Linking Metabolic Abnormalities to Apoptotic Pathways in Beta Cells in Type 2 Diabetes

{kind=link}
{kind=link}
Abstract
:1. Introduction
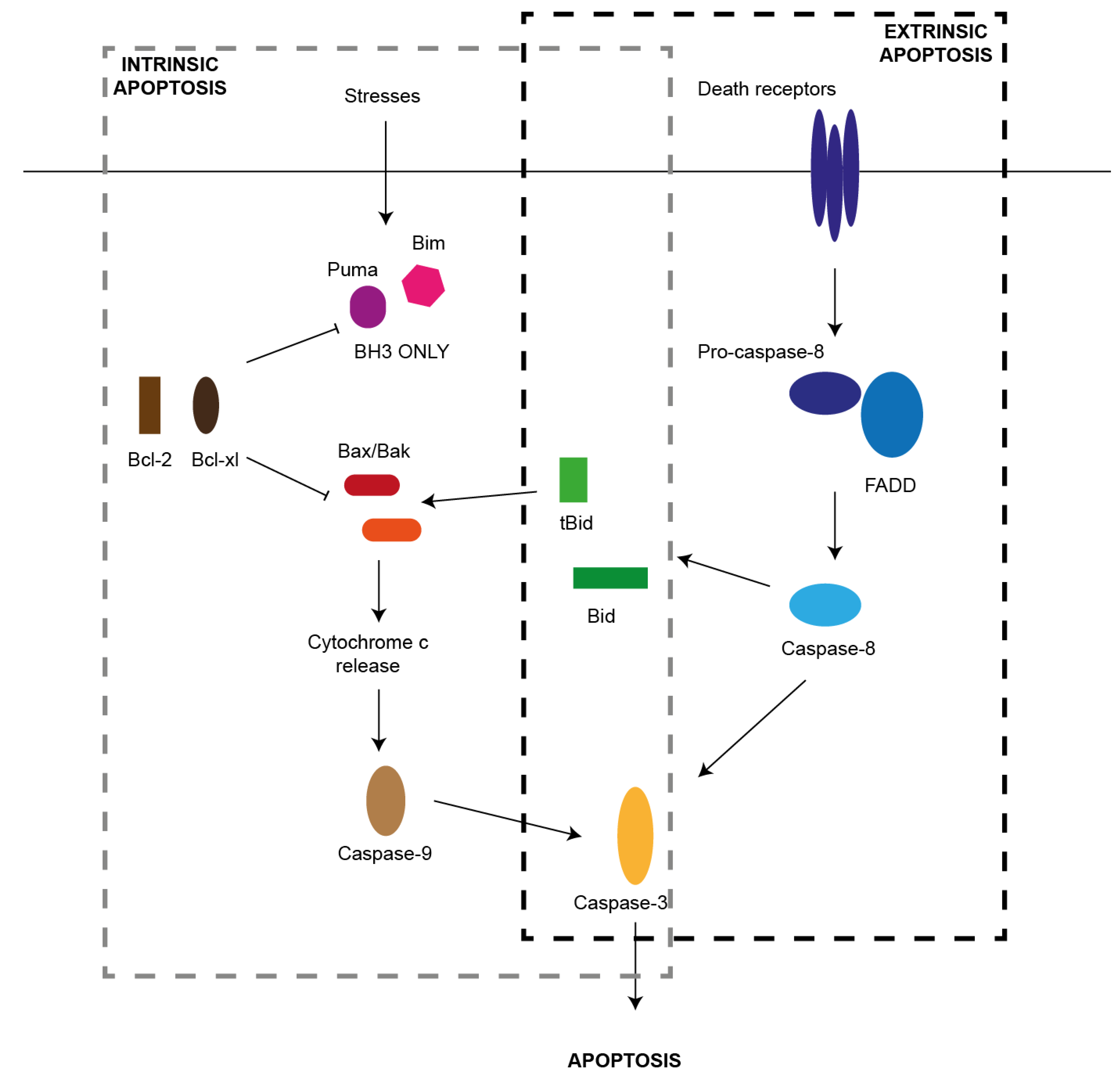
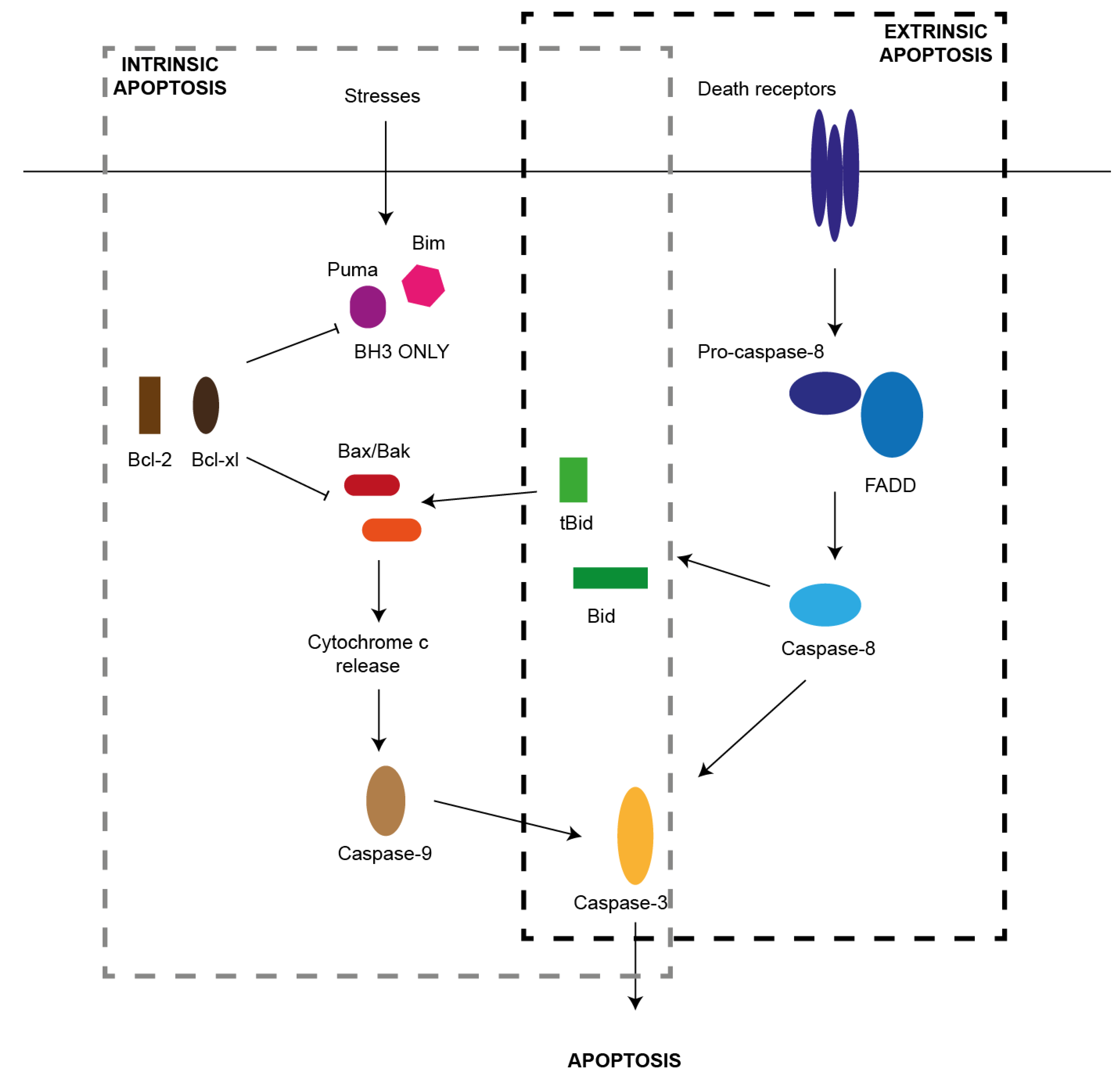
2. Pathways of Apoptosis
2.1. Extrinsic Pathway
2.2. Intrinsic Pathway
2.3. NLRP3 Inflammasome
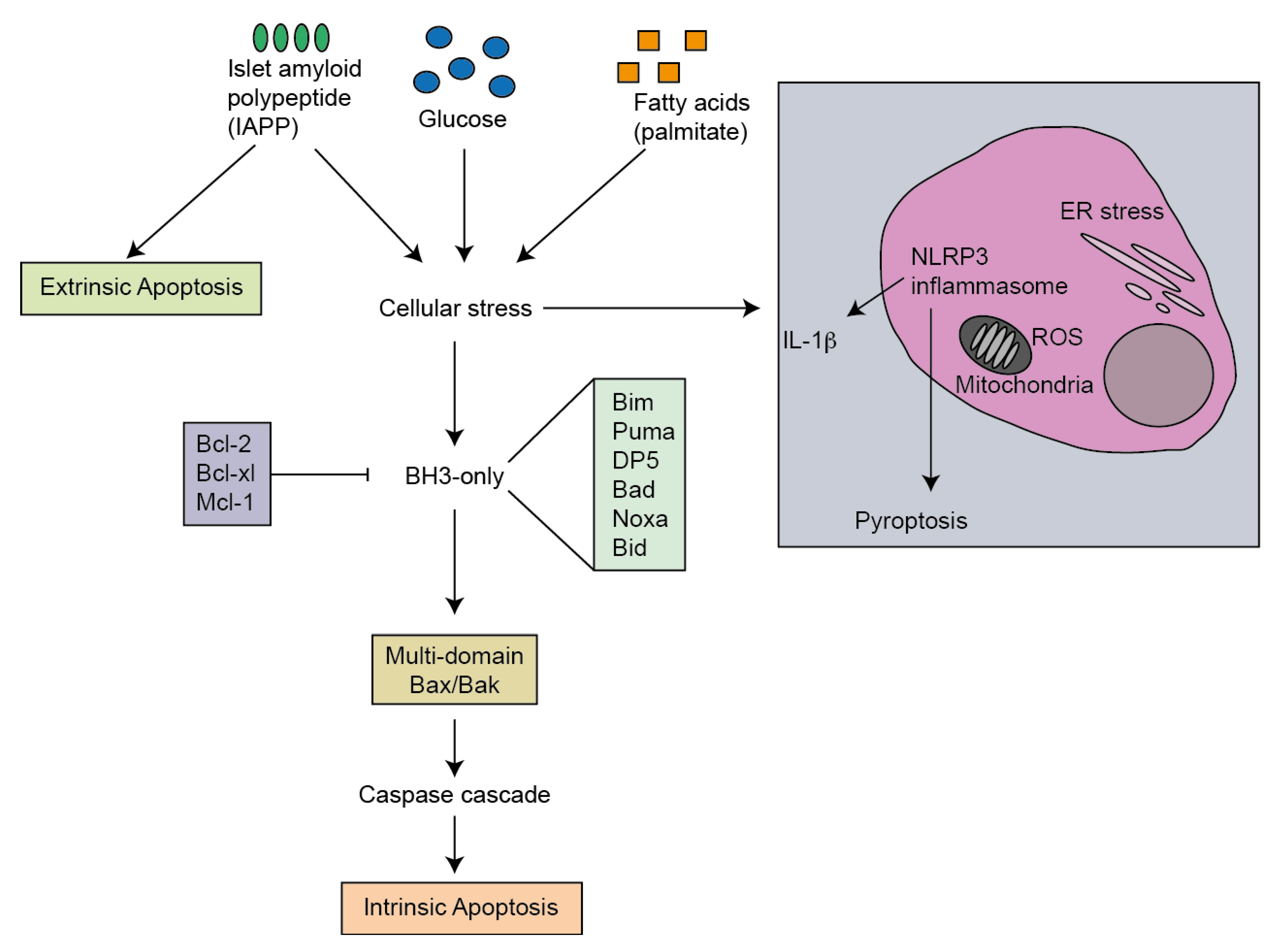
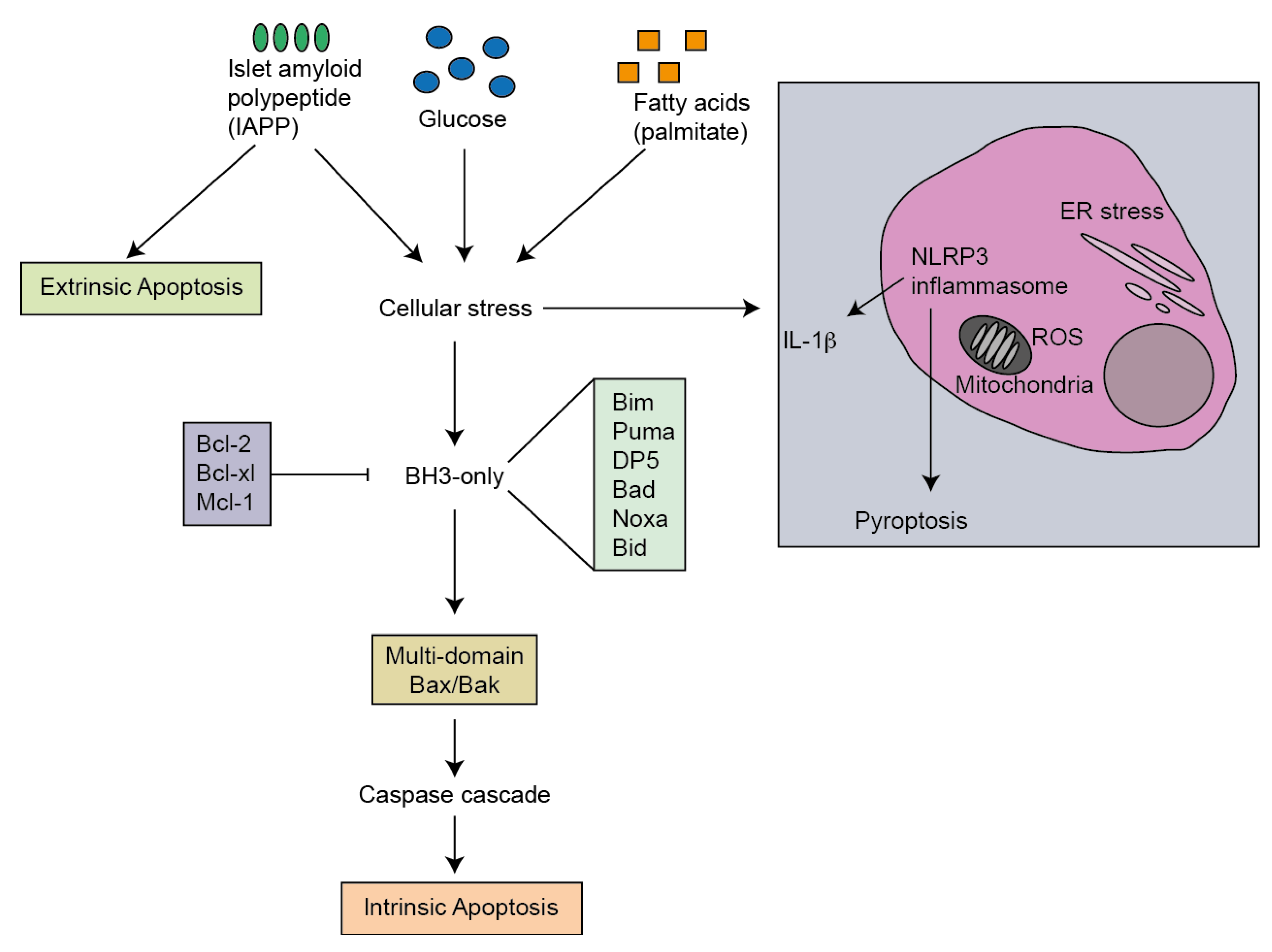
3. Mediators of Apoptosis in Type 2 Diabetes
4. Glucose
4.1. High Glucose Concentrations Cause Beta-cell Apoptosis
4.2. Glucose Induces Apoptosis through the Intrinsic Pathway
4.3. Glucose-Induced IL-1β Production and NLRP3 Inflammasome Activation
5. Palmitate
5.1. Excess Palmitate Causes Beta-Cell Apoptosis
5.2. Palmitate Induces Intrinsic Apoptosis
5.3. Palmitate-Induced NLRP3 Inflammasome Activation
6. Islet Amyloid Polypeptide
6.1. IAPP Induces Apoptosis
6.2. IAPP Induces Intrinsic and Extrinsic Apoptosis Pathways
6.3. IAPP Activates the NLRP3 Inflammasome
7. Activators of Apoptosis in T2D
8. ER Stress
8.1. ER Stress Induction by Glucose, Palmitate or IAPP
8.2. ER Stress Activates the Intrinsic Apoptosis Pathway
8.3. CHOP and JNK Activate Apoptosis Pathway Factors
8.4. ER Stress-Mediated Activation of the NLRP3 Inflammasome
9. Oxidative Stress
9.1. Glucose, Palmitate and IAPP Induce Oxidative Stress
9.2. Oxidative Stress Activates the Intrinsic Apoptosis Pathway
9.3. Oxidative Stress Activates NLRP3 Inflammasome
10. Conclusions
Acknowledgements
References
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef]
- Clark, A.; Wells, C.A.; Buley, I.D.; Cruickshank, J.K.; Vanhegan, R.I.; Matthews, D.R.; Cooper, G.J.; Holman, R.R.; Turner, R.C. Islet amyloid, Increased A-cells, Reduced B-cells and exocrine fibrosis: Quantitative changes in the pancreas in type 2 diabetes. Diabetes Res. 1988, 9, 151–159. [Google Scholar]
- Sakuraba, H.; Mizukami, H.; Yagihashi, N.; Wada, R.; Hanyu, C.; Yagihashi, S. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia 2002, 45, 85–96. [Google Scholar] [CrossRef]
- Song, B.; Scheuner, D.; Ron, D.; Pennathur, S.; Kaufman, R.J. Chop deletion reduces oxidative stress, Improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Invest. 2008, 118, 3378–3389. [Google Scholar]
- Oyadomari, S.; Koizumi, A.; Takeda, K.; Gotoh, T.; Akira, S.; Araki, E.; Mori, M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Invest. 2002, 109, 525–532. [Google Scholar]
