Abstract
Angiotensin-(1-7) [Ang-(1-7)]/Mas receptor is a counter-regulatory axis that counteracts detrimental renin-angiotensin system (RAS) effects, especially regarding systemic inflammation, vasopressin (AVP) release, and hypothalamic-pituitary-adrenal (HPA) activation. However, it is not completely understood whether this system may control centrally or systemically the late phase of systemic inflammation. Thus, the aim of this study was to determine whether intracerebroventricular (i.c.v.) administration of Ang-(1-7) can modulate systemic inflammation through the activation of humoral pathways in late phase of endotoxemia. Endotoxemia was induced by systemic injection of lipopolysaccharide (LPS) (1.5 mg/kg, i.v.) in Wistar rats. Ang-(1-7) (0.3 nmol in 2 µL) promoted the release of AVP and attenuated interleukin-6 (IL-6) and nitric oxide (NO) levels but increased interleukin-10 (IL-10) in the serum of the endotoxemic rats. The central administration of Mas receptor antagonist A779 (3 nmol in 2 µL, i.c.v.) abolished these anti-inflammatory effects in endotoxemic rats. Furthermore, Ang-(1-7) applied centrally restored mean arterial blood pressure (MABP) without affecting heart rate (HR) and prevented vascular hyporesponsiveness to norepinephrine (NE) and AVP in animals that received LPS. Together, our results indicate that Ang-(1-7) applied centrally promotes a systemic anti-inflammatory effect through the central Mas receptor and activation of the humoral pathway mediated by AVP.
1. Introduction
Endotoxemia, a classical model of systemic inflammation, is characterized by the amplified production of tumor necrosis factor alpha (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), and nitric oxide (NO) by immune cells and vascular endothelium [1]. The overproduction of inflammatory mediators has been involved in hypotension, hyporesponsiveness to vasoactive agents, and alterations in the hypothalamic-neurohypophyseal axis during systemic inflammation [2,3]. Our previous studies showed a marked decrease in vasopressin (AVP) plasma levels and increase in activation of the hypothalamic-pituitary-adrenal (HPA) axis during the late phase of endotoxemia, observed 6 h after lipopolysaccharide (LPS) administration [4,5,6]. It has been demonstrated that NO may play a role on the regulation of the HPA axis and AVP synthesis in the hypothalamic paraventricular nucleus (PVN) during endotoxemia [7,8,9,10,11]. However, besides the immune stimulus, there are other ways to control the hypothalamic-neurohypophyseal axis, including osmolality, hypotensive stimulus, and activation of the renin-angiotensin system (RAS) [12].
It is well known that the RAS plays a key role in the modulation of many functions in the body. Angiotensin-(1-7) [Ang-(1-7)]/Mas receptor, the counter-regulatory axis of the RAS, exerts beneficial effects against the pathophysiologic conditions [13,14]. The expression of Mas receptor was observed in neurons, glia cells, and endothelial cells of cerebral vessels [15]. As the agonist for the Mas receptor, Ang-(1-7) is also produced in the brain, in areas including the hypothalamus [16]. In PVN, this peptide is a potent secretagogue of AVP and may participate in controlling its release by magnocellular neurons [17]. In addition to neuroendocrine actions, the activation of Ang-(1-7)/Mas receptor axis attenuates inflammation in several experimental models, including polymicrobial sepsis and cerebral ischemia [18,19,20]. In our most recent study, it has been shown that central administration of Ang-(1-7) prevented LPS-induced vascular hyporesponsiveness and hypotension due to an anti-inflammatory effect via activation of sympathetic signaling during the initial phase of endotoxemia [21]. However, the central mechanisms of Ang-(1-7) to control systemic inflammation in the late phase of endotoxemia are not well known. Considering that Ang-(1-7) acts as a central neuropeptide controlling AVP release and the HPA axis as well as the importance of these hormones for hypotensive and inflammatory response during endotoxemia [22,23,24,25], in the present study we aimed to determine whether Ang-(1-7) can modulate systemic inflammation through the activation of humoral pathways in late phase of endotoxemia.
2. Material and Methods
2.1. Animal Experiments
Experiments were performed on adult male Wistar rats (215–220 g) obtained from the animal facility of the University of São Paulo, Ribeirão Preto Campus. The animals were housed at a controlled temperature (24.0 ± 2 °C) and exposed to a daily 12 h light–dark cycle (lights on from 6:00 to 18:00 h) and provided with food and water ad libitum. All experimental protocols were performed in accordance with the guidelines of the Ethics Committee on Animal Experimentation of the Ribeirão Preto College of Nursing, University of São Paulo (CEUA Protocol 14.1.872.53.4).
2.2. Stereotaxic Surgery
Seven days before the experiment, the rats were anesthetized with a mixture of ketamine and xylazine (90 mg/kg and 9 mg/kg, respectively, i.p., diluted in 0.9% isotonic saline) (Aldrich, Milwaukee, WI, USA) and immobilized in a stereotaxic frame. A stainless steel guide cannula (0.4 mm) was introduced into the right lateral ventricle (coordinates: A: −1.6 mm, L: 1 mm, D: 3.6 mm from the bregma) [26]. The displacement of the meniscus in a water manometer ensured the correct position of the cannula into the lateral ventricle. The cannula was fixed to the skull with stainless steel screws and dental acrylic cement. A tight-fitting stylet was kept inside the guide cannula to prevent occlusion and infection. At the end of the surgery, all animals received an injection of a polyvalent veterinary pentabiotic (24.000 UI/kg; Zoetis, Brazil). The rats were allowed to recover for seven days.
2.3. Cannulation Procedures
For intravenous (i.v.) drug administration, the rats were anesthetized on the day before the experiment with ketamine and xylazine, and a flexible catheter (PE-10, Silastic®, Dow Corning CO., Midland, MI, USA) was inserted into the right internal jugular vein. For direct mean arterial blood pressure (MABP) and heart rate (HR) measurements, an additional catheter (PE-10 heat-sealed to PE-50) was inserted into the femoral artery. The catheters were tunneled under the skin and exteriorized in the back of the neck, as described previously [5]. The animals were housed separately and allowed to recover for 24 h before taking measurements.
2.4. Drug Administration
Drugs were administered to the conscious rats, alone or in combination, according to the following chronology: (a) The rats received an intracerebroventricular (i.c.v.) A779 (3 nmol in 2 µL) or saline (0.9%), a potent and selective antagonist for receptor of Ang-(l-7), the Mas receptor; (b) After 30 min, the rats received an i.c.v. injection of Ang-(1-7) (0.3 nmol in 2 µL) or saline (NaCl 0.9%); (c) 1 min after Ang-(1-7) injection the rats received an i.v. bolus LPS injection (1.5 mg/kg) or saline (0.9%). Control animals were injected with the same volume of saline through the same routes. The doses of Ang-(1-7), A779, and LPS used were based on previous studies from our group [21,27]. Thus, the rats were divided into eight experimental groups according to analysis: (1) Saline (i.c.v.) + Saline (i.c.v.) + Saline (i.v.) (control group); (2) Saline (i.c.v.) + Ang-(1-7) (i.c.v.) + Saline (i.v.); (3) Saline (i.c.v.) + Saline (i.c.v.) + LPS (i.v.); (4) Saline (i.c.v.) + Ang-(1-7) (i.c.v.) + LPS (i.v.); (5) A779 (i.c.v.) + Saline (i.c.v.) + Saline (i.v.); (6) A779 (i.c.v.) + Ang-(1-7) (i.c.v.) + Saline (i.v.); (7) A779 (i.c.v.) + Saline (i.c.v.) + LPS (i.v.); (8) A779 (i.c.v.) + Ang-(1-7) (i.c.v.) + LPS (i.v.).
