The Inhibitor of Apoptosis (IAPs) in Adaptive Response to Cellular Stress



Abstract
:1. Introduction
2. The Inhibitor of Apoptosis (IAP) Family of Proteins
2.1. Structural Feature and Molecular Functions
2.2. IAPs as Inhibitor of Cell Death
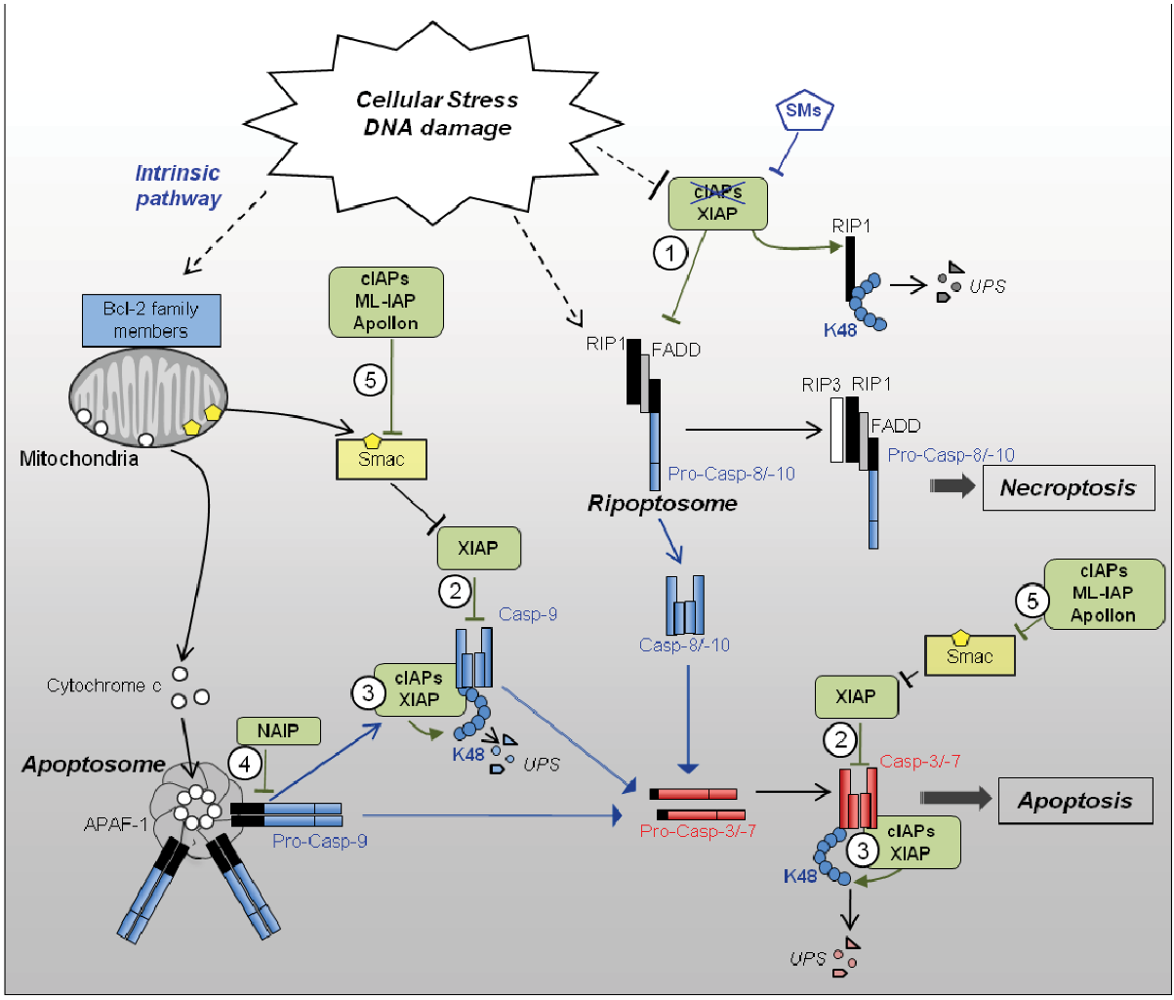
2.2.1. Apoptotic Signaling Pathways

2.2.2. Regulation of Caspases by IAPs (Figure 1)
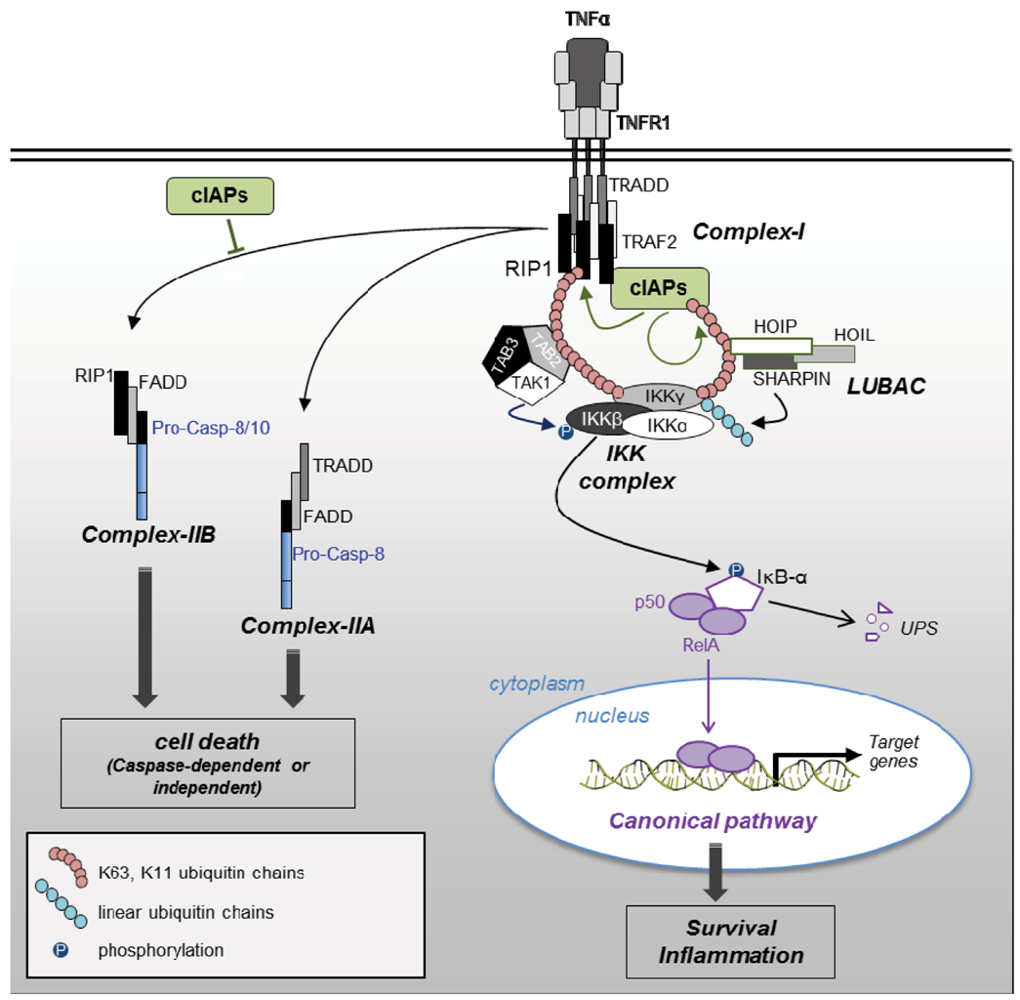
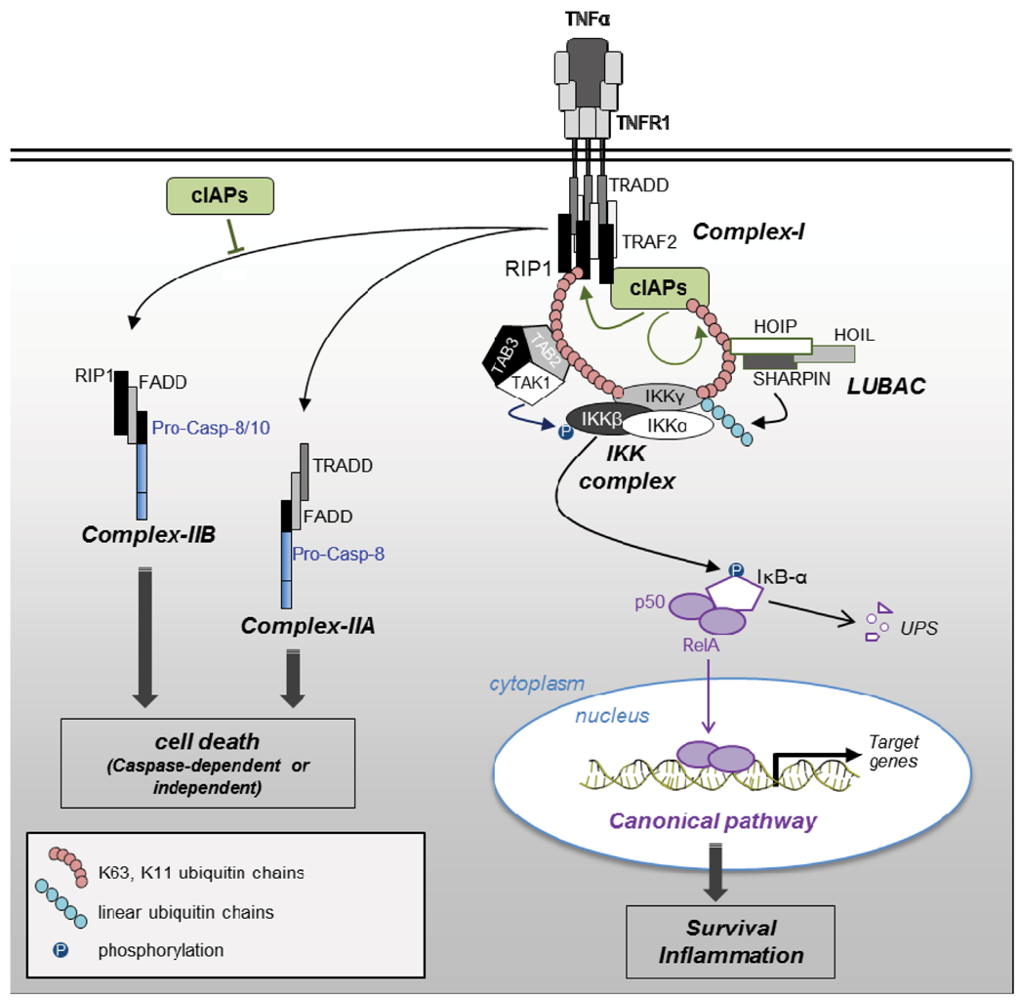
2.2.3. Blockade of RIP1-Containing Cell Death-Inducing Platform Formation
2.3. Pro-Survival Properties of IAPs
2.3.1. NF-κB Activating Signaling Pathways
2.3.2. Cell Proliferation
3. Role of IAPs in Adaptive Response to Cellular Stress (Summarized in Table 1)

{kind=link}
{kind=link}
{kind=link}
| Cellular Stress | IAP | Regulation (-) and effects (→) | Ref. | |
|---|---|---|---|---|
| UPR | cIAP1 | - | IRES-dependent translational up-regulation | [4] |
| - | PERK-dependent transcriptional and translational up-regulation | [84] | ||
| - | P3K dependent transcriptional up-regulation | [85] | ||
| ➨ Cell death protection | [84,85] | |||
| cIAP2 | - | PERK-dependent transcriptional and translational up-regulation | [84] | |
| ➨ Cell death protection | ||||
| XIAP | - | P3K dependent transcriptional up-regulation | [85] | |
| ➨ Cell death protection | ||||
| DNA damage | cIAP1 | - | IRES dependent translational up-regulation | [103] |
| ➨ Cell death protection | ||||
| ➨ Canonical NF-κB activation through ubiquitination of IKKγ | [71] | |||
| - | Auto-ubiquination and degradation | [14,25] | ||
| ➨ Ripoptosome formation and cell death | [25] | |||
| XIAP | - | MDM2 and IRES dependent up-regulation | [80,81] | |
| ➨ Cell death protection | [81] | |||
| ➨ Canonical NF-κB activation through bridging of IKK complex and TAK1 | [66] | |||
| - | Auto-ubiquination and degradation | [14,25] | ||
| ➨ Ripoptosome formation and cell death | ||||
| Pro-inflammatory environment | cIAP2 | - | Up-regulation upon activation by LPS | [109,110,111] |
| ➨ Macrophages survival in pro-inflammatory environment | [109] | |||
| cIAPs | ➨ Protective effect | [113] | ||
| ➨ Cell fate decision in response to TNFα or pathogen recognition | Reviewed in [2,3] | |||
| Hypoxia and ischemia | XIAP | ➨ Neuro-protective effect | [88,89] | |
| NAIP | ➨ Neuro-protective effect | [101,102] | ||
| Oxidative stress | XIAP | ➨ Regulates the level of expression of anti-oxidant enzymes →reduced intracellular ROS | [94,95] | |
| ➨ Ubiquitination of copper transporters →regulate copper homeostasis | [97,98,99] | |||
3.1. The Unfolded Protein Response (UPR)
3.2. Role of IAPs as Neuroprotective Molecules
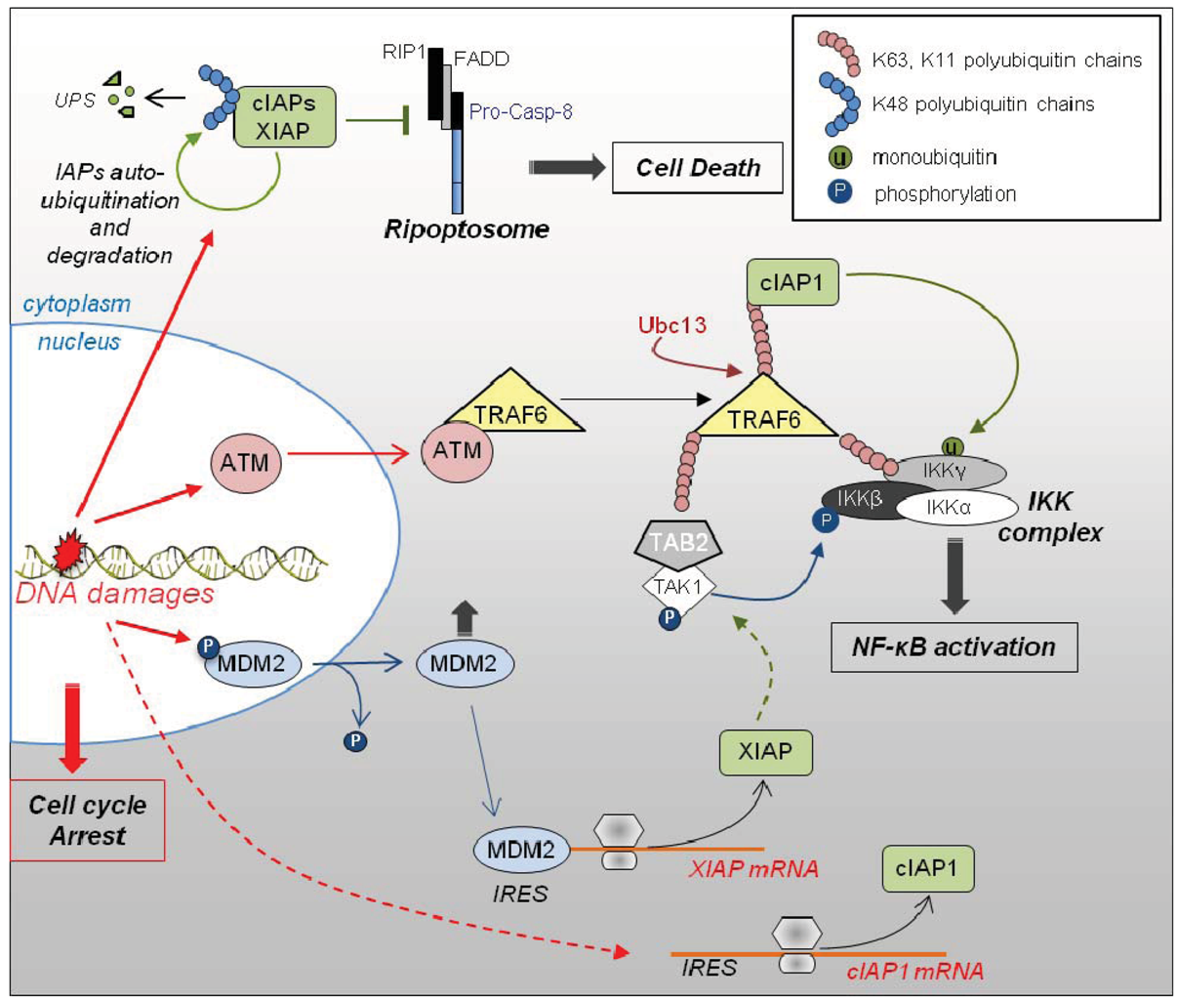
3.3. Role of IAPs in DNA Damage Response (Figure 3)
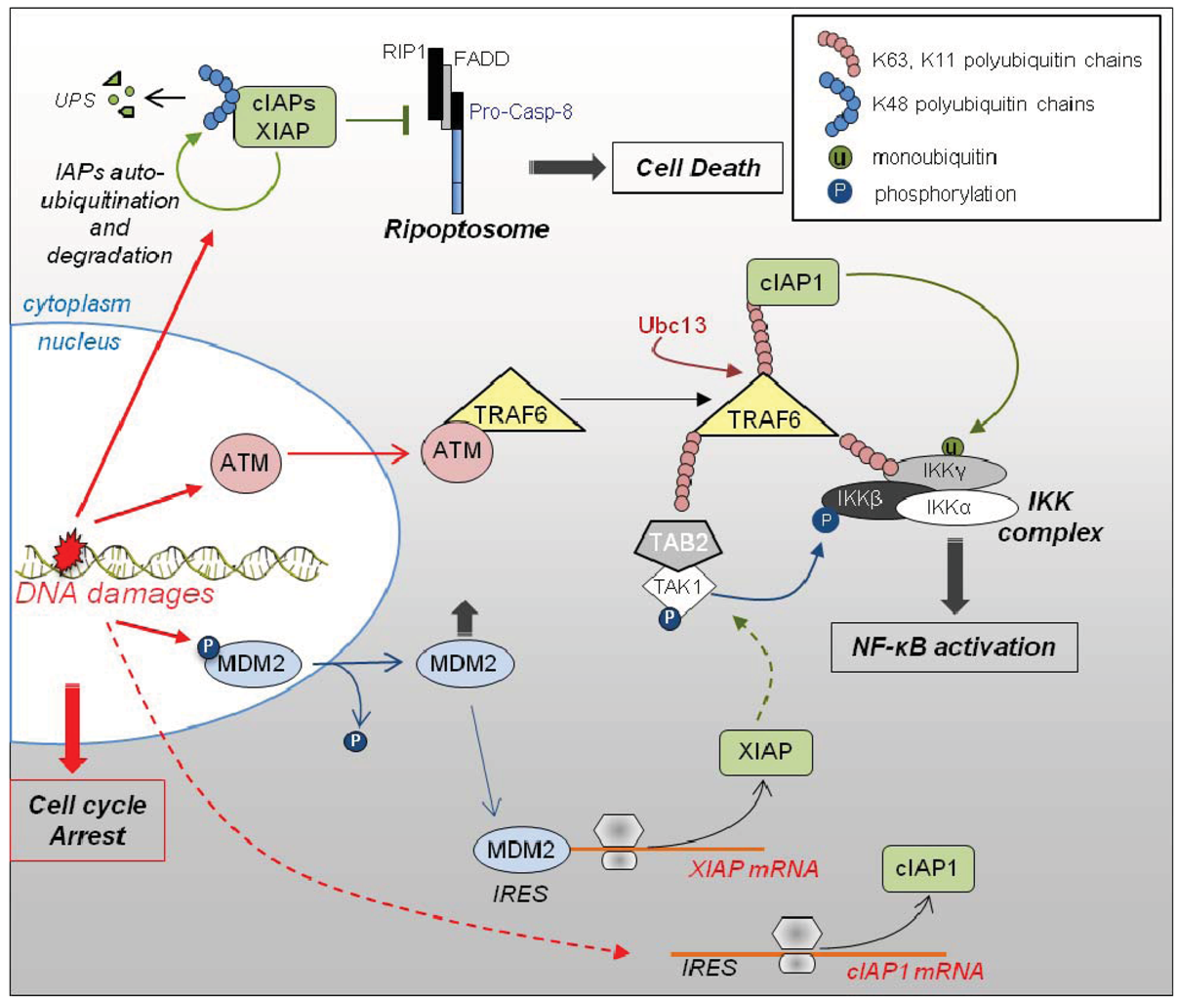
3.4. Role of IAPs in Adaptive Response of Cells to Pro-Inflammatory Environment
4. IAPs and Disease
5. Conclusion
Acknowledgments
Conflict of Interest
References and Notes
- Dubrez-Daloz, L.; Dupoux, A.; Cartier, J. IAPs: More than just inhibitors of apoptosis proteins. Cell. Cycle 2008, 7, 1036–1046. [Google Scholar] [CrossRef]
- Gyrd-Hansen, M.; Meier, P. IAPs: From caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat. Rev. Cancer 2010, 10, 561–574. [Google Scholar] [CrossRef]
- Beug, S.T.; Cheung, H.H.; Lacasse, E.C.; Korneluk, R.G. Modulation of immune signalling by inhibitors of apoptosis. Trends Immunol. 2012, in press. [Google Scholar]
- Warnakulasuriyarachchi, D.; Cerquozzi, S.; Cheung, H.H.; Holcik, M. Translational induction of the inhibitor of apoptosis protein HIAP2 during endoplasmic reticulum stress attenuates cell death and is mediated via an inducible internal ribosome entry site element. J. Biol. Chem. 2004, 279, 17148–17157. [Google Scholar] [CrossRef]
- Van Eden, M.E.; Byrd, M.P.; Sherrill, K.W.; Lloyd, R.E. Translation of cellular inhibitor of apoptosis protein 1 (c-IAP1) mRNA is IRES mediated and regulated during cell stress. RNA 2004, 10, 469–481. [Google Scholar] [CrossRef]
- Holcik, M.; Lefebvre, C.; Yeh, C.; Chow, T.; Korneluk, R.G. A new internal-ribosome-entry-site motif potentiates XIAP-mediated cytoprotection. Nat. Cell. Biol. 1999, 1, 190–192. [Google Scholar] [CrossRef]
- Eckelman, B.P.; Drag, M.; Snipas, S.J.; Salvesen, G.S. The mechanism of peptide-binding specificity of IAP BIR domains. Cell. Death Differ. 2008, 15, 920–928. [Google Scholar] [CrossRef]
