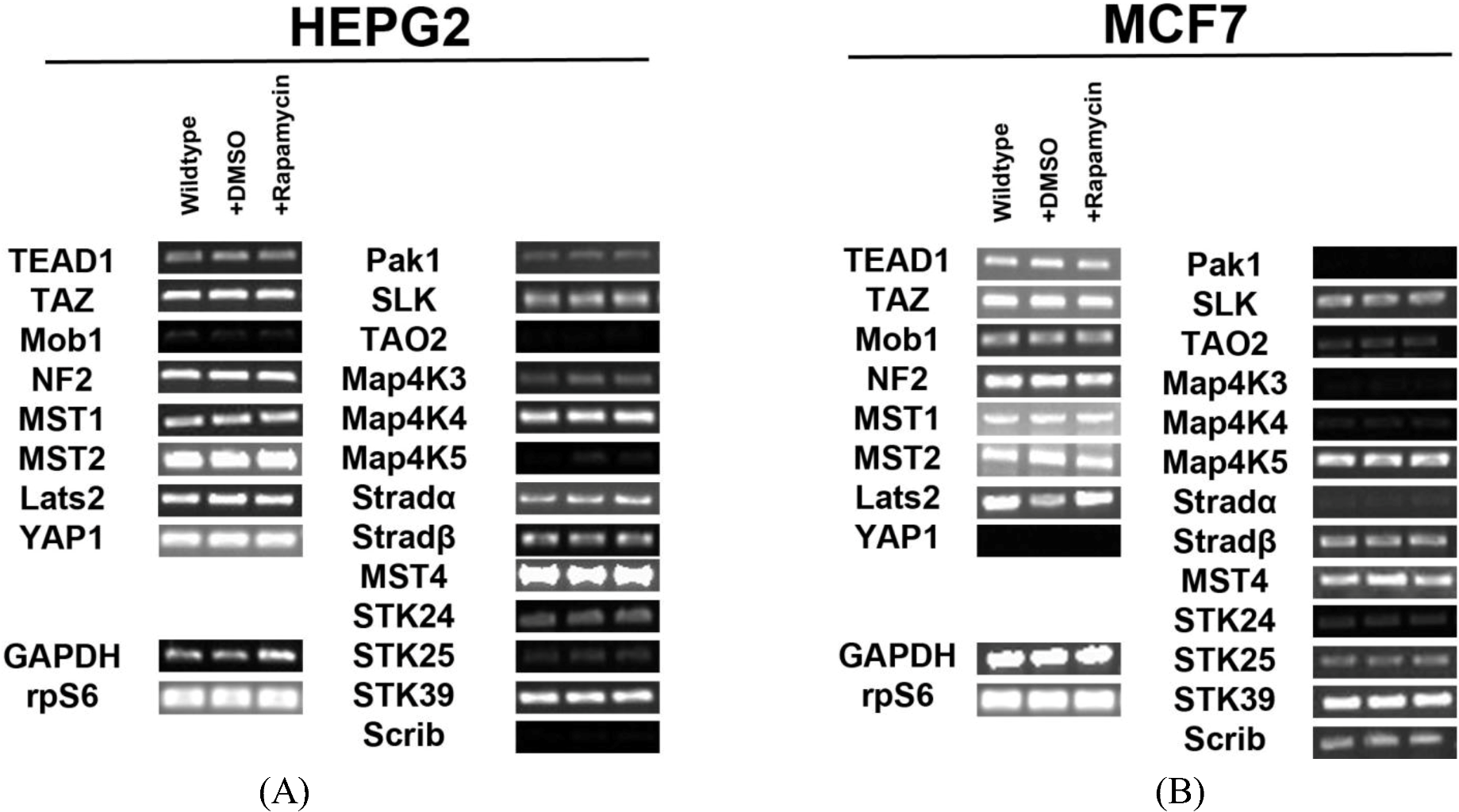
2.1. Hippo Pathway Components and STK Kinases Are Expressed in HepG2 and MCF7 Cell Lines
In order to determine the regulation of the SWH pathway in the HepG2 cell line, we first tested for expression of transcripts for the core components of the pathway and Ste20-like kinases. We identified expression of all the components involved in Hippo signaling (Mob1, NF2, MST1/2, Lats2) and their downstream effectors (TEAD1, TAZ, YAP1) in HepG2 cells. We also found that several Ste20-like kinases are expressed in HepG2 cells. These kinases include: Pak1, SLK, TAO2, MAP4K3, MAP4K4, MAP4K5, Stradα/β, MST4, STK24 (MST3), STK25, STK39 and Scrib (
Figure 1A). Other genes tested for but not expressed include: Fat2, YSK4 and TAO3. Pak1, TAO2, MAP4K3, MAP4K5, STK24 (MST3), STK25, and Scrib are expressed at low levels.
Figure 1.
Expression of Hippo pathway components and Ste20-like kinases in HepG2 and MCF7 cell lines. (A) Transcript expression levels of Hippo pathway components (left) and Ste20-like kinases (right) do not change in response to rapamycin treatment in HepG2 cells. Mob1, TAO2, Map4K5 and Scrib are expressed at very low levels. (B) Expression levels of Hippo pathway components and Ste20-like kinases transcripts do not change in response to rapamycin treatment in MCF7 cells. Pak1, TAO2, Map4K3, Map4K4, Stradα and STK24 are expressed at low levels. YAP1 is not expressed. Note: Mob1, Map4K5, STK25 and Scrib are expressed at higher levels in MCF7 cells than in HepG2 cells, while Map4K4 and Stradα are expressed at lower levels in MCF7 cells than in HepG2 cells. For both cell lines, GAPDH and the ribosomal protein rpS6 are included as housekeeping gene controls.
Figure 1.
Expression of Hippo pathway components and Ste20-like kinases in HepG2 and MCF7 cell lines. (A) Transcript expression levels of Hippo pathway components (left) and Ste20-like kinases (right) do not change in response to rapamycin treatment in HepG2 cells. Mob1, TAO2, Map4K5 and Scrib are expressed at very low levels. (B) Expression levels of Hippo pathway components and Ste20-like kinases transcripts do not change in response to rapamycin treatment in MCF7 cells. Pak1, TAO2, Map4K3, Map4K4, Stradα and STK24 are expressed at low levels. YAP1 is not expressed. Note: Mob1, Map4K5, STK25 and Scrib are expressed at higher levels in MCF7 cells than in HepG2 cells, while Map4K4 and Stradα are expressed at lower levels in MCF7 cells than in HepG2 cells. For both cell lines, GAPDH and the ribosomal protein rpS6 are included as housekeeping gene controls.
Similar findings were observed for the MCF7 cell line (
Figure 1B). We identified the expression of Hippo signaling components (Mob1, NF2, MST1/2, Lats2) and their targets TEAD1 and TAZ in these cells. Importantly, we noted the absence of YAP transcripts. This result was further confirmed through immunocytochemical analysis (
Figure S1). Ste20-like kinases expressed include: Pak1, SLK, TAO2, MAP4K3, MAP4K4, MAP4K5, Stradα/β, STK24 (MST3), MST4, STK25, STK39, and Scrib. Other genes tested for but not expressed include: Fat2, YSK4 and TAO3. Mob1, MAP4K5, STK25 and Scrib are expressed at higher levels in MCF7 cells than in HepG2 cells, while MAP4K4 and Stradα are expressed at lower levels in MCF7 cells than in HepG2 cells.
2.2.Inhibition of mTOR Signaling Does Not Affect Transcription of Hippo and Ste20-like Kinases in HepG2 and MCF7 Cell LinesSignaling through mTOR occurs through two different complexes: mTORC1 and mTORC2 [
14]. Rapamycin inhibits mTORC1 by inhibiting the interaction of Raptor with mTOR [
15,
16]. To study the effect of mTOR on Hippo signaling, we blocked mTOR activity by adding rapamycin to HepG2 cells and re-examined expression of Hippo components and Ste20-like kinases. We observed no changes in expression of pathway components and Ste20-like kinases when cells were treated with rapamycin (
Figure 1A). Similar studies in MCF7 cells confirmed the same results (
Figure 1B). Taken altogether, these results demonstrate that mTOR signaling does not regulate Hippo or Ste20-like kinase levels at the transcriptional level.
