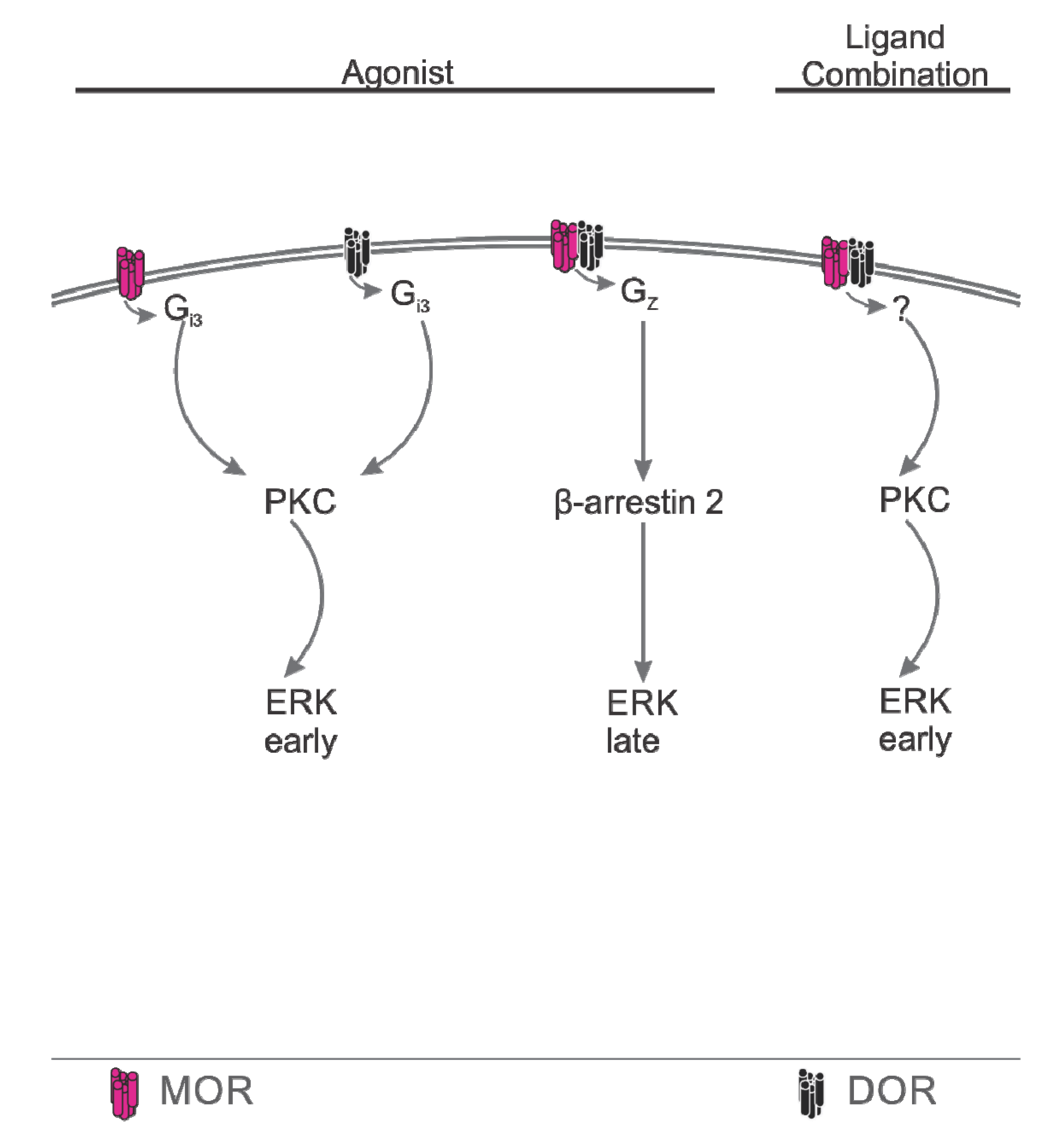
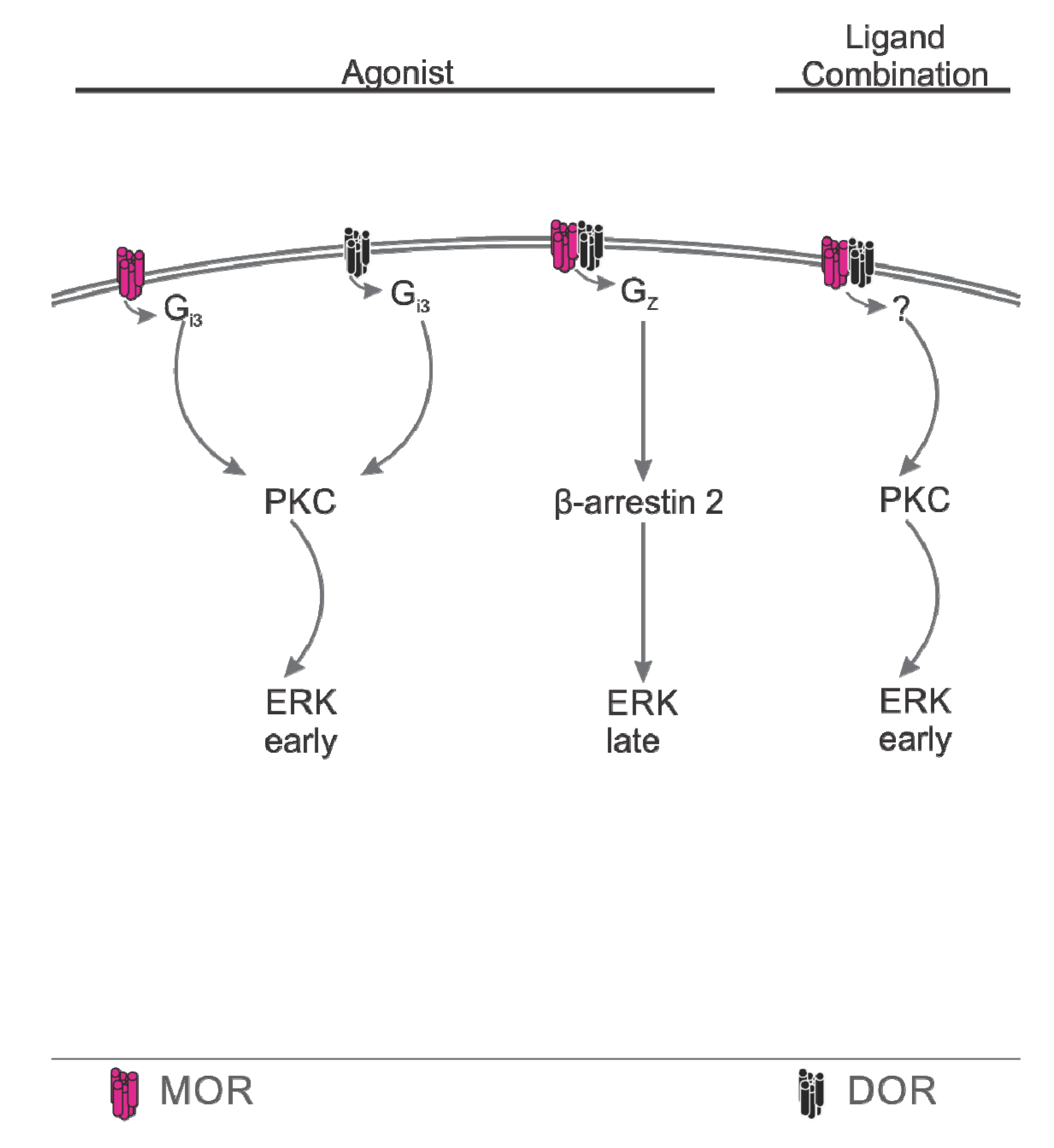
4.3. Downstream Coupling
Summary, Downstream Coupling
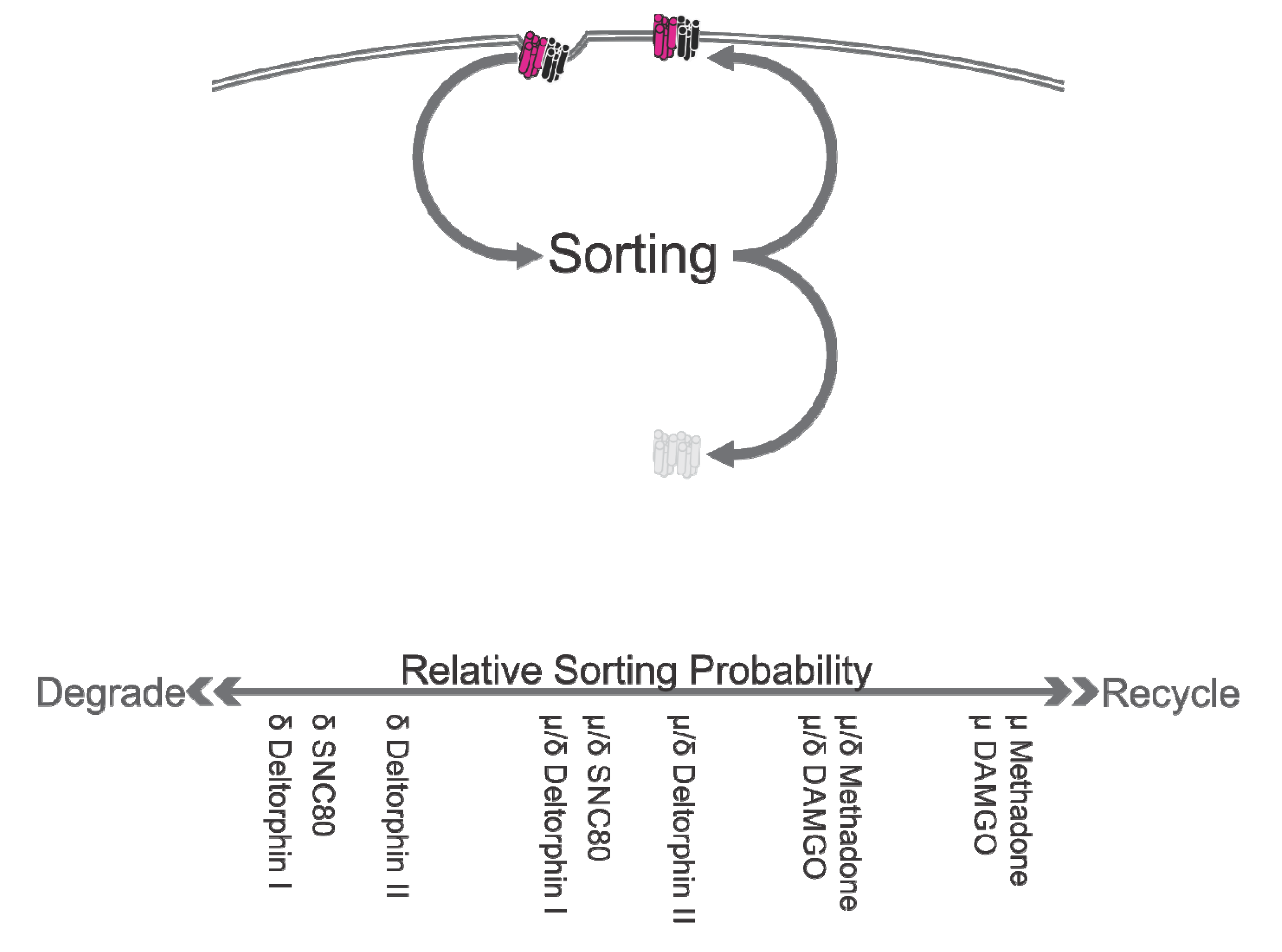
5. Inwards Trafficking
| Ligand | Internalization | Degradation | Recycling | Source |
|---|---|---|---|---|
| Deltorphin I | Increased | Increased | - | [27] |
| Deltorphin II | Increased | - | - | [27] |
| SNC80 | Increased | - | - | [27] |
| DAMGO | Increased | No change | - | [27] |
| DAMGO | Increased | - | - | [55] |
| DAMGO & Naltriben | No change | - | - | [55] |
| Methadone | Increased | Increased | - | [55] |
| Methadone & Naltriben | No change | - | - | [55] |
Summary, Inwards Trafficking
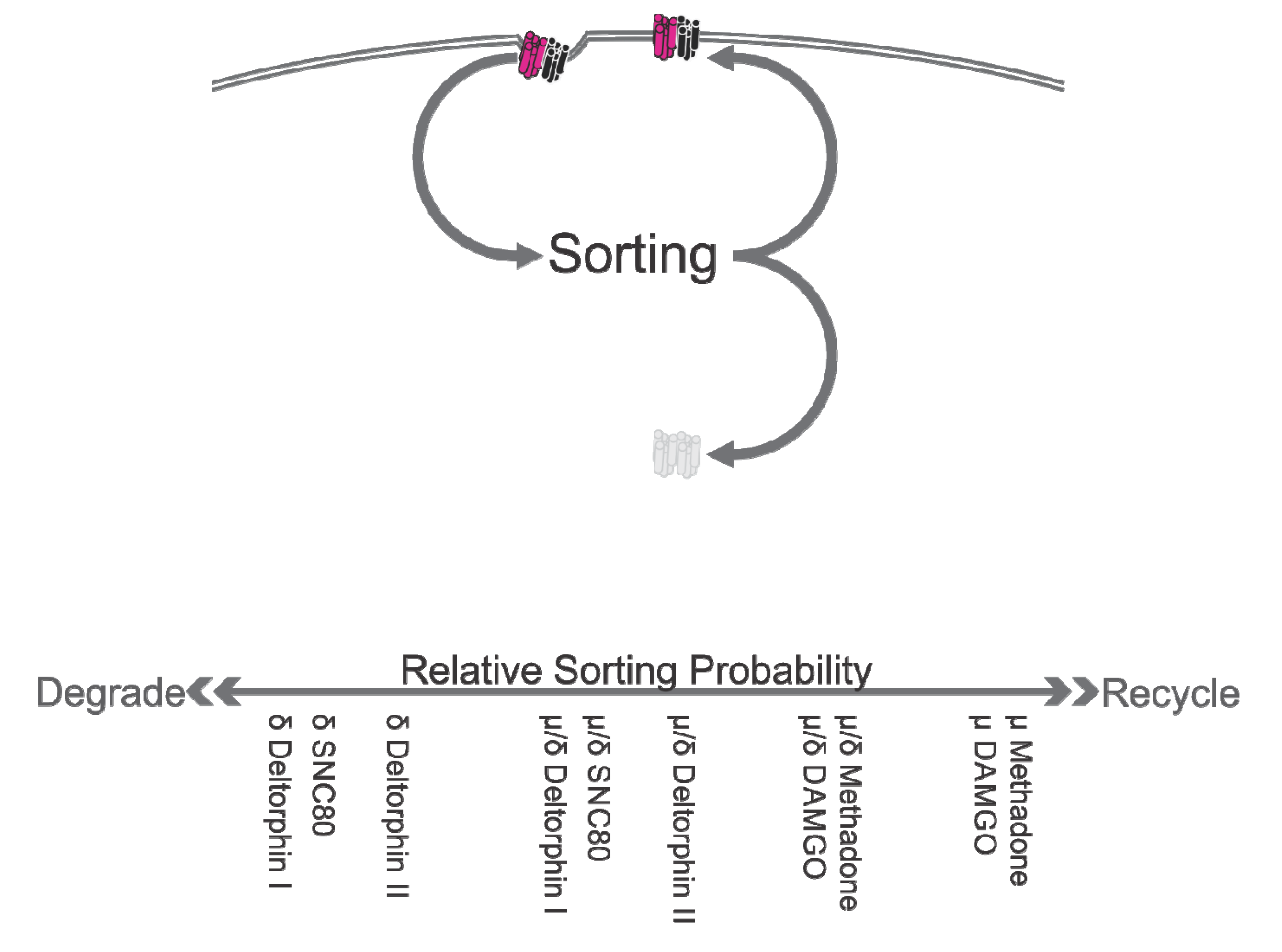
6. Conclusions and Outlooks
Conflicts of Interest
References
- Pasternak, G.W. Multiple opiate receptors: déjà vu all over again. Neuropharmacology 2004, 47 (Suppl. 1), 312–323. [Google Scholar] [CrossRef]
- Gris, P.; Gauthier, J.; Cheng, P.; Gibson, D.G.; Gris, D.; Laur, O.; Pierson, J.; Wentworth, S.; Nackley, A.G.; Maixner, W.; et al. A novel alternatively spliced isoform of the mu-opioid receptor: Functional antagonism. Mol. Pain 2010, 6, 33. [Google Scholar] [CrossRef]
- Costantino, C.M.; Gomes, I.; Stockton, S.D.; Lim, M.P.; Devi, L.A. Opioid receptor heteromers in analgesia. Expert Rev. Mol. Med. 2012, 14, e9. [Google Scholar] [CrossRef]
- Bouvier, M. Oligomerization of G-protein-coupled transmitter receptors. Nat. Rev. Neurosci. 2001, 2, 274–286. [Google Scholar] [CrossRef]