- Vucic, D.; Dixit, V.M.; Wertz, I.E. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat. Rev. Mol. Cell. Biol. 2011, 12, 439–452. [Google Scholar] [CrossRef]
- Silke, J.; Kratina, T.; Chu, D.; Ekert, P.G.; Day, C.L.; Pakusch, M.; Huang, D.C.; Vaux, D.L. Determination of cell survival by RING-mediated regulation of inhibitor of apoptosis (IAP) protein abundance. Proc. Natl. Acad. Sci. USA 2005, 102, 16182–16187. [Google Scholar]
- Mace, P.D.; Linke, K.; Feltham, R.; Schumacher, F.R.; Smith, C.A.; Vaux, D.L.; Silke, J.; Day, C.L. Structures of the cIAP2 RING domain reveal conformational changes associated with ubiquitin-conjugating enzyme (E2) recruitment. J. Biol. Chem. 2008, 283, 31633–31640. [Google Scholar]
- Cheung, H.H.; Plenchette, S.; Kern, C.J.; Mahoney, D.J.; Korneluk, R.G. The RING domain of cIAP1 mediates the degradation of RING-bearing inhibitor of apoptosis proteins by distinct pathways. Mol. Biol. Cell. 2008, 19, 2729–2740. [Google Scholar] [CrossRef]
- Rajalingam, K.; Sharma, M.; Paland, N.; Hurwitz, R.; Thieck, O.; Oswald, M.; Machuy, N.; Rudel, T. IAP-IAP complexes required for apoptosis resistance of C. trachomatis-infected cells. PLoS Pathog. 2006, 2, e114. [Google Scholar] [CrossRef]
- Dohi, T.; Okada, K.; Xia, F.; Wilford, C.E.; Samuel, T.; Welsh, K.; Marusawa, H.; Zou, H.; Armstrong, R.; Matsuzawa, S.; et al. An IAP-IAP complex inhibits apoptosis. J. Biol. Chem. 2004, 279, 34087–34090. [Google Scholar]
- Yang, Y.; Fang, S.; Jensen, J.P.; Weissman, A.M.; Ashwell, J.D. Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science 2000, 288, 874–877. [Google Scholar] [CrossRef]
- Hao, Y.; Sekine, K.; Kawabata, A.; Nakamura, H.; Ishioka, T.; Ohata, H.; Katayama, R.; Hashimoto, C.; Zhang, X.; Noda, T.; et al. Apollon ubiquitinates SMAC and caspase-9, and has an essential cytoprotection function. Nat. Cell. Biol. 2004, 6, 849–860. [Google Scholar] [CrossRef]
- Blankenship, J.W.; Varfolomeev, E.; Goncharov, T.; Fedorova, A.V.; Kirkpatrick, D.S.; Izrael-Tomasevic, A.; Phu, L.; Arnott, D.; Aghajan, M.; Zobel, K.; et al. Ubiquitin binding modulates IAP antagonist-stimulated proteasomal degradation of c-IAP1 and c-IAP2(1). Biochem. J. 2009, 417, 149–160. [Google Scholar] [CrossRef]
- Gyrd-Hansen, M.; Darding, M.; Miasari, M.; Santoro, M.M.; Zender, L.; Xue, W.; Tenev, T.; da Fonseca, P.C.; Zvelebil, M.; Bujnicki, J.M.; et al. IAPs contain an evolutionarily conserved ubiquitin-binding domain that regulates NF-kappaB as well as cell survival and oncogenesis. Nat. Cell. Biol. 2008, 10, 1309–1317. [Google Scholar] [CrossRef]
- Lopez, J.; John, S.W.; Tenev, T.; Rautureau, G.J.; Hinds, M.G.; Francalanci, F.; Wilson, R.; Broemer, M.; Santoro, M.M.; Day, C.L.; et al. CARD-mediated autoinhibition of cIAP1's E3 ligase activity suppresses cell proliferation and migration. Mol. Cell. 2011, 42, 569–583. [Google Scholar] [CrossRef]
- Roy, N.; Mahadevan, M.S.; McLean, M.; Shutler, G.; Yaraghi, Z.; Farahani, R.; Baird, S.; Besner-Johnston, A.; Lefebvre, C.; Kang, X.; et al. The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell 1995, 80, 167–178. [Google Scholar] [CrossRef]
- Fulda, S.; Vucic, D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012, 11, 109–124. [Google Scholar] [CrossRef]
- Pop, C.; Salvesen, G.S. Human caspases: Activation, specificity, and regulation. J. Biol. Chem. 2009, 284, 21777–21781. [Google Scholar] [CrossRef]
- Mace, P.D.; Riedl, S.J. Molecular cell death platforms and assemblies. Curr. Opin. Cell. Biol. 2010, 22, 828–836. [Google Scholar] [CrossRef]
- Wurstle, M.L.; Laussmann, M.A.; Rehm, M. The central role of initiator caspase-9 in apoptosis signal transduction and the regulation of its activation and activity on the apoptosome. Exp. Cell. Res. 2012, 318, 121–320. [Google Scholar]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Hacker, G.; Leverkus, M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell. 2011, 43, 449–463. [Google Scholar] [CrossRef]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell. 2011, 43, 432–448. [Google Scholar]
- Bertrand, M.J.; Vandenabeele, P. The Ripoptosome: Death decision in the cytosol. Mol. Cell. 2011, 43, 323–325. [Google Scholar] [CrossRef]
