
Impact of SOCE Abolition by ORAI1 Knockout on the Proliferation, Adhesion, and Migration of HEK-293 Cells

 , and
, and
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Culture
2.2. Transfections
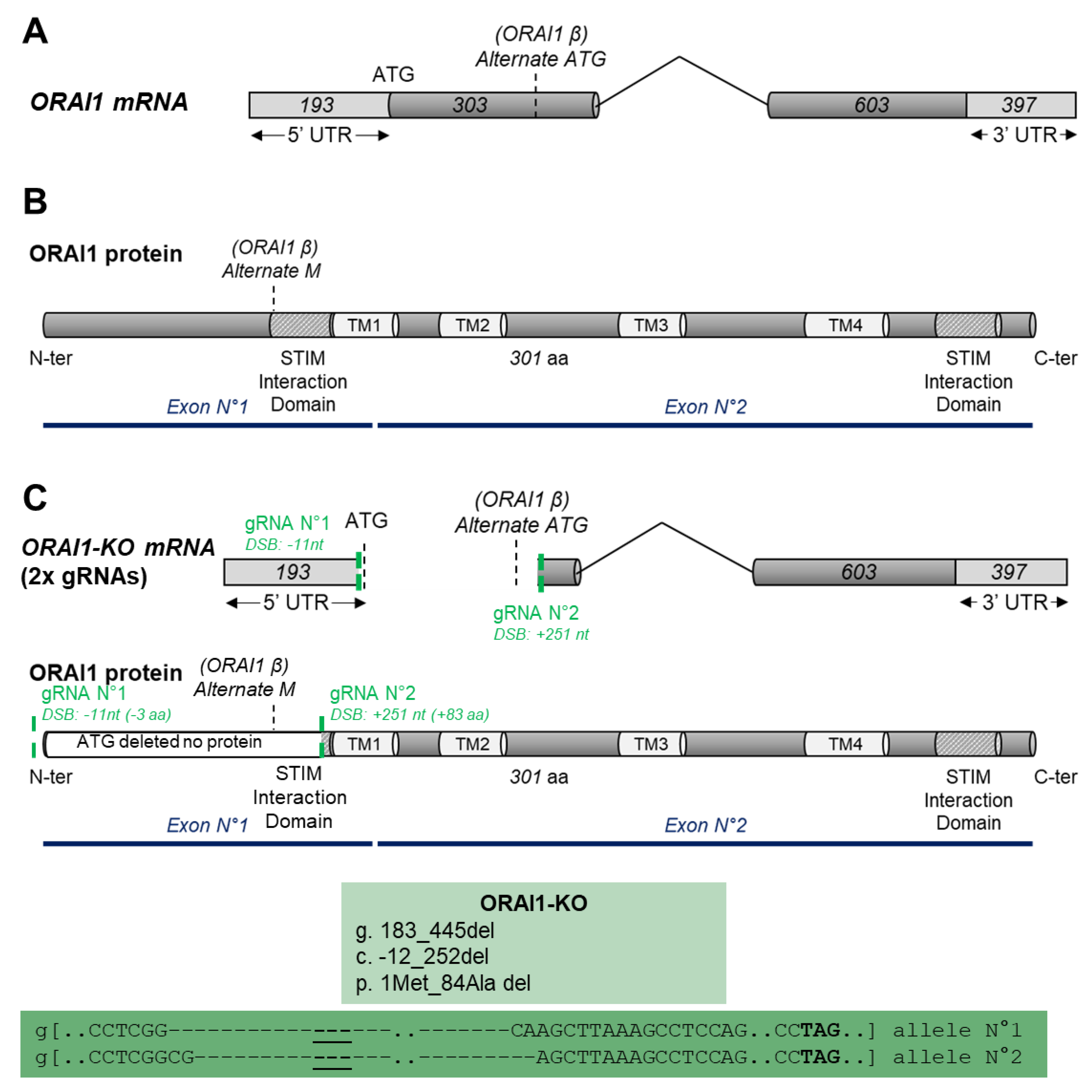
2.3. CRISPR/Cas9 KO Generation
2.4. Genomic DNA (gDNA) Extraction
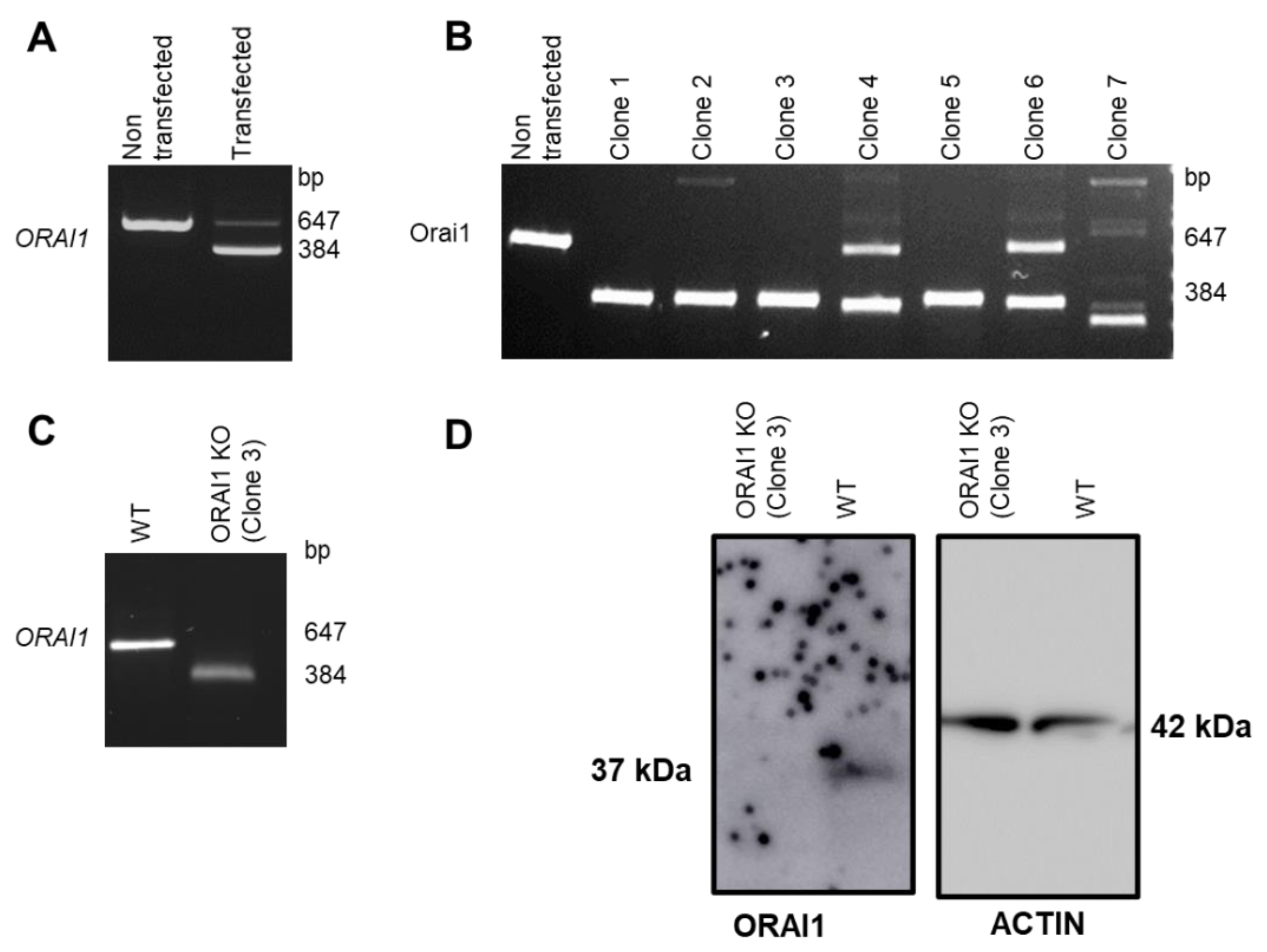
2.5. PCR and Genomic Sequencing
2.6. Quantitative Reverse Transcription PCR (RT-qPCR)
2.7. Immunobloting
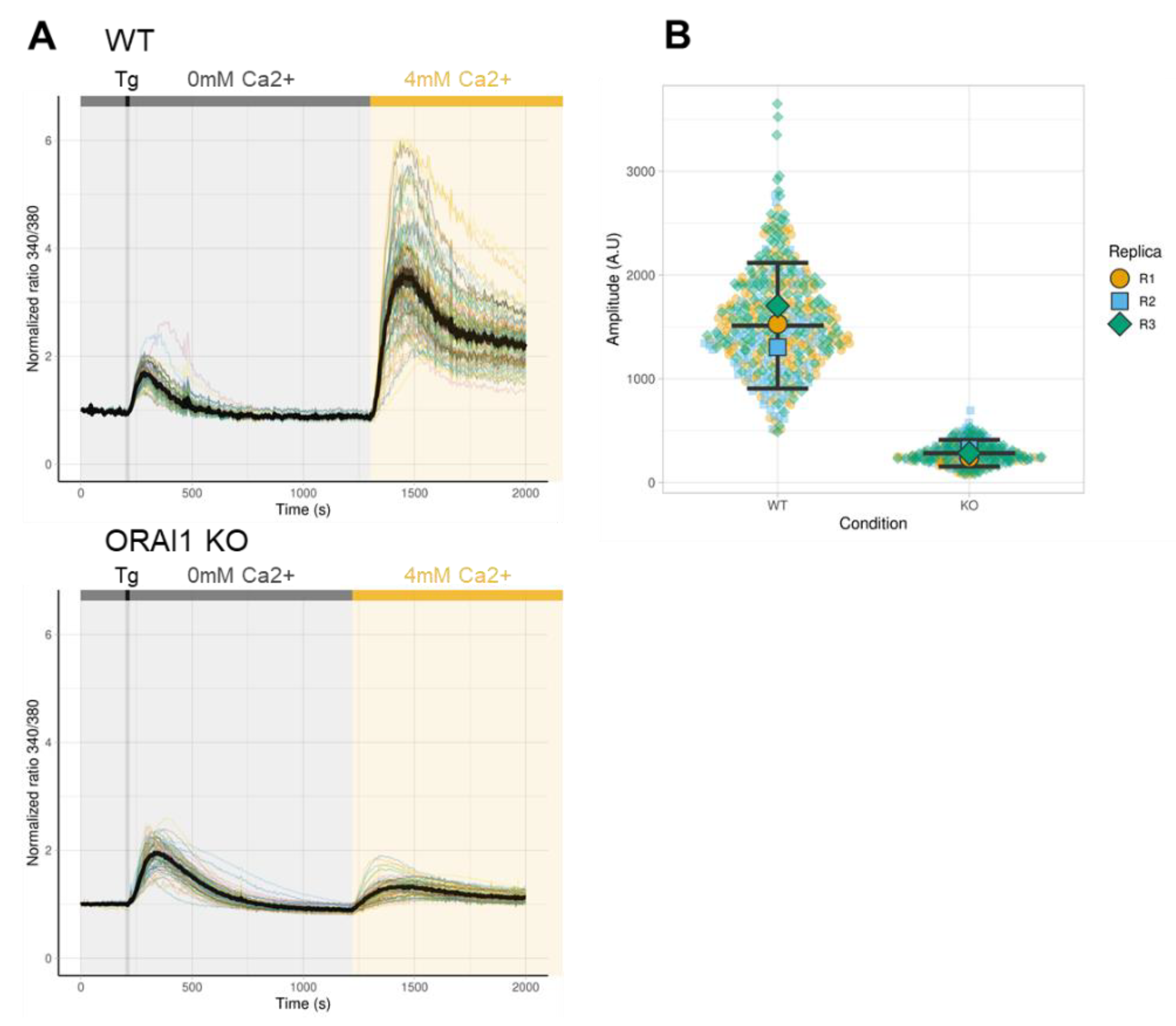
2.8. Calcium Imaging
- -
- For the 0 mM Ca2+ HBSS: NaCl 150 mM, KCl 5 mM, MgCl2 3 mM, CaCl2 0 mM, D-glucose 10 mM, HEPES 10 mM, EGTA 0.1 mM; pH 7.4 with NaOH;
- -
- For the 2 mM Ca2+ HBSS: NaCl 150 mM, KCl 5 mM, MgCl2 1 mM, CaCl2 2 mM, D-glucose 10 mM, HEPES 10 mM; pH 7.4 with NaOH;
- -
- For the 8 mM Ca2+ HBSS: NaCl 150 mM, KCl 5 mM, MgCl2 0 mM, CaCl2 8 mM, D-glucose 10 mM, HEPES 10 mM; pH 7.4 with NaOH;
2.9. Cell Cycle
2.10. Direct Cell Counting
2.11. Sulforhodamine B (SRB) Assay
2.12. Adhesion Assay
2.13. Wound Healing Assay
2.14. Boyden Chamber Migration Assay
2.15. Data Representation and Statistical Analysis
3. Results
3.1. Generation of ORAI1 KO in HEK-293 Cells
3.2. Effect of ORAI1 KO on SOCE
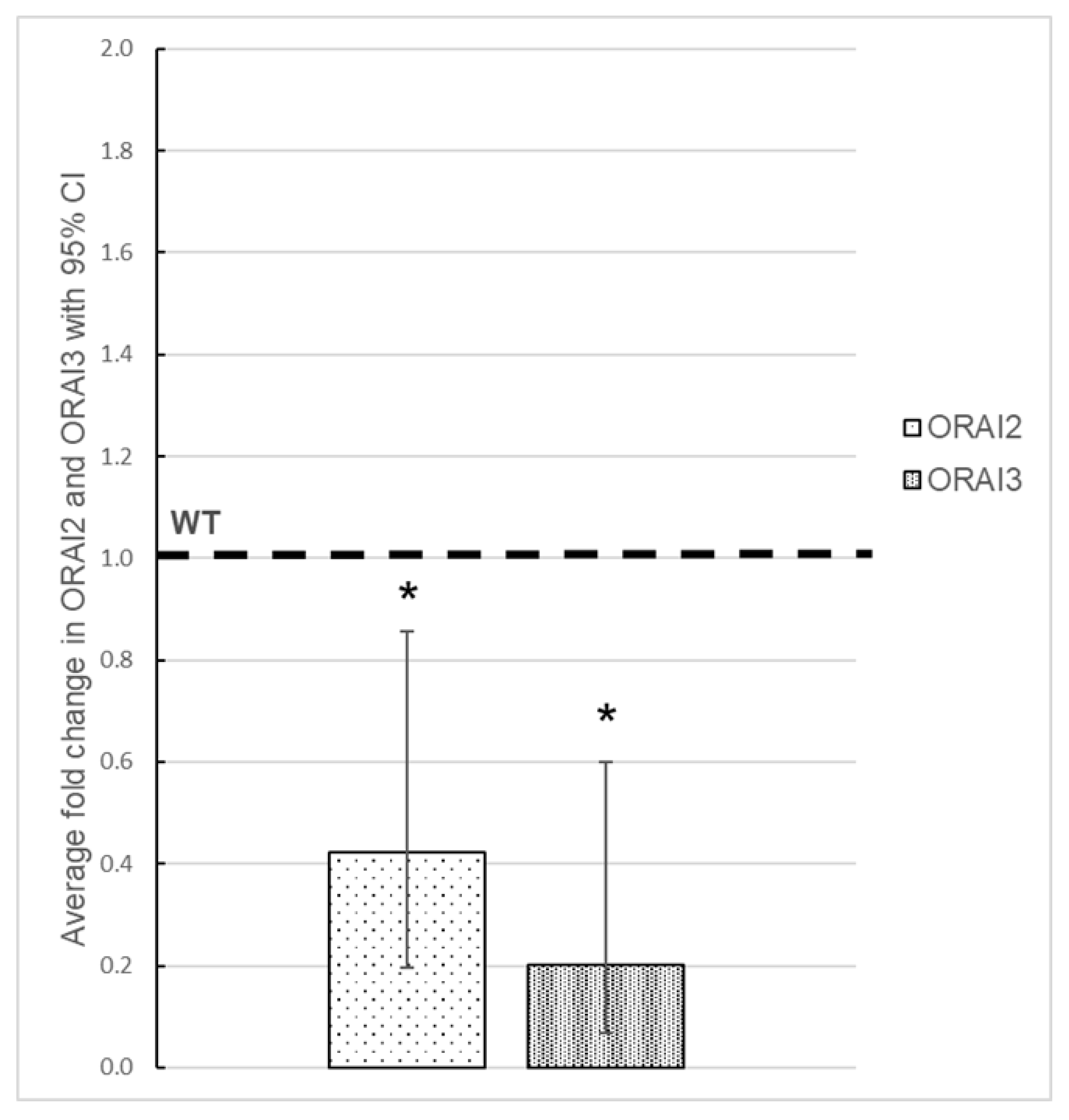
3.3. Effect of ORAI1 KO on the Expression of ORAI2 and ORAI3
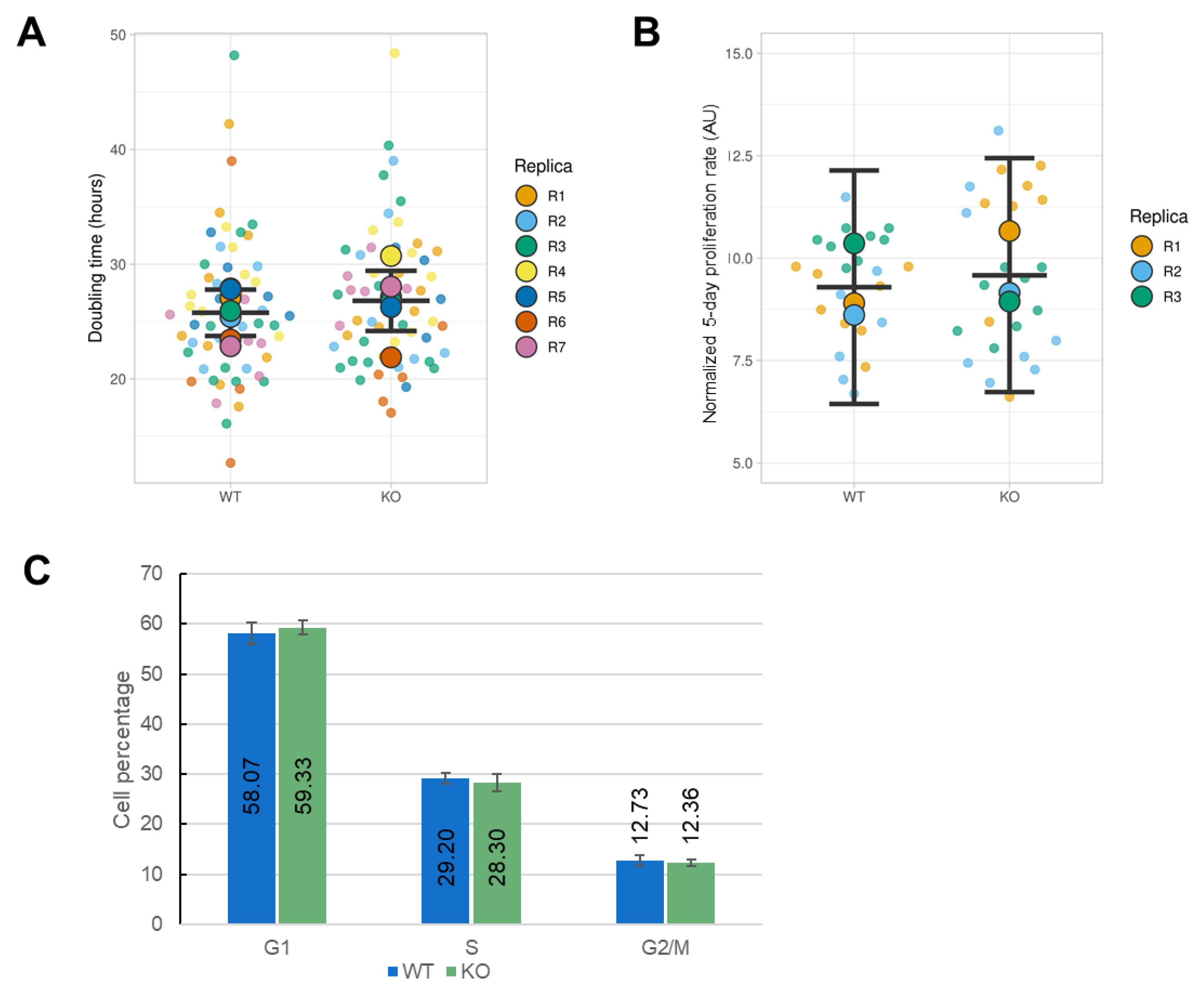
3.4. Effect of ORAI1 KO on the Proliferation of HEK-293 Cells
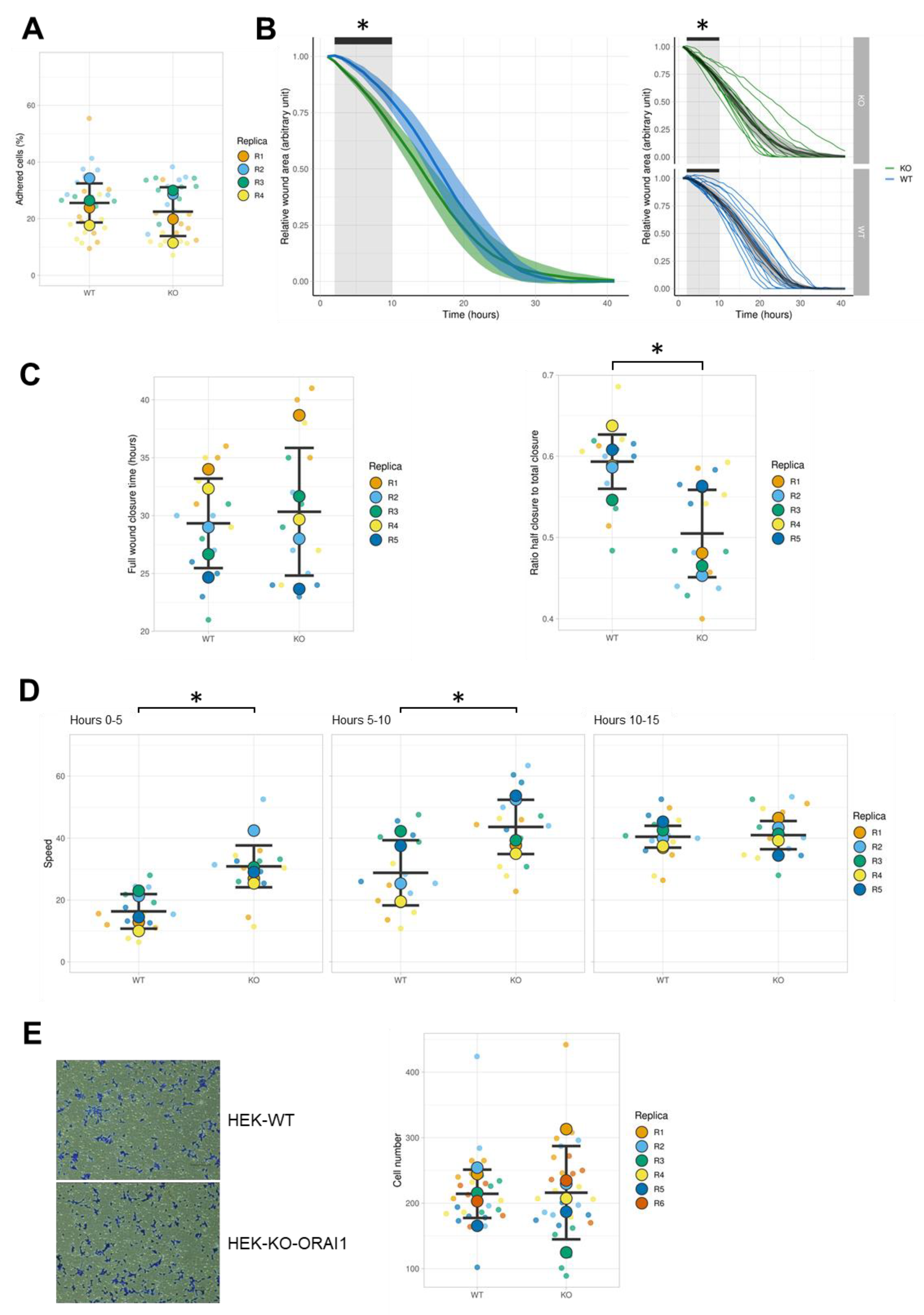
3.5. Effect of ORAI1 KO on the Migration of HEK-293 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Parekh, A.B. Ca2+ microdomains near plasma membrane Ca2+ channels: Impact on cell function. J. Physiol. 2008, 586, 3043–3054. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakriya, M.; Lewis, R.S. Store-operated calcium channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putney, J.W. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef]
