Regulation of Autophagy by Glucose in Mammalian Cells

Abstract
: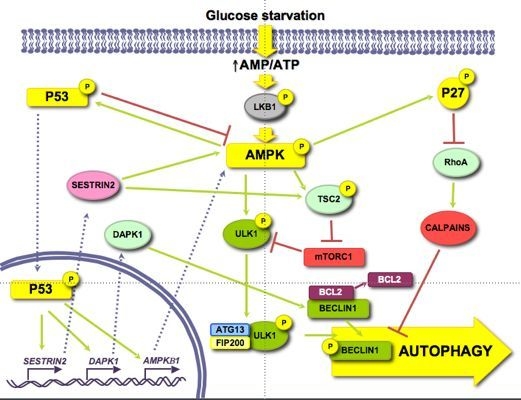
{kind=link}
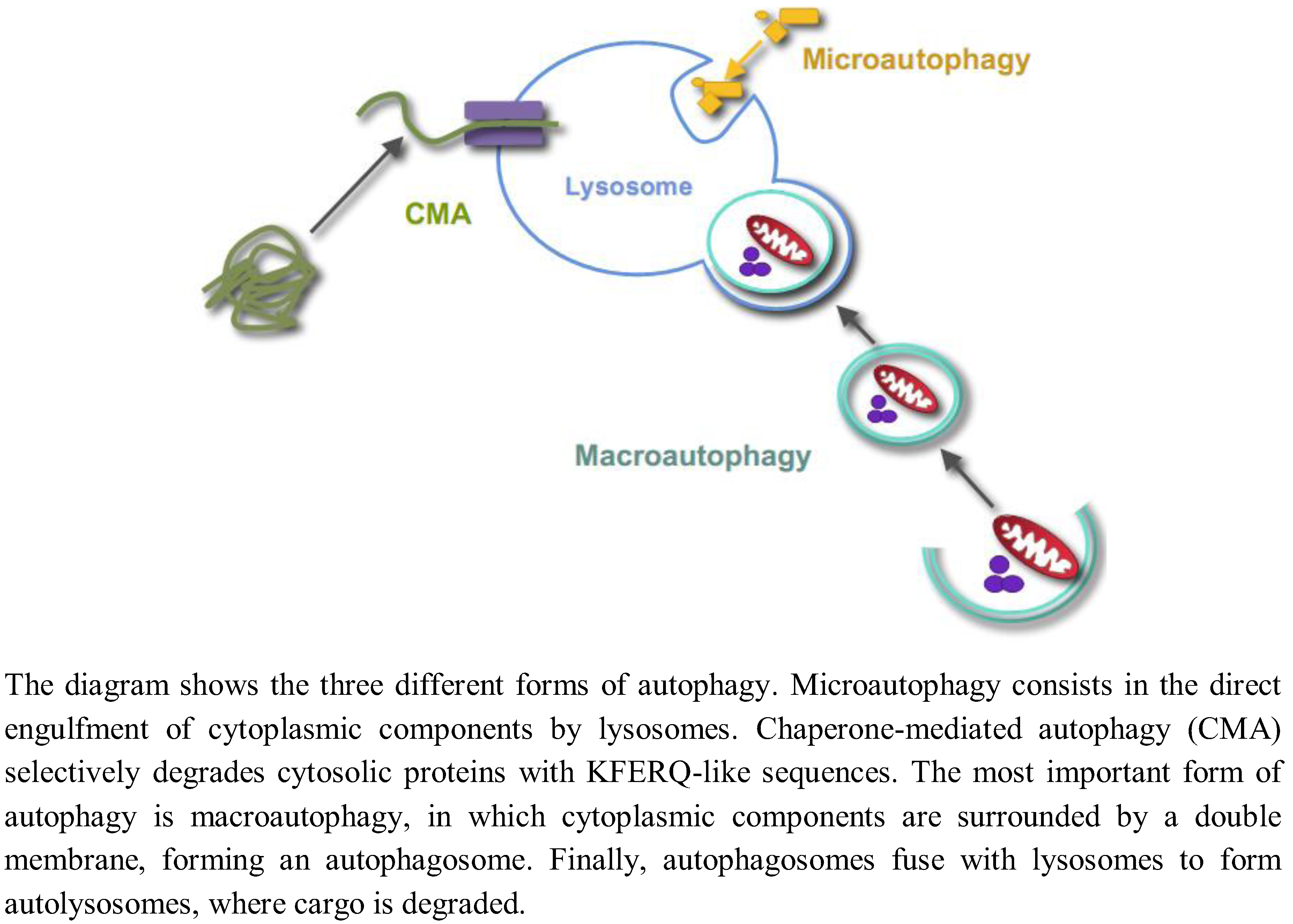{kind=link}
{kind=link}
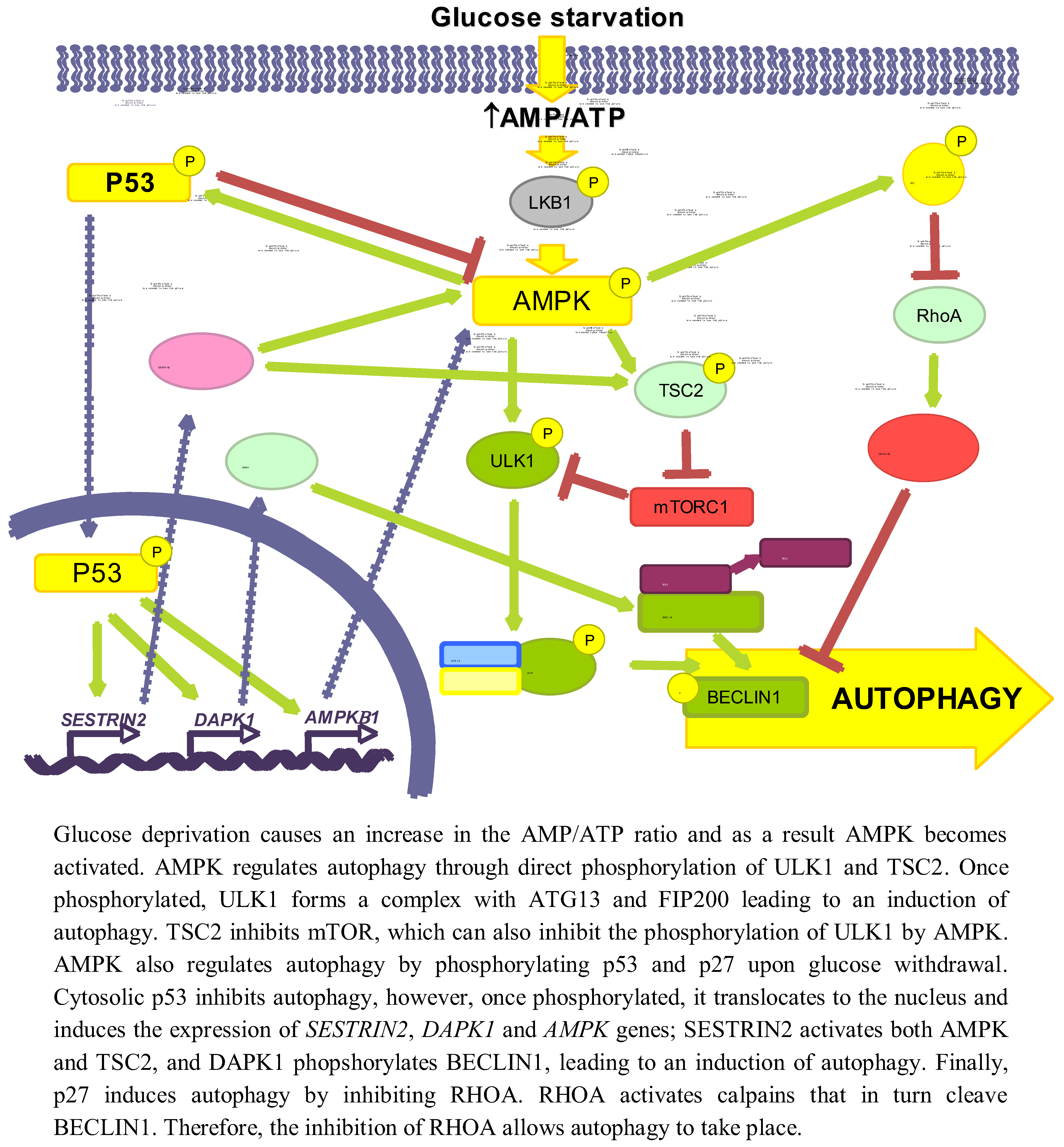
{kind=link}
{kind=link}
Abbreviations
| 2-DG | 2-deoxy-D-glucose |
| AICAR | 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside |
| AMPK | AMP-activated protein kinase |
| ATG | autophagy-related |
| ATP | adenosine triphosphate |
| ER | endoplasmic reticulum |
| ERK | extracellular signal-regulated protein kinase |
| FOXO | forkhead class O |
| IKK | IκB kinase |
| JNK | c-Jun amino-terminal kinase |
| PAS | preautophagosomal structure |
| PI3K | phospahtidylinositol 3-kinase |
| PKA | cAMP-dependent protein kinase A |
| ROS | reactive oxygen species |
| TAB | TAK-binding protein |
| V-ATPase | vacuolar ATPase |
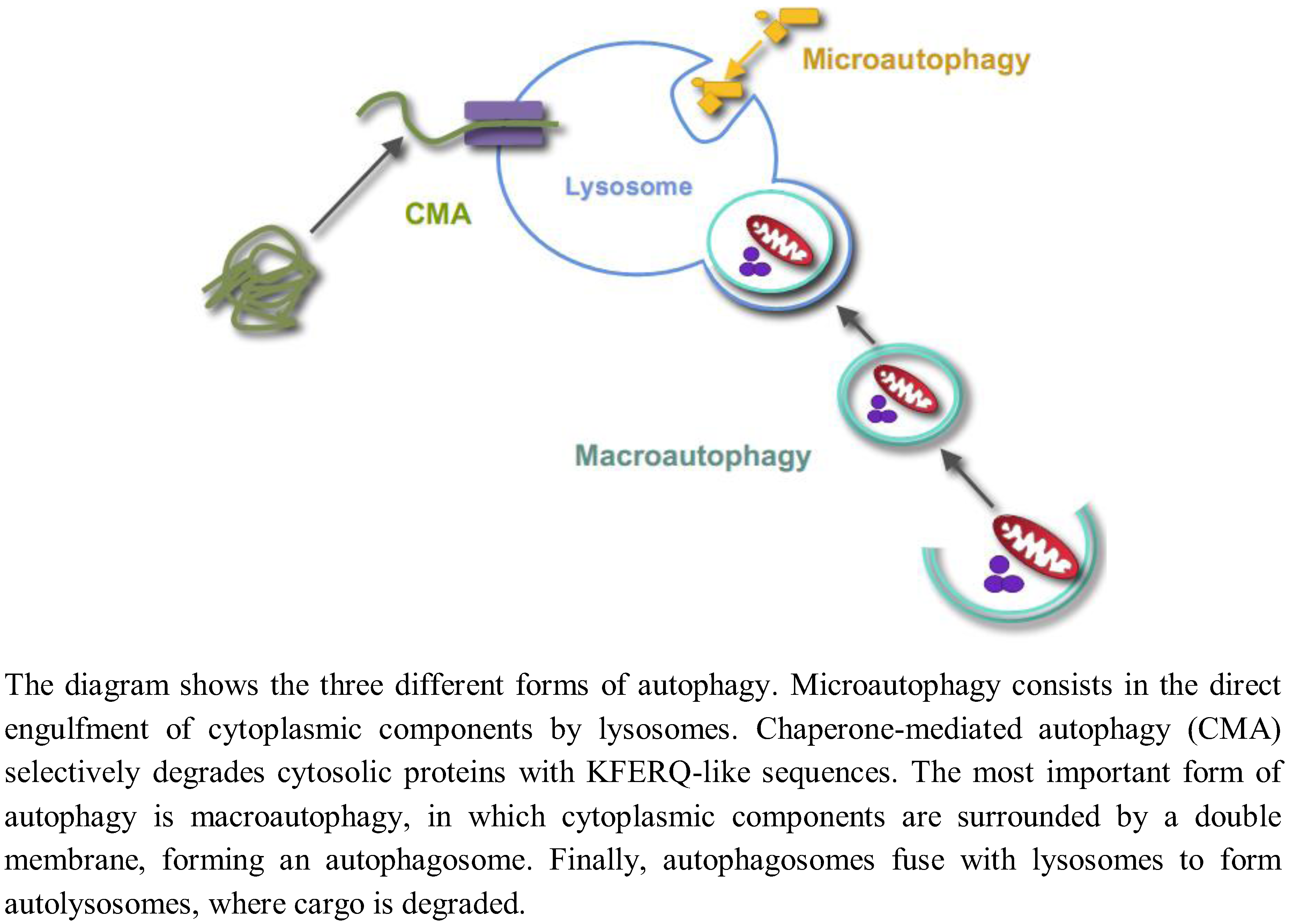
1. Introduction
2. Regulation of Autophagy by Glucose in Yeast
3. Regulation of Autophagy by Glucose in Mammalian Cells
3.1. Role of Energy-Dependent Pathways
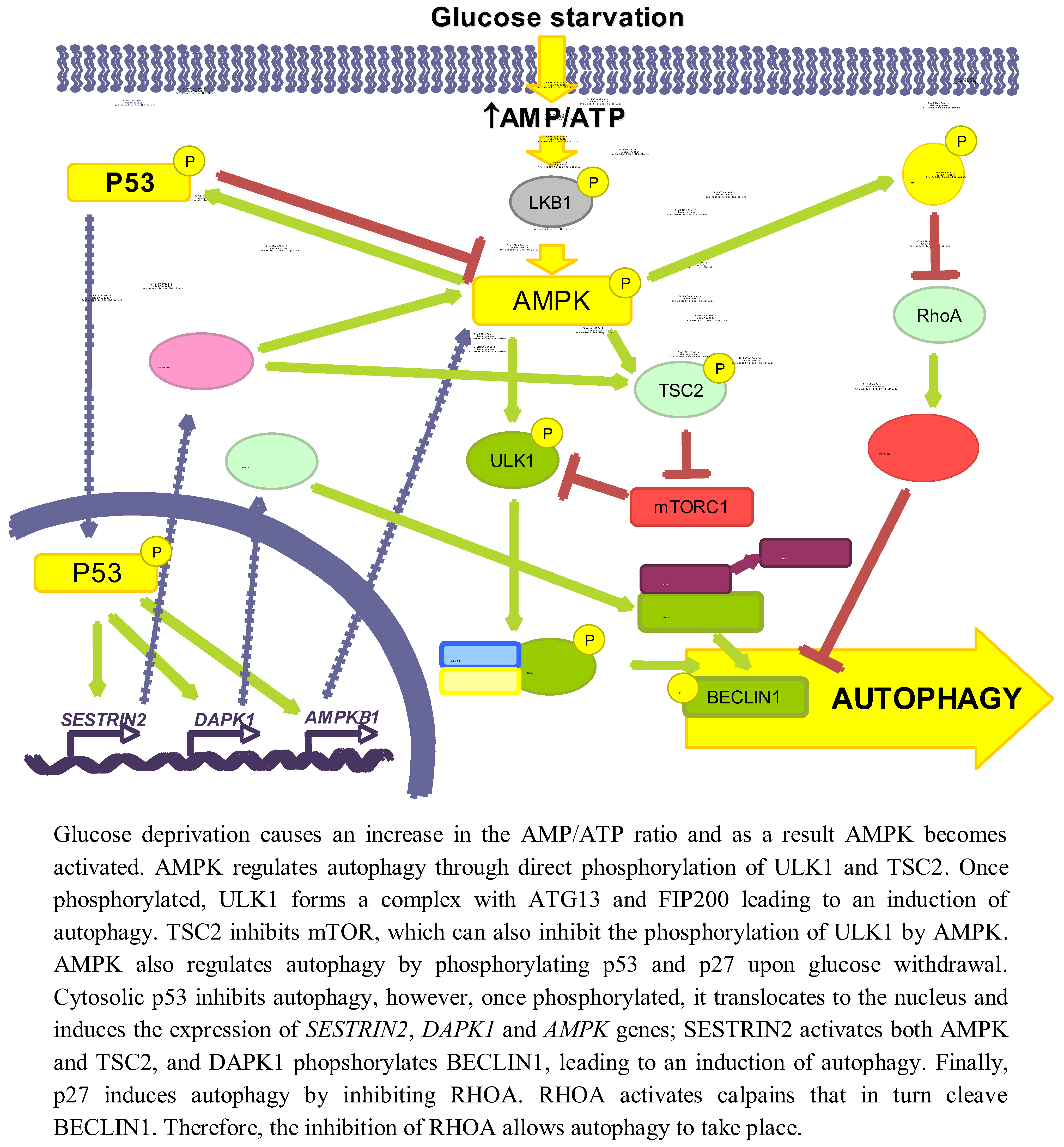
3.1.1. AMPK/mTOR
3.1.2. p53
3.1.3. p27KIP1
3.2. Role of Oxidative Stress-Dependent Pathways
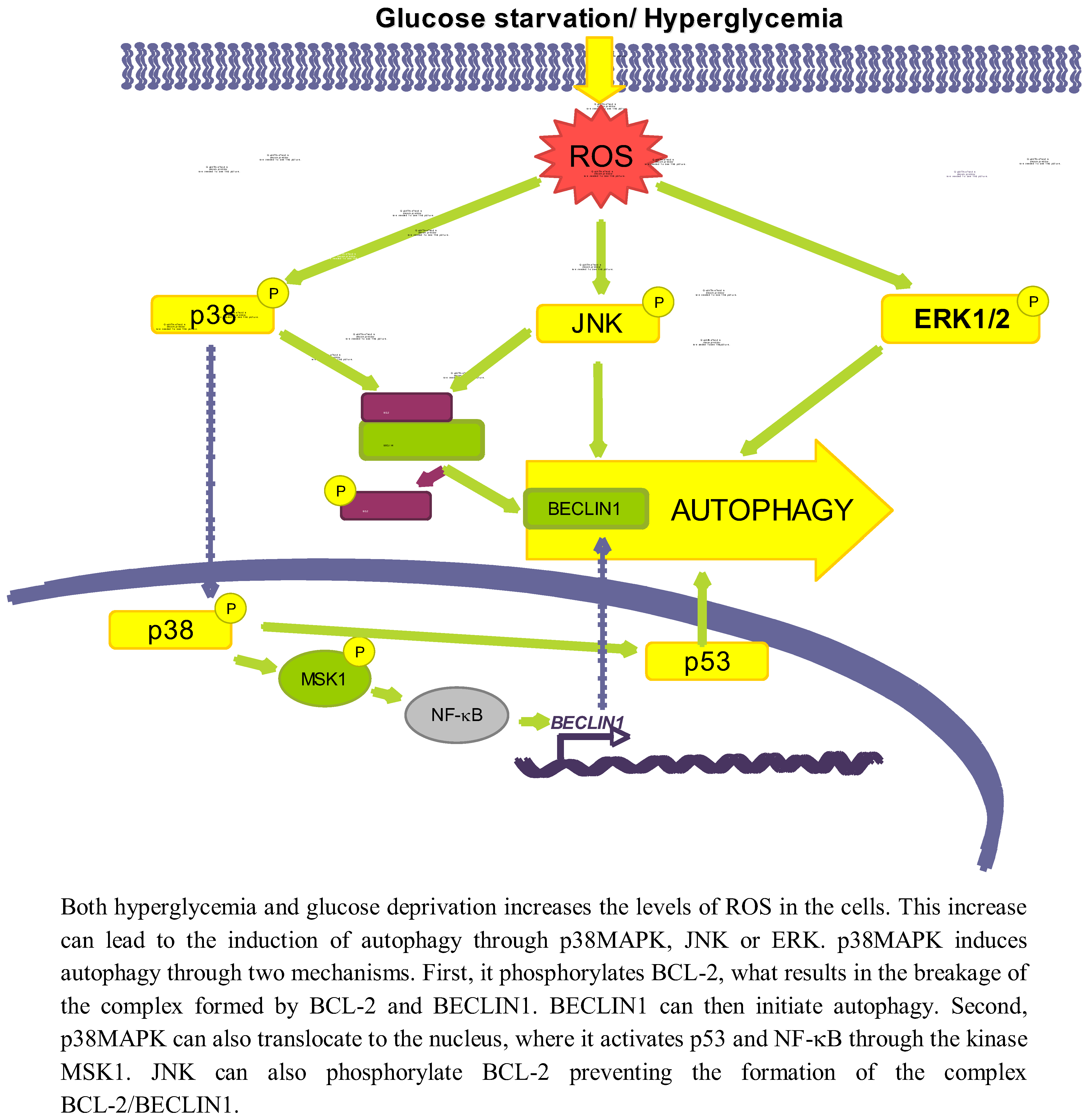
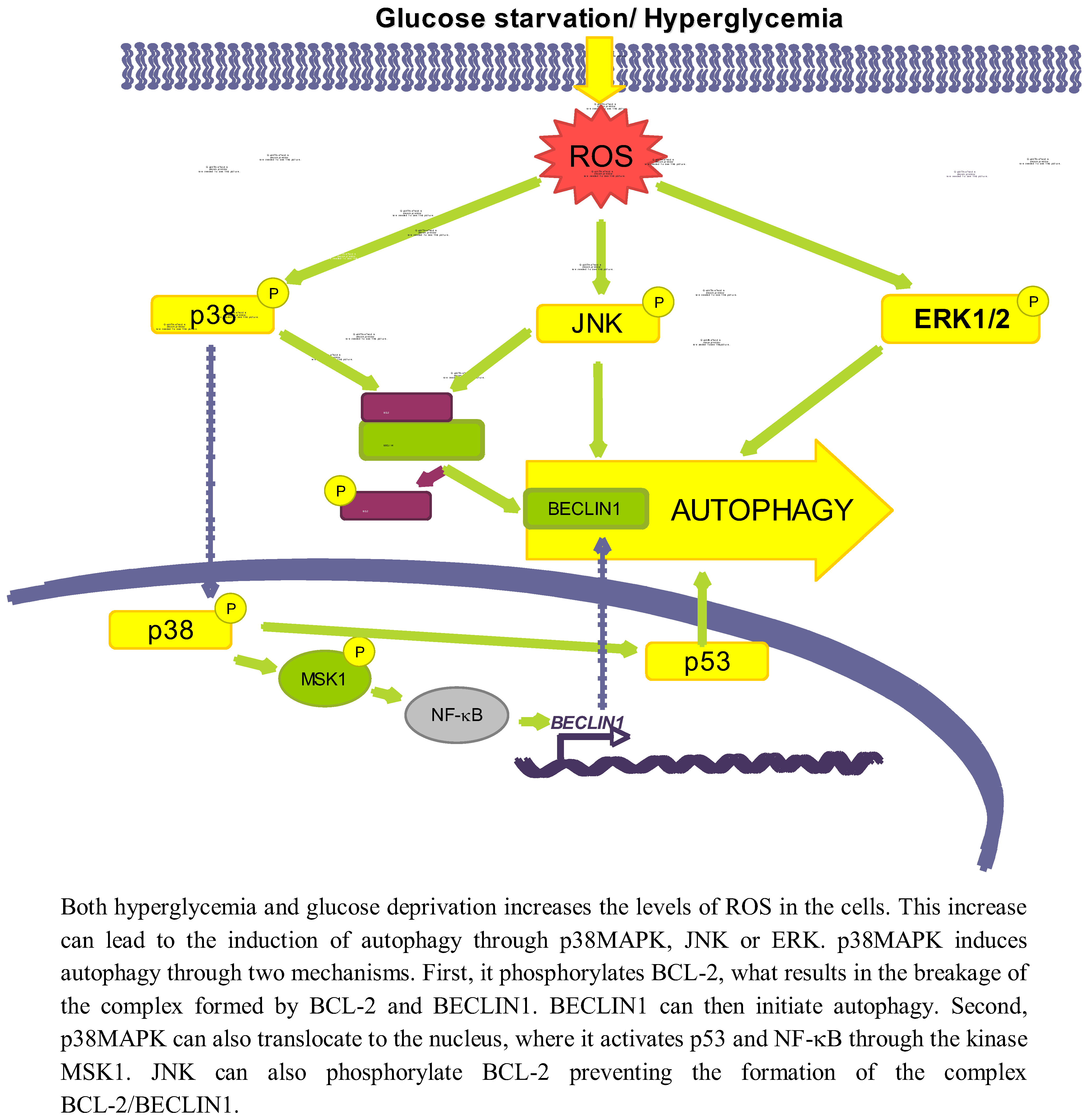
3.2.1. p38MAPK
3.2.2. JNK
3.2.3. ERK
3.3. Role of Other Pathways
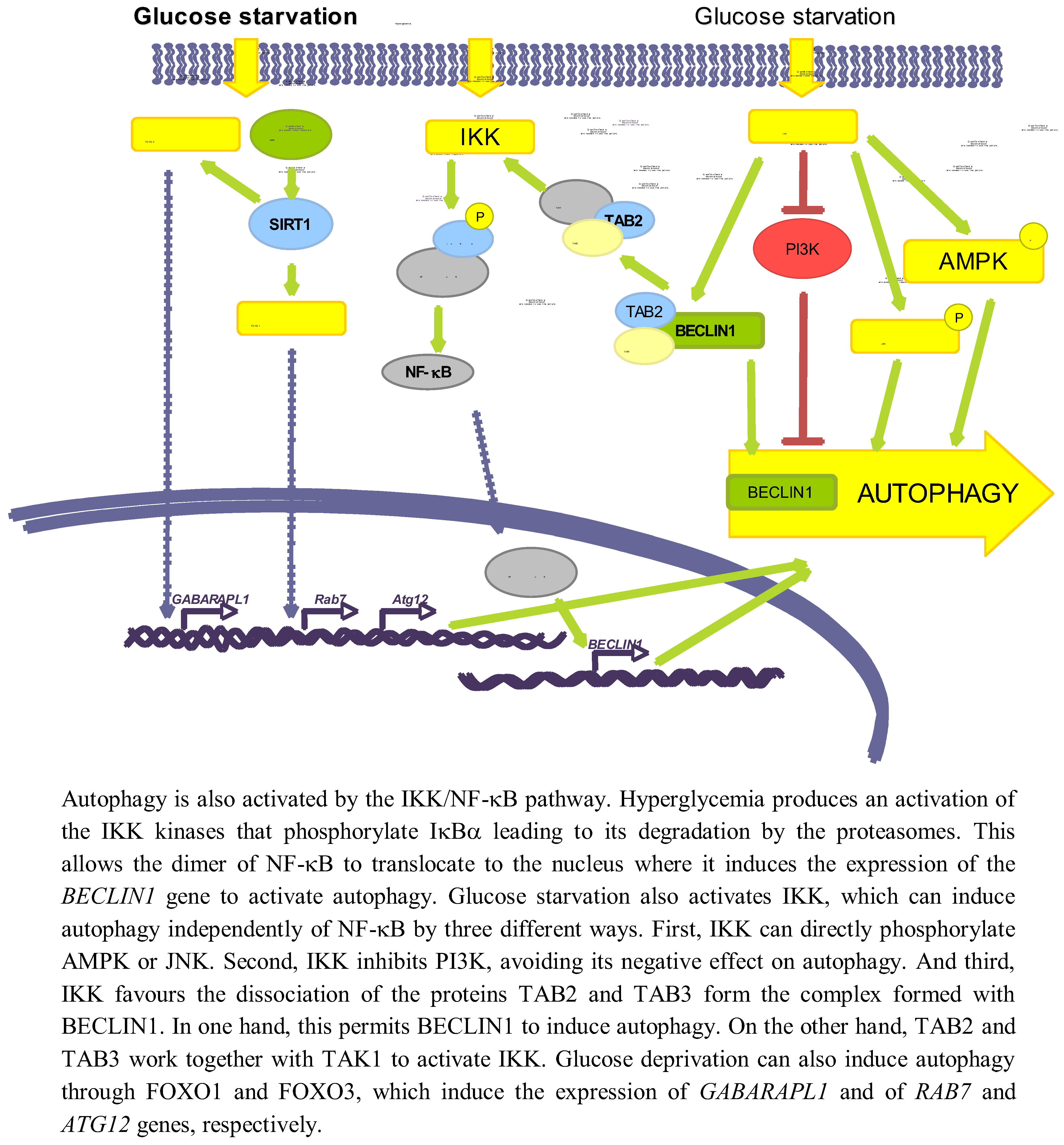
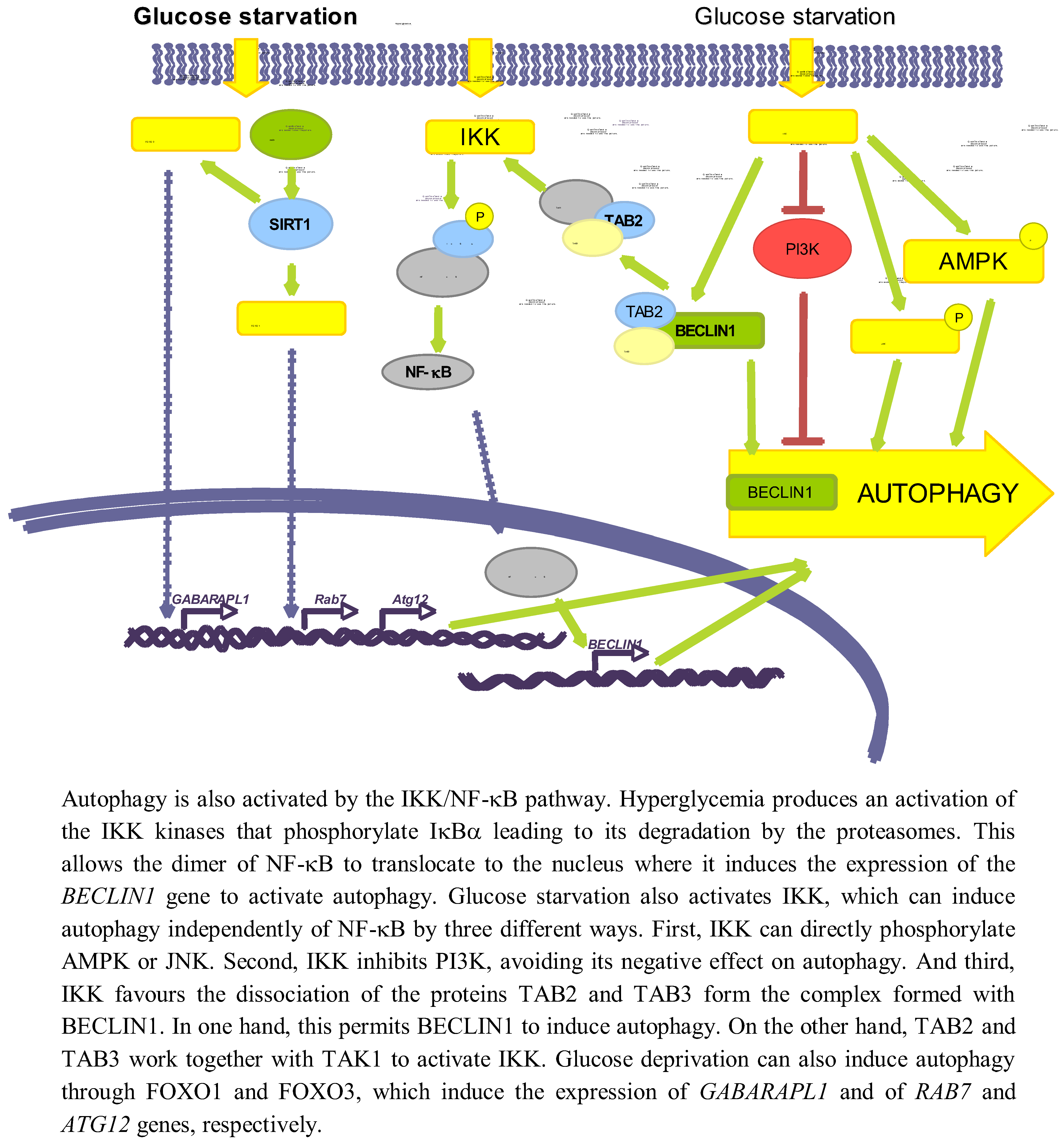
3.3.1. NF-κB
3.3.2. SIRT1 and FOXO
4. Pathological Implications of Autophagy Regulation by Glucose
5. Concluding Remarks
Acknowledgments
Conflict of interest
References
- Knecht, E.; Aguado, C.; Carcel, J.; Esteban, I.; Esteve, J.M.; Ghislat, G.; Moruno, J.F.; Vidal, J.M.; Saez, R. Intracellular protein degradation in mammalian cells: Recent developments. Cell Mol. Life Sci. 2009, 66, 2427–2443. [Google Scholar]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar]
- Esteban, I.; Aguado, C.; Sanchez, M.; Knecht, E. Regulation of various proteolytic pathways by insulin and amino acids in human fibroblasts. FEBS Lett. 2007, 581, 3415–3421. [Google Scholar]
- Simpson, M.V. The release of labeled amino acids from the proteins of rat liver slices. J. Biol. Chem. 1953, 201, 143–154. [Google Scholar]
- Plomp, P.J.; Wolvetang, E.J.; Groen, A.K.; Meijer, A.J.; Gordon, P.B.; Seglen, P.O. Energy dependence of autophagic protein degradation in isolated rat hepatocytes. Eur. J. Biochem. 1987, 164, 197–203. [Google Scholar]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Inoue, Y.; Klionsky, D.J. Regulation of macroautophagy in Saccharomyces cerevisiae. Semin. Cell Dev. Biol. 2010, 21, 664–670. [Google Scholar]
- Baba, M.; Takeshige, K.; Baba, N.; Ohsumi, Y. Ultrastructural analysis of the autophagic process in yeast: detection of autophagosomes and their characterization. J. Cell Biol. 1994, 124, 903–913. [Google Scholar]
- Farre, J.C.; Krick, R.; Subramani, S.; Thumm, M. Turnover of organelles by autophagy in yeast. Curr. Opin. Cell Biol. 2009, 21, 522–530. [Google Scholar] [CrossRef]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar]
- Brown, C.R.; Chiang, H.L. A selective autophagy pathway that degrades gluconeogenic enzymes during catabolite inactivation. Commun. Integr. Biol. 2009, 2, 177–183. [Google Scholar]
- Chiang, H.L.; Schekman, R.; Hamamoto, S. Selective uptake of cytosolic, peroxisomal, and plasma membrane proteins into the yeast lysosome for degradation. J. Biol. Chem. 1996, 271, 9934–9941. [Google Scholar]
- Tuttle, D.L.; Dunn, W.A., Jr. Divergent modes of autophagy in the methylotrophic yeast Pichia pastoris. J. Cell Sci. 1995, 108, 25–35. [Google Scholar]