- Donath, M.Y.; Gross, D.J.; Cerasi, E.; Kaiser, N. Hyperglycemia-induced beta-cell apoptosis in pancreatic islets of Psammomys obesus during development of diabetes. Diabetes 1999, 48, 738–744. [Google Scholar] [CrossRef]
- Pick, A.; Clark, J.; Kubstrup, C.; Levisetti, M.; Pugh, W.; Bonner-Weir, S.; Polonsky, K.S. Role of apoptosis in failure of beta-cell mass compensation for insulin resistance and beta-cell defects in the male Zucker diabetic fatty rat. Diabetes 1998, 47, 358–364. [Google Scholar]
- Zini, E.; Osto, M.; Franchini, M.; Guscetti, F.; Donath, M.Y.; Perren, A.; Heller, R.S.; Linscheid, P.; Bouwman, M.; Ackermann, M.; et al. Hyperglycaemia but not hyperlipidaemia causes beta cell dysfunction and beta cell loss in the domestic cat. Diabetologia 2009, 52, 336–346. [Google Scholar] [CrossRef] [Green Version]
- Hotchkiss, R.S.; Strasser, A.; McDunn, J.E.; Swanson, P.E. Cell death. New Engl. J. Med. 2009, 361, 1570–1583. [Google Scholar] [CrossRef]
- Strasser, A. The role of BH3-only proteins in the immune system. Nat. Rev. Immunol. 2005, 5, 189–200. [Google Scholar] [CrossRef]
- Thomas, H.E.; McKenzie, M.D.; Angstetra, E.; Campbell, P.D.; Kay, T.W. Beta cell apoptosis in diabetes. Apoptosis 2009, 14, 1389–1404. [Google Scholar] [CrossRef]
- Bergmann, L.; Kroncke, K.D.; Suschek, C.; Kolb, H.; Kolb-Bachofern, V. Cytotoxic action of IL-1 beta against pancreatic islets is mediated via nitric oxide formation and is inhibited by NG-monomethyl-L-arginine. FEBS Lett. 1992, 299, 103–106. [Google Scholar]
- Maedler, K.; Sergeev, P.; Ris, F.; Oberholzer, J.; Joller-Jemelka, H.I.; Spinas, G.A.; Kaiser, N.; Halban, P.A.; Donath, M.Y. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J. Clin. Invest. 2002, 110, 851–860. [Google Scholar]
- Masters, S.L.; Dunne, A.; Subramanian, S.L.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z.; et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat. Immunol. 2010, 11, 897–904. [Google Scholar] [CrossRef]
- Ichinohe, T.; Pang, I.K.; Kumamoto, Y.; Peaper, D.R.; Ho, J.H.; Murray, T.S.; Iwasaki, A. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc. Natl. Acad. Sci. USA 2011, 108, 5354–5359. [Google Scholar]
- Joosten, L.A.; Netea, M.G.; Mylona, E.; Koenders, M.I.; Malireddi, R.K.; Oosting, M.; Stienstra, R.; van de Veerdonk, F.L.; Stalenhoef, A.F.; Giamarellos-Bourboulis, E.J.; et al. Engagement of fatty acids with Toll-like receptor 2 drives interleukin-1beta production via the ASC/caspase 1 pathway in monosodium urate monohydrate crystal-induced gouty arthritis. Arthritis Rheum. 2010, 62, 3237–3248. [Google Scholar] [CrossRef]