- Williams, R.T.; Manji, S.S.M.; Parker, N.J.; Hancock, M.S.; Van Stekelenburg, L.; Eid, J.P.; Senior, P.V.; Kazenwadel, J.S.; Shandala, T.; Saint, R.; et al. Identification and characterization of the STIM (stromal interaction molecule) gene family: Coding for a novel class of transmembrane proteins. Biochem. J. 2001, 357, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, M.; Grabmayr, H.; Romanin, C. Mechanism of STIM activation. Curr. Opin. Physiol. 2020, 17, 74–79. [Google Scholar] [CrossRef]
- Lacruz, R.S.; Feske, S. Diseases caused by mutations in ORAI1 and STIM1. Ann. N. Y. Acad. Sci. 2015, 1356, 45–79. [Google Scholar] [CrossRef] [Green Version]
- Abdullaev, I.F.; Bisaillon, J.M.; Potier, M.; Gonzalez, J.C.; Motiani, R.K.; Trebak, M. Stim1 and orai1 mediate crac currents and store-operated calcium entry important for endothelial cell proliferation. Circ. Res. 2008, 103, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Darbellay, B.; Arnaudeau, S.; König, S.; Jousset, H.; Bader, C.; Demaurex, N.; Bernheim, L. STIM1- and Orai1-dependent store-operated calcium entry regulates human myoblast differentiation. J. Biol. Chem. 2009, 284, 5370–5380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diez-Bello, R.; Jardin, I.; Salido, G.M.; Rosado, J.A. Orai1 and Orai2 mediate store-operated calcium entry that regulates HL60 cell migration and FAK phosphorylation. Biochim. Biophys. Acta-Mol. Cell Res. 2017, 1864, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Rosado, J.A. STIM and calcium channel complexes in cancer. Biochim. Biophys. Acta-Mol. Cell Res. 2016, 1863, 1418–1426. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ren, Y.; Wang, L.; Zhao, W.; Dong, X.; Pan, J.; Gao, H.; Tian, Y. Orai1 and Stim1 Mediate the Majority of Store-Operated Calcium Entry in Multiple Myeloma and Have Strong Implications for Adverse Prognosis. Cell. Physiol. Biochem. 2018, 48, 2273–2285. [Google Scholar] [CrossRef]
- Dragoni, S.; Turin, I.; Laforenza, U.; Potenza, D.M.; Bottino, C.; Glasnov, T.N.; Prestia, M.; Ferulli, F.; Saitta, A.; Mosca, A.; et al. Store-Operated Ca2+ Entry Does Not Control Proliferation in Primary Cultures of Human Metastatic Renal Cellular Carcinoma. Biomed. Res. Int. 2014, 2014, 739494. [Google Scholar] [CrossRef] [Green Version]
