Targeting Neutrophil Apoptosis for Enhancing the Resolution of Inflammation


Abstract
:1. Introduction
2. Characteristic Features of Neutrophil Apoptosis
3. Therapeutic Induction of Neutrophil Apoptosis for Enhancing Resolution of Inflammation
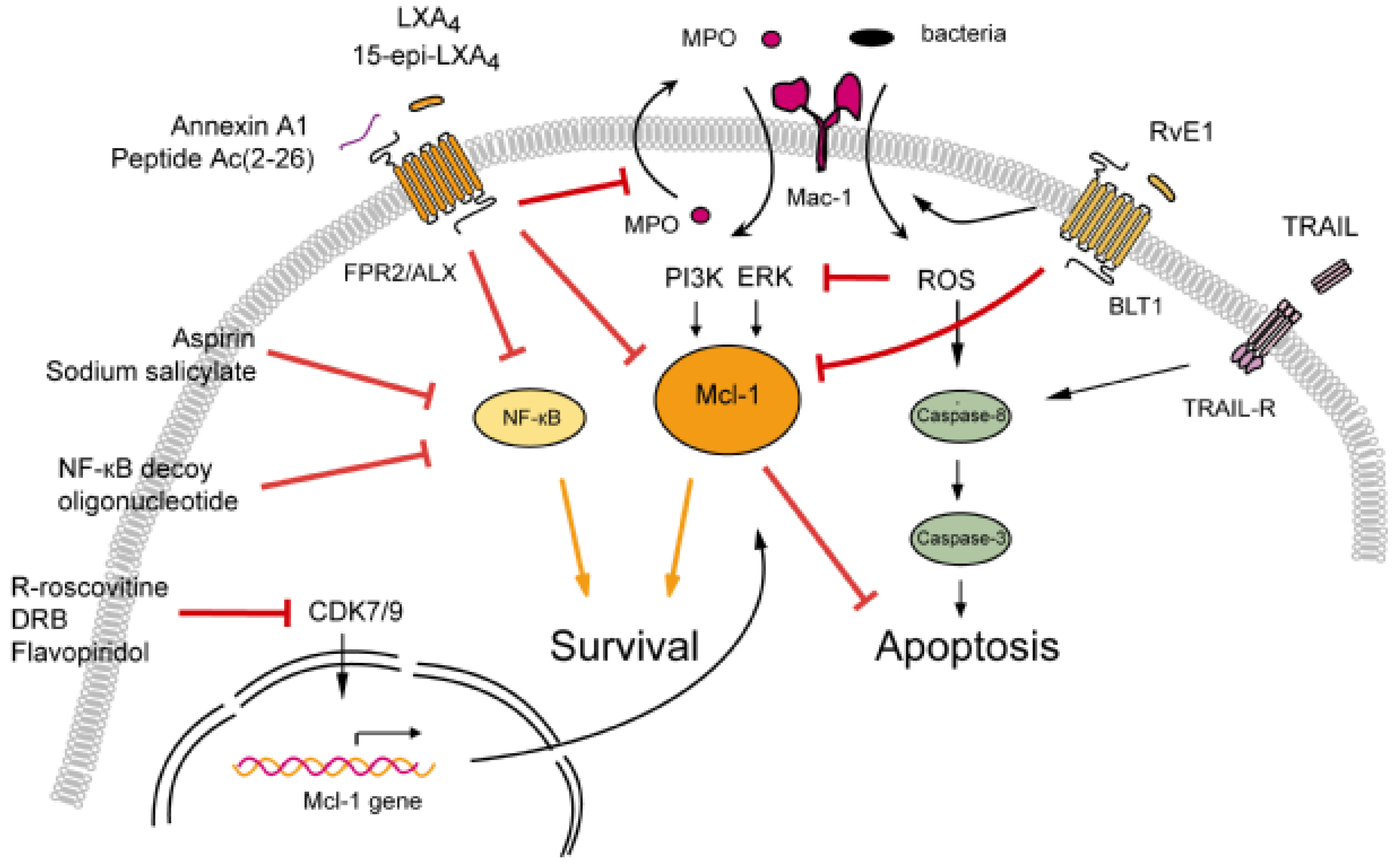
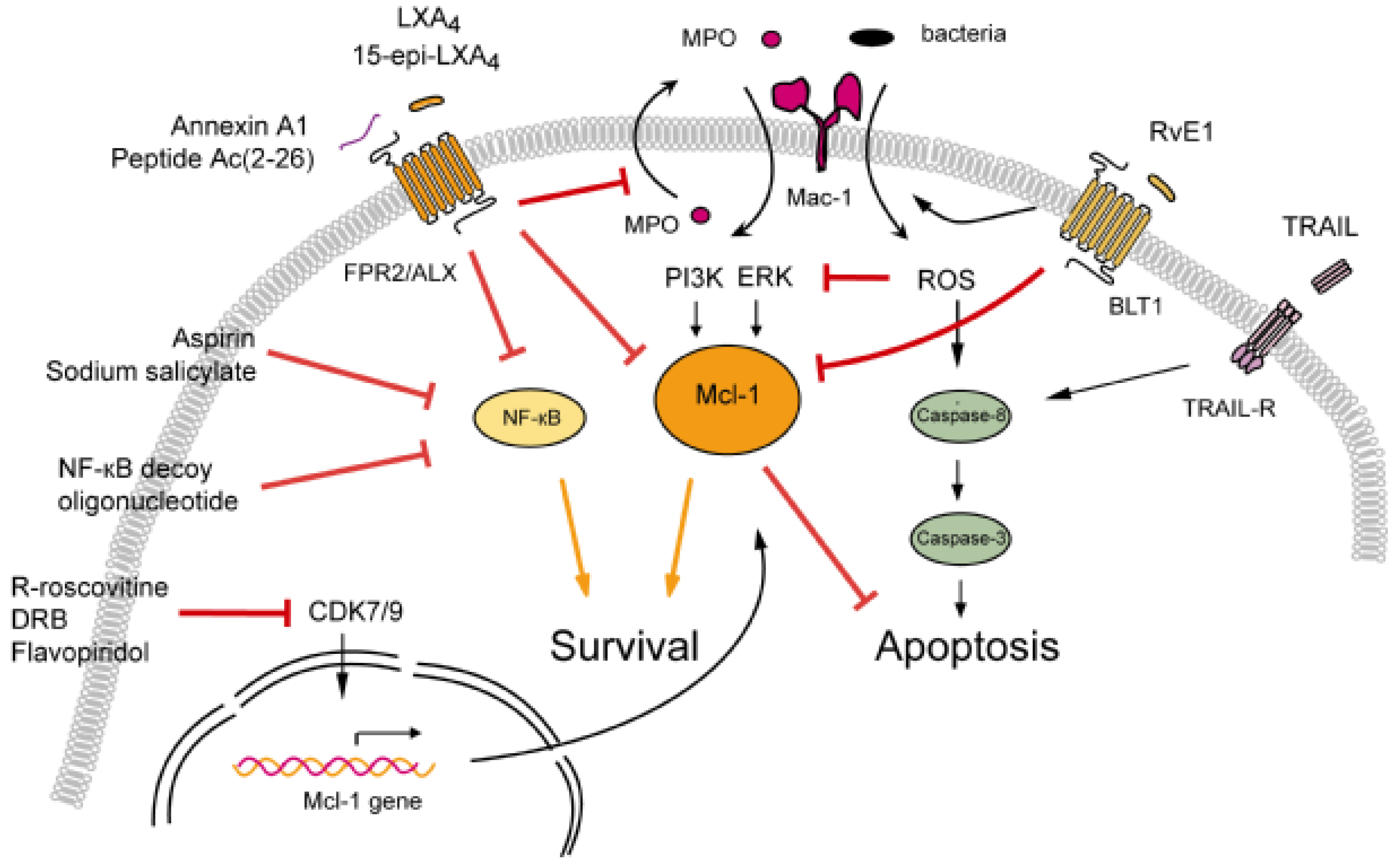
3.1. Modulation of Neutrophil Apoptosis by Outside-In Signaling through Mac-1
3.1.1. Lipoxins Inhibit Myeloperoxidase Signaling through Mac-1
3.1.2. Resolvin E1 Promotes Phagocytosis-Induced Neutrophil Apoptosis
3.2. Annexin A1-Mediated Neutrophil Apoptosis
3.3. The Death Receptor Ligand TRAIL: A Physiological Brake to Restrain Inflammation?
3.4. Cyclin-Dependent Kinase Inhibitors
3.5. NF-κB Inhibitors
4. Conclusions

{kind=link}
| Disease model | Species | Compound | Effects | Pathway | Refs |
|---|---|---|---|---|---|
| Carrageenan-induced pleurisy | Mouse | R-roscovitine | Enhanced PMN apoptosis and efferocytosis Reduced lung PMNs and monocytes | n.d. | [33] |
| Rat | IkBα repressor | Enhanced leukocyte apoptosis Reduced tissue inflammatory cells | Increased caspases-3 activity | [125] | |
| Carrageenan plus MPO-induced ALI | Mouse | 15-epi-LXA4 | Enhanced PMN apoptosis and efferocytosis Decreased PMN accumulation Increased lung monocytes/macrophages | Reduced Mcl-1, ERK and PI3K | [61] |
| Mouse | Resolvin E1 | Enhanced PMN apoptosis and efferocytosis Decreased PMN accumulation Increased lung monocytes/macrophages | Reduced Mcl-1 Enhanced phagocytosis | [88] | |
| E. coli peritonitis-associated ALI | Mouse | 15-epi-LXA4 | Enhanced PMN apoptosis and efferocytosis Decreased PMN accumulation | Reduced Mcl-1, ERK and PI3K | [61] |
| Resolvin E1 | Enhanced PMN apoptosis and efferocytosis Decreased PMN accumulation | Reduced Mcl-1 Enhanced phagocytosis | [88] | ||
| E. coli-induced pneumonia | Mouse | Resolvin E1 | Enhanced PMN apoptosis and efferocytosis Decreased PMN accumulationIncreased lung monocytes/macrophages | Reduced Mcl-1 expression | [88] |
| LPS-induced ALI | Mouse | MetforminRotenone | Decreased PMN accumulation | Decreased NF-κB activation | [126] |
| Mouse | Nutlin-3a | Enhanced PMN apoptosis | Increased p53 expression | [127] | |
| Mouse | rTRAIL | Enhanced PMN apoptosisReduced PMN accumulation No effect on macrophage number | Activation of caspases-8 | [114] | |
| LPS-induced pleurisy | Mouse | Rolipram | Enhanced PMN apoptosis Reduced lung PMNs | Enhanced PDE4 activityReduced PI3K/Akt | [124] |
| Mouse | Annexin A1 andpeptide Ac(2-26) | Enhanced PMN apoptosis Reduced PMN accumulation | Reduced Mcl-1, ERK and NF-κB | [97] | |
| Mouse | Cleavage-resistant annexin A1 | Reduced PMN accumulation | n.d. | [105] | |
| Bleomycin-induced lung injury | Mouse | R-roscovitine | Enhanced PMN apoptosis | Decreased Mcl-1 | [33] |
| Mouse | CDK7/9 inhibitor DRB | Enhanced PMN apoptosis | Decreased Mcl-1 transcription | [35] | |
| Collagen-induced arthritis | Mouse | Flavopiridol | Reduced joint infection Cellular targets were not identified | n.d. | [119] |
| Passive arthritis | Mouse | R-roscovitine | Improved clinical scores | n.d. | [33] |
| Thyoglycollate-induced peritonitis | Mouse | Aspirin Sodium salicylate | Enhanced PMN apoptosis and efferocytosis | Inhibition of NF-κB | [78] |
| Zymosan-induced peritonitis | Mouse | rTRAIL | Enhanced PMN apoptosis Reduced PMN accumulation No effect on macrophage number | Activation of caspases-8 | [114] |
| Pneumococcal meningitis | Mouse | R-roscovitine | Enhanced PMN apoptosis Alleviated brain damage | Reduced Bcl-2 expression | [128] |
| Subcutaneous sponge-implant | Rat | NF-κB decoy oligonucleotide | Enhanced PMN apoptosis and efferocytosis | Increased Bax, reduced Bcl2 | [123] |
Acknowledgments
Conflict of Interest
References
- Nathan, C.; Ding, A. Nonresolving inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef]
- Filep, J.G.; El Kebir, D. Role of neutrophil apoptosis in the resolution of inflammation. The Scientific World J. 2010, 10, 1731–1748. [Google Scholar] [CrossRef]
- Geering, B.; Simon, H.-U. Peculiarities of cell death mechanisms in neutrophils. Cell Death Differ. 2011, 18, 1457–1469. [Google Scholar] [CrossRef]