- Lerner, A.G.; Upton, J.P.; Praveen, P.V.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell. Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar]
- Oslowski, C.M.; Hara, T.; O'Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell. Metab. 2012, 16, 265–273. [Google Scholar] [CrossRef]
- Mahr, S.; Neumayer, N.; Gerhard, M.; Classen, M.; Prinz, C. IL-1beta-induced apoptosis in rat gastric enterochromaffin-like cells is mediated by iNOS, NF-kappaB, and Bax protein. Gastroenterology 2000, 118, 515–524. [Google Scholar]
- Storling, J.; Binzer, J.; Andersson, A.K.; Zullig, R.A.; Tonnesen, M.; Lehmann, R.; Spinas, G.A.; Sandler, S.; Billestrup, N.; Mandrup-Poulsen, T. Nitric oxide contributes to cytokine-induced apoptosis in pancreatic beta cells via potentiation of JNK activity and inhibition of Akt. Diabetologia 2005, 48, 2039–2050. [Google Scholar] [CrossRef]
- Thomas, H.E.; Darwiche, R.; Corbett, J.A.; Kay, T.W. Interleukin-1 plus gamma-interferon-induced pancreatic beta-cell dysfunction is mediated by beta-cell nitric oxide production. Diabetes 2002, 51, 311–316. [Google Scholar]
- Kepp, O.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Pyroptosis — a cell death modality of its kind? Eur. J. Immunol. 2010, 40, 627–630. [Google Scholar] [CrossRef]
- McKenzie, M.D.; Jamieson, E.; Jansen, E.S.; Scott, C.L.; Huang, D.C.; Bouillet, P.; Allison, J.; Kay, T.W.; Strasser, A.; Thomas, H.E. Glucose induces pancreatic islet cell apoptosis that requires the BH3-only proteins bim and puma and multi-BH domain protein Bax. Diabetes 2010, 59, 644–652. [Google Scholar] [CrossRef]
- Mellado-Gil, J.M.; Aguilar-Diosdado, M. High glucose potentiates cytokine- and streptozotocin-induced apoptosis of rat islet cells: Effect on apoptosis-related genes. J. Endocrinol. 2004, 183, 155–162. [Google Scholar] [CrossRef]
- Piro, S.; Anello, M.; Di Pietro, C.; Lizzio, M.N.; Patane, G.; Rabuazzo, A.M.; Vigneri, R.; Purrello, M.; Purrello, F. Chronic exposure to free fatty acids or high glucose induces apoptosis in rat pancreatic islets: possible role of oxidative stress. Metabolism 2002, 51, 1340–1347. [Google Scholar] [CrossRef]
- Federici, M.; Hribal, M.; Perego, L.; Ranalli, M.; Caradonna, Z.; Perego, C.; Usellini, L.; Nano, R.; Bonini, P.; Bertuzzi, F.; et al. High glucose causes apoptosis in cultured human pancreatic islets of Langerhans: a potential role for regulation of specific Bcl family genes toward an apoptotic cell death program. Diabetes 2001, 50, 1290–1301. [Google Scholar] [CrossRef]