- van Zutphen, T.; van der Klei, I.J.; Kiel, J.A. Pexophagy in Hansenula polymorpha. Methods Enzymol. 2008, 451, 197–215. [Google Scholar]
- Fry, M.R.; Thomson, J.M.; Tomasini, A.J.; Dunn, W.A., Jr. Early and late molecular events of glucose-induced pexophagy in Pichia pastoris require Vac8. Autophagy 2006, 2, 280–288. [Google Scholar]
- Bellu, A.R.; Salomons, F.A.; Kiel, J.A.; Veenhuis, M.; Van Der Klei, I.J. Removal of Pex3p is an important initial stage in selective peroxisome degradation in Hansenula polymorpha. J. Biol. Chem. 2002, 277, 42875–42880. [Google Scholar]
- Farre, J.C.; Manjithaya, R.; Mathewson, R.D.; Subramani, S. PpAtg30 tags peroxisomes for turnover by selective autophagy. Dev. Cell 2008, 14, 365–376. [Google Scholar]
- Jiang, Y.; Davis, C.; Broach, J.R. Efficient transition to growth on fermentable carbon sources in Saccharomyces cerevisiae requires signaling through the Ras pathway. EMBO J. 1998, 17, 6942–6951. [Google Scholar]
- Nazarko, V.Y.; Thevelein, J.M.; Sibirny, A.A. G-protein-coupled receptor Gpr1 and G-protein Gpa2 of cAMP-dependent signaling pathway are involved in glucose-induced pexophagy in the yeast Saccharomyces cerevisiae. Cell Biol. Int. 2008, 32, 502–504. [Google Scholar]
- Wang, Z.; Wilson, W.A.; Fujino, M.A.; Roach, P.J. Antagonistic controls of autophagy and glycogen accumulation by Snf1p, the yeast homolog of AMP-activated protein kinase, and the cyclin-dependent kinase Pho85p. Mol. Cell. Biol. 2001, 21, 5742–5752. [Google Scholar] [CrossRef]
- Budovskaya, Y.V.; Stephan, J.S.; Deminoff, S.J.; Herman, P.K. An evolutionary proteomics approach identifies substrates of the cAMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 2005, 102, 13933–13938. [Google Scholar]
- Stephan, J.S.; Yeh, Y.Y.; Ramachandran, V.; Deminoff, S.J.; Herman, P.K. The Tor and PKA signaling pathways independently target the Atg1/Atg13 protein kinase complex to control autophagy. Proc. Natl. Acad. Sci. USA 2009, 106, 17049–17054. [Google Scholar]
- Gupta, D.R.; Paul, S.K.; Oowatari, Y.; Matsuo, Y.; Kawamukai, M. Complex formation, phosphorylation, and localization of protein kinase A of Schizosaccharomyces pombe upon glucose starvation. Biosci. Biotechnol. Biochem. 2011, 75, 1456–1465. [Google Scholar]
- Yorimitsu, T.; Zaman, S.; Broach, J.R.; Klionsky, D.J. Protein kinase A and Sch9 cooperatively regulate induction of autophagy in Saccharomyces cerevisiae. Mol. Biol. Cell. 2007, 18, 4180–4189. [Google Scholar]
- Stephan, J.S.; Yeh, Y.Y.; Ramachandran, V.; Deminoff, S.J.; Herman, P.K. The Tor and cAMP-dependent protein kinase signaling pathways coordinately control autophagy in Saccharomyces cerevisiae. Autophagy 2010, 6, 294–295. [Google Scholar]
- Kane, P.M. The where, when, and how of organelle acidification by the yeast vacuolar H+-ATPas. Microbiol. Mol. Biol. Rev. 2006, 70, 177–191. [Google Scholar]
- Kane, P.M. Disassembly and reassembly of the yeast vacuolar H(+)-ATPase in vivo. J. Biol. Chem. 1995, 270, 17025–17032. [Google Scholar]
- Kane, P.M.; Parra, K.J. Assembly and regulation of the yeast vacuolar H(+)-ATPase. J. Exp. Biol. 2000, 203, 81–87. [Google Scholar]
- Martinez-Munoz, G.A.; Kane, P. Vacuolar and plasma membrane proton pumps collaborate to achieve cytosolic pH homeostasis in yeast. J. Biol. Chem. 2008, 283, 20309–20319. [Google Scholar]
- Bond, S.; Forgac, M. The Ras/cAMP/protein kinase A pathway regulates glucose-dependent assembly of the vacuolar (H+)-ATPase in yeast. J. Biol. Chem. 2008, 283, 36513–36521. [Google Scholar]