- El Boustany, C.; Katsogiannou, M.; Delcourt, P.; Dewailly, E.; Prevarskaya, N.; Borowiec, A.S.; Capiod, T. Differential roles of STIM1, STIM2 and Orai1 in the control of cell proliferation and SOCE amplitude in HEK-293 cells. Cell Calcium 2010, 47, 350–359. [Google Scholar] [CrossRef]
- Borowiec, A.S.; Bidaux, G.; Tacine, R.; Dubar, P.; Pigat, N.; Delcourt, P.; Mignen, O.; Capiod, T. Are Orai1 and Orai3 channels more important than calcium influx for cell proliferation? Biochim. Biophys. Acta-Mol. Cell Res. 2014, 1843, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 Are Critical for Breast Tumor Cell Migration and Metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Lu, F.; He, H.; Shen, J.; Messina, J.; Mathew, R.; Wang, D.; Sarnaik, A.A.; Chang, W.C.; Kim, M.; et al. STIM1- and Orai1-mediated Ca2+ oscillation orchestrates invadopodium formation and melanoma invasion. J. Cell Biol. 2014, 207, 535–548. [Google Scholar] [CrossRef] [Green Version]
- Zuccolo, E.; Laforenza, U.; Ferulli, F.; Pellavio, G.; Scarpellino, G.; Tanzi, M.; Turin, I.; Faris, P.; Lucariello, A.; Maestri, M.; et al. Stim and Orai mediate constitutive Ca2+ entry and control endoplasmic reticulum Ca2+ refilling in primary cultures of colorectal carcinoma cells. Oncotarget 2018, 9, 31098–31119. [Google Scholar] [CrossRef] [Green Version]
- Concordet, J.-P.; Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Haeussler, M.; Schönig, K.; Eckert, H.; Eschstruth, A.; Mianné, J.; Renaud, J.B.; Schneider-Maunoury, S.; Shkumatava, A.; Teboul, L.; Kent, J.; et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016, 17, 148. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganger, M.T.; Dietz, G.D.; Ewing, S.J. A common base method for analysis of qPCR data and the application of simple blocking in qPCR experiments. BMC Bioinform. 2017, 18, 534. [Google Scholar] [CrossRef]
- Goedhart, J. PlotTwist: A web app for plotting and annotating continuous data. PLoS Biol. 2020, 18, e3000581. [Google Scholar] [CrossRef] [Green Version]