2.5. Samples
Samples of PVN and supraoptic nucleus (SON) and blood were collected 6 h after LPS administration. After harvest, PVN and SON samples were stored in RNase-free microcentrifuge tubes in a freezer at −80 °C. A blood sample was collected with EDTA (1 mmol/L) and immediately centrifuged (3100 rpm, 4 °C, 15 min) to obtain plasma, which was stored in a freezer at −80 °C. Another blood sample was collected without anticoagulant and centrifuged (3500 rpm, 4 °C, 10 min) to obtain serum, which was also stored in a freezer at −80 °C.
2.6. Plasma Osmolality, Sodium, and Lactate Measurements
The determination of plasma osmolality was performed using an osmometer (Fiske OS Osmometer, Advanced Instruments, Norwood, MA, USA). Plasma sodium levels were analyzed using a quantitative electrode quantification technique (9180 Electrolyte Analyzer, Roche Diagnostics GmbH, Mannheim, Germany). To determine the lactate level, a commercial enzyme immunoassay kit (Quibasa Química Básica, Belo Horizonte, Brazil) was used.
2.7. Real-Time Polymerase Chain Reaction (RT-PCR)
After decapitation, PVN and SON samples were harvested, stored in RNase-free microcentrifuge tubes and frozen in liquid nitrogen within a 5-min time frame. Samples were then disintegrated in the presence of Trizol® (Introvigen, Carlsbad, CA, USA), and RNA extraction followed the Trizol standard procedure with glicogen. RNA was quantified by spectrophotometry using nanodrop 1000 equipment (Thermo Fisher Scientific, Wilmington, DE, USA). The analysis of the genes of interest was performed by RT-PCR using TaqMan assays (AVP Rn00690189_g1; corticotropin-releasing hormone-CRH Rn01462137_m1; TNF Rn99999017_m1; IL10 Rn01483988_g1; inducible nitric oxide synthase (NOS2) Rn00561646_m1; GAPDH Rn01775763_g1) as previously described in Faim et al. [28]. The results were expressed as normalized relative quantities (NRQ).
2.8. Corticosterone and Vasopressin (AVP) Measurements
The corticosterone and AVP radioimmunoassay were performed as previously described by Vecsei [29]. Plasma samples (25 μL) were extracted using ethanol, lyophilized, and stored at −20 °C until analysis of corticosterone. Plasma samples (0.5 mL) were extracted using the acetone/petroleum ether method, lyophilized, and stored at −20 °C until analysis of AVP. Assay sensitivity and intra- and inter-assay coefficients of variation were 0.4 μg/dL; 3.3% and 10.0% for corticosterone; and 0.7 pg/mL, 7.6%, and 12% for AVP. The results were expressed as μg/dL for corticosterone and pg/mL for AVP.
2.9. Cytokine Measurements
Levels of interleukin (IL)-1β (catalog # RLB00), IL-6 (catalog # R6000B), and interleukin-10 (IL-10) (catalog # R1000) were quantified by enzyme-linked immunosorbent assay (ELISA) using commercial kits from R&D Systems (Minneapolis, MN, USA) according to the user manual. The TNF level was quantified by an ELISA kit from Biolegend (catalog 438206) (San Diego, CA, USA) according to the user manual. The results were expressed as cytokine concentration in pg/mL based on standard curves.
2.10. Plasma Nitrite/Nitrate (NOx) Measurement
Plasma samples were deproteinized with 100 µL of absolute ethanol at 4 °C for 30 min. The samples then were centrifuged at 10,000 rpm for 5 min. After deproteinization, 5 µL of samples were injected into a reaction vessel containing vanadium trichloride. The NOx produced was detected as ozone-induced chemiluminescence using the Sievers Instruments NO analyzer (NOA model 280i; Boulder, Colorado). The results were expressed as NOx concentration in μM/L.
2.11. Thiobarbituric Acid Reactive Substance (TBARS) Measurement
Levels of TBARS were quantified by colorimetric assay using a commercial kit from Cayman chemical (#10009055, Michigan, MI, USA) according to the user manual. The results were expressed as TBARS concentration in nmol/L based on standard curve.
2.12. Mean Blood Pressure (MABP) and Heart Rate (HR) Measurements
On the day of the experiment, an arterial catheter was connected to a pressure transducer (TSD104A) and a data acquisition unit (MP100 System; BIOPAC Systems Inc, Santa Barbara, CA, USA) to record the MABP and HR of conscious and freely moving rats. The data were converted and analyzed using AcqKnowledge v.3.9.0 software (BIOPAC Systems Inc, Santa Barbara, CA, USA). A quiet environment was maintained to avoid stress, and the rats had pulsatile arterial pressure recorded at baseline conditions for 30 min. After LPS or saline injection, MABP and HR were then measured as a single time point at 360 min. The results were expressed as the difference from baseline.
2.13. Vascular Reactivity on Thoracic Aorta
After 6 h of LPS administration, the thoracic aorta of each animal of experimental groups (Saline (i.c.v.) + Saline (i.c.v.) + Saline (i.v.) (control group); (2) Saline (i.c.v.) + Ang-(1-7) (i.c.v.) + Saline (i.v.); (3) Saline (i.c.v.) + Saline (i.c.v.) + LPS (i.v.); (4) Saline (i.c.v.) + Ang-(1-7) (i.c.v.) + LPS (i.v.)) was sectioned and cut into four rings of the same size (4 mm) to normalize contractile forces. The rings were kept in two stainless steel stirrups and connected to an isometric force transducer (Letica Scientific Instruments, Barcelona, Spain) in a chamber containing Krebs solution (composition in mmol/L: NaCl 130.0; KCl 4.7; KH2PO4 1.2; MgSO4 1.2; NaHCO3 14.9; C6H12O6 5.5; CaCl2 1.6), pH 7.4, supplied with a gas containing 95% O2 and 5% CO2 at 37 °C. Each ring was stretched to a resting tension of 1.5 g, which was maintained for 60 min for stabilization. In vitro, the rings were subsequently stimulated with phenylephrine (0.1 μmol/L), a selective α1-adrenergic receptor agonist, and the presence or absence of endothelium was verified using acetylcholine (1 μmol/L). Cumulative concentration-effect curves were constructed for norepinephrine (NE) (0.1 μmol/L–10 μmol/L) in the presence or absence of the aminoguanidine, NOS2 selective inhibitor (100 μmol/L), and AVP (1 nmol/L).
2.14. Statistical Analysis
Statistical analyses were performed using Prism 6.0 (GraphPad) software. Osmolality, sodium, lactate, cytokines, NOx, corticosterone, AVP, MABP, HR, and RT-PCR measurements were statistically analyzed by two-way ANOVA followed by the Bonferroni post-hoc test. The maximum constrictor effect (Emax) was considered as the maximal amplitude response reached in the concentration-effect curves for the contractile agent. The concentration of agents that produced half-maximal relaxation amplitude was determined after logit transformation of the normalized concentration-response curves and reported as negative logarithm (pD2) of the mean of individual values. The experimental sample n refers to the number of animals, and data are expressed as the means ± standard error of the mean (SEM). Differences were considered statistically significant when p < 0.05.
3. Results
3.1. Central Ang-(1-7) Attenuated the Lactate Level and Did Not Change Osmolality and Sodium Plasma Levels in Endotoxemia
LPS increased the lactate plasma level as compared with the control group (3.94 ± 0.15 versus 2.01 ± 0.28 mM; F = 20.34, p < 0.0001, respectively). Central administration of Ang-(1-7) attenuated the lactate plasma level in the presence of LPS (2.71 ± 0.22 versus 3.94 ± 0.15 mM; F = 0.0654, p < 0.05, respectively). The statistical analysis showed significant endotoxemia and treatment interaction (F = 6.92, p = 0.0133) for lactate. Neither central Ang-(1-7) nor LPS affected the plasma osmolality and levels of sodium (317.88 ± 5.95 and 311.90 ± 6.70 versus 298.75 ± 5.69 mOsm/Kg; 144.75 ± 0.75 and 144.60 ± 1.06 versus 144.33 ± 0.80 mEq/L, respectively) (Table 1).