- Cheung, H.H.; Lynn Kelly, N.; Liston, P.; Korneluk, R.G. Involvement of caspase-2 and caspase-9 in endoplasmic reticulum stress-induced apoptosis: A role for the IAPs. Exp. Cell. Res. 2006, 312, 2347–2357. [Google Scholar] [CrossRef]
- Imre, G.; Heering, J.; Takeda, A.N.; Husmann, M.; Thiede, B.; zu Heringdorf, D.M.; Green, D.R.; van der Goot, F.G.; Sinha, B. Caspase-2 is an initiator caspase responsible for pore-forming toxin-mediated apoptosis. EMBO J. 2012, 31, 2615–2628. [Google Scholar] [CrossRef]
- Upton, J.P.; Austgen, K.; Nishino, M.; Coakley, K.M.; Hagen, A.; Han, D.; Papa, F.R.; Oakes, S.A. Caspase-2 cleavage of BID is a critical apoptotic signal downstream of endoplasmic reticulum stress. Mol. Cell. Biol. 2008, 28, 3943–3951. [Google Scholar] [CrossRef]
- Shi, Y. A conserved tetrapeptide motif: Potentiating apoptosis through IAP-binding. Cell. Death Differ. 2002, 9, 93–95. [Google Scholar]
- Hu, S.; Yang, X. Cellular inhibitor of apoptosis 1 and 2 are ubiquitin ligases for the apoptosis inducer Smac/DIABLO. J. Biol. Chem. 2003, 278, 10055–10060. [Google Scholar] [CrossRef]
- Vucic, D.; Franklin, M.C.; Wallweber, H.J.; Das, K.; Eckelman, B.P.; Shin, H.; Elliott, L.O.; Kadkhodayan, S.; Deshayes, K.; Salvesen, G.S.; et al. Engineering ML-IAP to produce an extraordinarily potent caspase 9 inhibitor: implications for Smac-dependent anti-apoptotic activity of ML-IAP. Biochem. J. 2005, 385, 11–20. [Google Scholar] [CrossRef]
- Wu, G.; Chai, J.; Suber, T.L.; Wu, J.W.; Du, C.; Wang, X.; Shi, Y. Structural basis of IAP recognition by Smac/DIABLO. Nature 2000, 408, 1008–1012. [Google Scholar]
- Liu, Z.; Sun, C.; Olejniczak, E.T.; Meadows, R.P.; Betz, S.F.; Oost, T.; Herrmann, J.; Wu, J.C.; Fesik, S.W. Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature 2000, 408, 1004–1008. [Google Scholar] [CrossRef]
- Eckelman, B.P.; Salvesen, G.S.; Scott, F.L. Human inhibitor of apoptosis proteins: Why XIAP is the black sheep of the family. EMBO Rep. 2006, 7, 988–994. [Google Scholar] [CrossRef]
- Scott, F.L.; Denault, J.B.; Riedl, S.J.; Shin, H.; Renatus, M.; Salvesen, G.S. XIAP inhibits caspase-3 and -7 using two binding sites: evolutionarily conserved mechanism of IAPs. EMBO J. 2005, 24, 645–655. [Google Scholar] [CrossRef]
- Shiozaki, E.N.; Chai, J.; Rigotti, D.J.; Riedl, S.J.; Li, P.; Srinivasula, S.M.; Alnemri, E.S.; Fairman, R.; Shi, Y. Mechanism of XIAP-mediated inhibition of caspase-9. Mol. Cell. 2003, 11, 519–527. [Google Scholar] [CrossRef]
- Riedl, S.J.; Renatus, M.; Schwarzenbacher, R.; Zhou, Q.; Sun, C.; Fesik, S.W.; Liddington, R.C.; Salvesen, G.S. Structural basis for the inhibition of caspase-3 by XIAP. Cell 2001, 104, 791–800. [Google Scholar] [CrossRef]
- Suzuki, Y.; Nakabayashi, Y.; Takahashi, R. Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc. Natl. Acad. Sci. USA 2001, 98, 8662–8667. [Google Scholar]
- Morizane, Y.; Honda, R.; Fukami, K.; Yasuda, H. X-linked inhibitor of apoptosis functions as ubiquitin ligase toward mature caspase-9 and cytosolic Smac/DIABLO. J. Biochem. 2005, 137, 125–132. [Google Scholar] [CrossRef]
- Broemer, M.; Tenev, T.; Rigbolt, K.T.; Hempel, S.; Blagoev, B.; Silke, J.; Ditzel, M.; Meier, P. Systematic in vivo RNAi analysis identifies IAPs as NEDD8-E3 ligases. Mol. Cell 2010, 40, 810–822. [Google Scholar] [CrossRef]
- Schile, A.J.; Garcia-Fernandez, M.; Steller, H. Regulation of apoptosis by XIAP ubiquitin-ligase activity. Genes Dev. 2008, 22, 2256–2266. [Google Scholar] [CrossRef]
- Ho, A.T.; Li, Q.H.; Okada, H.; Mak, T.W.; Zacksenhaus, E. XIAP activity dictates Apaf-1 dependency for caspase 9 activation. Mol. Cell. Biol. 2007, 27, 5673–5685. [Google Scholar] [CrossRef]
- Wright, K.M.; Linhoff, M.W.; Potts, P.R.; Deshmukh, M. Decreased apoptosome activity with neuronal differentiation sets the threshold for strict IAP regulation of apoptosis. J. Cell. Biol. 2004, 167, 303–313. [Google Scholar] [CrossRef]
- Potts, M.B.; Vaughn, A.E.; McDonough, H.; Patterson, C.; Deshmukh, M. Reduced Apaf-1 levels in cardiomyocytes engage strict regulation of apoptosis by endogenous XIAP. J. Cell. Biol. 2005, 171, 925–930. [Google Scholar] [CrossRef]
- Choi, Y.E.; Butterworth, M.; Malladi, S.; Duckett, C.S.; Cohen, G.M.; Bratton, S.B. The E3 ubiquitin ligase cIAP1 binds and ubiquitinates caspase-3 and -7 via unique mechanisms at distinct steps in their processing. J. Biol. Chem. 2009, 284, 12772–12782. [Google Scholar]