- Sautin, Y.Y.; Lu, M.; Gaugler, A.; Zhang, L.; Gluck, S.L. Phosphatidylinositol 3-kinase-mediated effects of glucose on vacuolar H+-ATPase assembly, translocation, and acidification of intracellular compartments in renal epithelial cells. Mol. Cell. Biol. 2005, 25, 575–589. [Google Scholar]
- Ravikumar, B.; Stewart, A.; Kita, H.; Kato, K.; Duden, R.; Rubinsztein, D.C. Raised intracellular glucose concentrations reduce aggregation and cell death caused by mutant huntingtin exon 1 by decreasing mTOR phosphorylation and inducing autophagy. Hum. Mol. Genet. 2003, 12, 985–994. [Google Scholar]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar]
- Casarejos, M.J.; Solano, R.M.; Gomez, A.; Perucho, J.; de Yebenes, J.G.; Mena, M.A. The accumulation of neurotoxic proteins, induced by proteasome inhibition, is reverted by trehalose, an enhancer of autophagy, in human neuroblastoma cells. Neurochem. Int. 2011, 58, 512–520. [Google Scholar]
- Maruyama, R.; Goto, K.; Takemura, G.; Ono, K.; Nagao, K.; Horie, T.; Tsujimoto, A.; Kanamori, H.; Miyata, S.; Ushikoshi, H.; et al. Morphological and biochemical characterization of basal and starvation-induced autophagy in isolated adult rat cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1599–1607. [Google Scholar]
- Williams, T.; Forsberg, L.J.; Viollet, B.; Brenman, J.E. Basal autophagy induction without AMP-activated protein kinase under low glucose conditions. Autophagy 2009, 5, 1155–1165. [Google Scholar] [CrossRef]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; Depinho, R.A.; Sadoshima, J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ. Res. 2010, 107, 1470–1482. [Google Scholar]
- Kobayashi, S.; Xu, X.; Chen, K.; Liang, Q. Suppression of autophagy is protective in high glucose-induced cardiomyocyte injury. Autophagy 2012, 8, 577–592. [Google Scholar]
- Singh, R.; Cuervo, A.M. Autophagy in the cellular energetic balance. Cell Metab. 2011, 13, 495–504. [Google Scholar]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar]
- Xi, H.; Kurtoglu, M.; Liu, H.; Wangpaichitr, M.; You, M.; Liu, X.; Savaraj, N.; Lampidis, T.J. 2-Deoxy-D-glucose activates autophagy via endoplasmic reticulum stress rather than ATP depletion. Cancer Chemother. Pharmacol. 2011, 67, 899–910. [Google Scholar] [CrossRef]
- Marambio, P.; Toro, B.; Sanhueza, C.; Troncoso, R.; Parra, V.; Verdejo, H.; Garcia, L.; Quiroga, C.; Munafo, D.; Diaz-Elizondo, J.; et al. Glucose deprivation causes oxidative stress and stimulates aggresome formation and autophagy in cultured cardiac myocytes. Biochim. Biophys. Acta 1802, 509–518. [Google Scholar]
- Younce, C.W.; Kolattukudy, P.E. MCP-1 causes cardiomyoblast death via autophagy resulting from ER stress caused by oxidative stress generated by inducing a novel zinc-finger protein, MCPIP. Biochem. J. 2010, 426, 43–53. [Google Scholar]
- Wang, Q.; Liang, B.; Shirwany, N.A.; Zou, M.H. 2-Deoxy-D-glucose treatment of endothelial cells induces autophagy by reactive oxygen species-mediated activation of the AMP-activated protein kinase. PLoS One 2011, 6, e17234. [Google Scholar]
- Hardie, D.G. AMP-activated protein kinase: An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef]
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 2007, 100, 914–922. [Google Scholar]
- Duan, X.; Ponomareva, L.; Veeranki, S.; Choubey, D. IFI16 induction by glucose restriction in human fibroblasts contributes to autophagy through activation of the ATM/AMPK/p53 pathway. PLoS One 2011, 6, e19532. [Google Scholar]
- Liang, J.; Shao, S.H.; Xu, Z.X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 2007, 9, 218–224. [Google Scholar]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar]