- Watson, R.W.G.; Rotstein, O.D.; Nathens, A.B.; Parodo, J.; Marshall, J.C. Neutrophil apoptosis is modulated by endothelial transmigration and adhesion molecule engagement. J. Immunol. 1997, 158, 945–953. [Google Scholar]
- Savill, J.; Dransfield, I.; Gregory, C.; Haslett, C. A blast from the past: Clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2002, 2, 965–975. [Google Scholar] [CrossRef]
- Serhan, C.N.; Savill, J. Resolution of inflammation: the beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar]
- Ariel, A.; Fredman, G.; Sun, Y.P.; Kantarci, A.; Van Dyke, T.E.; Luster, A.D.; Serhan, C.N. Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat. Immunol. 2006, 7, 1209–1216. [Google Scholar] [CrossRef]
- Ren, Y.; Xie, Y.; Jiang, G.; Fan, J.; Yeung, J.; Li, W.; Tam, P.K.H.; Savill, J. Apoptotic cells protect mice against lipopolysaccharide-induced shock. J. Immunol. 2008, 180, 4978–4985. [Google Scholar]
- Fadok, V.A.; Bratton, D.L.; Konowal, A.; Freed, P.W.; Westcott, J.Y.; Henson, P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-β, PGE2, and PAF. J. Clin. Invest. 1998, 101, 890–898. [Google Scholar] [CrossRef]
- Stables, M.J.; Shah, S.; Camon, E.B.; Lovering, R.C.; Newson, J.; Bystrom, J.; Farrow, S.; Gilroy, D. Transcriptomic analyses of murine resolution-phase macrophages. Blood 2011, 118, e192–e208. [Google Scholar] [CrossRef]
- Spite, M.; Serhan, C.N. Novel lipid mediators promote resolution of acute inflammation. Impact of aspirin and statins. Circ. Res. 2010, 107, 1170–1184. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Van Dyke, T. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef]
- Serhan, C.N. The resolution of inflammation: the devil in the flask and in the details. FASEB J. 2011, 25, 1441–1448. [Google Scholar] [CrossRef]
- Filep, J.G.; El Kebir, D. Neutrophil apoptosis: A target for enhancing the resolution of inflammation. J. Cell. Biochem. 2009, 108, 1039–1046. [Google Scholar] [CrossRef]
- Fox, S.; Leitch, A.; Duffin, R.; Haslett, C.; Rossi, A.G. Neutrophil apoptosis: Relevance to the innate immune response and inflammatory diseases. J. Innate Immunol. 2012, 2, 216–227. [Google Scholar]
- Godson, C.; Mitchell, S.; Harvey, K.; Petasis, N.A.; Hogg, N.; Brady, H.R. Cutting edge: Lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J. Immunol. 2000, 164, 1663–1667. [Google Scholar]
- Pillay, J.; den Braber, I.; Vrisekoop, N.; Kwast, L.M.; de Boer, R.J.; Borghans, J.A.; Tessalaar, K.; Koenderman, L. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood 2010, 116, 625–627. [Google Scholar] [CrossRef]
- Furze, R.C.; Rankin, S.M. The role of bone marrow in neutrophil clearance under homeostatic conditions in the mouse. FASEB J. 2008, 22, 3111–3119. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Soehnlein, O. Multiple roles for neutrophils in atherosclerosis. Circ. Res. 2012, 110, 875–888. [Google Scholar] [CrossRef]
- Allen, L.; Dockrell, D.H.; Pattery, T.; Lee, D.G.; Cornelis, P.; Hellewell, P.G.; Whyte, M.K.B. Pyocyanin production by Pseudomonas aeruginosa induces neutrophil apoptosis and impairs neutrophil-mediated host defenses in vivo. J. Immunol. 2005, 174, 3643–3649. [Google Scholar]
- Elbim, C.; Katsikis, P.D.; Estaquier, J. Neutrophil apoptosis during viral infections. Open Virology J. 2009, 3, 52–59. [Google Scholar] [CrossRef]
- Matute-Bello, G.; Liles, W.C.; Radella, F., 2nd; Steinberg, K.P.; Ruzinski, J.T.; Jonas, M.; Chi, E.Y.; Hudson, L.D.; Martin, T.R. Neutrophil apoptosis in the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1997, 156, 1969–1977. [Google Scholar] [CrossRef]
- Garlichs, C.D;, Eskafi; Cicha, I.; Schmeisser, A.; Walzog, B.; Raaz, D.; Stumpf, C.; Yilmaz, A.; Bremer, J.; Ludwig, J.; et al. Delay of neutrophil apoptosis in acute coronary syndromes. J. Leukoc. Biol. 2004, 75, 828–835. [Google Scholar]
- Wong, S.H.; Francis, N.; Chahal, H.; Raza, K.; Salmon, M.; Scheel-Toellner, D.; Lord, J.M. Lactoferrin is a survival factor for neutrophils in rheumatoid synovial fluid. Rheumatology 2009, 48, 39–44. [Google Scholar]