- Huang, H.; Joazeiro, C.A.; Bonfoco, E.; Kamada, S.; Leverson, J.D.; Hunter, T. The inhibitor of apoptosis, cIAP2, functions as a ubiquitin-protein ligase and promotes in vitro monoubiquitination of caspases 3 and 7. J. Biol. Chem. 2000, 275, 26661–26664. [Google Scholar]
- Lee, T.V.; Fan, Y.; Wang, S.; Srivastava, M.; Broemer, M.; Meier, P.; Bergmann, A. Drosophila IAP1-mediated ubiquitylation controls activation of the initiator caspase DRONC independent of protein degradation. PLoS Genet. 2011, 7, e1002261. [Google Scholar] [CrossRef]
- Davoodi, J.; Ghahremani, M.H.; Es-Haghi, A.; Mohammad-Gholi, A.; Mackenzie, A. Neuronal apoptosis inhibitory protein, NAIP, is an inhibitor of procaspase-9. Int. J. Biochem. Cell. Biol. 2010, 42, 958–964. [Google Scholar] [CrossRef]
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J.; et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007, 131, 669–681. [Google Scholar] [CrossRef]
- Dueber, E.C.; Schoeffler, A.J.; Lingel, A.; Elliott, J.M.; Fedorova, A.V.; Giannetti, A.M.; Zobel, K.; Maurer, B.; Varfolomeev, E.; Wu, P.; et al. Antagonists induce a conformational change in cIAP1 that promotes autoubiquitination. Science 2011, 334, 376–380. [Google Scholar]
- Vince, J.E.; Wong, W.W.; Khan, N.; Feltham, R.; Chau, D.; Ahmed, A.U.; Benetatos, C.A.; Chunduru, S.K.; Condon, S.M.; McKinlay, M.; et al. IAP Antagonists Target cIAP1 to Induce TNFalpha-Dependent Apoptosis. Cell 2007, 131, 682–693. [Google Scholar] [CrossRef]
- Varfolomeev, E.; Goncharov, T.; Fedorova, A.V.; Dynek, J.N.; Zobel, K.; Deshayes, K.; Fairbrother, W.J.; Vucic, D. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J. Biol. Chem. 2008, 283, 24295–24299. [Google Scholar]
- Vanlangenakker, N.; Vanden Berghe, T.; Bogaert, P.; Laukens, B.; Zobel, K.; Deshayes, K.; Vucic, D.; Fulda, S.; Vandenabeele, P.; Bertrand, M.J. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell. Death Differ. 2011, 18, 656–665. [Google Scholar] [CrossRef]
- Petersen, S.L.; Wang, L.; Yalcin-Chin, A.; Li, L.; Peyton, M.; Minna, J.; Harran, P.; Wang, X. Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell. 2007, 12, 445–456. [Google Scholar] [CrossRef]
- Gaither, A.; Porter, D.; Yao, Y.; Borawski, J.; Yang, G.; Donovan, J.; Sage, D.; Slisz, J.; Tran, M.; Straub, C.; et al. A Smac mimetic rescue screen reveals roles for inhibitor of apoptosis proteins in tumor necrosis factor-alpha signaling. Cancer Res. 2007, 67, 11493–11498. [Google Scholar]
- Vince, J.E.; Chau, D.; Callus, B.; Wong, W.W.; Hawkins, C.J.; Schneider, P.; McKinlay, M.; Benetatos, C.A.; Condon, S.M.; Chunduru, S.K.; et al. TWEAK-FN14 signaling induces lysosomal degradation of a cIAP1-TRAF2 complex to sensitize tumor cells to TNFalpha. J. Cell. Biol. 2008, 182, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, M.J.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell. 2008, 30, 689–700. [Google Scholar] [CrossRef]
- Park, S.M.; Yoon, J.B.; Lee, T.H. Receptor interacting protein is ubiquitinated by cellular inhibitor of apoptosis proteins (c-IAP1 and c-IAP2) in vitro. FEBS Lett. 2004, 566, 151–156. [Google Scholar] [CrossRef]
- Geserick, P.; Hupe, M.; Moulin, M.; Wong, W.W.; Feoktistova, M.; Kellert, B.; Gollnick, H.; Silke, J.; Leverkus, M. Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J. Cell. Biol. 2009, 187, 1037–1054. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, M.J.; Lippens, S.; Staes, A.; Gilbert, B.; Roelandt, R.; De Medts, J.; Gevaert, K.; Declercq, W.; Vandenabeele, P. cIAP1/2 are direct E3 ligases conjugating diverse types of ubiquitin chains to receptor interacting proteins kinases 1 to 4 (RIP1–4). PLoS One 2011, 6, e22356. [Google Scholar]
- Dynek, J.N.; Goncharov, T.; Dueber, E.C.; Fedorova, A.V.; Izrael-Tomasevic, A.; Phu, L.; Helgason, E.; Fairbrother, W.J.; Deshayes, K.; Kirkpatrick, D.S.; et al. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J. 2010, 29, 4198–4209. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell. Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Moulin, M.; Anderton, H.; Voss, A.K.; Thomas, T.; Wong, W.W.; Bankovacki, A.; Feltham, R.; Chau, D.; Cook, W.D.; Silke, J.; et al. IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J. 2012, 31, 1679–1691. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.S.; Ghosh, S. NF-kappaB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef]