- Maedler, K.; Spinas, G.A.; Lehmann, R.; Sergeev, P.; Weber, M.; Fontana, A.; Kaiser, N.; Donath, M.Y. Glucose induces beta-cell apoptosis via upregulation of the Fas receptor in human islets. Diabetes 2001, 50, 1683–1690. [Google Scholar] [CrossRef]
- McKenzie, M.D.; Carrington, E.M.; Kaufmann, T.; Strasser, A.; Huang, D.C.; Kay, T.W.; Allison, J.; Thomas, H.E. Proapoptotic BH3-only protein Bid is essential for death receptor-induced apoptosis of pancreatic beta-cells. Diabetes 2008, 57, 1284–1292. [Google Scholar]
- Westerbacka, J.; Lammi, K.; Hakkinen, A.M.; Rissanen, A.; Salminen, I.; Aro, A.; Yki-Jarvinen, H. Dietary fat content modifies liver fat in overweight nondiabetic subjects. J. Clin. Endocrinol. Metab. 2005, 90, 2804–2809. [Google Scholar] [CrossRef]
- Cunha, D.A.; Hekerman, P.; Ladriere, L.; Bazarra-Castro, A.; Ortis, F.; Wakeham, M.C.; Moore, F.; Rasschaert, J.; Cardozo, A.K.; Bellomo, E.; et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J. Cell Sci. 2008, 121, 2308–2318. [Google Scholar] [CrossRef]
- Johnson, J.D.; Han, Z.; Otani, K.; Ye, H.; Zhang, Y.; Wu, H.; Horikawa, Y.; Misler, S.; Bell, G.I.; Polonsky, K.S. RyR2 and calpain-10 delineate a novel apoptosis pathway in pancreatic islets. J. Biol. Chem. 2004, 279, 24794–24802. [Google Scholar] [CrossRef]
- Laybutt, D.R.; Preston, A.M.; Akerfeldt, M.C.; Kench, J.G.; Busch, A.K.; Biankin, A.V.; Biden, T.J. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 2007, 50, 752–763. [Google Scholar] [CrossRef]
- Cunha, D.A.; Igoillo-Esteve, M.; Gurzov, E.N.; Germano, C.M.; Naamane, N.; Marhfour, I.; Fukaya, M.; Vanderwinden, J.M.; Gysemans, C.; Mathieu, C.; et al. Death protein 5 and p53-upregulated modulator of apoptosis mediate the endoplasmic reticulum stress-mitochondrial dialog triggering lipotoxic rodent and human beta-cell apoptosis. Diabetes 2012, 61, 2763–2775. [Google Scholar] [CrossRef]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef]
- Montane, J.; Klimek-Abercrombie, A.; Potter, K.J.; Westwell-Roper, C.; Bruce Verchere, C. Metabolic stress, IAPP and islet amyloid. Diabetes Obes. Metab. 2012, 14 (Suppl. 3), 68–77. [Google Scholar] [CrossRef]
- Westermark, P.; Andersson, A.; Westermark, G.T. Islet amyloid polypeptide, Islet amyloid, and diabetes mellitus. Physiol. Rev. 2011, 91, 795–826. [Google Scholar] [CrossRef]