Table 1.
Effect of central Ang-(1-7) administration in osmolality, sodium, and lactate plasma levels of the endotoxemic rats.
3.2. Central Administration of Ang-(1-7) Did Not Attenuate Neuroinflammation in Endotoxemia
Neuroinflammatory analysis showed significant effects of LPS on TNF, NOS2, and IL10 gene expression in PVN (F = 54.09, p < 0.0001; F = 6.69, p = 0.8169; F = 14.39, p = 0.0008, respectively) and SON (F = 11.34, p = 0.0022; F = 2.11, p = 0.1582; F = 20.86, p < 0.0001, respectively) (Figure 1A–F). Central administration of Ang-(1-7) did not change the TNF, NOS2, and IL10 gene expression in the hypothalamic nuclei of endotoxemic rats, but there was a tendency to mitigate TNF and elevate IL10 gene expression in PVN (F = 0.1492, p = 0.7025; F = 4.181, p = 0.0515, respectively) (Figure 1A,C).
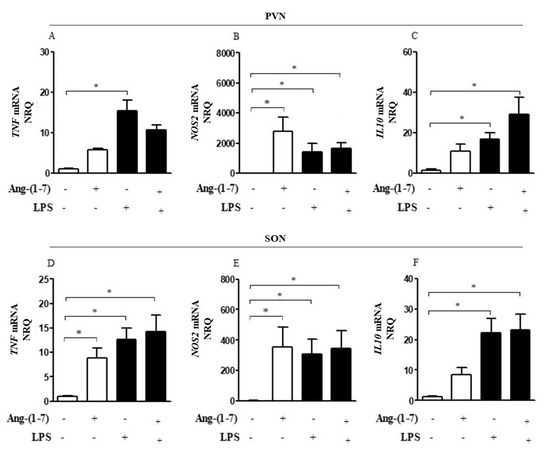
Figure 1.
Central administration of Ang-(1-7) did not attenuate neuroinflammation in endotoxemia. Effect of central Ang-(1-7) on the TNF (A,D), NOS2 (B,E), and IL10 (C,F) gene expression in PVN and SON of rats 6 h after LPS administration (1.5 mg/Kg, i.v.). Ang-(1-7) (0.3 nmol in 2 µL, i.c.v.) was injected 1 min before LPS. Data are expressed as the mean ± SEM, n = 7–9. ANOVA, followed by Bonferroni post-hoc test. * p < 0.05 versus Saline (i.c.v.) + Saline (i.v.) group.
3.3. Central Administration of Ang-(1-7) Attenuated Systemic Inflammation and Restored Plasma AVP Levels in Endotoxemic Rats via Mas Receptor
LPS significantly increased the production of inflammatory markers analyzed including serum and plasma levels of TNF-α, IL-1β, IL-6, IL-10, NOx, and TBARS as compared with the control group (F = 70.12, p < 0.0001; F = 171.70, p < 0.0001; F = 60.02, p < 0.0001; F = 181.60, p < 0.0001; F = 297.90, p < 0.0001; F = 33.99, p < 0.0001, respectively). Central administration of Ang-(1-7) specifically decreased IL-6, NOx, and TBARS, and increased serum IL-10 levels in endotoxemic rats (F = 1.47, p < 0.05; F = 2.88, p = 0.0429; F = 22.75, p < 0.0001; F = 4.99, p = 0.0044, respectively); however, administration of A779 abrogated these peripheral anti-inflammatory responses (Figure 2A–F). Statistical analysis showed significant endotoxemia and central administration of Ang-(1-7) interaction (F = 1.43, p < 0.05; F = 5.18, p = 0.0036; F = 3.08, p = 0.0339; F = 16.55, p < 0.0001) for IL-6, IL-10, NOx, and TBARS levels, respectively.
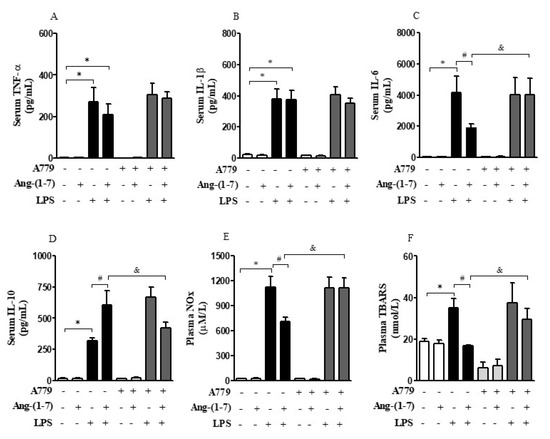
Figure 2.
Central administration of Ang-(1-7) attenuated systemic inflammation in endotoxemic rats via Mas receptor. Effect of central Ang-(1-7) on the TNF-α (A), IL-1β (B), IL-6 (C), IL-10 (D), NOx (E), and TBARS (F) levels in rats 6 h after LPS administration (1.5 mg/Kg, i.v.). A779 (3 nmol in 2 µL, i.c.v.), a selective antagonist for Mas receptor, was injected 30 min before Ang-(1-7) administration (0.3 nmol in 2 µL, i.c.v.). Data are expressed as the mean ± SEM, n = 7–12. ANOVA, followed by Bonferroni post-hoc test. * p < 0.05 versus Saline (i.c.v.) + Saline (i.v.) group, # p < 0.05 versus Saline (i.c.v) + LPS (i.v.) group, and & p < 0.05 versus Ang-(1-7) (i.c.v) + LPS (i.v.) group.
Considering that the central nervous system (CNS) may activate neuroimmune pathways in order to control peripheral inflammation, we analyzed whether central administration of Ang-(1-7) affected the production of corticosterone or AVP (Figure 3A–D). Systemic administration of LPS decreased (F = 0.15, p < 0.05) AVP mRNA in PVN, and Ang-(1-7) prevented this decrease (F = 2.61, p < 0.05) in PVN. Statistical analysis showed significant endotoxemia and central administration of Ang-(1-7) interaction (F = 4.79, p = 0.0382) for AVP mRNA in PVN (Figure 3A). Moreover, central administration of Ang-(1-7) increased AVP plasma levels in endotoxemic rats (F = 3.90, p = 0.0130), and this effect was blocked by central injection of A779. Neither central Ang-(1-7) nor A779 administration affected the levels of corticosterone (F = 0.67, p = 0.5768) in the presence of LPS (Figure 4A,B).
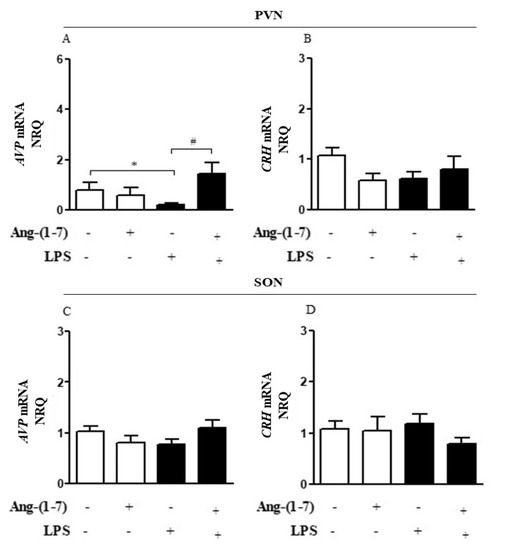
Figure 3.
Central administration of Ang-(1-7) increased AVP gene expression in endotoxemic rats. Effect of central Ang-(1-7) on the AVP (A,C) and CRH (B,D) gene expression in PVN and SON of rats 6 h after LPS administration (1.5 mg/Kg, i.v.). Ang-(1-7) (0.3 nmol in 2 µL, i.c.v.) was injected 1 min before LPS. Data are expressed as the mean ± SEM, n = 7-9. ANOVA, followed by Bonferroni post-hoc test. * p < 0.05 versus Saline (i.c.v.) + Saline (i.v.) group, # p < 0.05 versus Saline (i.c.v) + LPS (i.v.) group.