- George, S.R.; O’Dowd, B.F.; Lee, S.P. G-Protein-coupled receptor oligomerization and its potential for drug discovery. Nat. Rev. Drug Discov. 2002, 1, 808–820. [Google Scholar] [CrossRef]
- Lee, S. Homo- and hetero-oligomerization of G protein-coupled receptors. Life Sci. 2003, 74, 173–180. [Google Scholar] [CrossRef]
- Terrillon, S.; Bouvier, M. Roles of G-protein-coupled receptor dimerization. EMBO Rep. 2004, 5, 30–34. [Google Scholar] [CrossRef]
- Gurevich, V.V; Gurevich, E.V. How and why do GPCRs dimerize? Trends Pharmacol. Sci. 2008, 29, 234–240. [Google Scholar] [CrossRef]
- Vidi, P.; Chemel, B.R.; Hu, C.; Watts, V.J. Ligand-dependent oligomerization of dopamine D(2) and adenosine A(2A) receptors in living neuronal cells. Mol. Pharmacol. 2008, 74, 544–551. [Google Scholar] [CrossRef]
- Jordan, B.A.; Devi, L.A. G-protein-coupled receptor heterodimerization modulates receptor function. Nature 1999, 399, 697–700. [Google Scholar] [CrossRef]
- Gomes, I.; Jordan, B.A.; Gupta, A.; Trapaidze, N.; Nagy, V.; Devi, L.A. Heterodimerization of mu and delta opioid receptors: A role in opiate synergy. J. Neurosci. 2000, 20, RC110. [Google Scholar]
- George, S.R.; Fan, T.; Xie, Z.; Tse, R.; Tam, V.; Varghese, G.; O’Dowd, B.F. Oligomerization of mu- and delta-opioid receptors. Generation of novel functional properties. J. Biol. Chem. 2000, 275, 26128–26135. [Google Scholar]
- Gomes, I.; Filipovska, J.; Jordan, B.A.; Devi, L.A. Oligomerization of opioid receptors. Methods 2002, 27, 358–365. [Google Scholar] [CrossRef]