- Fialkow, L.; Filho, L.F.; Bozzetti, M.C.; Milani, A.R.; Filho, E.M.R.; Ladniuk, R.M.; Pierozan, P.; de Moura, R.M.; Prolla, J.C.; Vachon, E.; et al. Neutrophil apoptosis: A marker of disease severity in sepsis and sepsis-induced acute respiratory distress syndrome. Crit. Care 2006, 10, R155. [Google Scholar] [CrossRef]
- Edwards, S.W.; Derouet, M.; Howse, M.; Moots, R.J. Regulation of neutrophil apoptosis by Mcl-1. Biochem. Soc. Trans. 2004, 32, 489–492. [Google Scholar] [CrossRef]
- Dzhagalov, I.; St. John, A.; He, Y.W. The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood 2007, 109, 1620–1626. [Google Scholar] [CrossRef]
- Wardle, D.J.; Burgon, J.; Sabroe, I.; Bingle, C.D.; Whyte, M.K.B.; Renshaw, S.A. Effective caspase inhibition blocks neutrophil apoptosis and reveals Mcl-1 as both a regulator and a target of neutrophil caspase activation. PLoS ONE 2011, 6, e15768. [Google Scholar]
- Maianski, N.A.; Geissler, J.; Srinivasula, S.M.; Alnemri, E.S.; Roos, D.; Kuijpers, T.W. Functional characterization of mitochondria in neutrophils: a role restricted to apoptosis. Cell Death Differ. 2004, 11, 143–153. [Google Scholar] [CrossRef]
- Kasahara, Y.; Iwai, K.; Yachie, A.; Ohta, K.; Konno, A.; Seki, H.; Miyawaki, T.; Taniguchi, N. Involvement of reactive oxygen intermediates in spontaneous and CD95 (Fas/APO-1)-mediated apoptosis of neutrophils. Blood 1997, 89, 1748–1753. [Google Scholar]
- Xu, Y.; Loison, F.; Luo, H.R. Neutrophil spontaneous death is mediated by down-regulation of autocrine signaling through GPCR, PI3kγ, ROS, and actin. Proc. Natl. Acad. Sci. USA 2010, 107, 2950–2955. [Google Scholar]
- Rossi, A.G.; Sawatzky, D.A.; Walker, A.; Ward, C.; Sheldrake, T.A.; Riley, N.A.; Caldicott, A.; Martinez-Losa, M.; Walker, T.R.; Duffin, R.; et al. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat. Med. 2006, 12, 1056–1064. [Google Scholar] [CrossRef]
- Wang, K.; Hampson, P.; Hazeldine, J.; Krystof, V.; Strnad, M.; Pechan, P.M.J. Cyclin-dependent kinase 9 activity regulates neutrophil spontaneous apoptosis. PLoS One 2012, 7, e30128. [Google Scholar]
- Leitch, A.E.; Lucas, C.D.; Marwick, J.A.; Duffin, R.; Haslett, C.; Rossi, A.G. Cyclin-dependent kinases 7 and 9 specifically regulate neutrophil transcription and their inhibition drives apoptosis to promote resolution of inflammation. Cell Death Differ. 2012, 19, 1950–1961. [Google Scholar]
- Colotta, F.; Re, F.; Polentarutti, N.; Sozzani, S.; Mantovani, A. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood 1992, 80, 2012–2020. [Google Scholar]
- Lee, A.; Whyte, M.K.; Haslett, C. Inhibition of apoptosis and prolongation of neutrophil functional longevity by inflammatory mediators. J. Leukoc. Biol. 1993, 54, 283–288. [Google Scholar]
- El Kebir, D.; József, L.; Pan, W.; Petasis, N.A.; Serhan, C.N.; Filep, J.G. Aspirin-triggered lipoxins override the apoptosis-delaying action of serum amyloid A in human neutrophils: A novel mechanism for resolution of inflammation. J. Immunol. 2007, 179, 616–622. [Google Scholar]
- Christenson, K.; Björkman, L.; Tängemo, C.; Bylund, J. Serum amyloid A inhibits apoptosis of human neutrophils via a P2X7-sensitive pathway independent of formyl peptide receptor-like 1. J. Leukoc. Biol. 2008, 83, 139–148. [Google Scholar]
- József, L.; Khreiss, T.; Filep, J.G. CpG motifs in bacterial DNA delay apoptosis of neutrophil granulocytes. FASEB J. 2004, 18, 1776–1778. [Google Scholar]
- Klein, J.B.; Rane, M.J.; Scherzer, J.A.; Coxon, P.Y.; Kettritz, R.; Mathiesen, J.M.; Buridi, A.; McLeish, K.R. Granulocyte-macrophage colony-stimulating factor delays neutrophil constitutive apoptosis through phosphoinositide 3-kinase and extracellular signal-regulated kinase pathways. J. Immunol. 2000, 164, 4286–4291. [Google Scholar]
- Epling-Burnette, P.K.; Zhong, B.; Bai, F.; Jiang, K.; Bailey, R.D.; Garcia, R.; Jove, R.; Djeu, J.Y.; Loughran, T.P., Jr.; Wei, S. Cooperative regulation of Mcl-1 by Janus kinase/stat and phosphatidylinositol 3-kinase contribute to granulocyte-macrophage colony-stimulating factor-delayed apoptosis in human neutrophils. J. Immunol. 2001, 166, 7486–7495. [Google Scholar]