- Goedhart, J. SuperPlotsOfData—a web app for the transparent display and quantitative comparison of continuous data from different conditions. Mol. Biol. Cell 2021, 32, 470–474. [Google Scholar] [CrossRef]
- Lord, S.J.; Velle, K.B.; Dyche Mullins, R.; Fritz-Laylin, L.K. SuperPlots: Communicating reproducibility and variability in cell biology. J. Cell Biol. 2020, 219, e202001064. [Google Scholar] [CrossRef]
- Cumming, G.; Fidler, F.; Vaux, D.L. Error bars in experimental biology. J. Cell Biol. 2007, 177, 7–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukushima, M.; Tomita, T.; Janoshazi, A.; Putney, J.W. Alternative translation initiation gives rise to two isoforms of Orai1 with distinct plasma membrane mobilities. J. Cell Sci. 2012, 125, 4354–4361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcnally, B.A.; Somasundaram, A.; Jairaman, A.; Yamashita, M.; Prakriya, M. The C- and N-terminal STIM1 binding sites on Orai1 are required for both trapping and gating CRAC channels. J. Physiol. 2013, 591, 2833–2850. [Google Scholar] [CrossRef]
- Zheng, Q.; Cai, X.; Tan, M.H.; Schaffert, S.; Arnold, C.P.; Gong, X.; Chen, C.Z.; Huang, S. Precise gene deletion and replacement using the CRISPR/Cas9 system in human cells. Biotechniques 2014, 57, 115–124. [Google Scholar] [CrossRef]
- Neldeborg, S.; Lin, L.; Stougaard, M.; Luo, Y. Rapid and Efficient Gene Deletion by CRISPR/Cas9. In CRISPR Gene Editing; Humana Press: New York, NY, USA, 2019; Volume 100, pp. 233–247. ISBN 9781452289830. [Google Scholar]
- Bauer, D.E.; Canver, M.C.; Orkin, S.H. Generation of Genomic Deletions in Mammalian Cell Lines via CRISPR/Cas9. J. Vis. Exp. 2015, 95, e52118. [Google Scholar] [CrossRef] [PubMed]
- Lis, A.; Peinelt, C.; Beck, A.; Parvez, S.; Monteilh-Zoller, M.; Fleig, A.; Penner, R. CRACM1, CRACM2, and CRACM3 Are Store-Operated Ca2+ Channels with Distinct Functional Properties. Curr. Biol. 2007, 17, 794–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kar, P.; Lin, Y.P.; Bhardwaj, R.; Tucker, C.J.; Bird, G.S.; Hediger, M.A.; Monico, C.; Amin, N.; Parekh, A.B. The N terminus of Orai1 couples to the AKAP79 signaling complex to drive NFAT1 activation by local Ca2+ entry. Proc. Natl. Acad. Sci. USA 2021, 118, e2012908118. [Google Scholar] [CrossRef] [PubMed]