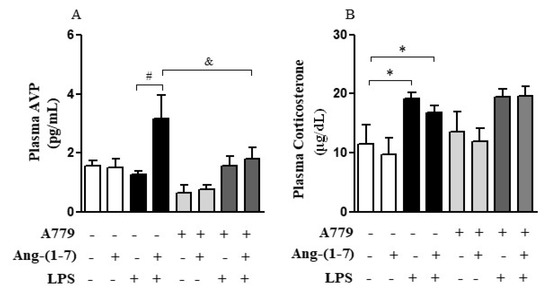
Figure 4.
Central administration of Ang-(1-7) restored plasma AVP levels in endotoxemic rats via Mas receptor. Effect of central Ang-(1-7) on the AVP (A) and corticosterone (B) plasma levels in rats 6 h after LPS administration (1.5 mg/Kg, i.v.). A779 (3 nmol in 2 µL, i.c.v.), a selective antagonist for Mas receptor, was injected 30 min before central Ang-(1-7) administration (0.3 nmol in 2 µL, i.c.v.). Data are expressed as the mean ± SEM, n = 7–10. ANOVA, followed by Bonferroni post-hoc test. * p < 0.05 versus Saline (i.c.v.) + Saline (i.v.) group, # p < 0.05 versus Saline (i.c.v) + LPS (i.v.) group, and & p < 0.05 versus Ang-(1-7) (i.c.v) + LPS (i.v.) group.
3.4. Central Ang-(1-7) Restored Vascular Hyporesponsiveness and Prevented LPS-Induced Hypotension
Systemic administration of LPS decreased the contractile response to NE in the Emax as compared with the control group, and central administration of Ang-(1-7) restored vascular hyporesponsiveness to NE in endotoxemic rats (Figure 5A and Table 2). The addition of aminoguanidine and AVP, in vitro, restored vascular hyporesponsiveness to NE induced by LPS (Figure 5B,C and Table 3 and Table 4). Thus, these results suggest that LPS effects on vascular function were dependent on the production of NO by NOS2 and reduction in the AVP plasma level during endotoxemia.
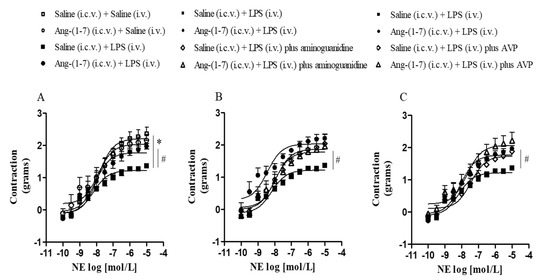
Figure 5.
Central Ang-(1-7) restored vascular hyporesponsiveness in endotoxemic rats. Effect of central Ang-(1-7) (0.3 nmol in 2 µL, i.c.v.) in vascular reactivity of the thoracic aorta to NE in the absence (A) or presence of aminoguanidine, a NOS2 selective inhibitor (100 μmol/L) (B), and AVP (1 nmol/L) (C) in rats 6 h after LPS administration (1.5 mg/Kg, i.v.). Data are expressed as the mean ± SEM, n = 5–7. ANOVA, followed by Bonferroni post-hoc test. * p < 0.05 versus Saline (i.c.v.) + Saline (i.v.) group and # p < 0.05 versus Saline (i.c.v) + LPS (i.v.) group.
Table 2.
Values of maximum constrictor effect (Emax) and pD2 obtained from concentration-response curves in response to NE in thoracic aorta rings of control or endotoxemic rats treated or not with Ang-(1-7).
Table 3.
Values of Emax and pD2 obtained from concentration-response curves in response to NE, in the presence or absence of aminoguanidine (in vitro), in thoracic aorta rings of endotoxemic rats treated or not with Ang-(1-7).
Table 4.
Values of Emax and pD2 obtained from concentration-response curves in response to NE, in the presence or absence of AVP (in vitro), in thoracic aorta rings of endotoxemic rats treated or not with Ang-(1-7).
In our experimental model, LPS induced hypotension (F = 37.11, p < 0.0001) without promoting tachycardia (F = 0.76, p = 0.3935) (Figure 6A,B). Considering that the hyporesponsiveness to vasoconstrictor agents, such as AVP lead to vasoplegia and finally to hypotension, we analyzed whether Ang-(1-7) controls pressor response through the restoration of vascular responsiveness. Central administration of Ang-(1-7) prevented LPS-induced hypotension without affecting the HR (F = 15.20, p = 0.0010; F = 0.92, p = 0.3496, respectively) (Figure 6A,B). The statistical analysis showed significant endotoxemia and central administration of Ang-(1-7) interaction (F = 6.40, p = 0.0205) for MABP measurement.
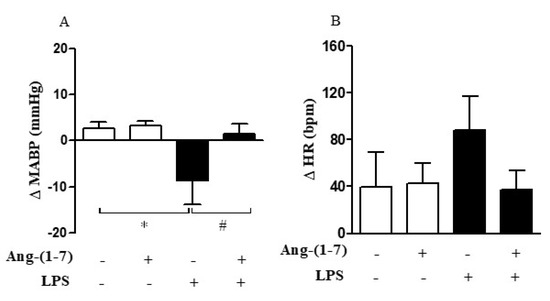
Figure 6.
Central Ang-(1-7) prevented LPS-induced hypotension in endotoxemic rats. Effect of central Ang-(1-7) (0.3 nmol in 2 µL, i.c.v.) on mean arterial blood pressure (MABP) (A) and heart rate (HR) (B) in rats submitted to endotoxemia (LPS, 1.5 mg/Kg, i.v.) for a 6-h period. Data are expressed as the mean ± SEM, n = 6–9. ANOVA, followed by Bonferroni post-hoc test. * p < 0.05 versus Saline (i.c.v.) + Saline (i.v.) group, and # p < 0.05 versus Saline (i.c.v) + LPS (i.v.) group.
4. Discussion
The main findings obtained in the present study were that Ang-(1-7) applied centrally prevented vascular hyporesponsiveness and hypotension by improving AVP impairment and systemic inflammation in endotoxemic rats. This effect is mediated by activation of Mas receptor located in the CNS, and it appears to be dependent on humoral pathway mediated by AVP.
The endotoxemia has been used as a powerful model to study the mechanisms involved in the pathophysiology of systemic inflammation [30]. During systemic inflammation, LPS activates peripheral immune cells causing synthesis and release of relatively high amounts of proinflammatory mediators. Furthermore, peripheral LPS challenge activates microglia, the major active immune cells in the CNS, leading to increased proinflammatory mediator levels in the brain, including elevated expression of TNF-α [31].
Evidence indicates that the RAS could promote pro-inflammatory effects within the hypothalamus, including microglial activation and production of pro-inflammatory mediators [32,33]. However, Ang-(1-7), a protective component of the RAS, exerts direct actions at the microglia to counteract these pro-inflammatory effects, via Mas receptor [34,35]. Mas receptor is expressed on neurons and there is evidence that this receptor is also expressed on microglia, astrocytes, and neurons, including the hypothalamus [15,19,35]. In the present study, we observed the elevated expression of TNF-α in PVN and SON during systemic inflammation, and central Ang-(1-7) administration showed a very clear tendency to promote anti-inflammatory effects particularly on PVN. Although it cannot be considered as being statistically significant, our results have the same anti-inflammatory profile as previous findings [13,19]. Although we haven’t found a potent central anti-inflammatory effect, we reported the systemic anti-inflammatory effect promoted by central Ang-(1-7), via Mas receptor, in endotoxemic rats.