- Zraika, S.; Hull, R.L.; Verchere, C.B.; Clark, A.; Potter, K.J.; Fraser, P.E.; Raleigh, D.P.; Kahn, S.E. Toxic oligomers and islet beta cell death: Guilty by association or convicted by circumstantial evidence? Diabetologia 2010, 53, 1046–1056. [Google Scholar] [CrossRef]
- Jurgens, C.A.; Toukatly, M.N.; Fligner, C.L.; Udayasankar, J.; Subramanian, S.L.; Zraika, S.; Aston-Mourney, K.; Carr, D.B.; Westermark, P.; Westermark, G.T.; et al. beta-cell loss and beta-cell apoptosis in human type 2 diabetes are related to islet amyloid deposition. Am. J. Pathol. 2011, 178, 2632–2640. [Google Scholar] [CrossRef]
- Ritzel, R.A.; Meier, J.J.; Lin, C.Y.; Veldhuis, J.D.; Butler, P.C. Human islet amyloid polypeptide oligomers disrupt cell coupling, Induce apoptosis, and impair insulin secretion in isolated human islets. Diabetes 2007, 56, 65–71. [Google Scholar] [CrossRef]
- Janson, J.; Ashley, R.H.; Harrison, D.; McIntyre, S.; Butler, P.C. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes 1999, 48, 491–498. [Google Scholar] [CrossRef]
- Subramanian, S.L.; Hull, R.L.; Zraika, S.; Aston-Mourney, K.; Udayasankar, J.; Kahn, S.E. cJUN N-terminal kinase (JNK) activation mediates islet amyloid-induced beta cell apoptosis in cultured human islet amyloid polypeptide transgenic mouse islets. Diabetologia 2012, 55, 166–174. [Google Scholar] [CrossRef]
- Park, Y.J.; Lee, S.; Kieffer, T.J.; Warnock, G.L.; Safikhan, N.; Speck, M.; Hao, Z.; Woo, M.; Marzban, L. Deletion of Fas protects islet beta cells from cytotoxic effects of human islet amyloid polypeptide. Diabetologia 2012, 55, 1035–1047. [Google Scholar] [CrossRef]
- Westwell-Roper, C.; Dai, D.L.; Soukhatcheva, G.; Potter, K.J.; van Rooijen, N.; Ehses, J.A.; Verchere, C.B. IL-1 blockade attenuates islet amyloid polypeptide-induced proinflammatory cytokine release and pancreatic islet graft dysfunction. J. Immunol. 2011, 187, 2755–2765. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Cardozo, A.K.; Cnop, M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr. Rev. 2008, 29, 42–61. [Google Scholar] [CrossRef]
- Fonseca, S.G.; Burcin, M.; Gromada, J.; Urano, F. Endoplasmic reticulum stress in beta-cells and development of diabetes. Curr. Opin. Pharmacol. 2009, 9, 763–770. [Google Scholar]
- Fonseca, S.G.; Gromada, J.; Urano, F. Endoplasmic reticulum stress and pancreatic beta-cell death. Trends Endocrinol. Metab. 2011, 22, 266–274. [Google Scholar]