- Wang, D.; Sun, X.; Bohn, L.M.; Sade, W. Opioid receptor homo- and heterodimerization in living cells by quantitative bioluminescence resonance energy transfer. Mol. Pharmacol. 2005, 67, 2173–2184. [Google Scholar] [CrossRef]
- Fan, T.; Varghese, G.; Nguyen, T.; Tse, R.; O’Dowd, B.F.; George, S.R. A role for the distal carboxyl tails in generating the novel pharmacology and G protein activation profile of mu and delta opioid receptor hetero-oligomers. J. Biol. Chem. 2005, 280, 38478–38488. [Google Scholar]
- Hasbi, A.; Nguyen, T.; Fan, T.; Cheng, R.; Rashid, A.; Alijaniaram, M.; Rasenick, M.M.; O’Dowd, B.F.; George, S.R. Trafficking of preassembled opioid mu-delta heterooligomer-Gz signaling complexes to the plasma membrane: Coregulation by agonists. Biochemistry 2007, 46, 12997–13009. [Google Scholar] [CrossRef]
- Décaillot, F.M.; Rozenfeld, R.; Gupta, A.; Devi, L.A. Cell surface targeting of mu-delta opioid receptor heterodimers by RTP4. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 16045–16050. [Google Scholar]
- Stockton, S.D.; Devi, L.A. Functional relevance of μ-δ opioid receptor heteromerization: A role in novel signaling and implications for the treatment of addiction disorders: From a symposium on new concepts in mu-opioid pharmacology. Drug Alcohol Depend. 2012, 121, 167–172. [Google Scholar] [CrossRef]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef]
- Granier, S.; Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Weis, W.I.; Kobilka, B.K. Structure of the δ-opioid receptor bound to naltrindole. Nature 2012, 485, 400–404. [Google Scholar] [CrossRef]