- Yoast, R.E.; Emrich, S.M.; Zhang, X.; Xin, P.; Johnson, M.T.; Fike, A.J.; Walter, V.; Hempel, N.; Yule, D.I.; Sneyd, J.; et al. The native ORAI channel trio underlies the diversity of Ca2+ signaling events. Nat. Commun. 2020, 11, 2444. [Google Scholar] [CrossRef]
- Cai, X.; Zhou, Y.; Nwokonko, R.M.; Loktionova, N.A.; Wang, X.; Xin, P.; Trebak, M.; Wang, Y.; Gill, D.L. The Orai1 store-operated calcium channel functions as a hexamer. J. Biol. Chem. 2016, 291, 25764–25775. [Google Scholar] [CrossRef] [Green Version]
- Alansary, D.; Peckys, D.B.; Niemeyer, B.A.; De Jonge, N. Detecting single ORAI1 proteins within the plasma membrane reveals higher-order channel complexes. J. Cell Sci. 2020, 133, jcs240358. [Google Scholar] [CrossRef]
- Mercer, J.C.; DeHaven, W.I.; Smyth, J.T.; Wedel, B.; Boyles, R.R.; Bird, G.S.; Putney, J.W. Large Store-operated Calcium Selective Currents Due to Co-expression of Orai1 or Orai2 with the Intracellular Calcium Sensor, Stim1. J. Biol. Chem. 2006, 281, 24979–24990. [Google Scholar] [CrossRef] [Green Version]
- Grimes, D.; Johnson, R.; Pashos, M.; Cummings, C.; Kang, C.; Sampedro, G.R.; Tycksen, E.; McBride, H.J.; Sah, R.; Lowell, C.A.; et al. ORAI1 and ORAI2 modulate murine neutrophil calcium signaling, cellular activation, and host defense. Proc. Natl. Acad. Sci. USA 2020, 117, 24403–24414. [Google Scholar] [CrossRef]
- Vaeth, M.; Yang, J.; Yamashita, M.; Zee, I.; Eckstein, M.; Knosp, C.; Kaufmann, U.; Jani, P.K.; Lacruz, R.S.; Flockerzi, V.; et al. ORAI2 modulates store-operated calcium entry and T cell-mediated immunity. Nat. Commun. 2017, 8, 14714. [Google Scholar] [CrossRef] [Green Version]