- Scott, J.W.; Hawley, S.A.; Green, K.A.; Anis, M.; Stewart, G.; Scullion, G.A.; Norman, D.G.; Hardie, D.G. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Invest. 2004, 113, 274–284. [Google Scholar]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005, 1, 15–25. [Google Scholar]
- Hawley, S.A.; Boudeau, J.; Reid, J.L.; Mustard, K.J.; Udd, L.; Makela, T.P.; Alessi, D.R.; Hardie, D.G. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2003, 2, 28. [Google Scholar]
- Meijer, A.J.; Codogno, P. AMP-activated protein kinase and autophagy. Autophagy 2007, 3, 238–240. [Google Scholar]
- Viana, R.; Aguado, C.; Esteban, I.; Moreno, D.; Viollet, B.; Knecht, E.; Sanz, P. Role of AMP-activated protein kinase in autophagy and proteasome function. Biochem. Biophys. Res. Commun. 2008, 369, 964–968. [Google Scholar] [CrossRef]
- Vucicevic, L.; Misirkic, M.; Janjetovic, K.; Vilimanovich, U.; Sudar, E.; Isenovic, E.; Prica, M.; Harhaji-Trajkovic, L.; Kravic-Stevovic, T.; Bumbasirevic, V.; et al. Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy 2011, 7, 40–50. [Google Scholar]
- Han, D.; Yang, B.; Olson, L.K.; Greenstein, A.; Baek, S.H.; Claycombe, K.J.; Goudreau, J.L.; Yu, S.W.; Kim, E.K. Activation of autophagy through modulation of 5'-AMP-activated protein kinase protects pancreatic beta-cells from high glucose. Biochem. J. 2010, 425, 541–551. [Google Scholar] [CrossRef]
- Meley, D.; Bauvy, C.; Houben-Weerts, J.H.; Dubbelhuis, P.F.; Helmond, M.T.; Codogno, P.; Meijer, A.J. AMP-activated protein kinase and the regulation of autophagic proteolysis. J. Biol. Chem. 2006, 281, 34870–34879. [Google Scholar]
- Shaw, R.J.; Bardeesy, N.; Manning, B.D.; Lopez, L.; Kosmatka, M.; DePinho, R.A.; Cantley, L.C. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 2004, 6, 91–99. [Google Scholar]
- Choo, A.Y.; Kim, S.G.; Vander Heiden, M.G.; Mahoney, S.J.; Vu, H.; Yoon, S.O.; Cantley, L.C.; Blenis, J. Glucose addiction of TSC null cells is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Mol. Cell 2010, 38, 487–499. [Google Scholar] [CrossRef]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar]
- Gao, D.; Inuzuka, H.; Tan, M.K.; Fukushima, H.; Locasale, J.W.; Liu, P.; Wan, L.; Zhai, B.; Chin, Y.R.; Shaik, S.; et al. mTOR drives its own activation via SCF(betaTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol. Cell 2011, 44, 290–303. [Google Scholar]
- Zhao, Y.; Xiong, X.; Sun, Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(betaTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol. Cell 2011, 44, 304–316. [Google Scholar]
- Yin, L.; Kharbanda, S.; Kufe, D. MUC1 oncoprotein promotes autophagy in a survival response to glucose deprivation. Int. J. Oncol. 2009, 34, 1691–1699. [Google Scholar]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar]
- Shieh, S.Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar]
- Lee, S.M.; Kim, J.H.; Cho, E.J.; Youn, H.D. A nucleocytoplasmic malate dehydrogenase regulates p53 transcriptional activity in response to metabolic stress. Cell Death Differ. 2009, 16, 738–748. [Google Scholar] [CrossRef]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef]
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009, 10, 285–292. [Google Scholar]
- Maiuri, M.C.; Galluzzi, L.; Morselli, E.; Kepp, O.; Malik, S.A.; Kroemer, G. Autophagy regulation by p53. Curr. Opin. Cell Biol. 2010, 22, 181–185. [Google Scholar]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Marino, G.; Michaud, M.; Vitale, I.; Maiuri, M.C.; Kroemer, G. Oncosuppressive functions of autophagy. Antioxid. Redox Signal. 2011, 14, 2251–2269. [Google Scholar]