- Jin, H.S.; Lee, D.H.; Kim, D.H.; Chung, J.H.; Lee, S.J.; Lee, T.H. cIAP1, cIAP2, and XIAP act cooperatively via nonredundant pathways to regulate genotoxic stress-induced nuclear factor-kappaB activation. Cancer Res. 2009, 69, 1782–1791. [Google Scholar] [CrossRef]
- Niu, J.; Shi, Y.; Iwai, K.; Wu, Z.H. LUBAC regulates NF-kappaB activation upon genotoxic stress by promoting linear ubiquitination of NEMO. EMBO J. 2011, 30, 3741–3753. [Google Scholar] [CrossRef]
- Gerlach, B.; Cordier, S.M.; Schmukle, A.C.; Emmerich, C.H.; Rieser, E.; Haas, T.L.; Webb, A.I.; Rickard, J.A.; Anderton, H.; Wong, W.W.; et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 2011, 471, 591–596. [Google Scholar]
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 2009, 36, 831–844. [Google Scholar] [CrossRef]
- Lu, M.; Lin, S.C.; Huang, Y.; Kang, Y.J.; Rich, R.; Lo, Y.C.; Myszka, D.; Han, J.; Wu, H. XIAP induces NF-kappaB activation via the BIR1/TAB1 interaction and BIR1 dimerization. Mol. Cell 2007, 26, 689–702. [Google Scholar] [CrossRef]
- Hinz, M.; Stilmann, M.; Arslan, S.C.; Khanna, K.K.; Dittmar, G.; Scheidereit, C. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-kappaB activation. Mol. Cell. 2010, 40, 63–74. [Google Scholar] [CrossRef]
- Zarnegar, B.J.; Wang, Y.; Mahoney, D.J.; Dempsey, P.W.; Cheung, H.H.; He, J.; Shiba, T.; Yang, X.; Yeh, W.C.; Mak, T.W.; et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 2008, 9, 1371–1378. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Matsuzawa, A.; Zhang, W.; Tseng, P.H.; Keats, J.J.; Wang, H.; Vignali, D.A.; Bergsagel, P.L.; Karin, M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat. Immunol. 2008, 9, 1364–1370. [Google Scholar] [CrossRef]
- van der Waal, M.S.; Hengeveld, R.C.; van der Horst, A.; Lens, S.M. Cell division control by the Chromosomal Passenger Complex. Exp. Cell. Res. 2012, 318, 1407–1420. [Google Scholar] [CrossRef]
- Cartier, J.; Berthelet, J.; Marivin, A.; Gemble, S.; Edmond, V.; Plenchette, S.; Lagrange, B.; Hammann, A.; Dupoux, A.; Delva, L.; et al. Cellular inhibitor of apoptosis protein-1 (cIAP1) can regulate E2F1 transcription factor-mediated control of cyclin transcription. J. Biol. Chem. 2011, 286, 26406–26417. [Google Scholar]
- Plenchette, S.; Cathelin, S.; Rebe, C.; Launay, S.; Ladoire, S.; Sordet, O.; Ponnelle, T.; Debili, N.; Phan, T.H.; Padua, R.A.; et al. Translocation of the inhibitor of apoptosis protein c-IAP1 from the nucleus to the Golgi in hematopoietic cells undergoing differentiation: A nuclear export signal-mediated event. Blood 2004, 104, 2035–2043. [Google Scholar] [CrossRef]
- Didelot, C.; Lanneau, D.; Brunet, M.; Bouchot, A.; Cartier, J.; Jacquel, A.; Ducoroy, P.; Cathelin, S.; Decologne, N.; Chiosis, G.; et al. Interaction of heat-shock protein 90 beta isoform (HSP90 beta) with cellular inhibitor of apoptosis 1 (c-IAP1) is required for cell differentiation. Cell. Death Differ. 2008, 15, 859–866. [Google Scholar] [CrossRef]
- Luscher, B.; Vervoorts, J. Regulation of gene transcription by the oncoprotein MYC. Gene 2012, 494, 145–160. [Google Scholar] [CrossRef]
- Xu, L.; Zhu, J.; Hu, X.; Zhu, H.; Kim, H.T.; LaBaer, J.; Goldberg, A.; Yuan, J. c-IAP1 cooperates with Myc by acting as a ubiquitin ligase for Mad1. Mol. Cell 2007, 28, 914–922. [Google Scholar] [CrossRef]
- Holcik, M.; Yeh, C.; Korneluk, R.G.; Chow, T. Translational upregulation of X-linked inhibitor of apoptosis (XIAP) increases resistance to radiation induced cell death. Oncogene 2000, 19, 4174–4177. [Google Scholar] [CrossRef]
- Gu, L.; Zhu, N.; Zhang, H.; Durden, D.L.; Feng, Y.; Zhou, M. Regulation of XIAP translation and induction by MDM2 following irradiation. Cancer Cell. 2009, 15, 363–375. [Google Scholar] [CrossRef]
- Nevins, T.A.; Harder, Z.M.; Korneluk, R.G.; Holcík, M. Distinct regulation of internal ribosome entry site-mediated translation following cellular stress is mediated by apoptotic fragments of eIF4G translation initiation factor family members eIF4GI and p97/DAP5/NAT1. J. Biol. Chem. 2003, 278, 3572–3579. [Google Scholar]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell. Biol. 2012, 13, 89–102. [Google Scholar]