- Wu, H.; Wacker, D.; Mileni, M.; Katritch, V.; Han, G.W.; Vardy, E.; Liu, W.; Thompson, A.A.; Huang, X.-P.; Carroll, F.I.; et al. Structure of the human κ-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. [Google Scholar] [CrossRef]
- Thompson, A.A.; Liu, W.; Chun, E.; Katritch, V.; Wu, H.; Vardy, E.; Huang, X.-P.; Trapella, C.; Guerrini, R.; Calo, G.; et al. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 2012, 485, 395–399. [Google Scholar] [CrossRef]
- Filizola, M.; Devi, L.A. Structural biology: How opioid drugs bind to receptors. Nature 2012, 485, 314–317. [Google Scholar] [CrossRef]
- Filizola, M.; Devi, L.A. Grand opening of structure-guided design for novel opioids. Trends Pharmacol. Sci. 2013, 34, 6–12. [Google Scholar]
- Filizola, M.; Olmea, O.; Weinstein, H. Prediction of heterodimerization interfaces of G-protein coupled receptors with a new subtractive correlated mutation method. Protein Eng. 2002, 15, 881–885. [Google Scholar] [CrossRef]
- Liu, X.; Kai, M.; Jin, L.; Wang, R. Computational study of the heterodimerization between mu and delta receptors. J. Comput. Aided. Mol. Des. 2009, 23, 321–332. [Google Scholar] [CrossRef]
- He, S.-Q.; Zhang, Z.-N.; Guan, J.-S.; Liu, H.-R.; Zhao, B.; Wang, H.-B.; Li, Q.; Yang, H.; Luo, J.; Li, Z.-Y.; et al. Facilitation of μ-opioid receptor activity by preventing δ-opioid receptor-mediated codegradation. Neuron 2011, 69, 120–131. [Google Scholar] [CrossRef]