- Lopez, J.J.; Jardin, I.; Sanchez-Collado, J.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC Channels in the SOCE Scenario. Cells 2020, 9, 126. [Google Scholar] [CrossRef] [Green Version]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Gwack, Y.; Feske, S.; Srikanth, S.; Hogan, P.G.; Rao, A. Signalling to transcription: Store-operated Ca2+ entry and NFAT activation in lymphocytes. Cell Calcium 2007, 42, 145–156. [Google Scholar] [CrossRef]
- Vaeth, M.; Maus, M.; Klein-Hessling, S.; Freinkman, E.; Yang, J.; Eckstein, M.; Cameron, S.; Turvey, S.E.; Serfling, E.; Berberich-Siebelt, F.; et al. Store-Operated Ca2+ Entry Controls Clonal Expansion of T Cells through Metabolic Reprogramming. Immunity 2017, 47, 664–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mognol, G.P.; Carneiro, F.R.G.; Robbs, B.K.; Faget, D.V.; Viola, J.P.B. Cell cycle and apoptosis regulation by NFAT transcription factors: New roles for an old player. Cell Death Dis. 2016, 7, e2199. [Google Scholar] [CrossRef] [Green Version]
- Dubois, C.; Vanden Abeele, F.; Lehen’kyi, V.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of Channel-Forming ORAI Proteins Determines an Oncogenic Switch in Prostate Cancer. Cancer Cell 2014, 26, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Patergnani, S.; Danese, A.; Bouhamida, E.; Aguiari, G.; Previati, M.; Pinton, P.; Giorgi, C. Various aspects of calcium signaling in the regulation of apoptosis, autophagy, cell proliferation, and cancer. Int. J. Mol. Sci. 2020, 21, 8323. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell Migration: Integrating Signals from Front to Back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Wang, X.; Zheng, M.; Cheng, H. Calcium gradients underlying cell migration. Curr. Opin. Cell Biol. 2012, 24, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Sukumaran, P.; Varma, A.; Derry, S.; Sahmoun, A.E.; Singh, B.B. Cholesterol-induced activation of TRPM7 regulates cell proliferation, migration, and viability of human prostate cells. Biochim. Biophys. Acta-Mol. Cell Res. 2014, 1843, 1839–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umemura, M.; Baljinnyam, E.; Feske, S.; De Lorenzo, M.S.; Xie, L.H.; Feng, X.; Oda, K.; Makino, A.; Fujita, T.; Yokoyama, U.; et al. Store-operated Ca2+ entry (SOCE) regulates melanoma proliferation and cell migration. PLoS ONE 2014, 9, e89292. [Google Scholar] [CrossRef]