- Fabbro, M.; Henderson, B.R. Regulation of tumor suppressors by nuclear-cytoplasmic shuttling. Exp. Cell Res. 2003, 282, 59–69. [Google Scholar]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D'Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar]
- Short, J.D.; Houston, K.D.; Dere, R.; Cai, S.L.; Kim, J.; Johnson, C.L.; Broaddus, R.R.; Shen, J.; Miyamoto, S.; Tamanoi, F.; et al. AMP-activated protein kinase signaling results in cytoplasmic sequestration of p27. Cancer Res. 2008, 68, 6496–6506. [Google Scholar]
- Eto, I. Upstream molecular signaling pathways of p27(Kip1) expression in human breast cancer cells in vitro: Differential effects of 4-hydroxytamoxifen and deficiency of either D-(+)-glucose or L-leucine. Cancer Cell Int. 2011, 11, 31. [Google Scholar] [CrossRef]
- Nagano, Y.; Fukushima, T.; Okemoto, K.; Tanaka, K.; Bowtell, D.D.; Ronai, Z.; Reed, J.C.; Matsuzawa, S. Siah1/SIP regulates p27(kip1) stability and cell migration under metabolic stress. Cell Cycle 2011, 10, 2592–2602. [Google Scholar]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK inhibitors: Cell cycle regulators and beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar]
- Besson, A.; Gurian-West, M.; Schmidt, A.; Hall, A.; Roberts, J.M. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004, 18, 862–876. [Google Scholar]
- Hoshino, D.; Tomari, T.; Nagano, M.; Koshikawa, N.; Seiki, M. A novel protein associated with membrane-type 1 matrix metalloproteinase binds p27(kip1) and regulates RhoA activation, actin remodeling, and matrigel invasion. J. Biol. Chem. 2009, 284, 27315–27326. [Google Scholar]
- Yoo, B.H.; Wu, X.; Li, Y.; Haniff, M.; Sasazuki, T.; Shirasawa, S.; Eskelinen, E.L.; Rosen, K.V. Oncogenic ras-induced down-regulation of autophagy mediator Beclin-1 is required for malignant transformation of intestinal epithelial cells. J. Biol. Chem. 2010, 285, 5438–5449. [Google Scholar]
- Liu, Y.; Song, X.D.; Liu, W.; Zhang, T.Y.; Zuo, J. Glucose deprivation induces mitochondrial dysfunction and oxidative stress in PC12 cell line. J. Cell Mol. Med. 2003, 7, 49–56. [Google Scholar]
- Takeda, K.; Lin, J.; Okubo, S.; Akazawa-Kudoh, S.; Kajinami, K.; Kanemitsu, S.; Tsugawa, H.; Kanda, T.; Matsui, S.; Takekoshi, N. Transient glucose deprivation causes upregulation of heme oxygenase-1 and cyclooxygenase-2 expression in cardiac fibroblasts. J. Mol. Cell Cardiol. 2004, 36, 821–830. [Google Scholar]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar]
- Sedeek, M.; Callera, G.; Montezano, A.; Gutsol, A.; Heitz, F.; Szyndralewiez, C.; Page, P.; Kennedy, C.R.; Burns, K.D.; Touyz, R.M.; et al. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: Implications in type 2 diabetic nephropathy. Am. J. Physiol. Renal. Physiol. 2010, 299, F1348–F1358. [Google Scholar]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar]
- Cao, M.; Jiang, J.; Du, Y.; Yan, P. Mitochondria-targeted antioxidant attenuates high glucose-induced P38 MAPK pathway activation in human neuroblastoma cells. Mol. Med. Rep. 2012, 5, 929–934. [Google Scholar]
- Liu, B.; Cheng, Y.; Zhang, B.; Bian, H.J.; Bao, J.K. Polygonatum cyrtonema lectin induces apoptosis and autophagy in human melanoma A375 cells through a mitochondria-mediated ROS-p38-p53 pathway. Cancer Lett. 2009, 275, 54–60. [Google Scholar]
- Kim, D.S.; Kim, J.H.; Lee, G.H.; Kim, H.T.; Lim, J.M.; Chae, S.W.; Chae, H.J.; Kim, H.R. p38 Mitogen-activated protein kinase is involved in endoplasmic reticulum stress-induced cell death and autophagy in human gingival fibroblasts. Biol. Pharm. Bull. 2010, 33, 545–549. [Google Scholar]