2.2. Rapamycin Alters TEAD1 Protein Expression in MCF7 Cells
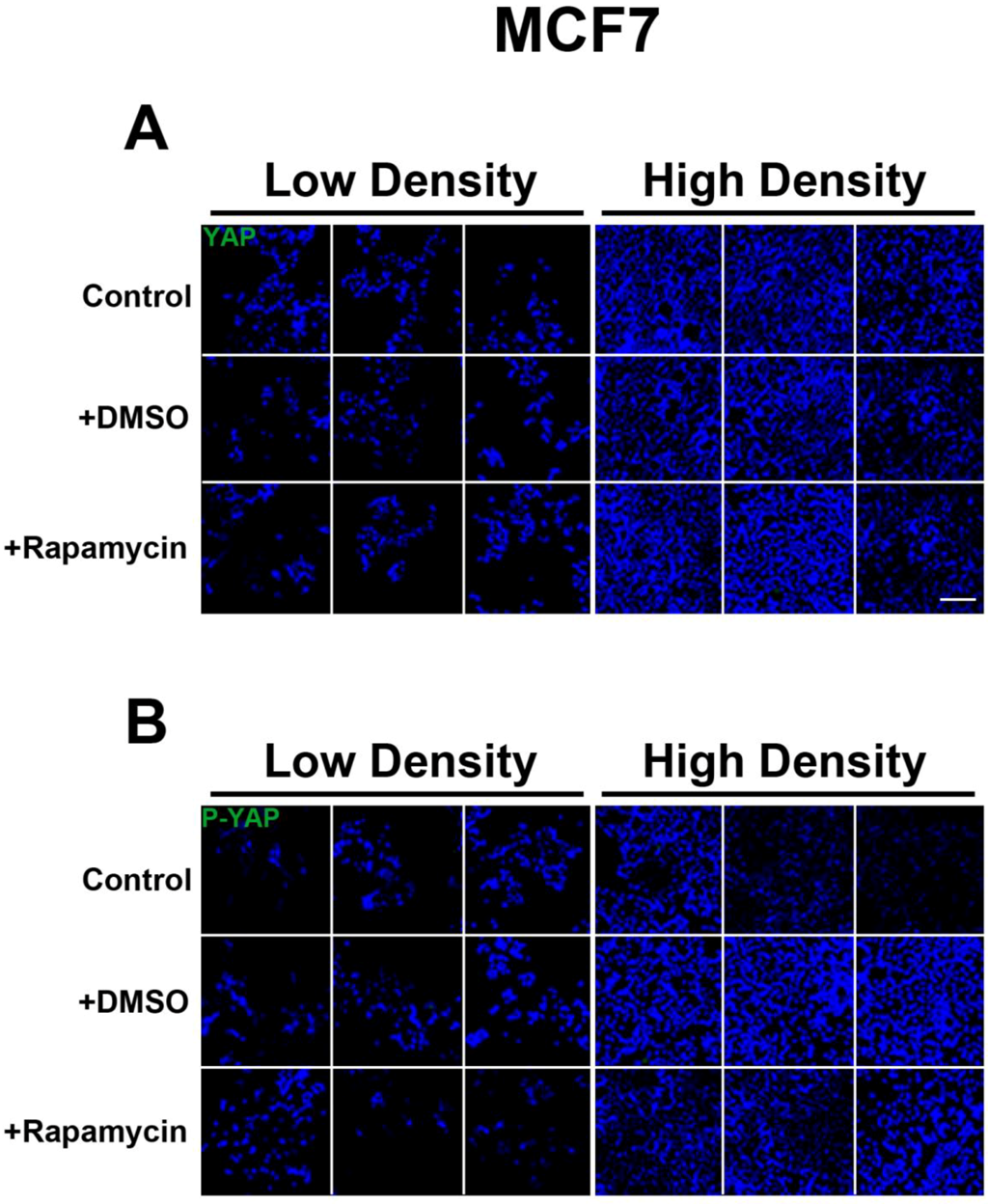
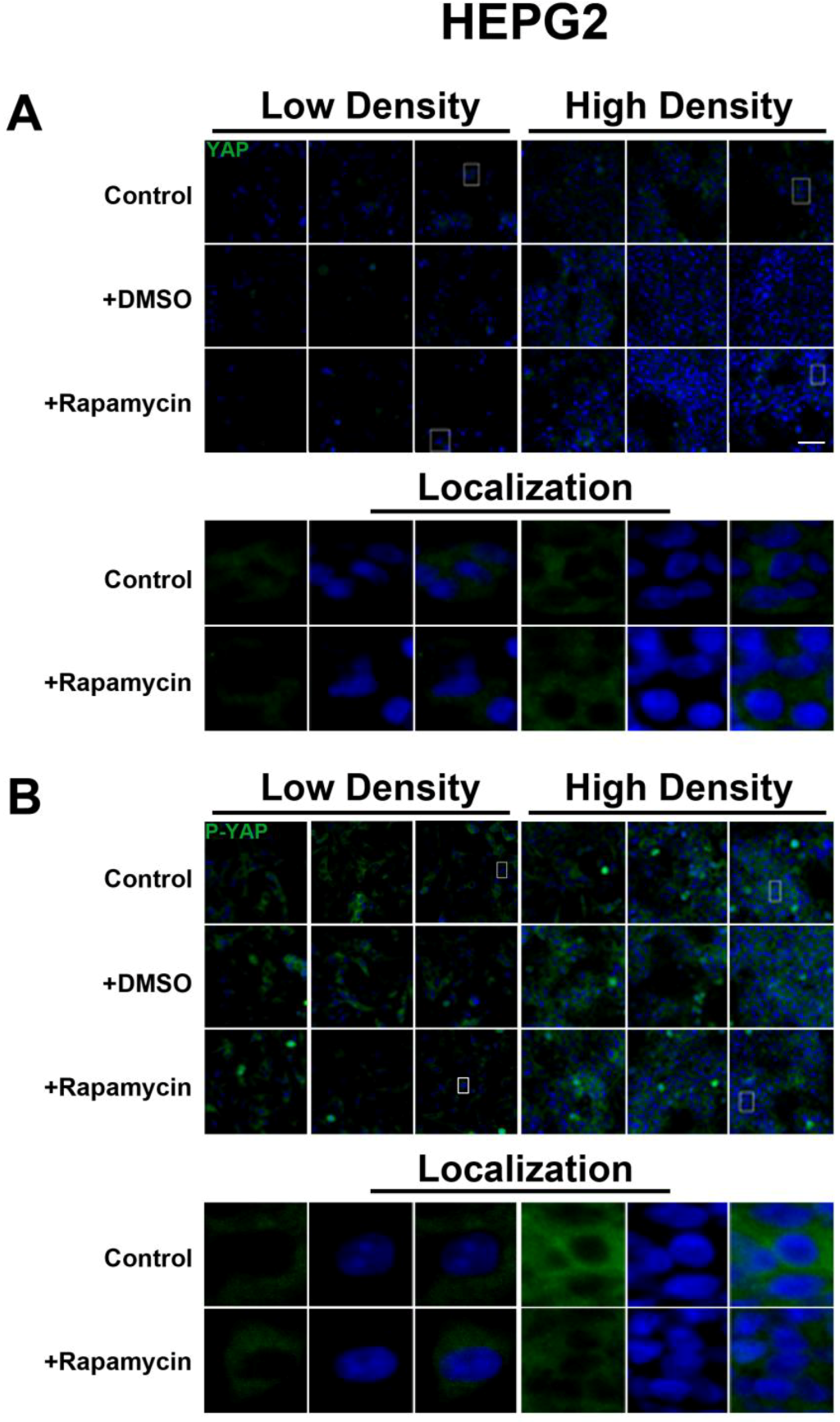
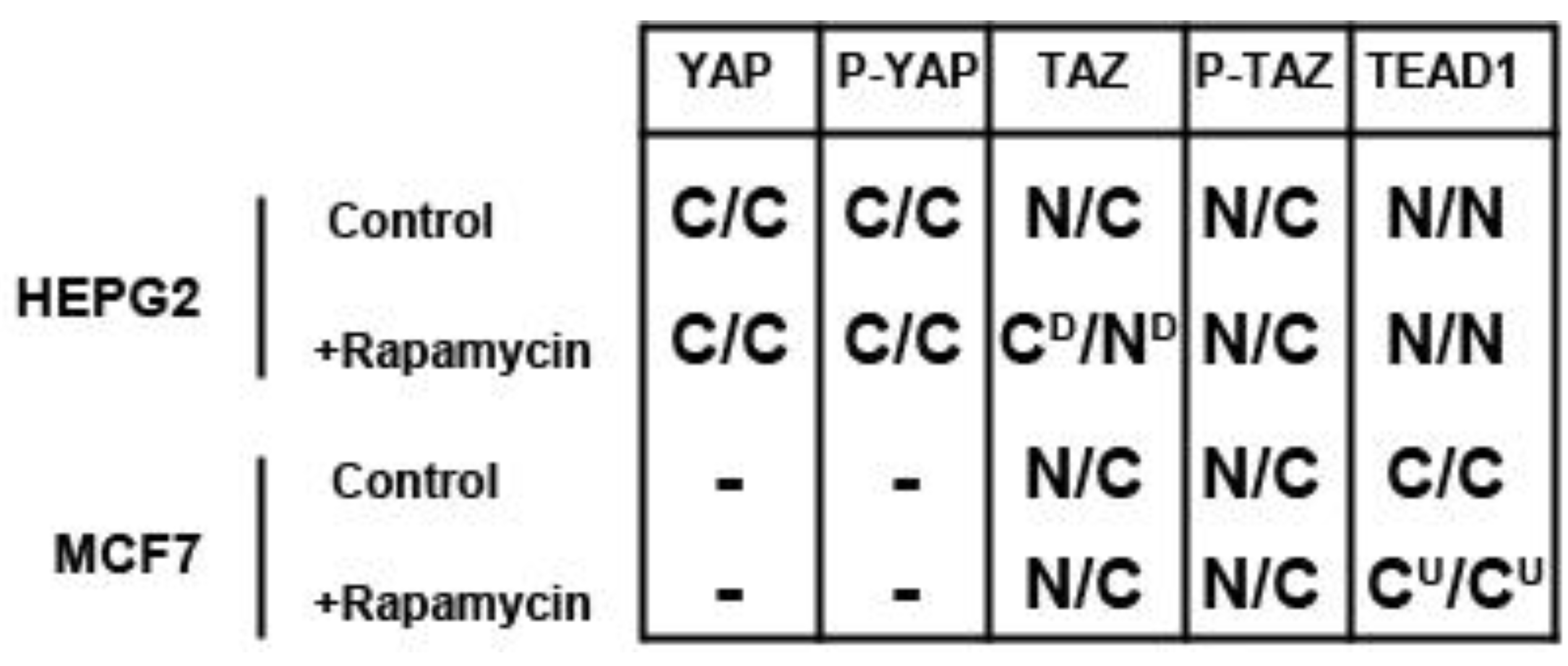
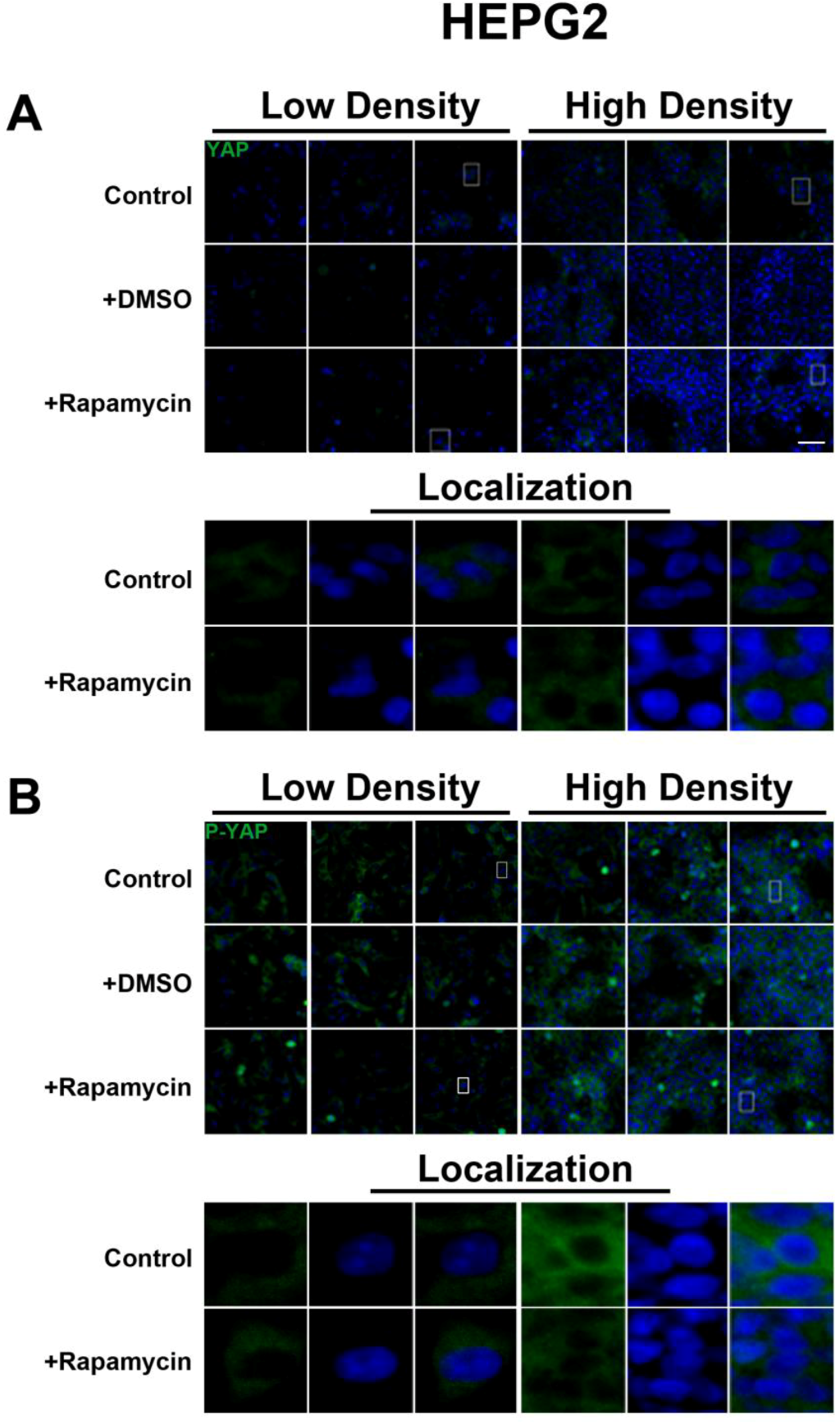
We determined the intracellular localization and expression levels of the Hippo target YAP, for both its phosphorylated and unphosphorylated forms, in HepG2 cells, via immunocytochemistry in low and high cell density conditions (
Figure S2). We observed that YAP and P-YAP proteins are mainly cytosolic and their expression levels or