Recent studies have reported elaborate neuroimmune interactions, in which the CNS controls the innate immune system and promotes efferent anti-inflammatory signals to regulate the excessive activation of the immune system. From a neuroimmune perspective, the CNS can regulate the activation of PVN resulting in the stimulation of humoral routes, mainly HPA axis activation and AVP release [36].
The activation of the HPA axis and secretion of corticosterone are clearly important in the control of inflammatory response during the late phase of endotoxemia [37]. Corticosterone exerts anti-inflammatory effects by inhibiting the function of nuclear factor kappa B and consequently modifying at both transcriptional and post-transcriptional levels of the pro-inflammatory genes on peripheral immune cells [38,39]. However, cytokines as well as NO production in hypothalamic nuclei are also critical for the activation of the HPA axis. Evidence has suggested that NO may be involved regulating the activity of the HPA axis, although it remains controversial as to whether NO has a stimulatory or an inhibitory effect on the release of CRH [10,40,41]. LPS-induced endotoxemia causes HPA axis activation through the parvocellular neurons activation that synthesize and secrete CRH in the hypothalamus [42]. Subsequently, CRH as well as AVP induces secretion of adrenocorticotropic hormone by the anterior pituitary and finally glucocorticoids from the adrenal cortex [43]. Our study corroborates the findings of previous studies. As expected, we observed an increase in plasma corticosterone levels after LPS treatment in rats, although no increase in CRH mRNA was observed in our model.
In addition, the increase in AVP mRNA induced by Ang-(1-7) did not potentiate the secretion of CRH, nor the plasma concentration of corticosterone in the rats in which LPS was administered. The use of A779 did not alter the plasma corticosterone level indicating that central Ang-(1-7) administration does not seem to participate in the activation of the HPA axis in our study. In accordance with our results, a recent study showed that ACE2 overexpression mice had no effect on plasma corticosterone under stress conditions [44]. Thus, our data suggest that other humoral pathways, such as AVP release, may mediate the anti-inflammatory induced by Ang-(1-7), in an experimental model of endotoxemia.
HPA axis activation is associated with peripheral anti-inflammatory effects of AVP, composing the humoral network for the control of systemic inflammation. During physiological conditions, the magnocellular neurons of PVN and SON in the hypothalamus synthesize and release AVP [45,46]. During endoxemia, the increase of plasma AVP levels was observed after LPS administration (early phase), followed by a rapid decrease over the next few hours, despite the presence of persisting hypotension [6,11]. In addition, no changes in the AVP stocks were seen in the neurohypophysis [7]. “AVP impairment” refers to the inappropriate decrease of AVP concentration seen in the late phase of a sepsis or endotoxemia model despite the persistent hypotension that can lead to shock and eventually to death [22]. In endotoxemia, AVP impairment in late phase of endotoxemia is associated with an increase in the synthesis of pro-inflammatory mediators during and the consequent late production of NO by NOS2 [47]. The data of the current study showed that the systemic administration of LPS decreased AVP mRNA in PVN and did not change the plasma AVP concentration even in the face of hypotension and exacerbated systemic inflammatory response, suggesting the occurrence of AVP impaired in our experimental model.
NO has been reported to participate in the modulation of AVP secretion; however, studies to date have produced contradictory evidence regarding the effect of NO in secretion of this hormone during systemic inflammation [11,39,48]. Ota et al. [49] and Yamaguchi, Watanabe, and Yamaya [50] showed that central injection of an NO donor caused an increase in plasma AVP concentration; whereas Carnio et al. [51] and Reis et al. [52] suggest that NO arising from NOS2 plays an important inhibitory role in AVP release during endotoxemia. The mechanism by which NO can modulate AVP release is not well established. It has been shown that NO can act directly or not inhibiting the postsynaptic activity of neurohypophysial neurons [53]. The data of the current study showed that Ang-(1-7) applied centrally has not reduced the increase in gene expression of NOS2 in PVN and SON induced by systemic LPS. Interestingly, we observed that expression of NOS2 in PVN did not affect the Ang-(1-7) effect on AVP secretion, suggesting that NO showed no inhibitory effect in our experimental model. Similarly, Nomura et al. [9] did not observe any effect of the NOS2 gene disruption on AVP mRNA levels in the mouse hypothalamus. In addition, we also cannot exclude the possibility that the NOS2 posttranslational modifications have occurred, resulting in an impairment in NOS.
In this context, classic studies revealed that Ang-(1-7) induced AVP release in rat hypothalamus–pituitary explants with a potency equal to Ang II [17], whereas other studies indicated that central Ang-(1-7) administration did not change AVP release in basal conditions in rats [54]. In PVN, this peptide is a potent secretagogue of AVP and may participate in controlling its release by magnocellular neurons [17]. In fact, Qadri et al. [55] confirmed the first study performed in neurohypophysial explants by Schiavone et al. [16], showing that Ang-(1-7) microinjections into the PVN induce a release of AVP. The excitatory action of Ang-(l-7) on magnocellular neurons of the PVN provides evidence at a cellular level for a modulatory action of this heptapeptide on the regulation of vasopressin secretion [56]. Although these studies point to contrasting effects of Ang-(1-7) on the release of AVP, together they strengthen the concept that distinct Ang-(1-7) effects can be observed in specific brain areas depending on the existing pathophysiological condition [56]. Moriguchi et al. [57], evaluating the effect of i.c.v. Ang-(1-7) on the synthesis of AVP in the PVN of hypertensive rats, observed that the exogenous infusion of Ang-(1-7) proved to be as potent as to Ang II in stimulating AVP synthesis, reinforcing the role of this heptapeptide in the hydro-electrolytic control carried out by the PVN. Moreover, although there are classic studies demonstrating the effect of the release of AVP by Ang-(1-7), the role in which this heptapeptide promotes this action is not clear. Specifically in PVN, Ambuhl et al. [58] demonstrated that applications by Ang-(1-7) microiontophoresis resulted in an increase in the excitability of neurons in this region, this effect being blocked by A779. In our study, the pharmacological blockage of central Mas receptor blocked the rise in AVP plasma levels induced by central administration of Ang-(1-7) in endotoxemic rats. Considering the previously demonstrated expression of the Mas receptor in the hypothalamus [19], we suggest that Ang-(1-7) may have affected the electrical activity of the paraventricular magnocellular neurons to promote AVP secretion in our current study. However, we cannot exclude the possibility that other neuroimmune routes participate in Ang-(1-7) anti-inflammatory effects in our experimental model. Our group recently demonstrated that in early stage of endotoxemia, Ang-(1-7) elicits anti-inflammatory effects through the activation of a neuroimmune pathway involving central activation of Mas receptors and subsequent sympathetic autonomic signaling [21].
Although the number of studies demonstrating the systemic anti-inflammatory effects of AVP is still limited, AVP has been shown to have direct action on immune cells to control systemic inflammation. In cultured rat mesangial cells, AVP inhibits LPS- and IL-1β-stimulated NO and cGMP via V1 receptor [23], whereas in murine macrophages [25,59] AVP promotes anti-inflammatory effects by the inhibition of CD14 expression, endotoxin binding, and subsequent NF-κB activation. Regarding the peripheral anti-inflammatory effects of PVA, previous data highlighted the involvement of the V2 receptor in the reduction of sepsis-induced lung inflammation [60]. In the present study, we do not use AVP receptor antagonists as a pharmacological tool; however, we suggest that AVP may have mediated the attenuation of the systemic inflammatory response during endotoxemia by direct action on its receptors expressed in the cells of the immune system.