- Webber, J.L.; Tooze, S.A. Coordinated regulation of autophagy by p38alpha MAPK through mAtg9 and p38IP. EMBO J. 2010, 29, 27–40. [Google Scholar] [CrossRef]
- De Chiara, G.; Marcocci, M.E.; Torcia, M.; Lucibello, M.; Rosini, P.; Bonini, P.; Higashimoto, Y.; Damonte, G.; Armirotti, A.; Amodei, S.; et al. Bcl-2 Phosphorylation by p38 MAPK: Identification of target sites and biologic consequences. J. Biol. Chem. 2006, 281, 21353–21361. [Google Scholar]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef]
- Comes, F.; Matrone, A.; Lastella, P.; Nico, B.; Susca, F.C.; Bagnulo, R.; Ingravallo, G.; Modica, S.; Lo Sasso, G.; Moschetta, A.; et al. A novel cell type-specific role of p38alpha in the control of autophagy and cell death in colorectal cancer cells. Cell Death Differ. 2007, 14, 693–702. [Google Scholar] [CrossRef]
- Thyagarajan, A.; Jedinak, A.; Nguyen, H.; Terry, C.; Baldridge, L.A.; Jiang, J.; Sliva, D. Triterpenes from Ganoderma Lucidum induce autophagy in colon cancer through the inhibition of p38 mitogen-activated kinase (p38 MAPK). Nutr. Cancer 2010, 62, 630–640. [Google Scholar]
- Haussinger, D.; Schliess, F.; Dombrowski, F.; Vom Dahl, S. Involvement of p38MAPK in the regulation of proteolysis by liver cell hydration. Gastroenterology 1999, 116, 921–935. [Google Scholar]
- Menon, M.B.; Kotlyarov, A.; Gaestel, M. SB202190-Induced Cell Type-Specific Vacuole Formation and Defective Autophagy Do Not Depend on p38 MAP Kinase Inhibition. PLoS One 2011, 6, e23054. [Google Scholar]
- hang, H.; Chen, G.G.; Zhang, Z.; Chun, S.; Leung, B.C.; Lai, P.B. Induction of autophagy in hepatocellular carcinoma cells by SB203580 requires activation of AMPK and DAPK but not p38 MAPK. Apoptosis 2012, 17, 325–334. [Google Scholar] [CrossRef]
- Tsai, K.H.; Wang, W.J.; Lin, C.W.; Pai, P.; Lai, T.Y.; Tsai, C.Y.; Kuo, W.W. NADPH oxidase-derived superoxide anion-induced apoptosis is mediated via the JNK-dependent activation of NF-kappaB in cardiomyocytes exposed to high glucose. J. Cell. Physiol. 2012, 227, 1347–1357. [Google Scholar]
- Song, J.J.; Lee, Y.J. Cross-talk between JIP3 and JIP1 during glucose deprivation: SEK1-JNK2 and Akt1 act as mediators. J. Biol. Chem. 2005, 280, 26845–26855. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, H.Y.; Lee, Y.K.; Yoon, Y.S.; Xu, W.G.; Yoon, J.K.; Choi, S.E.; Ko, Y.G.; Kim, M.J.; Lee, S.J.; et al. Involvement of mitophagy in oncogenic K-Ras-induced transformation: Overcoming a cellular energy deficit from glucose deficiency. Autophagy 2011, 7, 1187–1198. [Google Scholar]
- Yan, J.; Feng, Z.; Liu, J.; Shen, W.; Wang, Y.; Wertz, K.; Weber, P.; Long, J. Enhanced autophagy plays a cardinal role in mitochondrial dysfunction in type 2 diabetic Goto-Kakizaki (GK) rats: ameliorating effects of (−)-epigallocatechin-3-gallate. J. Nutr. Biochem. 2011, 23, 716–724. [Google Scholar]
- Wang, J.; Whiteman, M.W.; Lian, H.; Wang, G.; Singh, A.; Huang, D.; Denmark, T. A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J. Biol. Chem. 2009, 284, 21412–21424. [Google Scholar]
- Sivaprasad, U.; Basu, A. Inhibition of ERK attenuates autophagy and potentiates tumour necrosis factor-alpha-induced cell death in MCF-7 cells. J. Cell Mol. Med. 2008, 12, 1265–1271. [Google Scholar] [CrossRef]
- Ogier-Denis, E.; Couvineau, A.; Maoret, J.J.; Houri, J.J.; Bauvy, C.; De Stefanis, D.; Isidoro, C.; Laburthe, M.; Codogno, P. A heterotrimeric Gi3-protein controls autophagic sequestration in the human colon cancer cell line HT-29. J. Biol. Chem. 1995, 270, 13–16. [Google Scholar]