- Ward, C.; Walker, A.; Dransfield, I.; Haslett, C.; Rossi, A.G. Regulation of granulocyte apoptosis by NF-κB. Biochem. Soc. Trans. 2004, 32, 465–467. [Google Scholar] [CrossRef]
- Zhang, B.; Hirahashi, J.; Cullere, X.; Mayadas, T.N. Elucidation of molecular events leading to neutrophils apoptosis following phagocytosis. J. Biol. Chem. 2003, 278, 28443–28454. [Google Scholar]
- Alvarado-Kristensson, M.; Melander, F.; Leandersson, K.; Rónnstrand, L.; Wernstedt, C.; Anderson, T. p38-MAPK signals survival by phosphorylation of caspase-8 and caspase-3 in human neutrophils. J. Exp. Med. 2003, 199, 449–458. [Google Scholar]
- Derouet, M.; Thomas, L.; Moulding, D.A.; Akgul, C.; Cross, A.; Moots, R.J.; Edwards, S.W. Sodium salicylate promotes neutrophil apoptosis by stimulating caspase-dependant turn over of Mcl-1. J. Immunol. 2006, 176, 957–965. [Google Scholar]
- Luo, B.H.; Carman, C.V.; Springer, T.A. Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 2007, 25, 619–647. [Google Scholar] [CrossRef]
- Abram, C.L.; Lowell, C.A. The ins and outs of leukocyte integrin signaling. Annu. Rev. Immunol. 2009, 27, 339–362. [Google Scholar] [CrossRef]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef]
- Ross, G.D. Regulation of the adhesion versus cytotoxic functions of Mac-1/CR-3/α-m β-2 integrin glycoprotein. Crit. Rev. Immunol. 2000, 20, 197–222. [Google Scholar]
- Whitlock, B.B.; Gardai, S.; Fadok, V.; Bratton, D.; Henson, P.M. Differential roles for α(M)β(2) integrin clustering or activation in the control of apoptosis via regulation of Akt and ERK survival mechanisms. J. Cell Biol. 2000, 151, 1305–1320. [Google Scholar] [CrossRef]
- Rubel, C.; Gomez, S.; Fernandez, G.C.; Isturiz, M.A.; Caamano, J.; Palermo, M.S. Fibrinogen-CD11b/CD18 interaction activates the NF-κB pathway and delays apoptosis in human neutrophils. Eur. J. Immunol. 2003, 33, 1429–1438. [Google Scholar] [CrossRef]
- Johansson, M.W.; Patarroyo, M.; Oberg, F.; Siegbahn, A.; Nilson, K. Myeloperoxidase mediates cell adhesion via the αMβ2 integrin (Mac-1, CD11b/CD18). J. Cell Sci. 1997, 110, 1133–1139. [Google Scholar]
- Lau, D.; Mollnau, H.; Eiserich, J.P.; Freeman, B.A.; Daiber, A.; Gehling, U.M.; Brummer, J.; Rudolph, V.; Munzel, T.; Heitzer, T.; et al. Myeloperoxidase mediates neutrophil activation by association with CD11b/CD18 integrins. Proc. Natl. Acad. Sci. USA 2005, 102, 431–436. [Google Scholar] [CrossRef]
- Klebanoff, S.J. Myeloperoxidase: friend and foe. J. Leukoc. Biol. 2005, 77, 598–625. [Google Scholar] [CrossRef]
- Nauseef, W.M. How human neutrophils kill and degrade microbes: An integrated view. Immunol. Rev. 2007, 219, 88–102. [Google Scholar] [CrossRef]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulus, V.; Zychlinsky, A. Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef]
- Parker, H.; Albrett, A.M.; Kettle, A.J.; Winterbourn, C.C. Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J. Leukoc. Biol. 2012, 91, 369–376. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef]
- El Kebir, D.; József, L.; Pan, W.; Filep, J.G. Myeloperoxidase delays neutrophil apoptosis through CD11b/CD18 integrins and prolongs inflammation. Circ. Res. 2008, 103, 352–359. [Google Scholar] [CrossRef]
- El Kebir, D.; József, L.; Pan, W.; Wang, L.; Petasis, N.A.; Serhan, C.N.; Filep, J.G. 15-epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am. J. Respir. Crit. Care Med. 2009, 180, 311–319. [Google Scholar] [CrossRef]
- Brovkovych, V.; Gao, X.P.; Ong, E.; Brovkovych, S.; Brennan, M.L.; Su, X.; Hazen, S.L.; Malik, A.B.; Skidgel, R.A. Augmented iNOS expression and increased NO production reduce sepsis-induced lung injury and mortality in myeloperoxidase-null mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L96–L103. [Google Scholar] [CrossRef]
- Matthijsen, R.A.; Huugen, D.; Hoebers, N.T.; de Vries, B.; Peutz-Kootstra, C.J.; Aratani, Y.; Daha, M.R.; Tervaert, J.W.C.; Buuman, W.A.; Heeringa, P. Myeloperoxidase is critically involved in the induction of organ damage after renal ischemia reperfusion. Am. J. Pathol. 2007, 171, 1743–1752. [Google Scholar] [CrossRef]