- Hamanaka, R.B.; Bobrovnikova-Marjon, E.; Ji, X.; Liebhaber, S.A.; Diehl, J.A. PERK-dependent regulation of IAP translation during ER stress. Oncogene 2009, 28, 910–920. [Google Scholar] [CrossRef]
- Hu, P.; Han, Z.; Couvillon, A.D.; Exton, J.H. Critical role of endogenous Akt/IAPs and MEK1/ERK pathways in counteracting endoplasmic reticulum stress-induced cell death. J. Biol. Chem. 2004, 279, 49420–49429. [Google Scholar]
- Hegde, R.; Srinivasula, S.M.; Datta, P.; Madesh, M.; Wassell, R.; Zhang, Z.; Cheong, N.; Nejmeh, J.; Fernandes-Alnemri, T.; Hoshino, S.; et al. The polypeptide chain-releasing factor GSPT1/eRF3 is proteolytically processed into an IAP-binding protein. J. Biol. Chem. 2003, 278, 38699–38706. [Google Scholar]
- Nakamura, T.; Cho, D.H.; Lipton, S.A. Redox regulation of protein misfolding, mitochondrial dysfunction, synaptic damage, and cell death in neurodegenerative diseases. Exp. Neurol. 2012, 238, 12–21. [Google Scholar] [CrossRef]
- Russell, J.C.; Whiting, H.; Szuflita, N.; Hossain, M.A. Nuclear translocation of X-linked inhibitor of apoptosis (XIAP) determines cell fate after hypoxia ischemia in neonatal brain. J. Neurochem. 2008, 106, 1357–1370. [Google Scholar] [CrossRef]
- West, T.; Stump, M.; Lodygensky, G.; Neil, J.J.; Deshmukh, M.; Holtzman, D.M. Lack of X-linked inhibitor of apoptosis protein leads to increased apoptosis and tissue loss following neonatal brain injury. ASN Neuro 2009, 1, 43–53. [Google Scholar]
- Hill, C.A.; Fitch, R.H. Sex differences in mechanisms and outcome of neonatal hypoxia-ischemia in rodent models: Implications for sex-specific neuroprotection in clinical neonatal practice. Neurol. Res. Int. 2012, 2012, 867531–867539. [Google Scholar]
- Guegan, C.; Braudeau, J.; Couriaud, C.; Dietz, G.P.; Lacombe, P.; Bahr, M.; Nosten-Bertrand, M.; Onteniente, B. PTD-XIAP protects against cerebral ischemia by anti-apoptotic and transcriptional regulatory mechanisms. Neurobiol. Dis. 2006, 22, 177–186. [Google Scholar] [CrossRef]
- Li, T.; Fan, Y.; Luo, Y.; Xiao, B.; Lu, C. In vivo delivery of a XIAP (BIR3-RING) fusion protein containing the protein transduction domain protects against neuronal death induced by seizures. Exp. Neurol. 2006, 197, 301–308. [Google Scholar] [CrossRef]
- Fan, Y.F.; Lu, C.Z.; Xie, J.; Zhao, Y.X.; Yang, G.Y. Apoptosis inhibition in ischemic brain by intraperitoneal PTD-BIR3-RING (XIAP). Neurochem. Int. 2006, 48, 50–59. [Google Scholar] [CrossRef]
- Zhu, C.; Xu, F.; Fukuda, A.; Wang, X.; Fukuda, H.; Korhonen, L.; Hagberg, H.; Lannering, B.; Nilsson, M.; Eriksson, P.S.; et al. X chromosome-linked inhibitor of apoptosis protein reduces oxidative stress after cerebral irradiation or hypoxia-ischemia through up-regulation of mitochondrial antioxidants. Eur. J. Neurosci. 2007, 26, 3402–3410. [Google Scholar] [CrossRef]
- Kairisalo, M.; Korhonen, L.; Blomgren, K.; Lindholm, D. X-linked inhibitor of apoptosis protein increases mitochondrial antioxidants through NF-kappaB activation. Biochem. Biophys. Res. Commun. 2007, 364, 138–144. [Google Scholar] [CrossRef]
- Resch, U.; Schichl, Y.M.; Sattler, S.; de Martin, R. XIAP regulates intracellular ROS by enhancing antioxidant gene expression. Biochem. Biophys. Res. Commun. 2008, 375, 156–161. [Google Scholar] [CrossRef]
- Maine, G.N.; Mao, X.; Muller, P.A.; Komarck, C.M.; Klomp, L.W.; Burstein, E. COMMD1 expression is controlled by critical residues that determine XIAP binding. Biochem. J. 2009, 417, 601–609. [Google Scholar] [CrossRef]
- Burstein, E.; Ganesh, L.; Dick, R.D.; van De Sluis, B.; Wilkinson, J.C.; Klomp, L.W.; Wijmenga, C.; Brewer, G.J.; Nabel, G.J.; Duckett, C.S. A novel role for XIAP in copper homeostasis through regulation of MURR1. EMBO J. 2004, 23, 244–254. [Google Scholar] [CrossRef]
- Brady, G.F.; Galban, S.; Liu, X.; Basrur, V.; Gitlin, J.D.; Elenitoba-Johnson, K.S.; Wilson, T.E.; Duckett, C.S. Regulation of the copper chaperone CCS by XIAP-mediated ubiquitination. Mol. Cell. Biol. 2010, 30, 1923–1936. [Google Scholar] [CrossRef]
- Mufti, A.R.; Burstein, E.; Csomos, R.A.; Graf, P.C.; Wilkinson, J.C.; Dick, R.D.; Challa, M.; Son, J.K.; Bratton, S.B.; Su, G.L.; et al. XIAP Is a copper binding protein deregulated in Wilson's disease and other copper toxicosis disorders. Mol. Cell 2006, 21, 775–785. [Google Scholar] [CrossRef]