- Lipson, K.L.; Ghosh, R.; Urano, F. The role of IRE1alpha in the degradation of insulin mRNA in pancreatic beta-cells. PLoS One 2008, 3, e1648. [Google Scholar] [CrossRef]
- Hou, Z.Q.; Li, H.L.; Gao, L.; Pan, L.; Zhao, J.J.; Li, G.W. Involvement of chronic stresses in rat islet and INS-1 cell glucotoxicity induced by intermittent high glucose. Mol. Cell. Endocrinol. 2008, 291, 71–78. [Google Scholar]
- Maedler, K.; Schulthess, F.T.; Bielman, C.; Berney, T.; Bonny, C.; Prentki, M.; Donath, M.Y.; Roduit, R. Glucose and leptin induce apoptosis in human beta-cells and impair glucose-stimulated insulin secretion through activation of c-Jun N-terminal kinases. FASEB J. 2008, 22, 1905–1913. [Google Scholar] [CrossRef] [Green Version]
- Hull, R.L.; Zraika, S.; Udayasankar, J.; Aston-Mourney, K.; Subramanian, S.L.; Kahn, S.E. Amyloid formation in human IAPP transgenic mouse islets and pancreas, and human pancreas, is not associated with endoplasmic reticulum stress. Diabetologia 2009, 52, 1102–1111. [Google Scholar] [CrossRef]
- Matveyenko, A.V.; Gurlo, T.; Daval, M.; Butler, A.E.; Butler, P.C. Successful versus failed adaptation to high-fat diet-induced insulin resistance: the role of IAPP-induced beta-cell endoplasmic reticulum stress. Diabetes 2009, 58, 906–916. [Google Scholar] [CrossRef]
- Huang, C.J.; Lin, C.Y.; Haataja, L.; Gurlo, T.; Butler, A.E.; Rizza, R.A.; Butler, P.C. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 2007, 56, 2016–2027. [Google Scholar] [CrossRef]
- Cnop, M.; Foufelle, F.; Velloso, L.A. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol. Med. 2012, 18, 59–68. [Google Scholar] [CrossRef]
- Puthalakath, H.; O'Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef]
- Li, J.; Lee, B.; Lee, A.S. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J. Biol. Chem. 2006, 281, 7260–7270. [Google Scholar] [CrossRef]
- Kieran, D.; Woods, I.; Villunger, A.; Strasser, A.; Prehn, J.H. Deletion of the BH3-only protein puma protects motoneurons from ER stress-induced apoptosis and delays motoneuron loss in ALS mice. Proc. Natl. Acad. Sci. USA 2007, 104, 20606–20611. [Google Scholar]
- Cazanave, S.C.; Elmi, N.A.; Akazawa, Y.; Bronk, S.F.; Mott, J.L.; Gores, G.J. CHOP and AP-1 cooperatively mediate PUMA expression during lipoapoptosis. Am. J. Physiol.-Gastr. L. 2010, 299, G236–G243. [Google Scholar]
- Giam, M.; Huang, D.C.; Bouillet, P. BH3-only proteins and their roles in programmed cell death. Oncogene 2008, 27 (Suppl. 1), S128–S136. [Google Scholar] [CrossRef]