- Weidinger, C.; Shaw, P.J.; Feske, S. STIM1 and STIM2-mediated Ca2+ influx regulates antitumour immunity by CD8+ T cells. EMBO Mol. Med. 2013, 5, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Price, R.D.; Yamaji, T.; Matsuoka, N. FK506 potentiates NGF-induced neurite outgrowth via the Ras/Raf/MAP kinase pathway. Br. J. Pharmacol. 2003, 140, 825–829. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, K.; Yamamoto, T.S.; Ueno, N. Intracellular calcium signal at the leading edge regulates mesodermal sheet migration during Xenopus gastrulation. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Cho, J.H.; Reid, B.; Tseng, C.C.; He, L.; Tan, P.; Yeh, C.Y.; Wu, P.; Li, Y.; Widelitz, R.B.; et al. Calcium oscillations coordinate feather mesenchymal cell movement by SHH dependent modulation of gap junction networks. Nat. Commun. 2018, 9, 5377. [Google Scholar] [CrossRef]
- Catacuzzeno, L.; Franciolini, F. Role of KCa3.1 channels in modulating ca2+ oscillations during glioblastoma cell migration and invasion. Int. J. Mol. Sci. 2018, 19, 2970. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Wu, S.; Liu, Q.; Xie, J.; Zhang, J.; Han, D.; Lu, Q.; Lu, Q. Mertk deficiency affects macrophage directional migration via disruption of cytoskeletal organization. PLoS ONE 2015, 10, e0117787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, J.N.; Platoshyn, O.; Golovina, V.A.; Liu, L.; Zou, T.; Marasa, B.S.; Turner, D.J.; Yuan, J.X.J.; Wang, J.Y. TRPC1 functions as a store-operated Ca2+ channel in intestinal epithelial cells and regulates early mucosal restitution after wounding. Am. J. Physiol.-Gastrointest. Liver Physiol. 2006, 290, 782–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elzamzamy, O.M.; Penner, R.; Hazlehurst, L.A. The Role of TRPC1 in Modulating Cancer Progression. Cells 2020, 9, 388. [Google Scholar] [CrossRef] [Green Version]