intracellular localization do not appear to be affected by cell density. Upon treatment with rapamycin, we observed that YAP and P-YAP remained cytosolic with drug treatment and that the expression levels of YAP and P-YAP did not change in response to cell density (
Figure S2).
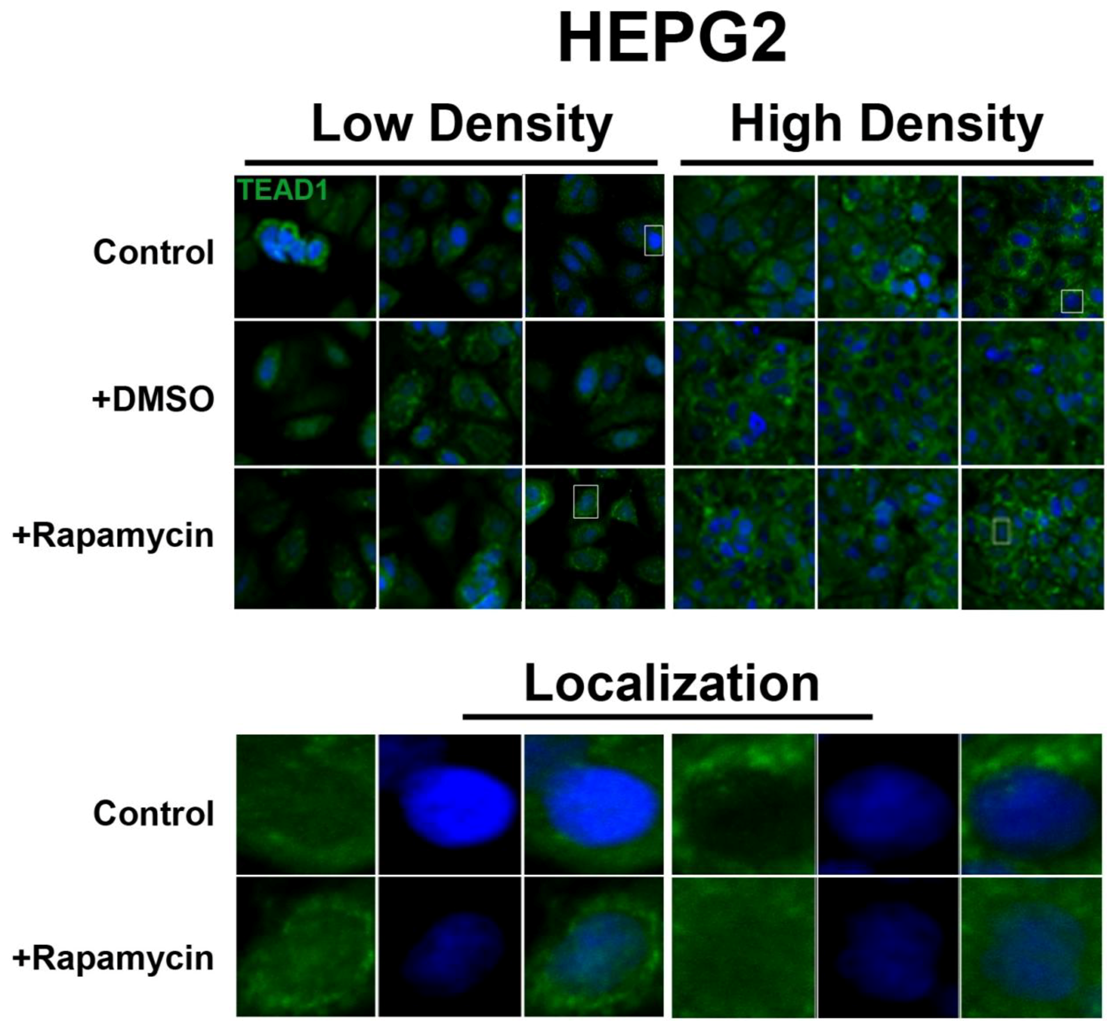
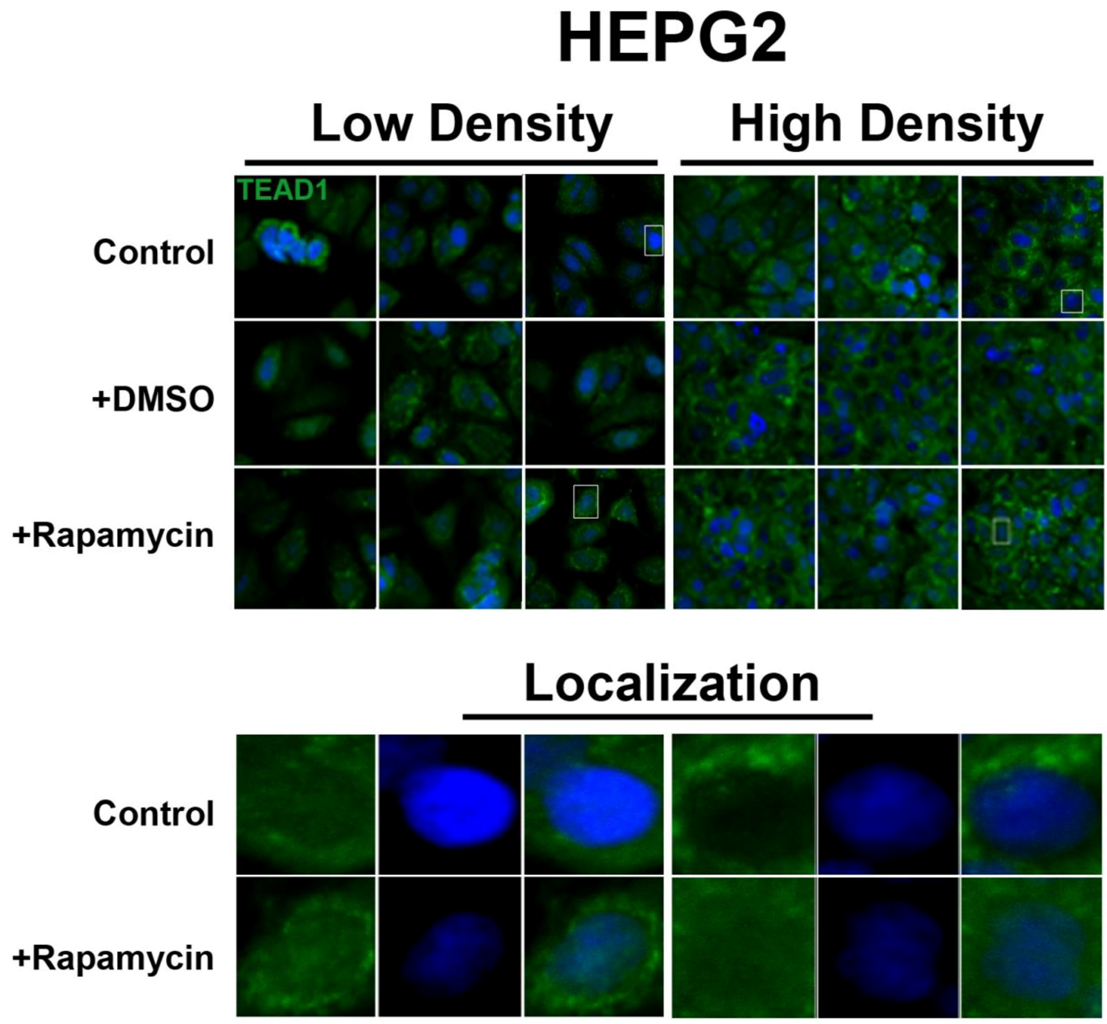
In contrast, the transcription factor TEAD1, a binding partner for YAP and target of the Hippo pathway, is mainly nuclear at low cell density conditions, but less nuclear at high density in HepG2 cells (