- Steckley, D.; Karajgikar, M.; Dale, L.B.; Fuerth, B.; Swan, P.; Drummond-Main, C.; Poulter, M.O.; Ferguson, S.S.; Strasser, A.; Cregan, S.P. Puma is a dominant regulator of oxidative stress induced Bax activation and neuronal apoptosis. J. Neurosci. 2007, 27, 12989–12999. [Google Scholar] [CrossRef]
- Ghosh, A.P.; Klocke, B.J.; Ballestas, M.E.; Roth, K.A. CHOP potentially co-operates with FOXO3a in neuronal cells to regulate PUMA and BIM expression in response to ER stress. PLoS One 2012, 7, e39586. [Google Scholar]
- Menu, P.; Mayor, A.; Zhou, R.; Tardivel, A.; Ichijo, H.; Mori, K.; Tschopp, J. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell. Death Dis. 2012, 3, e261. [Google Scholar]
- Chen, J.; Fontes, G.; Saxena, G.; Poitout, V.; Shalev, A. Lack of TXNIP protects against mitochondria-mediated apoptosis but not against fatty acid-induced ER stress-mediated beta-cell death. Diabetes 2010, 59, 440–447. [Google Scholar] [CrossRef]
- Mates, J.M. Effects of antioxidant enzymes in the molecular control of reactive oxygen species toxicology. Toxicology 2000, 153, 83–104. [Google Scholar] [CrossRef]
- Jonas, J.C.; Bensellam, M.; Duprez, J.; Elouil, H.; Guiot, Y.; Pascal, S.M. Glucose regulation of islet stress responses and beta-cell failure in type 2 diabetes. Diabetes Obes. Metab. 2009, 11 (Suppl. 4), 65–81. [Google Scholar] [CrossRef]
- Lenzen, S.; Drinkgern, J.; Tiedge, M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radical Biol. Med. 1996, 20, 463–466. [Google Scholar] [CrossRef]
- Moore, P.C.; Ugas, M.A.; Hagman, D.K.; Parazzoli, S.D.; Poitout, V. Evidence against the involvement of oxidative stress in fatty acid inhibition of insulin secretion. Diabetes 2004, 53, 2610–2616. [Google Scholar] [CrossRef]
- Tanaka, Y.; Tran, P.O.; Harmon, J.; Robertson, R.P. A role for glutathione peroxidase in protecting pancreatic beta cells against oxidative stress in a model of glucose toxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 12363–12368. [Google Scholar]
- Kaneto, H.; Kajimoto, Y.; Miyagawa, J.; Matsuoka, T.; Fujitani, Y.; Umayahara, Y.; Hanafusa, T.; Matsuzawa, Y.; Yamasaki, Y.; Hori, M. Beneficial effects of antioxidants in diabetes: possible protection of pancreatic beta-cells against glucose toxicity. Diabetes 1999, 48, 2398–2406. [Google Scholar] [CrossRef]