- Coxon, A.; Rieu, P.; Barkalow, F.J.; Askari, S.; Sharpe, A.H.; von Andrian, U.H.; Arnaout, M.A.; Mayadas, T.N. A novel role for the beta 2 integrin, CD11b/CD18, in neutrophil apoptosis: A homeostatic mechanism in inflammation. Immunity 1996, 5, 653–666. [Google Scholar] [CrossRef]
- DeLeo, F.R. Modulation of phagocyte apoptosis by bacterial pathogens. Apoptosis 2004, 9, 399–413. [Google Scholar] [CrossRef]
- Watson, R.W.G.; Redmond, H.P.; Wang, J.H.; Condron, C.; Bouchier-Hayes, D. Neutrophils undergo apoptosis following ingestion of Escherichia coli. J. Immunol. 1996, 156, 3986–3992. [Google Scholar]
- Perskvist, N.; Long, M.; Stendahl, O.; Zheng, L. Mycobacterium tuberculosis promotes apoptosis in human neutrophils by activating caspase-3 and altering expression of Bax/Bcl-xL via an oxygen-dependent pathway. J. Immunol. 2002, 168, 6358–6365. [Google Scholar]
- Karlsson, A.; Dahlgren, C. Assembly and activation of the neutrophil NADPH oxidase in granule membranes. Antioxid. Redox Signal. 2002, 4, 49–60. [Google Scholar] [CrossRef]
- Arroyo, A.; Modriansky, M.; Serinkan, F.B.; Bello, R.I.; Matsura, T.; Jiang, J.; Tyurin, V.A.; Tyurina, Y.Y.; Fadeel, B.; Kagan, V.E. NADPH oxidase-dependent oxidation and externalization of phosphatidylserine during apoptosis in Me2SO-differentiated HL-60 cells. Role in phagocytic clearance. J. Biol. Chem. 2002, 277, 49965–49975. [Google Scholar]
- Clària, J.; Serhan, C.N. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc. Natl. Acad. Sci. USA 1995, 92, 9475–9479. [Google Scholar] [CrossRef]
- Birnbaum, Y.; Ye, Y.; Lin, Y.; Freeberg, S.Y.; Nishi, S.P.; Martinez, J.D.; Huang, M.H.; Uretzky, B.F.; Perez-Polo, J.R. Augmentation of myocardial production of 15-epi-lipoxin A4 by pioglitazone and atorvastatin in the rat. Circulation 2006, 114, 929–935. [Google Scholar] [CrossRef]
- Machado, F.S.; Johndrow, J.E.; Esper, L.; Dias, A.; Bafica, A.; Serhan, C.N.; Aliberti, J. Anti-inflammatory actions of lipoxin A4 and aspirin-triggered lipoxin are socs-2-dependent. Nat. Med. 2006, 12, 330–334. [Google Scholar] [CrossRef]
- József, L.; Zouki, C.; Petasis, N.A.; Serhan, C.N.; Filep, J.G. Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 inhibit peroxynitrite formation, NF-κB and AP-1 activation, and IL-8 gene expression in human leukocytes. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 13266–13271. [Google Scholar]
- Mitchell, S.; Thomas, G.; Harvey, K.; Cottel, D.; Reville, K.; Berlasconi, G.; Petasis, N.A.; Erwig, L.; Rees, A.J.; Savill, J.; et al. Lipoxins, aspirin-triggered epi-lipoxins, lipoxin stable analogues, and the resolution of inflammation: Stimulation of macrophage phagocytosis of apoptotic neutrophils in vivo. J. Am. Soc. Nephrol. 2002, 13, 2497–2507. [Google Scholar] [CrossRef]
- Prieto, P.; Cuenca, J.; Través, P.G.; Fernández-Velasco, M.; Martin-Saenz, P.; Boscá, L. Lipoxin A4 impairment of apoptotic signaling in macrophages: implication of the PI3K/Akt and the ERK/Nrf-2 defense pathways. Cell Death Differ. 2010, 17, 1179–1188. [Google Scholar] [CrossRef]
- Fukunaga, K.; Kohli, P.; Bonnans, C.; Fredenburgh, L.E.; Levy, B.D. Cyclooxygenase 2 plays a pivotal role in the resolution of acute lung injury. J. Immunol. 2005, 174, 5033–5039. [Google Scholar]
- Planaguma, A.; Pfeffer, M.A.; Rubin, G.; Croze, R.; Uddin, M.; Serhan, C.N. Lovastatin decreases acute mucosal inflammation via 15-epi-lipoxin A4. Mucosal Immunol. 2010, 3, 270–279. [Google Scholar]
- Negrotto, S.; Malaver, E.; Alvarez, M.E.; Pacienza, N.; D’Atri, L.P.; Pozner, R.G.; Gomez, R.M.; Schattner, M. Aspirin and salicylate suppress polymorphonuclear apoptosis delay mediated by proinflammatory stimuli. J. Pharmacol. Exp. Ther. 2006, 319, 972–979. [Google Scholar] [CrossRef]
- Serhan, C.N.; Clish, C.B.; Brannon, J.; Colgan, S.P.; Chiang, N.; Gronert, C. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from ω-3 fatty acids via cyclooxygenase2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 2000, 192, 1197–1204. [Google Scholar] [CrossRef]