Figure 2). While no appreciable change in TEAD1 expression was found upon treatment with drug in both high and low cell density conditions, rapamycin enhances the nuclear localization of TEAD1 in both cell density conditions (
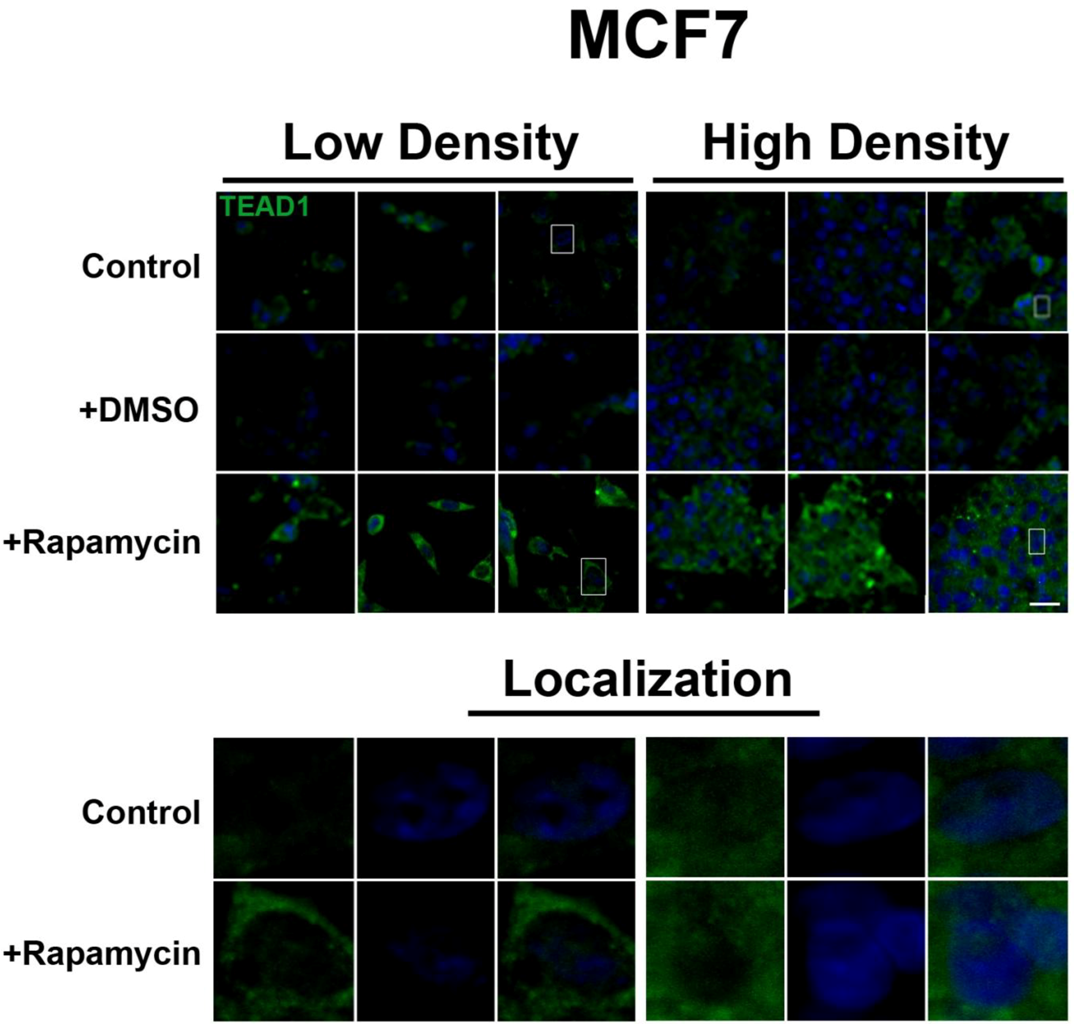
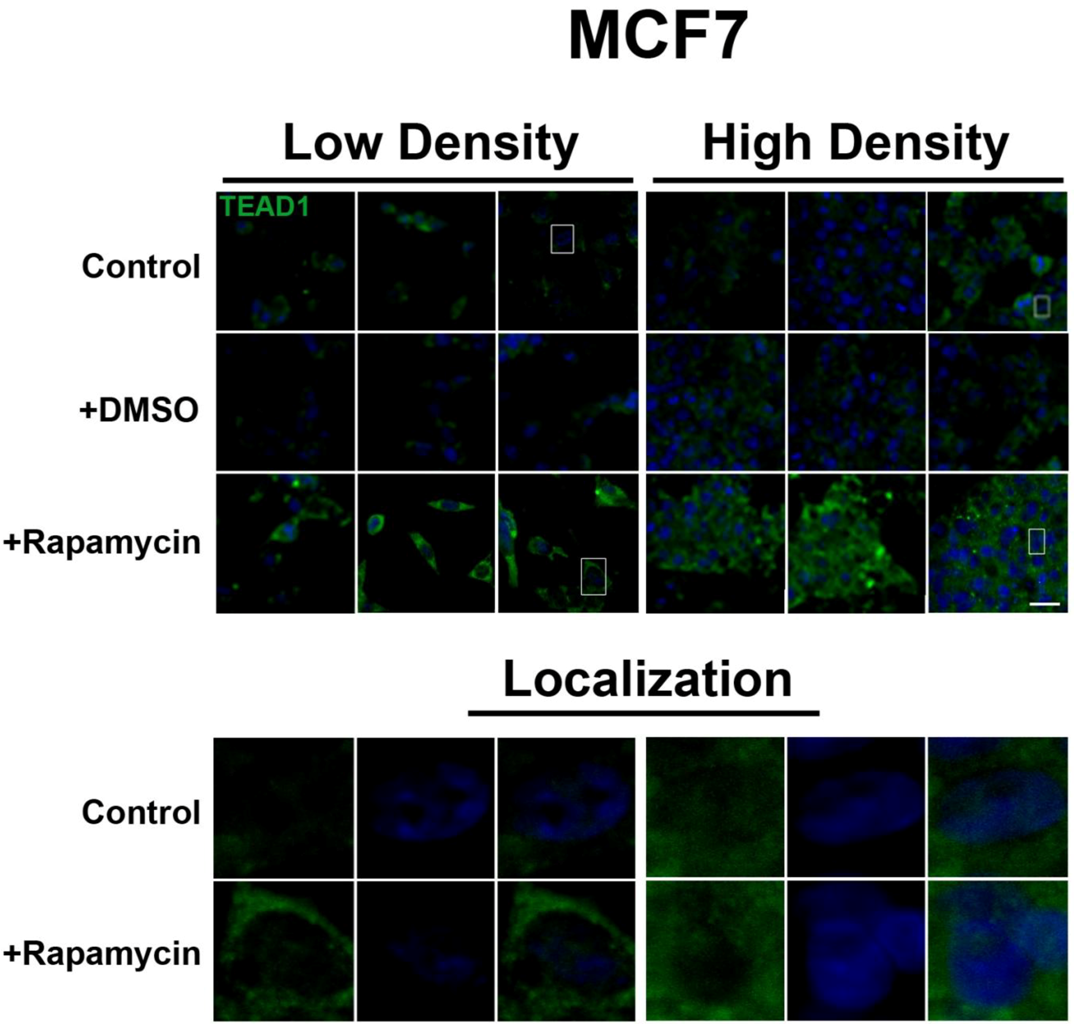
Figure 2). However, TEAD1 is mainly cytosolic in MCF7 cells and its expression levels increase upon treatment with rapamycin without affecting intracellular localization (
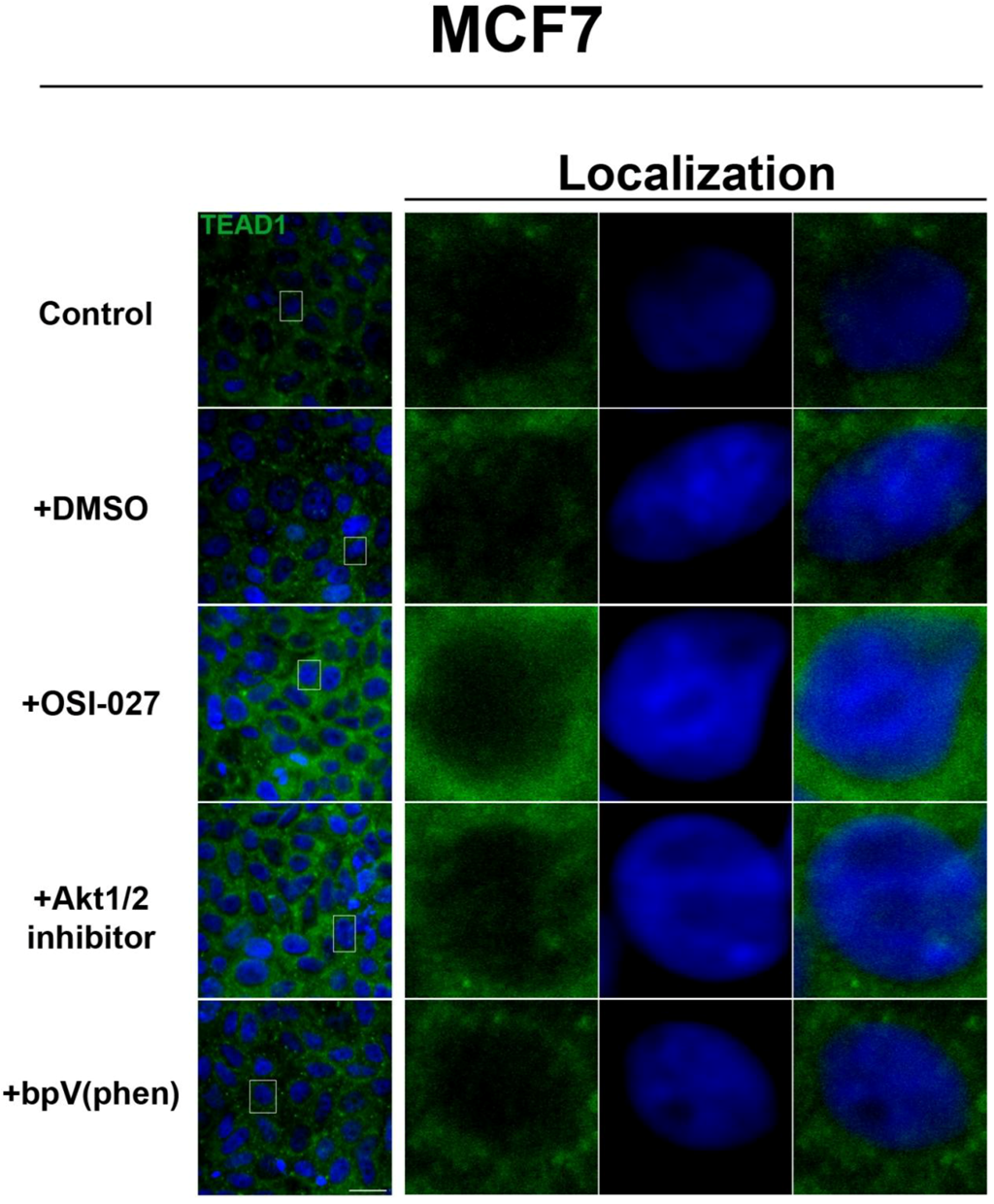
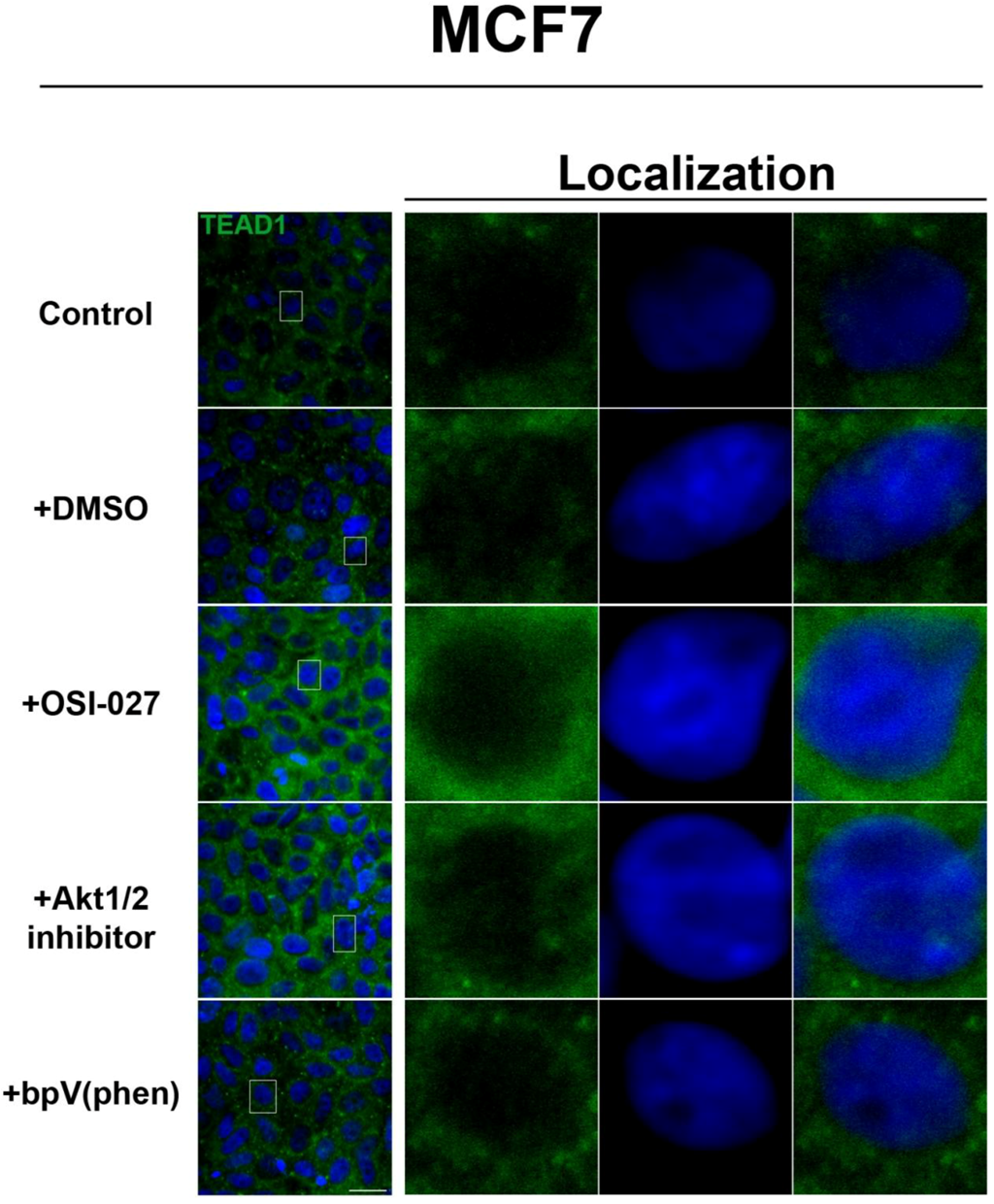
Figure 3). We further confirmed these findings by using an inhibitor of both mTORC1 and mTORC2, OSI-027 [
17] (
Figure 4). A similar increase in TEAD1 levels was observed upon Akt inhibition, but not with upregulation of Akt activity via treatment with a PTEN inhibitor (
Figure 4). These results demonstrate that mTORC1 signaling selectively regulates specific Hippo pathway components and their intracellular localization in a cell-type specific manner: in HepG2 cells, it regulates nuclear localization of TEAD1, while in MCF7 cells, it determines its expression levels.