- O’Dowd, B.F.; Ji, X.; O’Dowd, P.B.; Nguyen, T.; George, S.R. Disruption of the mu-delta opioid receptor heteromer. Biochem. Biophys. Res. Commun. 2012, 422, 556–560. [Google Scholar] [CrossRef]
- Cheng, P.Y.; Liu-Chen, L.Y.; Pickel, V.M. Dual ultrastructural immunocytochemical labeling of mu and delta opioid receptors in the superficial layers of the rat cervical spinal cord. Brain Res. 1997, 778, 367–380. [Google Scholar] [CrossRef]
- Scherrer, G.; Imamachi, N.; Cao, Y.-Q.; Contet, C.; Mennicken, F.; O’Donnell, D.; Kieffer, B.L.; Basbaum, A.I. Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell 2009, 137, 1148–1159. [Google Scholar] [CrossRef]
- Wang, H.-B.; Zhao, B.; Zhong, Y.-Q.; Li, K.-C.; Li, Z.-Y.; Wang, Q.; Lu, Y.-J.; Zhang, Z.-N.; He, S.-Q.; Zheng, H.-C.; et al. Coexpression of delta- and mu-opioid receptors in nociceptive sensory neurons. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 13117–13122. [Google Scholar] [CrossRef]
- Chieng, B.; Christie, M.J. Chronic morphine treatment induces functional delta-opioid receptors in amygdala neurons that project to periaqueductal grey. Neuropharmacology 2009, 57, 430–437. [Google Scholar] [CrossRef]
- Beaudry, H.; Dubois, D.; Gendron, L. Activation of spinal mu- and delta-opioid receptors potently inhibits substance P release induced by peripheral noxious stimuli. J. Neurosci. 2011, 31, 13068–13077. [Google Scholar] [CrossRef]
- Massotte, D. Co-localization of mu and delta opioid receptors in the nervous system using double fluorescent knock-in mice. In Proceedings of the International Narcotics Research Conference, Cairns, Australia, July 2013; p. 8.