During endotoxemia, the vascular reactivity and endothelial barrier function are impaired, and this contributes to the hypotensive response [21]. In the vascular endothelium, LPS can cause synthesis and release of relatively high amounts of pro-inflammatory cytokines, which in turn produces relaxation of vascular smooth muscle tone resulting in hypotension and reduction of vasoconstrictor response to catecholamines [2,3]. IL-6 and NO are two well-recognized inflammatory mediators upregulated in inflammatory models, whereas IL-10 plays an immunoregulatory role inhibiting vascular IL-6 production [61]. Interestingly, IL-6 increases NOS2 activity in aortic smooth muscle cells and is associated with a drop in MABP in septic patients [62,63]. Previous studies of our group also showed that hypotension induced by LPS is dependent on NO release [4,5,6]. In this context, glucocorticoids and AVP also regulate different aspects of endothelial physiology during systemic inflammation. Glucocorticoids act as a negative regulator of NO and prostacyclin release in endothelial cells, whereas in the vascular smooth muscle cells, it increases arterial contractile sensitivity to NE and vascular resistance [64,65]. In respect to endogenous AVP, in addition to their anti-inflammatory property, it exerts a potent vasoconstrictor effect during systemic inflammation. In fact, concomitant glucocorticoid and AVP therapy may be associated with a survival benefit in septic patients [66]. The central Ang-(1-7) administration in our study attenuated hypotension and vascular hyporesponsiveness by reduction of NO production in endotoxemic rats. Herein, we reported that Ang-(1-7) reestablished vascular responsiveness to NE and AVP in the endotoxemia model. Based on these data, we speculate that the effect of Ang-(1-7) on vascular responsiveness is the result of the plasma reduction of IL-6, the systemic increase of IL-10, and the consequent decrease in NO production. Furthermore, Ang-(1-7) applied centrally has not been shown to have an effect on HR. Tachycardia, in turn, has been reported as a reflex compensatory response of hypotension and alterations can indicate reduction in the spontaneous baroreflex sensitivity during systemic inflammation [30]. In our experimental model, despite the tendency to increase the HR of endotoxemic animals, this response was not significant. Studies have already shown that Ang-(1-7) in the CNS improves baroreflex sensitivity [34,67]. Thus, although we did not observe the tachycardic response, we found an improvement in MABP accompanied by the restoration of HR in endotoxemic rats, suggesting a possible effect of Ang- (1-7) on baroreflex sensitivity. Moreover, hyperlactatemia is also used as an important biochemical marker of the progression of the systemic inflammation and the associated hypotension in endotoxemia models [6]. The improvement in vascular responsiveness, in the presence of AVP, may have led to the attenuation of the hypotensive response in endotoxemic animals treated with Ang-(1-7). Therefore, possibly the improvement of LPS-induced hypotension by Ang-(1-7) provides adequate perfusion in the endotoxemic rats, controlling hyperlactatemia in our study.
In conclusion, our data demonstrate the participation of central Ang-(1-7), via Mas receptor, on modulation of peripheral inflammation and on pressor response during endotoxemia. Although we have demonstrated the importance of the Ang-(1-7)/Mas receptor axis for the control of the inflammatory response in the endotoxemic animal, the present study has limitations. First, as in classic studies, we demonstrated the stimulating effect of the Ang-(1-7)/Mas receptor axis on the release of AVP during endotoxemia; however, we did not evaluate the exact mechanism by which this effect occurred in our study. Further studies are needed to investigate the exact mechanism by which central administration of Ang-(1-7) induces the synthesis and release of AVP in PVN in the experimental model of endotoxemia. Second, we did not use AVP receptor antagonists as a pharmacological tool in our study to assess the participation of AVP by mediating the peripheral effects of central Ang-(1-7) administration. Thus, mechanisms of these effects are not precisely elucidated, but our results suggest a strong participation of the humoral pathway mediated by AVP regulating the effects resulting from the Ang-(1-7) applied centrally.
Author Contributions
P.P. and E.C.C. conceived, designed the research, and wrote the manuscript; P.P., F.d.L.F., and M.E.B. performed the experiments; R.L. performed RT-PCR assay and interpreted the results; P.P. and L.M.B. interpreted the vascular reactivity results; J.A.-R. kindly contributed to radioimmunoassay assays; P.P., F.d.L.F., A.M.S., R.L., and E.C.C. wrote the manuscript, which has been approved by all the authors; E.C.C. supervised the project and provided funding. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the São Paulo Research Foundation (FAPESP–Brazil), grants: 2011/20343-4, 2014/22477-6 and 2015/09857-7 and the Coordination of Improvement of Higher Education Personnel (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior -CAPES).
Institutional Review Board Statement
Experimental procedures were approved by Ethics Committee on Animal Experimentation of the Ribeirão Preto College of Nursing, University of São Paulo (CEUA Protocol 14.1.872.53.4).
Informed Consent Statement
Not applicable.
Data Availability Statement
We included individual values in each figure which show all the data in the paper itself. The raw data supporting the findings of this manuscript will be provided by the authors at any time to the reviewers and thereafter to any researcher after publication in Cells.
Acknowledgments
We thank Dra Terezila Machado Coimbra, who kindly allowed us to analyze osmolality and sodium of the samples in her laboratory, and Juliana A. Vercesi, Milene Mantovani, and Maria Valci dos Santos for their excellent technical assistance.
Conflicts of Interest
The authors have no financial conflict of interest.
References
- Andersson, U.; Tracey, K.J. Neural reflexes in inflammation and immunity. J. Exp. Med. 2012, 209, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Hallemeesch, M.M.; Janssen, B.J.; de Jonge, W.J.; Soeters, P.B.; Lamers, W.H.; Deutz, N.E. NO production by cNOS and iNOS reflects blood pressure changes in LPS-challenged mice. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E871–E875. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.J.; Li, S.Y.; Shih, C.C.; Liao, M.H.; Wu, C.C. NO contributes to abnormal vascular calcium regulation and reactivity induced by peritonitis-associated septic shock in rats. Shock 2010, 33, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Stabile, A.M.; Moreto, V.; Antunes-Rodrigues, J.; Carnio, E.C. Participation of the inducible nitric oxide synthase on atrial natriuretic peptide plasma concentration during endotoxemic shock. Regul. Pept. 2007, 140, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Batalhão, M.E.; Moreto, V.; Stabile, A.M.; Antunes-Rodrigues, J.; Carnio, E.C. Role of dexamethasone on vasopressin release during endotoxemic shock. Regul. Pept. 2008, 147, 67–71. [Google Scholar] [CrossRef]