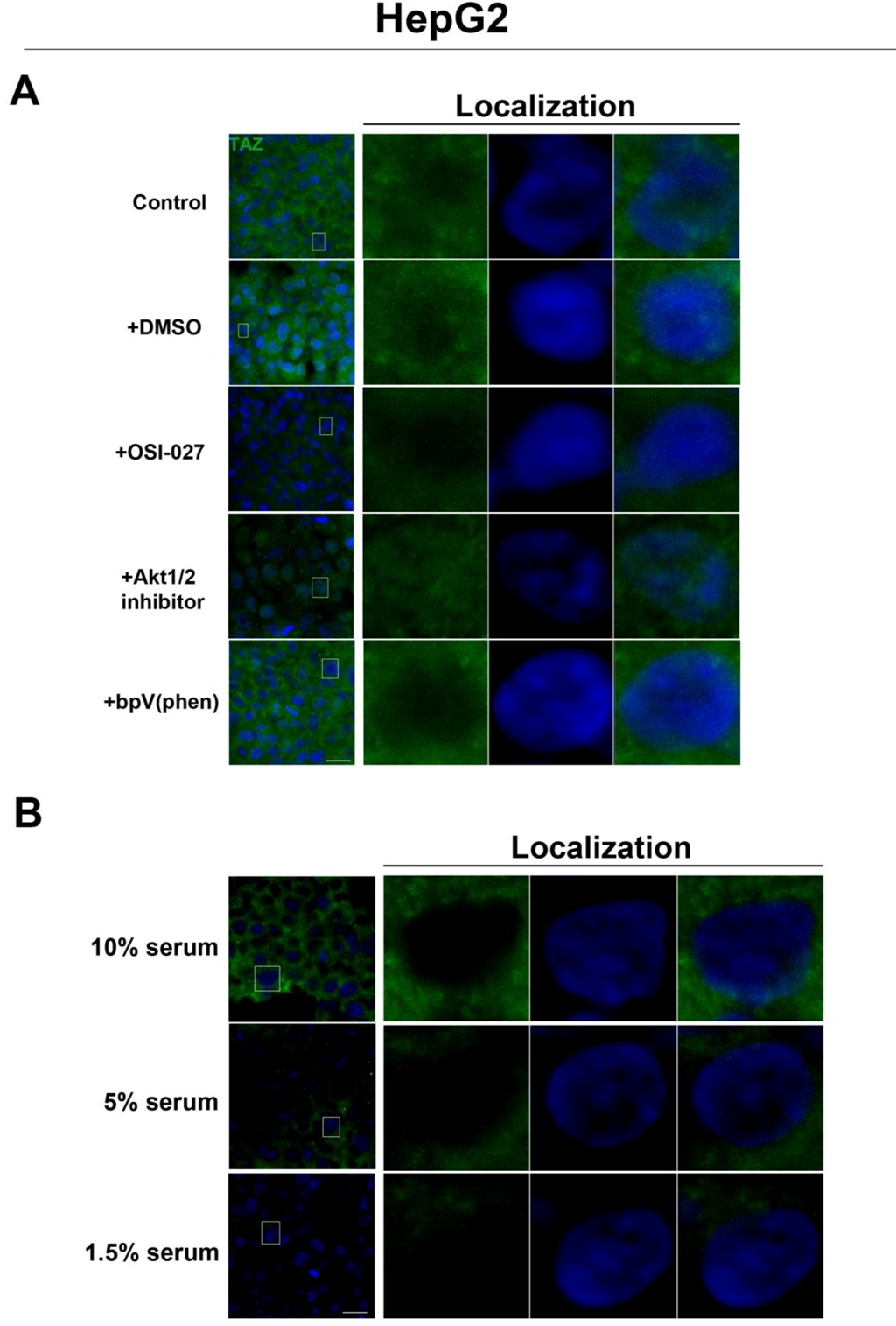
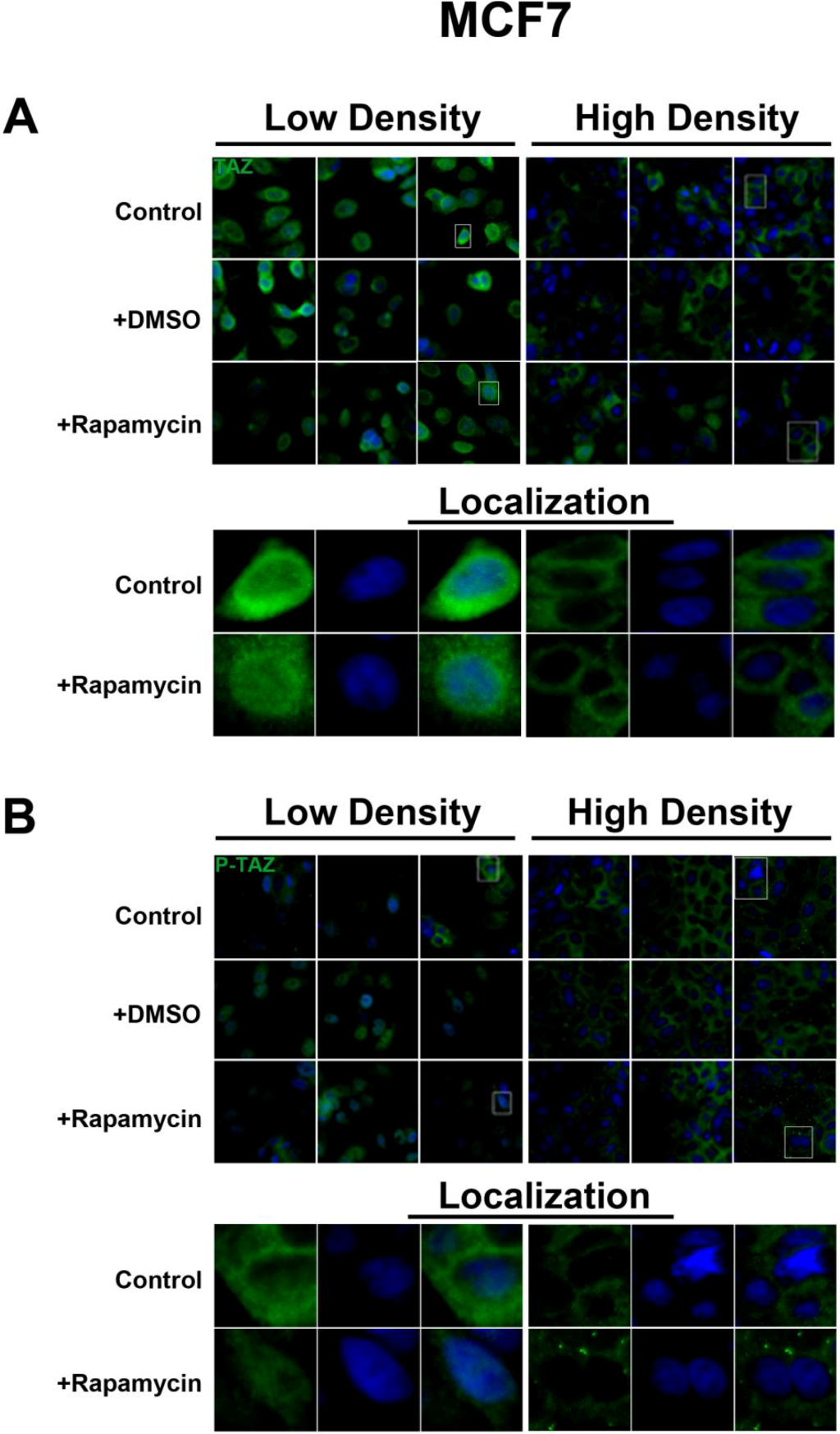
2.3. mTOR Inhibition Affects the Expression Levels of the Hippo Target TAZ in HepG2 Cells But Not MCF7 Cells
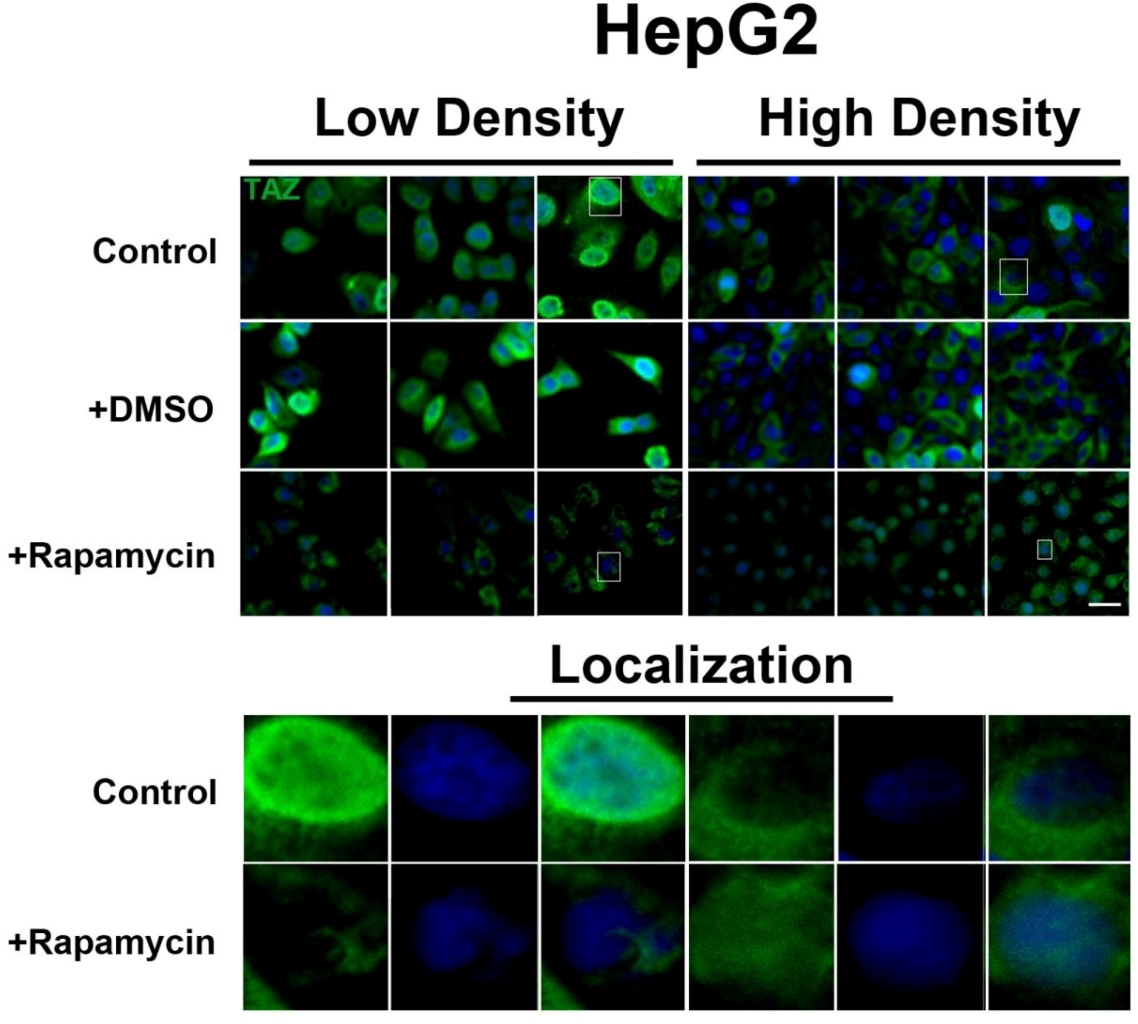
To further investigate the relationship between the Hippo and mTOR pathways, we next examined the effect of mTOR inhibition on the Hippo target TAZ. In both cell lines, TAZ and P-TAZ are localized in the nucleus at low cell density, but translocate to the cytosol in response to increased cell density (
Figure 5,
Figure S3,
Figure S4). However, inhibition of mTOR signaling via rapamycin treatment yielded no change in localization or expression of TAZ or P-TAZ in MCF7 cells (
Figure S3). In contrast, we found that inhibition of the mTOR pathway via rapamycin treatment in HepG2 cells resulted in decreased expression levels of TAZ without affecting levels of P-TAZ (
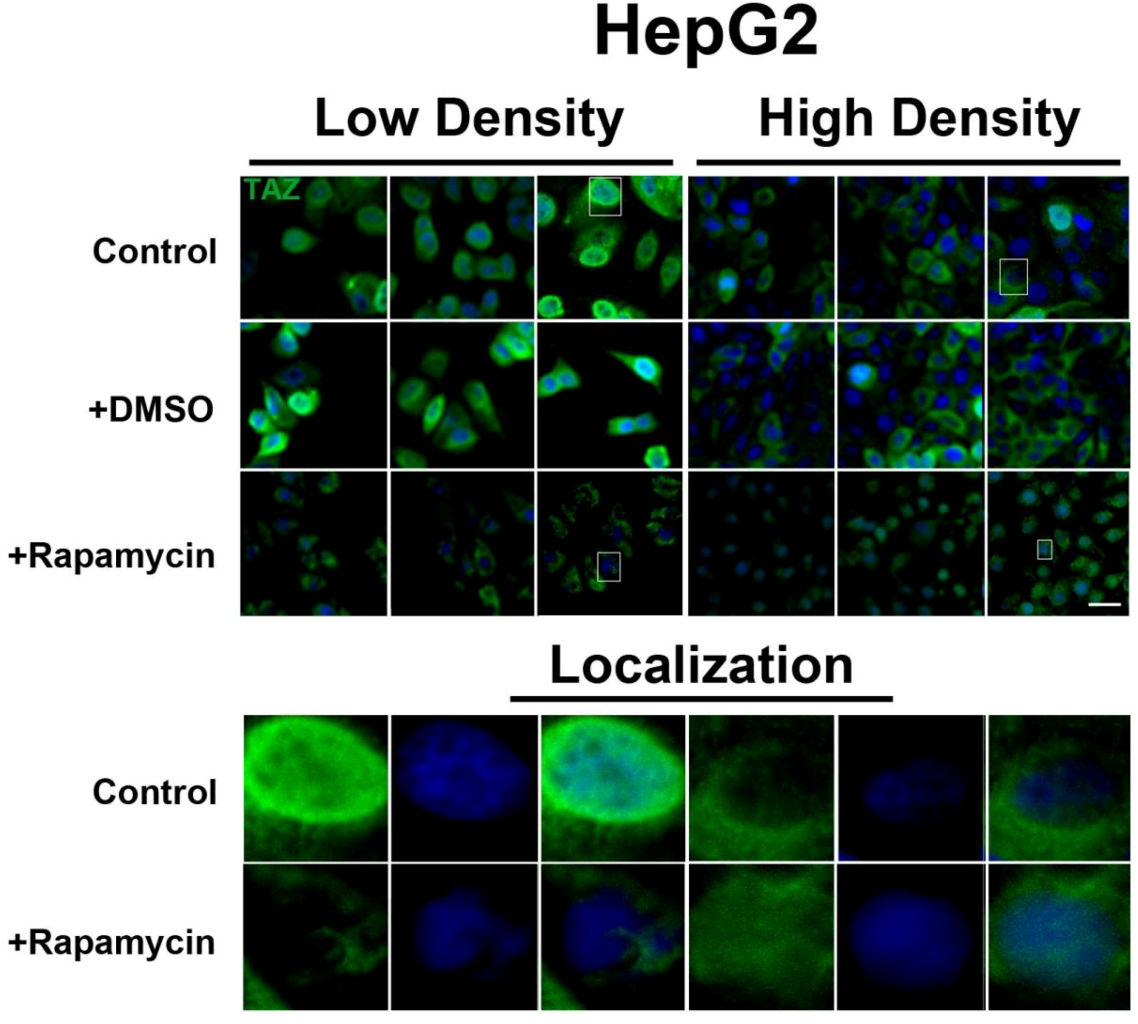
Figure 5 and
Figure S4). Additionally, TAZ in rapamycin-treated HepG2 cells at low cell density appears to translocate to the cytosol (
Figure 5), while TAZ in rapamycin-treated cells at high density appears to translocate to the nucleus, compared to controls (
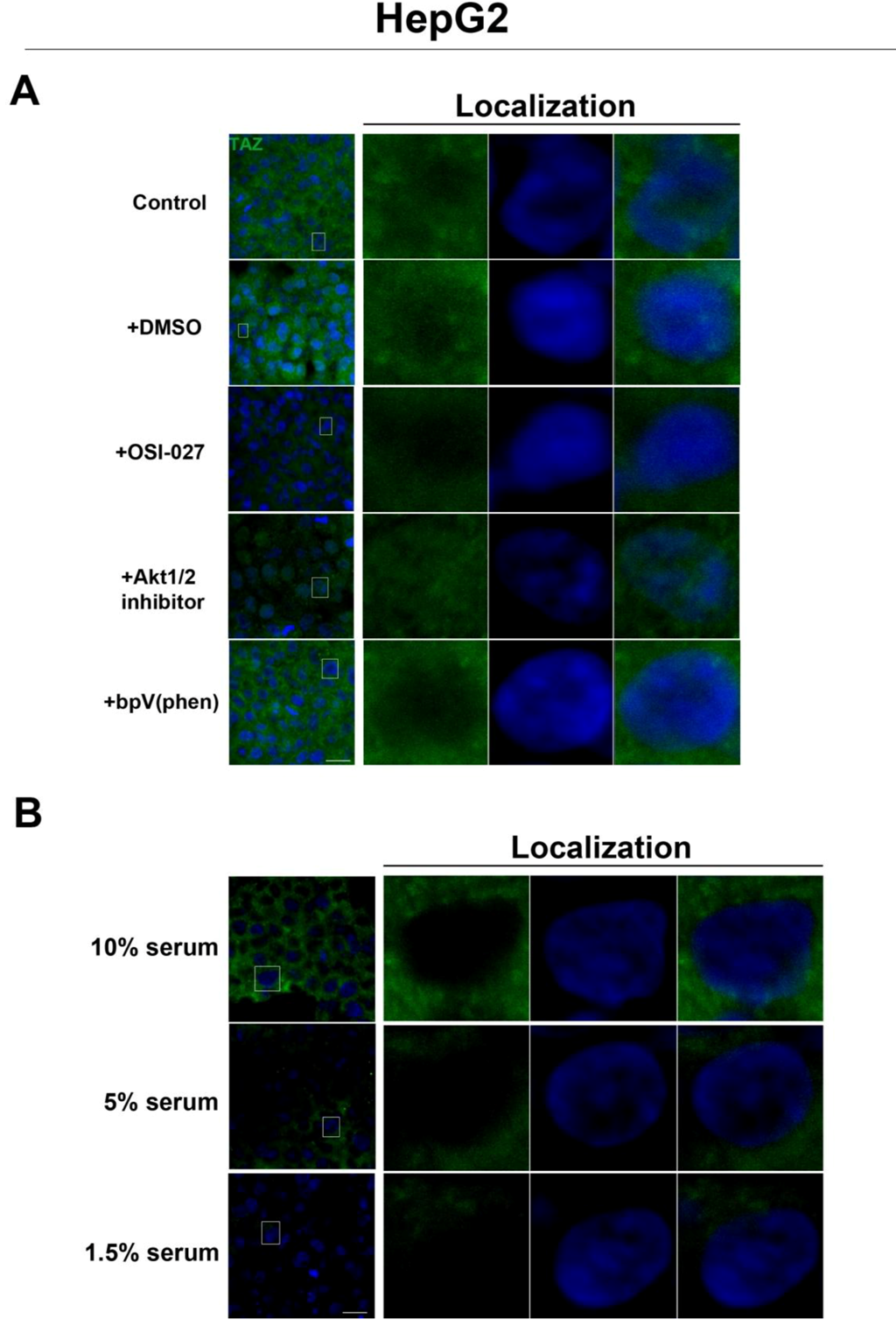
Figure 5). Interestingly, while inhibition of Akt decreases TAZ levels and causes an increase in its nuclear localization as is the case with rapamycin (mTORC1 inhibition with partial mTORC2 inhibition), to our surprise OSI-027 (mTORC1 + mTORC2 inhibition) treatment decreases TAZ levels but does not increase nuclear localization (
Figure 6A). A similar decrease in TAZ levels occurs upon reduction of serum levels (
Figure 6B). These results indicate that mTORC1 signaling regulates levels of TAZ, while its localization to the nucleus may be more dependent on mTORC2 in high cell density conditions.
Figure 2.
Rapamycin treatment does not affect TEAD1 protein expression levels but affects its nuclear localization in HepG2 cells. TEAD1 (green) expression does not change with and without rapamycin treatment at low and high cell density conditions. Bottom panels represent the boxes in top images: localization of TEAD1 at low and high cell density conditions demonstrates that, in contrast to MCF7 cells, it is mainly nuclear in low density conditions, while it is less nuclear at high density conditions. Rapamycin treatment increases nuclear localization at high density conditions. TOPRO3 (blue) labels nuclei. Scale bars, 20 µm.
Figure 2.