- Wang, H.-B.; Guan, J.-S.; Bao, L.; Zhang, X. Distinct subcellular distribution of delta-opioid receptor fused with various tags in PC12 cells. Neurochem. Res. 2008, 33, 2028–2034. [Google Scholar] [CrossRef]
- Scherrer, G.; Tryoen-Tóth, P.; Filliol, D.; Matifas, A.; Laustriat, D.; Cao, Y.Q.; Basbaum, A.I.; Dierich, A.; Vonesh, J.-L.; Gavériaux-Ruff, C.; et al. Knock in mice expressing fluorescent delta-opioid receptors uncover G protein-coupled receptor dynamics in vivo. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 9691–9696. [Google Scholar] [CrossRef]
- Gupta, A.; Mulder, J.; Gomes, I.; Rozenfeld, R.; Bushlin, I.; Ong, E.; Lim, M.; Maillet, E.; Junek, M.; Cahill, C.M.; et al. Increased abundance of opioid receptor heteromers after chronic morphine administration. Sci. Signal. 2010, 3, ra54. [Google Scholar]
- Cahill, C.M.; Morinville, A.; Lee, M.C.; Vincent, J.P.; Collier, B.; Beaudet, A. Prolonged morphine treatment targets delta opioid receptors to neuronal plasma membranes and enhances delta-mediated antinociception. J. Neurosci. 2001, 21, 7598–7607. [Google Scholar]
- Gendron, L.; Lucido, A.L.; Mennicken, F.; O’Donnell, D.; Vincent, J.-P.; Stroh, T.; Beaudet, A. Morphine and pain-related stimuli enhance cell surface availability of somatic delta-opioid receptors in rat dorsal root ganglia. J. Neurosci. 2006, 26, 953–962. [Google Scholar] [CrossRef]
- Morinville, A.; Cahill, C.M.; Esdaile, M.J.; Aibak, H.; Collier, B.; Kieffer, B.L.; Beaudet, A. Regulation of delta-opioid receptor trafficking via mu-opioid receptor stimulation: Evidence from mu-opioid receptor knock-out mice. J. Neurosci. 2003, 23, 4888–4898. [Google Scholar]
- Morinville, A.; Cahill, C.M.; Kieffer, B.; Collier, B.; Beaudet, A. Mu-opioid receptor knockout prevents changes in delta-opioid receptor trafficking induced by chronic inflammatory pain. Pain 2004, 109, 266–273. [Google Scholar] [CrossRef]
- Law, P.-Y.; Erickson-Herbrandson, L.J.; Zha, Q.Q.; Solberg, J.; Chu, J.; Sarre, A.; Loh, H.H. Heterodimerization of mu- and delta-opioid receptors occurs at the cell surface only and requires receptor-G protein interactions. J. Biol. Chem. 2005, 280, 11152–11564. [Google Scholar]
- Petaja-Repo, U.E.; Hogue, M.; Laperriere, A.; Bhalla, S.; Walker, P.; Bouvier, M. Newly synthesized human delta opioid receptors retained in the endoplasmic reticulum are retrotranslocated to the cytosol, deglycosylated, ubiquitinated, and degraded by the proteasome. J. Biol. Chem. 2001, 276, 4416–4423. [Google Scholar]
- Cahill, C.M.; McClellan, K.A.; Morinville, A.; Hoffert, C.; Hubatsch, D.; O’Donnell, D.; Beaudet, A. Immunohistochemical distribution of delta opioid receptors in the rat central nervous system: Evidence for somatodendritic labeling and antigen-specific cellular compartmentalization. J. Comp. Neurol. 2001, 440, 65–84. [Google Scholar] [CrossRef]
- Saito, H.; Kubota, M.; Roberts, R.W.; Chi, Q.; Matsunami, H. RTP family members induce functional expression of mammalian odorant receptors. Cell 2004, 119, 679–691. [Google Scholar] [CrossRef]
- Petäjä-Repo, U.E.; Hogue, M.; Bhalla, S.; Laperrière, A.; Morello, J.-P.; Bouvier, M. Ligands act as pharmacological chaperones and increase the efficiency of delta opioid receptor maturation. EMBO J. 2002, 21, 1628–1637. [Google Scholar] [CrossRef]
- Petäjä-Repo, U.E.; Hogue, M.; Leskelä, T.T.; Markkanen, P.M. H.; Tuusa, J.T.; Bouvier, M. Distinct subcellular localization for constitutive and agonist-modulated palmitoylation of the human delta opioid receptor. J. Biol. Chem. 2006, 281, 15780–15789. [Google Scholar] [CrossRef]
- Walwyn, W.; John, S.; Maga, M.; Evans, C.J.; Hales, T.G. Delta receptors are required for full inhibitory coupling of mu-receptors to voltage-dependent Ca(2+) channels in dorsal root ganglion neurons. Mol. Pharmacol. 2009, 76, 134–143. [Google Scholar] [CrossRef]