- Holcik, M.; Thompson, C.S.; Yaraghi, Z.; Lefebvre, C.A.; MacKenzie, A.E.; Korneluk, R.G. The hippocampal neurons of neuronal apoptosis inhibitory protein 1 (NAIP1)-deleted mice display increased vulnerability to kainic acid-induced injury. Proc. Natl. Acad. Sci. USA 2000, 97, 2286–2290. [Google Scholar]
- Masumu, M.; Hata, R. Recent advances in adenovirus-mediated gene therapy for cerebral ischemia. Curr. Gene Ther. 2003, 3, 43–48. [Google Scholar] [CrossRef]
- Lewis, S.M.; Holcik, M. IRES in distress: Translational regulation of the inhibitor of apoptosis proteins XIAP and HIAP2 during cell stress. Cell. Death Differ. 2005, 12, 547–553. [Google Scholar] [CrossRef]
- Parihar, A.; Eubank, T.D.; Doseff, A.I. Monocytes and macrophages regulate immunity through dynamic networks of survival and cell death. J. Innate Immun. 2010, 2, 204–215. [Google Scholar] [CrossRef]
- Lin, H.; Chen, C.; Chen, B.D. Resistance of bone marrow-derived macrophages to apoptosis is associated with the expression of X-linked inhibitor of apoptosis protein in primary cultures of bone marrow cells. Biochem. J. 2001, 353, 299–306. [Google Scholar] [CrossRef]
- Miranda, M.B.; Dyer, K.F.; Grandis, J.R.; Johnson, D.E. Differential activation of apoptosis regulatory pathways during monocytic vs granulocytic differentiation: A requirement for Bcl-X(L)and XIAP in the prolonged survival of monocytic cells. Leukemia 2003, 17, 390–400. [Google Scholar] [CrossRef]
- Hida, A.; Kawakami, A.; Nakashima, T.; Yamasaki, S.; Sakai, H.; Urayama, S.; Ida, H.; Nakamura, H.; Migita, K.; Kawabe, Y.; et al. Nuclear factor-kappaB and caspases co-operatively regulate the activation and apoptosis of human macrophages. Immunology 2000, 99, 553–560. [Google Scholar] [CrossRef]
- Lin, H.; Chen, C.; Li, X.; Chen, B.D. Activation of the MEK/MAPK pathway is involved in bryostatin1-induced monocytic differenciation and up-regulation of X-linked inhibitor of apoptosis protein. Exp. Cell. Res. 2002, 272, 192–198. [Google Scholar] [CrossRef]
- Conte, D.; Holcik, M.; Lefebvre, C.A.; Lacasse, E.; Picketts, D.J.; Wright, K.E.; Korneluk, R.G. Inhibitor of apoptosis protein cIAP2 is essential for lipopolysaccharide-induced macrophage survival. Mol. Cell. Biol. 2006, 26, 699–708. [Google Scholar] [CrossRef]
- Cui, X.; Imaizumi, T.; Yoshida, H.; Tanji, K.; Matsumiya, T.; Satoh, K. Lipopolysaccharide induces the expression of cellular inhibitor of apoptosis protein-2 in human macrophages. Biochim. Biophys. Acta 2000, 1524, 178–182. [Google Scholar] [CrossRef]
- Matsunaga, T.; Ishida, T.; Takekawa, M.; Nishimura, S.; Adachi, M.; Imai, K. Analysis of gene expression during maturation of immature dendritic cells derived from peripheral blood monocytes. Scand. J. Immunol. 2002, 56, 593–601. [Google Scholar] [CrossRef]
- Dupoux, A.; Cartier, J.; Cathelin, S.; Filomenko, R.; Solary, E.; Dubrez-Daloz, L. cIAP1-dependent TRAF2 degradation regulates the differentiation of monocytes into macrophages and their response to CD40 ligand. Blood 2009, 113, 175–185. [Google Scholar] [CrossRef]
- Busca, A.; Saxena, M.; Kumar, A. Critical role for antiapoptotic Bcl-xL and Mcl-1 in human macrophage survival and cellular IAP1/2 (cIAP1/2) in resistance to HIV-Vpr-induced apoptosis. J. Biol. Chem. 2012, 287, 15118–15133. [Google Scholar]
- Zender, L.; Spector, M.S.; Xue, W.; Flemming, P.; Cordon-Cardo, C.; Silke, J.; Fan, S.T.; Luk, J.M.; Wigler, M.; Hannon, G.J.; et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 2006, 125, 1253–1267. [Google Scholar] [CrossRef]
- Ma, O.; Cai, W.W.; Zender, L.; Dayaram, T.; Shen, J.; Herron, A.J.; Lowe, S.W.; Man, T.K.; Lau, C.C.; Donehower, L.A. MMP13, Birc2 (cIAP1), and Birc3 (cIAP2), amplified on chromosome 9, collaborate with p53 deficiency in mouse osteosarcoma progression. Cancer Res. 2009, 69, 2559–2567. [Google Scholar]
- Cheng, L.; Zhou, Z.; Flesken-Nikitin, A.; Toshkov, I.A.; Wang, W.; Camps, J.; Ried, T.; Nikitin, A.Y. Rb inactivation accelerates neoplastic growth and substitutes for recurrent amplification of cIAP1, cIAP2 and Yap1 in sporadic mammary carcinoma associated with p53 deficiency. Oncogene 2010, 29, 5700–5711. [Google Scholar] [CrossRef]
- Imoto, I.; Tsuda, H.; Hirasawa, A.; Miura, M.; Sakamoto, M.; Hirohashi, S.; Inazawa, J. Expression of cIAP1, a target for 11q22 amplification, correlates with resistance of cervical cancers to radiotherapy. Cancer Res. 2002, 62, 4860–4866. [Google Scholar]