- Asghar, M.Y.; Törnquist, K. Transient receptor potential canonical (TRPC) channels as modulators of migration and invasion. Int. J. Mol. Sci. 2020, 21, 1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bomben, V.C.; Turner, K.L.; Barclay, T.-T.C.; Sontheimer, H. Transient receptor potential canonical channels are essential for chemotactic migration of human malignant gliomas. J. Cell. Physiol. 2011, 226, 1879–1888. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment Type | Name | Sequence 5′→3′ |
|---|---|---|
| CRISPR gRNA cloning | gRNA1 | ATGCATGGAGCACGCCGCCG |
| gRNA2 | CGCCAAGCTTAAAGCCTCCA | |
| PCR Screening | ORAI1 screening forward | GGCACTTCTTCGACCTCGTC |
| ORAI1 screening reverse | CTTGTCACCACCCCAGATCG | |
| RT-qPCR | ORAI1 forward | ATGGTGGCAATGGTGGAG |
| ORAI1 reverse | CTGATCATGAGCGCAAACA | |
| ORAI2 forward | ACCTGGAACTGGTCACCTCT | |
| ORAI2 reverse | ATGGCCACCATGGCAAAGC | |
| ORAI3 forward | GGCCAAGCTCAAAGCTTCC | |
| ORAI3 reverse | CCTGGTGGGTACTCGTGGT | |
| GAPDH forward | ACCCACTCCTCCACCTTTG | |
| GAPDH reverse | CTCTTGTGCTCTTGCTGGG | |
| TBP forward | CTTGACCTAAAGACCATTGCACTTC | |
| TBP reverse | GTTCTTCACTCTTGGCTCCTGTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bokhobza, A.; Ziental-Gelus, N.; Allart, L.; Iamshanova, O.; Vanden Abeele, F. Impact of SOCE Abolition by ORAI1 Knockout on the Proliferation, Adhesion, and Migration of HEK-293 Cells. Cells 2021, 10, 3016. https://doi.org/10.3390/cells10113016
Bokhobza A, Ziental-Gelus N, Allart L, Iamshanova O, Vanden Abeele F. Impact of SOCE Abolition by ORAI1 Knockout on the Proliferation, Adhesion, and Migration of HEK-293 Cells. Cells. 2021; 10(11):3016. https://doi.org/10.3390/cells10113016
Chicago/Turabian StyleBokhobza, Alexandre, Nathalie Ziental-Gelus, Laurent Allart, Oksana Iamshanova, and Fabien Vanden Abeele. 2021. "Impact of SOCE Abolition by ORAI1 Knockout on the Proliferation, Adhesion, and Migration of HEK-293 Cells" Cells 10, no. 11: 3016. https://doi.org/10.3390/cells10113016