- Portoghese, P.S.; Lunzer, M.M. Identity of the putative delta1-opioid receptor as a delta-kappa heteromer in the mouse spinal cord. Eur. J. Pharmacol. 2003, 467, 233–234. [Google Scholar] [CrossRef]
- Ansonoff, M.A.; Portoghese, P.S.; Pintar, J.E. Consequences of opioid receptor mutation on actions of univalent and bivalent kappa and delta ligands. Psychopharmacology 2010, 210, 161–168. [Google Scholar] [CrossRef]
- Van Rijn, R.M.; Harvey, J.H.; Brissett, D.I.; DeFriel, J.N.; Whistler, J.L. Novel screening assay for the selective detection of G-protein-coupled receptor heteromer signaling. J. Pharmacol. Exp. Ther. 2013, 344, 179–188. [Google Scholar] [CrossRef]
- Yekkirala, A.S.; Kalyuzhny, A.E.; Portoghese, P.S. Standard opioid agonists activate heteromeric opioid receptors: Evidence for morphine and [d-Ala(2)-MePhe(4)-Glyol(5)]enkephalin as selective μ-δ agonists. ACS Chem. Neurosci. 2010, 1, 146–154. [Google Scholar] [CrossRef]
- Waldhoer, M.; Fong, J.; Jones, R.M.; Lunzer, M.M.; Sharma, S.K.; Kostenis, E.; Portoghese, P.S.; Whistler, J.L. A heterodimer-selective agonist shows in vivo relevance of G protein-coupled receptor dimers. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 9050–9055. [Google Scholar] [CrossRef]
- Metcalf, M.D.; Yekkirala, A.S.; Powers, M.D.; Kitto, K.F.; Fairbanks, C.A.; Wilcox, G.L.; Portoghese, P.S. The δ Opioid Receptor Agonist SNC80 Selectively Activates Heteromeric μ-δ Opioid Receptors. ACS Chem. Neurosci. 2012, 3, 505–509. [Google Scholar] [CrossRef]
- Milan-Lobo, L.; Whistler, J.L. Heteromerization of the μ- and δ-opioid receptors produces ligand-biased antagonism and alters μ-receptor trafficking. J. Pharmacol. Exp. Ther. 2011, 337, 868–875. [Google Scholar] [CrossRef]
- Gomes, I.; Gupta, A.; Filipovska, J.; Szeto, H.H.; Pintar, J.E.; Devi, L.A. A role for heterodimerization of mu and delta opiate receptors in enhancing morphine analgesia. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 5135–5139. [Google Scholar] [CrossRef]
- Szentirmay, A.K.; Király, K.P.; Lenkey, N.; Lackó, E.; Al-Khrasani, M.; Friedmann, T.; Timár, J.; Gyarmati, S.; Tóth, G.; Fürst, S.; et al. Spinal interaction between the highly selective μ agonist DAMGO and several δ opioid receptor ligands in naive and morphine-tolerant mice. Brain Res. Bull. 2013, 90, 66–71. [Google Scholar] [CrossRef]
- Yekkirala, A.S.; Banks, M.L.; Lunzer, M.M.; Negus, S.S.; Rice, K.C.; Portoghese, P.S. Clinically employed opioid analgesics produce antinociception via μ-δ opioid receptor heteromers in Rhesus monkeys. ACS Chem. Neurosci. 2012, 3, 720–727. [Google Scholar] [CrossRef]
- Daniels, D.J.; Lenard, N.R.; Etienne, C.L.; Law, P.-Y.; Roerig, S.C.; Portoghese, P.S. Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 19208–19213. [Google Scholar]
- Gomes, I.; Fujita, W.; Gupta, A.; Saldanha, A.S.; Negri, A.; Pinello, C.E.; Roberts, E.; Filizola, M.; Hodder, P.; Devi, L.A. Identification of a μ-δ opioid receptor heteromer-biased agonist with antinociceptive activity. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 12072–12077. [Google Scholar] [CrossRef]
- Rozenfeld, R.; Devi, L.A. Receptor heterodimerization leads to a switch in signaling: Beta-arrestin2-mediated ERK activation by mu-delta opioid receptor heterodimers. FASEB J. 2007, 21, 2455–2465. [Google Scholar] [CrossRef]
- Bhushan, R.G.; Sharma, S.K.; Xie, Z.; Daniels, D.J.; Portoghese, P.S. A bivalent ligand (KDN-21) reveals spinal delta and kappa opioid receptors are organized as heterodimers that give rise to delta(1) and kappa(2) phenotypes. Selective targeting of delta-kappa heterodimers. J. Med. Chem. 2004, 47, 2969–2972. [Google Scholar] [CrossRef]