- Oprescu, A.I.; Bikopoulos, G.; Naassan, A.; Allister, E.M.; Tang, C.; Park, E.; Uchino, H.; Lewis, G.F.; Fantus, I.G.; Rozakis-Adcock, M.; et al. Free fatty acid-induced reduction in glucose-stimulated insulin secretion: Evidence for a role of oxidative stress in vitro and in vivo. Diabetes 2007, 56, 2927–2937. [Google Scholar] [CrossRef]
- Carlsson, C.; Borg, L.A.; Welsh, N. Sodium palmitate induces partial mitochondrial uncoupling and reactive oxygen species in rat pancreatic islets in vitro. Endocrinology 1999, 140, 3422–3428. [Google Scholar] [CrossRef]
- Zraika, S.; Hull, R.L.; Udayasankar, J.; Aston-Mourney, K.; Subramanian, S.L.; Kisilevsky, R.; Szarek, W.A.; Kahn, S.E. Oxidative stress is induced by islet amyloid formation and time-dependently mediates amyloid-induced beta cell apoptosis. Diabetologia 2009, 52, 626–635. [Google Scholar] [CrossRef]
- Jiang, D.; Jha, N.; Boonplueang, R.; Andersen, J.K. Caspase 3 inhibition attenuates hydrogen peroxide-induced DNA fragmentation but not cell death in neuronal PC12 cells. J. Neuro. Chem. 2001, 76, 1745–1755. [Google Scholar]
- Takahashi, A.; Masuda, A.; Sun, M.; Centonze, V.E.; Herman, B. Oxidative stress-induced apoptosis is associated with alterations in mitochondrial caspase activity and Bcl-2-dependent alterations in mitochondrial pH (pHm). Brain Res. Bull. 2004, 62, 497–504. [Google Scholar] [CrossRef]
- Cai, Y.; Martens, G.A.; Hinke, S.A.; Heimberg, H.; Pipeleers, D.; Van de Casteele, M. Increased oxygen radical formation and mitochondrial dysfunction mediate beta cell apoptosis under conditions of AMP-activated protein kinase stimulation. Free Radical Biol. Med. 2007, 42, 64–78. [Google Scholar] [CrossRef]
- Wang, M.; Crager, M.; Pugazhenthi, S. Modulation of apoptosis pathways by oxidative stress and autophagy in beta cells. Exp. Diabetes Res. 2012, 2012, 647914. [Google Scholar]
- Petrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell. Death Differ. 2007, 14, 1583–1589. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wali, J.A.; Masters, S.L.; Thomas, H.E. Linking Metabolic Abnormalities to Apoptotic Pathways in Beta Cells in Type 2 Diabetes. Cells 2013, 2, 266-283. https://doi.org/10.3390/cells2020266
Wali JA, Masters SL, Thomas HE. Linking Metabolic Abnormalities to Apoptotic Pathways in Beta Cells in Type 2 Diabetes. Cells. 2013; 2(2):266-283. https://doi.org/10.3390/cells2020266
Chicago/Turabian StyleWali, Jibran A., Seth L. Masters, and Helen E. Thomas. 2013. "Linking Metabolic Abnormalities to Apoptotic Pathways in Beta Cells in Type 2 Diabetes" Cells 2, no. 2: 266-283. https://doi.org/10.3390/cells2020266