Rapamycin treatment does not affect TEAD1 protein expression levels but affects its nuclear localization in HepG2 cells. TEAD1 (green) expression does not change with and without rapamycin treatment at low and high cell density conditions. Bottom panels represent the boxes in top images: localization of TEAD1 at low and high cell density conditions demonstrates that, in contrast to MCF7 cells, it is mainly nuclear in low density conditions, while it is less nuclear at high density conditions. Rapamycin treatment increases nuclear localization at high density conditions. TOPRO3 (blue) labels nuclei. Scale bars, 20 µm.
Figure 3.
mTOR pathway inhibition increases TEAD1 levels in MCF7 cells. Elevated levels of TEAD1 (green) were observed at both high and low cell densities after treatment with rapamycin. Bottom panels represent the boxes in top images: localization of TEAD1 at low and high cell density conditions demonstrates cytoplasmic localization that does not change with rapamycin treatment. TOPRO3 (blue) labels nuclei. Scale bars, 20µm.
Figure 3.
mTOR pathway inhibition increases TEAD1 levels in MCF7 cells. Elevated levels of TEAD1 (green) were observed at both high and low cell densities after treatment with rapamycin. Bottom panels represent the boxes in top images: localization of TEAD1 at low and high cell density conditions demonstrates cytoplasmic localization that does not change with rapamycin treatment. TOPRO3 (blue) labels nuclei. Scale bars, 20µm.
2.4. Discussion
Previous studies have shown that the SWH signaling pathway is critical in regulating cellular development and homeostasis [
2,
7], and interference with pathway components results directly in dysregulation of tissue and organ size. Our results confirmed expression of the key SWH pathway machinery (Mob1, MST2, Lats2) transcripts in HepG2 and MCF7 cells. We also confirmed the transcriptional expression of several Ste20p-like kinases in HepG2 and MCF7 cells. Upon treatment with rapamycin, the transcript expression of both SWH pathway components and Ste20p-like kinases in these cell lines did not change, demonstrating that mTOR does not regulate these elements of the Hippo pathway at the transcriptional level. This data also suggests that select Ste20p-like kinases (Pak1, SLK, TAO2, MAP4K3, MAP4K4, MAP4K5, Stradα/β, MST4, STK24, STK25, STK39, and Scrib) are not regulated by mTORC1 signaling. Despite the lack of effect on transcription levels, the known role of mTOR in promoting protein translation suggested the possibility that it may regulate components of the Hippo pathway at the translational level.
Figure 4.
Akt pathway inhibition increases TEAD1 levels in MCF7 cells. Elevated levels of TEAD1 (green) were observed at high cell density conditions after treatment with an Akt inhibitor. The observed effects of rapamycin were confirmed with the mTORC1/2 inhibitor OSI-027. Panels on the right represent boxed images on the left: localization of TEAD1 at high cell density demonstrates cytoplasmic localization that does not change with drug treatment. TOPRO3 (blue) labels nuclei. Scale bars, 20µm.
Figure 4.
Akt pathway inhibition increases TEAD1 levels in MCF7 cells. Elevated levels of TEAD1 (green) were observed at high cell density conditions after treatment with an Akt inhibitor. The observed effects of rapamycin were confirmed with the mTORC1/2 inhibitor OSI-027. Panels on the right represent boxed images on the left: localization of TEAD1 at high cell density demonstrates cytoplasmic localization that does not change with drug treatment. TOPRO3 (blue) labels nuclei. Scale bars, 20µm.
Figure 5.
mTOR pathway inhibition affects expression and localization of TAZ in HepG2 cells. Decreased levels of TAZ (green) were observed at both high and low cell densities after treatment with rapamycin. Bottom panels represent the boxes in top images: localization of TAZ at low cell density conditions demonstrates mostly nuclear expression that becomes cytoplasmic upon rapamycin treatment, while in high density conditions, TAZ is mostly cytoplasmic in control cells but shifts to the nucleus upon rapamycin treatment. TOPRO3 (blue) labels nuclei. Scale bars, 20 µm.
Figure 5.
mTOR pathway inhibition affects expression and localization of TAZ in HepG2 cells. Decreased levels of TAZ (green) were observed at both high and low cell densities after treatment with rapamycin. Bottom panels represent the boxes in top images: localization of TAZ at low cell density conditions demonstrates mostly nuclear expression that becomes cytoplasmic upon rapamycin treatment, while in high density conditions, TAZ is mostly cytoplasmic in control cells but shifts to the nucleus upon rapamycin treatment. TOPRO3 (blue) labels nuclei. Scale bars, 20 µm.
TEA Domain family member 1 (TEAD1) is a known homologue of the
Drosophila melanogaster transcription factor Scalloped, a known effector of the Hippo pathway that interacts with WW-domain coactivators. In vertebrates, these coactivators are encoded by the
YAP and
TAZ genes and they promote cell cycle progression [
18], cell proliferation, and differentiation [
19]. TEAD1 has been shown to play a role in retaining TAZ in the nucleus to promote cell proliferation [
20], mediate YAP-dependent growth control [
21], and its knockdown has been shown to decrease cell proliferation [
22]. While increases in TEAD1 expression levels are associated with decreased survival in prostate cancer [
22], and in conjunction with TAZ induces epithelial-mesenchymal transition [
3], there is limited data on the role of TEAD1 in cancer. Our immunocytochemical results demonstrate a differential effect of rapamycin on increasing TEAD1 levels in MCF7 but not HepG2 cells, and this was confirmed with the mTORC1/2 inhibitor OSI-027. This difference is likely due to a cell-type specific effect of TEAD1 in breast cancer cells that awaits further study.
Figure 6.
Akt pathway inhibition and changes in serum levels affects expression and localization of TAZ in HepG2 cells. (A) Decreased levels of TAZ (green) were observed at high cell density conditions after treatment with an Akt inhibitor but not with the PTEN inhibitor bpV(phen). (B) Decreased levels of TAZ were also observed at high cell density conditions after a reduction in serum levels. Panels on the right represent boxed images on the left: localization of TAZ in high cell density conditions demonstrates mostly cytoplasmic expression that does not change upon OSI-027 treatment but shifts to the nucleus upon Akt1/2 inhibitor treatment. TOPRO3 (blue) labels nuclei. Scale bars, 20 µm.
Figure 6.
Akt pathway inhibition and changes in serum levels affects expression and localization of TAZ in HepG2 cells. (A) Decreased levels of TAZ (green) were observed at high cell density conditions after treatment with an Akt inhibitor but not with the PTEN inhibitor bpV(phen). (B) Decreased levels of TAZ were also observed at high cell density conditions after a reduction in serum levels. Panels on the right represent boxed images on the left: localization of TAZ in high cell density conditions demonstrates mostly cytoplasmic expression that does not change upon OSI-027 treatment but shifts to the nucleus upon Akt1/2 inhibitor treatment. TOPRO3 (blue) labels nuclei. Scale bars, 20 µm.
The lack of changes in P-YAP in response to rapamycin treatment suggests that there is no YAP-mediated interaction between the SWH and mTOR pathways both at the transcriptional and post-translational levels. However, our study was limited to the use of an antibody that recognizes phosphorylation at the Serine 127 and Serine 89 residues [
23]. This accounts for the higher levels of P-YAP compared to total YAP seen in HepG2 cells. Therefore, it remains to be determined if additional phosphorylation sites may potentially mediate cross-talk between the two pathways.
TAZ is a demonstrated transcriptional coactivator regulated by the Hippo pathway that promotes cell proliferation and epithelial-mesenchymal transition [
23,
24]. Phosphorylation at key residues by Hippo pathway kinases causes retention of TAZ in the cytoplasm and prevents its growth-promoting activity [
22]. We did not observe changes in P-TAZ levels or intracellular localization upon rapamycin treatment in both cell lines. TAZ can be phosphorylated at four different serine residues (Ser89, Ser66, Ser117, Ser311) [
22]. We examined only the Ser89 residue because it is most commonly dependent on Hippo signaling. It is possible that the expression levels of TAZ that has been phosphorylated at other sites in the protein change in response to rapamycin treatment. More importantly, our results demonstrate that the intracellular localization of TAZ is actively regulated in both MCF7 and HepG2 cell lines. In conditions of high cell density, TAZ translocates to the cytosol and this is associated with contact-inhibition of cell growth. However, inhibition of mTOR signaling via rapamycin had no effect in MCF7 cells but decreased TAZ levels in HepG2 cells, as do decreases in serum levels. This decrease in expression was accompanied by TAZ translocation to the nucleus at high density, likely the result of inhibition of translation of proteins required for its cytosolic retention. Similarly, Akt inhibition decreases its expression levels and promotes its nuclear localization, consistent with the changes observed with rapamycin. Interestingly, while mTORC1 inhibition with rapamycin in high cell density conditions promotes the nuclear localization of TAZ, the mTORC1/2 inhibitor OSI-027 has no effect on localization in HepG2 cells. This suggests a potential role for mTORC2 in the nuclear localization of TAZ upon mTOR inhibition.
Based on our findings, TAZ represents a novel biomarker for investigation in liver cancer, and its responsiveness to rapamycin specifically in this type of tumor suggests a potential therapeutic intervention for hepatic tumors in which TAZ is upregulated. Elevated levels of TAZ have been correlated with breast cancer cell migration, invasion, and tumorigenesis, while TAZ knockdown in breast cancer cell lines have impaired the tumorigenic ability of those cells [
25]. These findings have defined TAZ as an important regulator of breast cancer development and progression. However, the correlation of TAZ levels with other types of tumors has been limited to a few cases, including non-small cell lung cancer cell lines [
26] and papillary thyroid carcinoma [
27]. The decrease in TAZ levels upon rapamycin treatment suggests a potential role for the mTOR pathway in maintaining TAZ levels, and further studies of this mechanism in other cancers may help elucidate the role of Hippo signaling in tumorigenesis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}