- Saia, R.S.; Bertozi, G.; Mestriner, F.L.; Antunes-Rodrigues, J.; Queiróz Cunha, F.; Cárnio, E.C. Cardiovascular and inflammatory response to cholecystokinin during endotoxemic shock. Shock 2013, 39, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Giusti-Paiva, A.; Ruginsk, S.G.; de Castro, M.; Elias, L.L.; Carnio, E.C.; Antunes-Rodrigues, J. Role of nitric oxide in lipopolysaccharide-induced release of vasopressin in rats. Neurosci. Lett. 2003, 346, 21–24. [Google Scholar] [CrossRef]
- Stern, J.E. Nitric oxide and homeostatic control: An intercellular signalling molecule contributing to autonomic and neuroendocrine integration? Prog. Biophys. Mol. Biol. 2004, 84, 197–215. [Google Scholar] [CrossRef]
- Nomura, M.; Tsutsui, M.; Shimokawa, H.; Fujimoto, N.; Ueta, Y.; Morishita, T.; Yanagihara, N.; Matsumoto, T. Effects of nitric oxide synthase isoform deletion on oxytocin and vasopressin messenger RNA in mouse hypothalamus. Neuroreport 2005, 15, 413–417. [Google Scholar] [CrossRef]
- Akasaka, S.; Nomura, M.; Nishii, H.; Fujimoto, N.; Ueta, Y.; Tsutsui, M.; Shimokawa, H.; Yanagihara, N.; Matsumoto, T. The hypothalamo-pituitary axis responses to lipopolysaccharide-induced endotoxemia in mice lacking inducible nitric oxide synthase. Brain Res. 2006, 1089, 1–9. [Google Scholar] [CrossRef]
- Corrêa, P.B.; Pancoto, J.A.; de Oliveira-Pelegrin, G.R.; Cárnio, E.C.; Rocha, M.J. Participation of iNOS-derived NO in hypothalamic activation and vasopressin release during polymicrobial sepsis. J. Neuroimmunol. 2007, 183, 17–25. [Google Scholar] [CrossRef]
- Rotondo, F.; Butz, H.; Syro, L.V.; Yousef, G.M.; Di Ieva, A.; Restrepo, L.M.; Quintanar-Stephano, A.; Berczi, I.; Kovacs, K. Arginine vasopressin (AVP): A review of its historical perspectives, current research and multifunctional role in the hypothalamo-hypophysial system. Pituitary 2016, 19, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Gao, L.; Gun, J.; Lu, J.; Wang, Y.; Zhang, Y. Suppressing inflammation by inhibiting NF-κB pathway contributes to the neuroprotection of angiotensin-(1-7) in rats with permanent cerebral ischemia. Br. J. Pharmacol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues Prestes, T.R.; Rocha, N.P.; Miranda, A.S.; Teixeira, A.L.; Simoes-e-Silva, A.C. The Anti-Inflammatory Potential of ACE2/Angiotensin-(1-7)/Mas Receptor Axis: Evidence from Basic and Clinical Research. Curr. Drug Targets 2017, 18, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Becker, L.K.; Etelvino, G.M.; Walther, T.; Santos, R.A.S.; Campagnole-Santos, M.J. Immunofluorescence localization of the receptor Mas in cardiovascular-related areas of the rat brain. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1416–H1424. [Google Scholar] [CrossRef]
- Santos, R.A.S.; e Silva, A.C.S.; Maric, C.; Silva, D.M.R.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.B.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef]
- Schiavone, M.T.; Santos, R.A.; Brosnihan, K.B.; Khosla, M.C.; Ferrario, C.M. Release of vasopressin from the rat hypothalamo-neurohypophysial system by angiotensin-(1-7) heptapeptide. Proc. Natl. Acad. Sci. USA 1988, 85, 4095–4098. [Google Scholar] [CrossRef]
- Souza, L.L.; Costa-Neto, C.M. Angiotensin-(1-7) decreases LPS-induced inflammatory response in macrophages. J. Cell. Physiol. 2012, 227, 2117–2122. [Google Scholar] [CrossRef]
- Regenhardt, R.W.; Desland, F.; Mecca, A.P.; Pioquinto, D.J.; Afzal, A.; Mocco, J.; Sumners, C. Anti-inflammatory effects of angiotensin-(1-7) in ischemic stroke. Neuropharmacology 2013, 71, 154–163. [Google Scholar] [CrossRef]
- Souza, L.L.; Duchene, J.; Todiras, M.; Azevedo, L.C.P.; Costa-Neto, C.M.; Alenina, N.; Santos, R.A.; Bader, M. Receptor Mas Protects Mice Against Hypothermia and Mortality Induced By Endotoxemia. Shock 2014, 41, 331–336. [Google Scholar] [CrossRef]
- Passaglia, P.; Faim, F.L.; Batalhão, M.E.; Bendhack, L.M.; Antunes-Rodrigues, J.; Ulloa, L.; Kanashiro, A.; Carnio, E.C. Central angiotensin-(1-7) attenuates systemic inflammation via activation of sympathetic signaling in endotoxemic rats. Brain Behav. Immun. 2020, 88, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Landry, D.W.; Levin, H.R.; Gallant, E.M.; Ashton, R.C., Jr.; Seo, S.; D’Alessandro, D.; Oz, M.C.; Oliver, J.A. Vasopressin deficiency contributes to the vasodilation of septic shock. Circulation 1997, 95, 1122–1125. [Google Scholar] [CrossRef] [PubMed]
- Umino, T.; Kusano, E.; Muto, S.; Akimoto, T.; Yanagiba, S.; Ono, S.; Amemiya, M.; Ando, Y.; Homma, S.; Ikeda, U.; et al. AVP inhibits LPS- and IL-1beta-stimulated NO and cGMP via V1 receptor in cultured rat mesangial cells. Am. J. Physiol. 1999, 276, F433–F441. [Google Scholar] [CrossRef] [PubMed]
- Pittman, Q.J. A neuro-endocrine-immune symphony. J. Neuroendocrinol. 2011, 23, 1296–1297. [Google Scholar] [CrossRef]
- Jan, W.; Kao, M.; Yang, C.; Chang, Y.; Huang, C. Phosphoinositide 3-Kinase Is Involved in Mediating the Anti-inflammation Effects of Vasopressin. Inflammation 2017, 40, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates; Academic Press: New York, NY, USA, 2007. [Google Scholar]
- Saramago, E.A.; Borges, G.S.; Singolani, C.G., Jr.; Nogueira, J.E.; Soriano, R.N.; Cárnio, E.C.; Branco, L.G.S. Molecular hydrogen potentiates hypothermia and prevents hypotension and fever in LPS-induced systemic inflammation. Brain Behav. Immun. 2019, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Faim, F.; Passaglia, P.; Batalhao, M.; Lacchini, R.; Stabile, A.M.; Carnio, E.C. Role of ghrelin on growth hormone/insulin-like growth factor-1 axis during endotoxemia. Growth Horm. IGF Res. 2019, 48–49, 36–44. [Google Scholar] [CrossRef]
- Vecsei, P. Glucocorticoids: Cortisol, corticosterone and compounds. In Methods of Hormone Radioimmunoassay; Jaffe, B.M., Behrman, H.R., Eds.; Academic Press: New York, NY, USA, 1979; pp. 767–796. [Google Scholar]
- Amorim, M.R.; de Deus, J.L.; Cazuza, R.A.; Mota, C.M.D.; da Silva, L.E.V.; Borges, G.S.; Batalhão, M.E.; Cárnio, E.C.; Branco, L.G.S. Neuroinflammation in the NTS is associated with changes in cardiovascular reflexes during systemic inflammation. J. Neuroinflamm. 2019, 16, 125. [Google Scholar] [CrossRef]
- Hoogland, I.C.M.; Houbolt, C.; van Westerloo, D.J.; van Gool, W.A.; van de Beek, D. Systemic inflammation and microglial activation: Systematic review of animal experiments. J. Neuroinflamm. 2015, 12, 114. [Google Scholar] [CrossRef]
- Sriramula, S.; Cardinale, J.P.; Lazartigues, E.; Francis, J. ACE2 overexpression in the paraventricular nucleus attenuates angiotensin II-induced hypertension. Cardiovasc. Res. 2011, 92, 401–408. [Google Scholar] [CrossRef]
- Xia, H.; Suda, S.; Bindom, S.; Feng, Y.; Gurley, S.B.; Seth, D.; Navar, G.L.; Lazartigues, E. ACE2-mediated reduction of oxidative stress in the central nervous system is associated with improvement of autonomic function. PLoS ONE 2011, 6, e22682. [Google Scholar] [CrossRef] [PubMed]