- Oh, S.F.; Pillai, P.S.; Recchiuti, A.; Yang, R.; Serhan, C.N. Pro-resolving actions and stereoselective biosynthesis of 18S E-series resolvins in human leukocytes and murine inflammation. J. Clin. Invest. 2011, 121, 569–581. [Google Scholar] [CrossRef]
- Arita, M.; Bianchini, F.; Aliberti, J.; Sher, A.; Chiang, N.; Hong, S.; Yang, R.; Petasis, N.A.; Serhan, C.N. Stereochemical assignment, antiinflammatory properties, and receptor for the ω3 lipid mediator resolvin E1. J. Exp. Med. 2005, 201, 713–722. [Google Scholar] [CrossRef]
- Arita, M.; Ohira, T.; Sun, Y.P.; Elangovan, S.; Chiang, N.; Serhan, C.N. Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J. Immunol. 2007, 178, 3912–3917. [Google Scholar]
- Ohira, T.; Arita, M.; Omori, K.; Recchiuti, A.; van Dyke, T.E.; Serhan, C.N. Resolvin E1 receptor activation signals phosphorylation and phagocytosis. J. Biol. Chem. 2010, 285, 3451–3461. [Google Scholar]
- Schwab, J.M.; Chiang, N.; Arita, M.; Serhan, C.N. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 2007, 447, 869–874. [Google Scholar] [CrossRef]
- Seki, H.; Fukunaga, K.; Arita, M.; Arai, M.; Nakanishi, H.; Taguchi, R.; Miyasho, T.; Takamiya, R.; Asano, K.; Ishizaka, A.; et al. The anti-inflammatory and proresolving mediator resolvin E1 protects mice from bacterial pneumonia and acute lung injury. J. Immunol. 2010, 184, 836–843. [Google Scholar] [CrossRef]
- Haworth, O.; Cernadas, M.; Yang, R.; Serhan, C.N.; Levy, B.D. Resolvin E1 regulates interleukin 23, interferon gamma and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat. Immunol. 2008, 8, 873–879. [Google Scholar]
- Cash, J.L.; Hart, R.; Russ, A.; Dixon, J.P.; Colledge, W.H.; Doran, J.; Hendrick, A.G.; Carlton, M.B.; Greaves, D.R. Synthetic chemerin-derived peptides suppress inflammation through ChemR23. J. Exp. Med. 2008, 205, 767–775. [Google Scholar]
- El Kebir, D.; Gjorstrup, P.; Filep, J.G. Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc. Natl. Acad. Sci. USA 2012, 109, 14983–14988. [Google Scholar] [CrossRef]
- Perretti, M.; D’Acquisto, F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat. Rev. Immunol. 2009, 9, 62–70. [Google Scholar] [CrossRef]
- Babbin, B.A.; Laukoetter, M.G.; Nava, P.; Koch, S.; Lee, W.Y.; Gapaldo, C.T.; Peatman, E.; Severson, E.A.; Flower, R.J.; Perretti, M.; et al. Annexin A1 regulates intestinal mucosal injury, inflammation, and repair. J. Immunol. 2008, 181, 5035–5044. [Google Scholar]
- Hannon, R.; Croxtall, J.D.; Getting, S.J.; Roviezzo, F.; Yona, S.; Paul-Clark, M.J.; Gavins, F.N.; Perretti, M.; Morris, J.F.; Buckingham, J.C.; Flower, R.J. Aberrant inflammation and resistance to glucocorticoids in annexin-1-/- mouse. FASEB J. 2003, 17, 253–255. [Google Scholar]
- Yang, Y.H.; Morand, E.F.; Getting, S.J.; Paul-Clark, M.; Liu, D.L.; Yona, S.; Hannon, R.; Buckingham, J.C.; Perretti, M.; Flower, R.J. Modulation of inflammation in response to dexamethasone by annexin 1 in antigen-induced arthritis. Arthritis Rheum. 2004, 50, 976–984. [Google Scholar] [CrossRef]
- Perretti, M.; Croxtall, J.D.; Wheller, S.K.; Gouding, N.J.; Hannon, R.; Flower, R.J. Mobilizing lipocortin 1 in adherent human leukocytes downregulates their transmigration. Nat. Med. 1996, 22, 1259–1262. [Google Scholar]
- Peretti, M.; Chiang, N.; La, M.; Fiero, I.M.; Marullo, S.; Getting, S.J.; Solito, E.; Serhan, C.N. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat. Med. 2002, 8, 1296–1302. [Google Scholar] [CrossRef]
- Vergnolle, N.; Coméra, C.; More, J.; Alvinerie, M.; Buéno, L. Expression and secretion of lipocortin 1 in gut inflammation are not regulated by pituitary-adrenal axis. Am. J. Physiol. 1997, 273, R623–R629. [Google Scholar]
- Solito, E.; Kamal, A.M.; Russo-Marie, F.; Buckingham, J.C.; Marullo, S.; Perretti, M. A novel calcium-dependent proapoptotic effect of annexin 1 on human neutrophils. FASEB J. 2003, 17, 1544–1546. [Google Scholar]
- Vago, J.P.; Nogueira, C.R.C.; Tavares, L.P.; Soriani, F.M.; Lopes, F.; Russo, R.C.; Pinho, V.; Teixeira, M.M.; Sousa, L.P. Annexin A1 modulates natural and glucocorticoid-induced resolution of inflammation by enhancing neutrophil apoptosis. J. Leukoc. Biol. 2012, 92, 249–258. [Google Scholar] [CrossRef]