- Ogier-Denis, E.; Pattingre, S.; El Benna, J.; Codogno, P. Erk1/2-dependent phosphorylation of Galpha-interacting protein stimulates its GTPase accelerating activity and autophagy in human colon cancer cells. J. Biol. Chem. 2000, 275, 39090–39095. [Google Scholar]
- Corcelle, E.; Nebout, M.; Bekri, S.; Gauthier, N.; Hofman, P.; Poujeol, P.; Fenichel, P.; Mograbi, B. Disruption of autophagy at the maturation step by the carcinogen lindane is associated with the sustained mitogen-activated protein kinase/extracellular signal-regulated kinase activity. Cancer Res. 2006, 66, 6861–6870. [Google Scholar]
- Wong, C.H.; Iskandar, K.B.; Yadav, S.K.; Hirpara, J.L.; Loh, T.; Pervaiz, S. Simultaneous induction of non-canonical autophagy and apoptosis in cancer cells by ROS-dependent ERK and JNK activation. PLoS One 2010, 5, e9996. [Google Scholar]
- Yuan, Z.; Feng, W.; Hong, J.; Zheng, Q.; Shuai, J.; Ge, Y. p38MAPK and ERK promote nitric oxide production in cultured human retinal pigmented epithelial cells induced by high concentration glucose. Nitric Oxide 2009, 20, 9–15. [Google Scholar]
- Briaud, I.; Lingohr, M.K.; Dickson, L.M.; Wrede, C.E.; Rhodes, C.J. Differential activation mechanisms of Erk-1/2 and p70(S6K) by glucose in pancreatic beta-cells. Diabetes 2003, 52, 974–983. [Google Scholar]
- Wu, X.; Mahadev, K.; Fuchsel, L.; Ouedraogo, R.; Xu, S.Q.; Goldstein, B.J. Adiponectin suppresses IkappaB kinase activation induced by tumor necrosis factor-alpha or high glucose in endothelial cells: role of cAMP and AMP kinase signaling. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1836–E1844. [Google Scholar]
- Comb, W.C.; Hutti, J.E.; Cogswell, P.; Cantley, L.C.; Baldwin, A.S. p85alpha SH2 Domain Phosphorylation by IKK Promotes Feedback Inhibition of PI3K and Akt in Response to Cellular Starvation. Mol. Cell 2012, 45, 719–730. [Google Scholar]
- Ramana, K.V.; Friedrich, B.; Srivastava, S.; Bhatnagar, A.; Srivastava, S.K. Activation of nuclear factor-kappaB by hyperglycemia in vascular smooth muscle cells is regulated by aldose reductase. Diabetes 2004, 53, 2910–2920. [Google Scholar]
- Chen, Y.W.; Chenier, I.; Chang, S.Y.; Tran, S.; Ingelfinger, J.R.; Zhang, S.L. High glucose promotes nascent nephron apoptosis via NF-kappaB and p53 pathways. Am. J. Physiol. Renal. Physiol. 2011, 300, F147–156. [Google Scholar]
- Copetti, T.; Bertoli, C.; Dalla, E.; Demarchi, F.; Schneider, C. p65/RelA modulates BECN1 transcription and autophagy. Mol. Cell. Biol. 2009, 29, 2594–2608. [Google Scholar]
- Comb, W.C.; Cogswell, P.; Sitcheran, R.; Baldwin, A.S. IKK-dependent, NF-kappaB-independent control of autophagic gene expression. Oncogene 2011, 30, 1727–1732. [Google Scholar]
- Criollo, A.; Senovilla, L.; Authier, H.; Maiuri, M.C.; Morselli, E.; Vitale, I.; Kepp, O.; Tasdemir, E.; Galluzzi, L.; Shen, S.; et al. The IKK complex contributes to the induction of autophagy. EMBO J. 2010, 29, 619–631. [Google Scholar]
- Criollo, A.; Niso-Santano, M.; Malik, S.A.; Michaud, M.; Morselli, E.; Marino, G.; Lachkar, S.; Arkhipenko, A.V.; Harper, F.; Pierron, G.; et al. Inhibition of autophagy by TAB2 and TAB3. EMBO J. 2011, 30, 4908–4920. [Google Scholar]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar]
- Sengupta, A.; Molkentin, J.D.; Yutzey, K.E. FoxO transcription factors promote autophagy in cardiomyocytes. J. Biol. Chem. 2009, 284, 28319–28331. [Google Scholar]