- Daniels, D.J.; Kulkarni, A.; Xie, Z.; Bhushan, R.G.; Portoghese, P.S. A bivalent ligand (KDAN-18) containing delta-antagonist and kappa-agonist pharmacophores bridges delta2 and kappa1 opioid receptor phenotypes. J. Med. Chem. 2005, 48, 1713–1716. [Google Scholar] [CrossRef]
- Lenard, N.R.; Daniels, D.J.; Portoghese, P.S.; Roerig, S.C. Absence of conditioned place preference or reinstatement with bivalent ligands containing mu-opioid receptor agonist and delta-opioid receptor antagonist pharmacophores. Eur. J. Pharmacol. 2007, 566, 75–82. [Google Scholar] [CrossRef]
- Groer, C.E.; Tidgewell, K.; Moyer, R.A.; Harding, W.W.; Rothman, R.B.; Prisinzano, T.E.; Bohn, L.M. An opioid agonist that does not induce micro-opioid receptor--arrestin interactions or receptor internalization. Mol. Pharmacol. 2007, 71, 549–557. [Google Scholar]
- He, L.; Kim, J.A.; Whistler, J.L. Biomarkers of morphine tolerance and dependence are prevented by morphine-induced endocytosis of a mutant mu-opioid receptor. FASEB J. 2009, 23, 4327–4234. [Google Scholar] [CrossRef]
- Kim, J.A.; Bartlett, S.; He, L.; Nielsen, C.K.; Chang, A.M.; Kharazia, V.; Waldhoer, M.; Ou, C.J.; Taylor, S.; Ferwerda, M.; et al. Morphine-induced receptor endocytosis in a novel knockin mouse reduces tolerance and dependence. Curr. Biol. 2008, 18, 129–135. [Google Scholar] [CrossRef]
- Pradhan, A.A.A.; Walwyn, W.; Nozaki, C.; Filliol, D.; Erbs, E.; Matifas, A.; Evans, C.; Kieffer, B.L. Ligand-directed trafficking of the δ-opioid receptor in vivo: Two paths toward analgesic tolerance. J. Neurosci. 2010, 30, 16459–16468. [Google Scholar]
- Cahill, C.M.; Holdridge, S.V; Morinville, A. Trafficking of delta-opioid receptors and other G-protein-coupled receptors: Implications for pain and analgesia. Trends Pharmacol. Sci. 2007, 28, 23–31. [Google Scholar] [CrossRef]
- Law, P.Y.; Loh, H.H. Regulation of opioid receptor activities. J. Pharmacol. Exp. Ther. 1999, 289, 607–624. [Google Scholar]
- Martini, L.; Whistler, J.L. The role of mu opioid receptor desensitization and endocytosis in morphine tolerance and dependence. Curr. Opin. Neurobiol. 2007, 17, 556–564. [Google Scholar] [CrossRef]
- Zuo, Z. The role of opioid receptor internalization and beta-arrestins in the development of opioid tolerance. Anesth. Analg. 2005, 101, 728–734, table of contents. [Google Scholar] [CrossRef]
- Drake, M.T.; Shenoy, S.K.; Lefkowitz, R.J. Trafficking of G protein-coupled receptors. Circ. Res. 2006, 99, 570–582. [Google Scholar] [CrossRef]
- Tanowitz, M.; Von Zastrow, M. Ubiquitination-independent trafficking of G protein-coupled receptors to lysosomes. J. Biol. Chem. 2002, 277, 50219–50222. [Google Scholar] [CrossRef]
- Whistler, J.L.; Enquist, J.; Marley, A.; Fong, J.; Gladher, F.; Tsuruda, P.; Murray, S.R.; Von Zastrow, M. Modulation of postendocytic sorting of G protein-coupled receptors. Science 2002, 297, 615–620. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ong, E.W.; Cahill, C.M. Molecular Perspectives for mu/delta Opioid Receptor Heteromers as Distinct, Functional Receptors. Cells 2014, 3, 152-179. https://doi.org/10.3390/cells3010152
Ong EW, Cahill CM. Molecular Perspectives for mu/delta Opioid Receptor Heteromers as Distinct, Functional Receptors. Cells. 2014; 3(1):152-179. https://doi.org/10.3390/cells3010152
Chicago/Turabian StyleOng, Edmund W., and Catherine M. Cahill. 2014. "Molecular Perspectives for mu/delta Opioid Receptor Heteromers as Distinct, Functional Receptors" Cells 3, no. 1: 152-179. https://doi.org/10.3390/cells3010152
APA StyleOng, E. W., & Cahill, C. M. (2014). Molecular Perspectives for mu/delta Opioid Receptor Heteromers as Distinct, Functional Receptors. Cells, 3(1), 152-179. https://doi.org/10.3390/cells3010152