- Perretti, M. To resolve or not to resolve: Annexin A1 pushes resolution on track. J. Leukoc. Biol. 2012, 92, 245–247. [Google Scholar] [CrossRef]
- Liles, W.C.; Dale, D.C.; Klebanoff, S.J. Glucocorticoids inhibit apoptosis of human neutrophils. Blood 1995, 86, 3181–3188. [Google Scholar]
- De Coupade, C.; Ajuebor, M.N.; Russo-Marie, F.; Perretti, M.; Solito, E. Cytokine modulation of liver annexin 1 expression during experimental endotoxemia. Am. J. Pathol. 2001, 159, 1435–1443. [Google Scholar] [CrossRef]
- Arur, S.; Uche, U.E.; Rezaul, K.; Fong, M.; Scranton, V.; Cowan, A.E.; Mohler, W.; Han, D.K. Annexin I is an endogenous ligand that mediates apoptotic cell engulfment. Dev. Cell 2003, 4, 587–598. [Google Scholar] [CrossRef]
- Maderna, P.; Yona, S.; Perretti, M.; Godson, C. Modulation of phagocytosis of apoptotic neutrophils by supernatant from dexamethasone-treated macrophages and annexin-derived peptide Ac(2–26). J. Immunol. 2005, 174, 3727–3733. [Google Scholar]
- Scannell, M.; Flanagan, M.B.; de Stefani, A.; Wynne, K.J.; Cagney, G.; Godson, C.; Maderna, P. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J. Immunol. 2007, 178, 4595–4605. [Google Scholar]
- Dalli, J.; Jones, C.P.; Cavalcanti, D.M.; Farsky, S.H.; Perretti, M.; Rankin, S. Annexin A1 regulates neutrophil clearance by macrophages in the mouse bone marrow. FASEB J. 2011, 26, 387–396. [Google Scholar]
- Pederzoli-Ribeil, M.; Maione, F.; Cooper, D.; Al-Kashi, A.; Dalli, J.; Peretti, M.; D’Acquisto, F. Design and characterization of a cleavage—resistant Annexin A1 mutant to control inflammation in the microvasculature. Blood 2010, 116, 4288–4296. [Google Scholar] [CrossRef]
- El Kebir, D.; József, L.; Filep, J.G. Opposing regulation of neutrophil apoptosis through the formyl-peptide receptor-like 1/lipoxin A4 receptor: implications for the resolution of inflammation. J. Leukoc. Biol. 2008, 84, 600–606. [Google Scholar] [CrossRef]
- Bozinovski, S.; Uddin, M.; Vlahos, R.; Thompson, M.; McQualter, J.L.; Merritt, A.S.; Wark, P.A.B.; Hutchinson, A.; Irving, L.B.; Levy, B.D.; et al. Serum amyloid A opposes lipoxin A4 to mediate glucocorticoid refractory lung inflammation in chronic obstructive pulmonary disease. Proc. Natl. Acad. Sci. USA 2012, 109, 935–940. [Google Scholar] [CrossRef]
- Wilson, N.S.; Dixit, V.; Ashkenazi, A. Death receptor signal transducers: Nodes of coordination of immune signaling networks. Nat. Immunol. 2009, 10, 348–355. [Google Scholar] [CrossRef]
- Pan, G.; O’Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The receptor for the cytotoxic ligand TRAIL. Science 1997, 276, 111–113. [Google Scholar] [CrossRef]
- Chaudhary, P.M.; Eby, M.; Jasmin, A.; Bookwalter, A.; Murray, J.; Hood, L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-κB pathway. Immunity 1997, 7, 821–830. [Google Scholar] [CrossRef]
- Gonzalvez, F.; Ashkenazi, A. New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene 2010, 29, 4752–4765. [Google Scholar] [CrossRef]
- Cassatella, M.A.; Huber, V.; Calzetti, F.; Margotto, D.; Tamassia, N.; Peri, G.; Mantovani, A.; Rivoltini, L.; Tecchio, C. Interferon-activated neutrophils store a TNF-related apoptosis-inducing ligand (TRAIL/Apo-2 ligand) intracellular pool that is readily mobilizable following exposure to proinflammatory mediators. J. Leukoc. Biol. 2006, 79, 123–132. [Google Scholar]
- Tecchio, C.; Huber, V.; Scapini, P.; Calzetti, F.; Margotto, D.; Todeschini, G.; Pilla, L.; Martinelli, G.; Pizzolo, G.; Rivoltini, L.; et al. IFNα-stimulated neutrophils and monocytes release a soluble form of TNF-related apoptosis-inducing ligand (TRAIL/Apo-2 ligand) displaying apoptotic activity on leukemic cells. Blood 2004, 103, 3837–3844. [Google Scholar] [CrossRef]
- McGrath, E.E.; Marriott, H.M.; Lawrie, A.; Francis, S.E.; Sabroe, I.; Renshaw, S.A.; Dockrell, D.H.; Whyte, M.K. TNF-related apoptosis-inducing ligand (TRAIL) regulates inflammatory neutrophil apoptosis and enhances resolution of inflammation. J. Leukoc. Biol. 2011, 90, 855–865. [Google Scholar] [CrossRef]