- Esteve, J.M.; Knecht, E. Mechanisms of autophagy and apoptosis: Recent developments in breast cancer cells. World J. Biol. Chem. 2011, 2, 232–238. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Nakatani, Y.; Kaneto, H.; Kawamori, D.; Yoshiuchi, K.; Hatazaki, M.; Matsuoka, T.A.; Ozawa, K.; Ogawa, S.; Hori, M.; Yamasaki, Y.; et al. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J. Biol. Chem. 2005, 280, 847–851. [Google Scholar]
- Kaniuk, N.A.; Kiraly, M.; Bates, H.; Vranic, M.; Volchuk, A.; Brumell, J.H. Ubiquitinated-protein aggregates form in pancreatic beta-cells during diabetes-induced oxidative stress and are regulated by autophagy. Diabetes 2007, 56, 930–939. [Google Scholar]
- Jung, H.S.; Chung, K.W.; Won Kim, J.; Kim, J.; Komatsu, M.; Tanaka, K.; Nguyen, Y.H.; Kang, T.M.; Yoon, K.H.; Kim, J.W.; et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008, 8, 318–324. [Google Scholar]
- Gonzalez, C.D.; Lee, M.S.; Marchetti, P.; Pietropaolo, M.; Towns, R.; Vaccaro, M.I.; Watada, H.; Wiley, J.W. The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy 2011, 7, 2–11. [Google Scholar]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar]
- Sabio, G.; Davis, R.J. cJun NH2-terminal kinase 1 (JNK1): Roles in metabolic regulation of insulin resistance. Trends Biochem. Sci. 2010, 35, 490–496. [Google Scholar]
- Kaneto, H.; Nakatani, Y.; Miyatsuka, T.; Kawamori, D.; Matsuoka, T.A.; Matsuhisa, M.; Kajimoto, Y.; Ichijo, H.; Yamasaki, Y.; Hori, M. Possible novel therapy for diabetes with cell-permeable JNK-inhibitory peptide. Nat. Med. 2004, 10, 1128–1132. [Google Scholar]
- Stebbins, J.L.; De, S.K.; Machleidt, T.; Becattini, B.; Vazquez, J.; Kuntzen, C.; Chen, L.H.; Cellitti, J.F.; Riel-Mehan, M.; Emdadi, A.; et al. Identification of a new JNK inhibitor targeting the JNK-JIP interaction site. Proc. Natl. Acad. Sci. USA 2008, 105, 16809–16813. [Google Scholar]
- Luitse, M.J.; Biessels, G.J.; Rutten, G.E.; Kappelle, L.J. Diabetes, hyperglycaemia, and acute ischaemic stroke. Lancet Neurol. 2012, 11, 261–271. [Google Scholar] [CrossRef]
- Sciarretta, S.; Zhai, P.; Shao, D.; Maejima, Y.; Robbins, J.; Volpe, M.; Condorelli, G.; Sadoshima, J. Rheb is a critical regulator of autophagy during myocardial ischemia: Pathophysiological implications in obesity and metabolic syndrome. Circulation 2012, 125, 1134–1146. [Google Scholar]
- Hamacher-Brady, A.; Brady, N.R.; Gottlieb, R.A. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J. Biol. Chem. 2006, 281, 29776–29787. [Google Scholar]
- Buss, S.J.; Muenz, S.; Riffel, J.H.; Malekar, P.; Hagenmueller, M.; Weiss, C.S.; Bea, F.; Bekeredjian, R.; Schinke-Braun, M.; Izumo, S.; et al. Beneficial effects of Mammalian target of rapamycin inhibition on left ventricular remodeling after myocardial infarction. J. Am. Coll. Cardiol. 2009, 54, 2435–2446. [Google Scholar]
- Ma, X.; Liu, H.; Foyil, S.R.; Godar, R.J.; Weinheimer, C.J.; Hill, J.A.; Diwan, A. Impaired Autophagosome Clearance Contributes to Cardiomyocyte Death in Ischemia-Reperfusion Injury. Circulation 2012, 125, 3170–3181. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Moruno, F.; Pérez-Jiménez, E.; Knecht, E. Regulation of Autophagy by Glucose in Mammalian Cells. Cells 2012, 1, 372-395. https://doi.org/10.3390/cells1030372
Moruno F, Pérez-Jiménez E, Knecht E. Regulation of Autophagy by Glucose in Mammalian Cells. Cells. 2012; 1(3):372-395. https://doi.org/10.3390/cells1030372
Chicago/Turabian StyleMoruno, Félix, Eva Pérez-Jiménez, and Erwin Knecht. 2012. "Regulation of Autophagy by Glucose in Mammalian Cells" Cells 1, no. 3: 372-395. https://doi.org/10.3390/cells1030372
APA StyleMoruno, F., Pérez-Jiménez, E., & Knecht, E. (2012). Regulation of Autophagy by Glucose in Mammalian Cells. Cells, 1(3), 372-395. https://doi.org/10.3390/cells1030372