- Dai, Z.; Zhu, W.G.; Morrison, C.D.; Brena, R.M.; Smiraglia, D.J.; Raval, A.; Wu, Y.Z.; Rush, L.J.; Ross, P.; Molina, J.R.; et al. A comprehensive search for DNA amplification in lung cancer identifies inhibitors of apoptosis cIAP1 and cIAP2 as candidate oncogenes. Hum. Mol. Genet. 2003, 12, 791–801. [Google Scholar] [CrossRef]
- Snijders, A.M.; Schmidt, B.L.; Fridlyand, J.; Dekker, N.; Pinkel, D.; Jordan, R.C.; Albertson, D.G. Rare amplicons implicate frequent deregulation of cell fate specification pathways in oral squamous cell carcinoma. Oncogene 2005, 24, 4232–4242. [Google Scholar] [CrossRef]
- Imoto, I.; Yang, Z.Q.; Pimkhaokham, A.; Tsuda, H.; Shimada, Y.; Imamura, M.; Ohki, M.; Inazawa, J. Identification of cIAP1 as a candidate target gene within an amplicon at 11q22 in esophageal squamous cell carcinomas. Cancer Res. 2001, 61, 6629–6634. [Google Scholar]
- Varfolomeev, E.; Wayson, S.M.; Dixit, V.M.; Fairbrother, W.J.; Vucic, D. The inhibitor of apoptosis protein fusion c-IAP2.MALT1 stimulates NF-kappaB activation independently of TRAF1 AND TRAF2. J. Biol. Chem. 2006, 281, 29022–29029. [Google Scholar]
- Garrison, J.B.; Samuel, T.; Reed, J.C. TRAF2-binding BIR1 domain of c-IAP2/MALT1 fusion protein is essential for activation of NF-kappaB. Oncogene 2009, 28, 1584–1593. [Google Scholar] [CrossRef]
- Oberoi, T.K.; Dogan, T.; Hocking, J.C.; Scholz, R.P.; Mooz, J.; Anderson, C.L.; Karreman, C.; Meyer Zu Heringdorf, D.; Schmidt, G.; Ruonala, M.; et al. IAPs regulate the plasticity of cell migration by directly targeting Rac1 for degradation. Embo J. 2011, 31, 14–28. [Google Scholar] [CrossRef]
- Dogan, T.; Harms, G.S.; Hekman, M.; Karreman, C.; Oberoi, T.K.; Alnemri, E.S.; Rapp, U.R.; Rajalingam, K. X-linked and cellular IAPs modulate the stability of C-RAF kinase and cell motility. Nat. Cell. Biol. 2008, 10, 1447–1455. [Google Scholar] [CrossRef]
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.J.; Van Wier, S.; Tiedemann, R.; Shi, C.X.; Sebag, M.; et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007, 12, 131–144. [Google Scholar] [CrossRef]
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007, 12, 115–130. [Google Scholar] [CrossRef]
- Filipovich, A.H.; Zhang, K.; Snow, A.L.; Marsh, R.A. X-linked lymphoproliferative syndromes: brothers or distant cousins? Blood 2010, 116, 3398–3408. [Google Scholar] [CrossRef]
- Tsang, A.H.; Lee, Y.I.; Ko, H.S.; Savitt, J.M.; Pletnikova, O.; Troncoso, J.C.; Dawson, V.L.; Dawson, T.M.; Chung, K.K. S-nitrosylation of XIAP compromises neuronal survival in Parkinson's disease. Proc. Natl. Acad. Sci. USA 2009, 106, 4900–4905. [Google Scholar]
- Goffredo, D.; Rigamonti, D.; Zuccato, C.; Tartari, M.; Valenza, M.; Cattaneo, E. Prevention of cytosolic IAPs degradation: A potential pharmacological target in Huntington's Disease. Pharmacol. Res. 2005, 52, 140–150. [Google Scholar] [CrossRef]
- Watihayati, M.S.; Fatemeh, H.; Marini, M.; Atif, A.B.; Zahiruddin, W.M.; Sasongko, T.H.; Tang, T.H.; Zabidi-Hussin, Z.A.; Nishio, H.; Zilfalil, B.A. Combination of SMN2 copy number and NAIP deletion predicts disease severity in spinal muscular atrophy. Brain Dev. 2009, 31, 42–45. [Google Scholar] [CrossRef]
- Weiss, K.H.; Runz, H.; Noe, B.; Gotthardt, D.N.; Merle, U.; Ferenci, P.; Stremmel, W.; Fullekrug, J. Genetic analysis of BIRC4/XIAP as a putative modifier gene of Wilson disease. J. Inherit. Metab. Dis. 2010, 10, 1007–1014. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Marivin, A.; Berthelet, J.; Plenchette, S.; Dubrez, L. The Inhibitor of Apoptosis (IAPs) in Adaptive Response to Cellular Stress. Cells 2012, 1, 711-737. https://doi.org/10.3390/cells1040711
Marivin A, Berthelet J, Plenchette S, Dubrez L. The Inhibitor of Apoptosis (IAPs) in Adaptive Response to Cellular Stress. Cells. 2012; 1(4):711-737. https://doi.org/10.3390/cells1040711
Chicago/Turabian StyleMarivin, Arthur, Jean Berthelet, Stéphanie Plenchette, and Laurence Dubrez. 2012. "The Inhibitor of Apoptosis (IAPs) in Adaptive Response to Cellular Stress" Cells 1, no. 4: 711-737. https://doi.org/10.3390/cells1040711
APA StyleMarivin, A., Berthelet, J., Plenchette, S., & Dubrez, L. (2012). The Inhibitor of Apoptosis (IAPs) in Adaptive Response to Cellular Stress. Cells, 1(4), 711-737. https://doi.org/10.3390/cells1040711