- Lum, J.J.; Bren, G.; McClure, R.; Badley, A.D. Elimination of senescent neutrophils by TNF-related apoptosis-inducing ligand. J. Immunol. 2005, 175, 1232–1238. [Google Scholar]
- Falschlechner, C.; Schaefer, U.; Walczak, H. Following TRAIL’s path in the immune system. Immunology 2009, 127, 145–154. [Google Scholar] [CrossRef]
- Leu, S.W.; Shi, L.; Xu, C.; Zhao, Y.; Liu, B.; Li, Y.; Schiedlin, A.; Xiang, C.; Shen, H.; Quinn, D.A.; et al. TLR4 through IFN-β promotes low molecular mass hyaluronan-induced neutrophil apoptosis. J. Immunol. 2011, 186, 556–562. [Google Scholar] [CrossRef]
- Leitch, A.E.; Lucas, C.D.; Rossi, A.G. Neutrophil apoptosis: hot on the TRAIL of inflammatory resolution. J. Leukoc. Biol. 2011, 90, 841–843. [Google Scholar] [CrossRef]
- Sekine, C.; Sugihara, T.; Miyake, S.; Hirai, H.; Yoshida, M.; Miyasaka, N.; Kohsaka, H. Successful treatment of animal models of rheumatoid arthritis with small-molecule cyclin-dependent kinase inhibitors. J. Immunol. 2008, 180, 1954–1961. [Google Scholar]
- Moriceau, S.; Lenoir, G.; Witko-Sarsat, V. In cystic fibrosis homozygotes and heterozygotes, neutrophil apoptosis is delayed and modulated by diamide or roscovitine: Evidence for an innate neutrophil disturbance. J. Innate Immunol. 2009, 2, 260–266. [Google Scholar] [CrossRef]
- Gosh, S.; Hayden, M.S. New regulators of NF-κB in inflammation. Nat. Rev. Immunol. 2008, 8, 837–848. [Google Scholar] [CrossRef]
- Lawrence, T.; Gilroy, D.; Colville-Nash, V.P.R.; Willoughby, D.A. Possible new role for NF-κB in the resolution of inflammation. Nat. Med. 2001, 7, 1291–1297. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Tajana, G.; Iuvone, T.; De Stefano, D.; Mele, G.; Ribecco, M.T.; Cinelli, M.P.; Romano, M.F.; Turco, M.C.; Camuccio, R. Nuclear factor-κB regulates inflammatory cell apoptosis and phagocytosis in rat carrageenin-sponge implant model. Am. J. Pathol. 2004, 165, 115–126. [Google Scholar] [CrossRef]
- Sousa, L.P.; Lopes, F.; Silva, D.M.; Tavares, L.P.; Vieira, A.T.; Rezende, B.M.; Carmo, A.F.; Russo, R.C.; Garcia, C.C.; Bonjardim, C.A.; et al. PDE4 inhibition drives resolution of neutrophilic inflammation by inducing apoptosis in a PKA-PI3K/Akt-dependent and NF-κB-independent manner. J. Leukoc. Biol. 2010, 87, 895–904. [Google Scholar] [CrossRef]
- Blackwell, N.M.; Sembi, P.; Newson, J.S.; Lawrence, T.; Gilroy, D.W.; Kabouridis, P.S. Reduced infiltration and increased apoptosis of leukocytes at sites of inflammation by systemic administration of a membrane-permeable IκBα repressor. Arthr. Rheum. 2004, 50, 2675–2684. [Google Scholar] [CrossRef]
- Zmijewski, J.W.; Lorne, E.; Zhao, X.; Tsuruta, Y.; Sha, Y.; Liu, G.; Siegal, G.P.; Abraham, E. Mitochondrial respiratory complex I regulates neutrophil activation and severity of lung injury. Am. J. Respir. Crit. Care Med. 2008, 178, 168–179. [Google Scholar] [CrossRef]
- Liu, G.; Park, Y.J.; Tsuruta, Y.; Lorne, E.; Abraham, E. p53 attenuates lipopolysaccharide-induced NF-κB activation and acute lung injury. J. Immunol. 2009, 182, 5063–5071. [Google Scholar] [CrossRef]
- Koedel, U.; Frankenberg, T.; Kirschnek, S.; Obermaier, B.; Häcker, H.; Paul, R.; Häcker, G. Apoptosis is essential for neutrophil functional shutdown and determines tissue damage in experimental pneumococcal meningitis. PLoS Pathog. 2009, 5, e1000461. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
El Kebir, D.; Filep, J.G. Targeting Neutrophil Apoptosis for Enhancing the Resolution of Inflammation. Cells 2013, 2, 330-348. https://doi.org/10.3390/cells2020330
El Kebir D, Filep JG. Targeting Neutrophil Apoptosis for Enhancing the Resolution of Inflammation. Cells. 2013; 2(2):330-348. https://doi.org/10.3390/cells2020330
Chicago/Turabian StyleEl Kebir, Driss, and János G. Filep. 2013. "Targeting Neutrophil Apoptosis for Enhancing the Resolution of Inflammation" Cells 2, no. 2: 330-348. https://doi.org/10.3390/cells2020330
APA StyleEl Kebir, D., & Filep, J. G. (2013). Targeting Neutrophil Apoptosis for Enhancing the Resolution of Inflammation. Cells, 2(2), 330-348. https://doi.org/10.3390/cells2020330