- Gironacci, M.M.; Cerniello, F.M.; Longo Carbajosa, N.A.; Goldstein, J.; Cerrato, B.D. Protective axis of the renin-angiotensin system in the brain. Clin. Sci. 2014, 127, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Shi, P.; Sumners, C. Direct anti-inflammatory effects of angiotensin-(1-7) on microglia. J. Neurochem. 2016, 136, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, V.A.; Chavan, S.S.; Tracey, K.J. Molecular and Functional Neuroscience in Immunity. Annu. Rev. Immunol. 2018, 36, 783–812. [Google Scholar] [CrossRef]
- Vandewalle, J.; Libert, C. Glucocorticoids in Sepsis: To Be or Not to Be. Front. Immunol. 2020, 11, 1318. [Google Scholar] [CrossRef]
- Clark, A.R. Anti-inflammatory functions of glucocorticoid-induced genes. Mol. Cell Endocrinol. 2007, 275, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Bellavance, M.; Rivest, S. The HPA-Immune Axis and the Immunomodulatory Actions of Glucocorticoids in the Brain. Front Immunol. 2014, 5, 136. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.O.; Rivier, C. Microinfusion of a nitric oxide donor in discrete brain regions activates the hypothalamic-pituitary-adrenal axis. J. Neuroendocrinol. 2001, 13, 925–933. [Google Scholar] [CrossRef]
- Seo, D.O.; Lee, S.; Rivier, C. Comparison between the influence of the intravenous and intracerebroventricular injection of a nitric oxide donor on adrenocorticotropic hormone release and hypothalamic neuronal activity. J. Neuroendocrinol. 2002, 14, 568–573. [Google Scholar] [CrossRef]
- Rorato, R.; Castro, M.; Borges, B.C.; Benedetti, M.; Germano, C.M.; Antunes-Rodrigues, J.; Elias, L.L. Adrenalectomy enhances endotoxemia-induced hypophagia: Higher activation of corticotrophin-releasing-factor and proopiomelanocortin hypothalamic neurons. Horm. Behav. 2008, 54, 134–142. [Google Scholar] [CrossRef]
- Dantzer, R. Neuroimmune Interactions: From the Brain to the Immune System and Vice Versa. Physiol. Rev. 2018, 98, 477–504. [Google Scholar] [CrossRef] [PubMed]
- Kloet, A.D.; Cahill, K.M.; Scott, K.A.; Krause, E.G. Overexpression of angiotensin converting enzyme 2 reduces anxiety-like behavior in female mice. Physiol. Behav. 2020, 224, 113002. [Google Scholar] [CrossRef] [PubMed]
- Antunes-Rodrigues, J.; de Castro, M.; Elias, L.L.; Valença, M.M.; McCann, S.M. Neuroendocrine control of body fluid metabolism. Physiol. Rev. 2004, 84, 169–208. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, E.T., Jr.; Sawchenko, P.E. Reflex control of magnocellular vasopressin and oxytocin secretion. Trends Neurosci. 1991, 14, 406–411. [Google Scholar] [CrossRef]
- Costa, L.H.A.; Júnior, N.N.S.; Catalão, C.H.R.; Sharshar, T.; Chrétien, F.; Rocha, M.J.A. Vasopressin Impairment during Sepsis Is Associated with Hypothalamic Intrinsic Apoptotic Pathway and Microglial Activation. Mol. Neurobiol. 2017, 54, 5526–5533. [Google Scholar] [CrossRef]
- Kadekaro, M. Nitric oxide modulation of the hypothalamo-neurohypophyseal system. Braz. J. Med. Biol. Res. 2004, 37, 441–450. [Google Scholar] [CrossRef]
- Ota, M.; Crofton, J.T.; Festavan, G.T.; Share, L. Evidence that nitric oxide can act centrally to stimulate vasopressin release. Neuroendocrinology 1993, 57, 955–959. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Watanabe, K.; Yamaya, K. Evaluation for roles of nitric oxide generated in the anteroventral third ventricular region in controlling vasopressin secretion and cardiovascular system of conscious rats. Eur. J. Endocrinol. 2000, 143, 523–533. [Google Scholar] [CrossRef][Green Version]
- Carnio, E.C.; Stabile, A.M.; Batalhão, M.E.; Silva, J.S.; Antunes-Rodrigues, J.; Branco, L.G.; Magder, S. Vasopressin release during endotoxaemic shock in mice lacking inducible nitric oxide synthase. Pflug. Arch. 2005, 450, 390–394. [Google Scholar] [CrossRef]
- Reis, W.L.; Giusti-Paiva, A.; Ventura, R.R.; Margatho, L.O.; Gomes, D.A.; Elias, L.L.K.; Antunes-Rodrigues, J. Central nitric oxide blocks vasopressin, oxytocin and atrial natriuretic peptide release and antidiuretic and natriuretic responses induced by central angiotensin II in conscious rats. Exp. Physiol. 2007, 92, 903–911. [Google Scholar] [CrossRef]
- Zhang, J.; Snyder, S.H. Nitric oxide in the nervous system. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 213–233. [Google Scholar] [CrossRef] [PubMed]
- Mahon, J.M.; Allen, M.; Herbert, J.; Fitzsimons, J.T. The association of thirst, sodium appetite and vasopressin release with c-fos expression in the forebrain of the rat after intracerebroventricular injection of angiotensin II, angiotensin-(1-7) or carbachol. Neuroscience 1995, 69, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Qadri, F.; Wolf, A.; Waldmann, T.; Rascher, W.; Unger, T. Sensitivity of hypothalamic paraventricular nucleus to C- and N-terminal angiotensin fragments: Vasopressin release and drinking. J. Neuroendocrinol. 1998, 10, 275–281. [Google Scholar] [PubMed]
- Felix, D.; Khosla, M.C.; Barnes, K.L.; Imboden, H.; Montani, B.; Ferrario, C.M. Neurophysiological responses to angiotensin-(1-7). Hypertension 1991, 17, 1111–1114. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, A.; Ferrario, C.M.; Brosnihan, K.B.; Ganten, D.; Morris, M. Differential regulation of central vasopressin in transgenic rats harboring the mouse Ren-2 gene. Am. J. Physiol. 1994, 267, R786–R791. [Google Scholar] [CrossRef]
- Abdühl, P.; Felix, D.; Khosla, M.C. [7-D-Ala]-Angiotensin-(1-7): Selective antagonism of angiotensin-(1-7) in the rat paraventricular nucleus. Brain Res. Bull. 1994, 35, 289–291. [Google Scholar] [CrossRef]
- Chang, Y.; Yang, C.; Wang, S.; Kao, M.; Tsai, P.; Huang, C. Vasopressin inhibits endotoxin binding in activated macrophages. J. Surg. Res. 2015, 197, 412–418. [Google Scholar] [CrossRef]
- Boyd, J.H.; Holmes, C.L.; Wang, Y.; Roberts, H.; Walley, K.R. Vasopressin decreases sepsis-induced pulmonary inflammation through the V2R. Resuscitation 2008, 79, 325–331. [Google Scholar] [CrossRef]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar] [CrossRef]
- Meduri, G.U.; Headley, S.; Kohler, G. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS. Plasma IL-1 beta and IL-6 levels are consistent and efficient predictors of outcome over time. Chest 1995, 107, 1062–1073. [Google Scholar] [CrossRef]
- Recoquillon, S.; Carusio, N.; Lagrue-Lakhal, A.H. Interaction in endothelium of non-muscular myosin light-chain kinase and the NF-κB pathway is critical to lipopolysaccharide-induced vascular hyporeactivity. Clin. Sci. 2015, 129, 687–698. [Google Scholar] [CrossRef]
- Duma, D.; Silva-Santos, J.E.; Assreuy, J. Inhibition of glucocorticoid receptor binding by nitric oxide in endotoxemic rats. Crit. Care Med. 2004, 32, 2304–2310. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, L. Glucocorticoids and vascular reactivity. Curr. Vasc. Pharmacol. 2004, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Torgersen, C.; Luckner, G.; Schröder, D.C.H.; Schmittinger, C.A.; Rex, C.; Ulmer, H.; Dünser, M.W. Concomitant arginine-vasopressin and hydrocortisone therapy in severe septic shock: Association with mortality. Intensive Care Med. 2011, 37, 1432–1437. [Google Scholar] [CrossRef][Green Version]
- Britto, R.R.; Santos, R.A.; Fagundes-Moura, C.R.; Khosla, M.C.; Campagnole-Santos, M.J. Role of angiotensin-(1-7) in the modulation of the baroreflex in renovascular hypertensive rats. Hypertension 1997, 30, 549–556. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).