Characterization of Genetic Resistance to Fusarium Head Blight and Bacterial Leaf Streak in Intermediate Wheatgrass (Thinopyrum intermedium)

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Biparental Population for QTL Mapping
2.2. Breeding Population for Genomic Prediction
2.3. Genotyping
2.4. Linkage Map Construction and QTL Mapping
2.5. Phenotyping and Statistical Analysis
2.6. Statistical Analyses and Genomic Predictions
3. Results
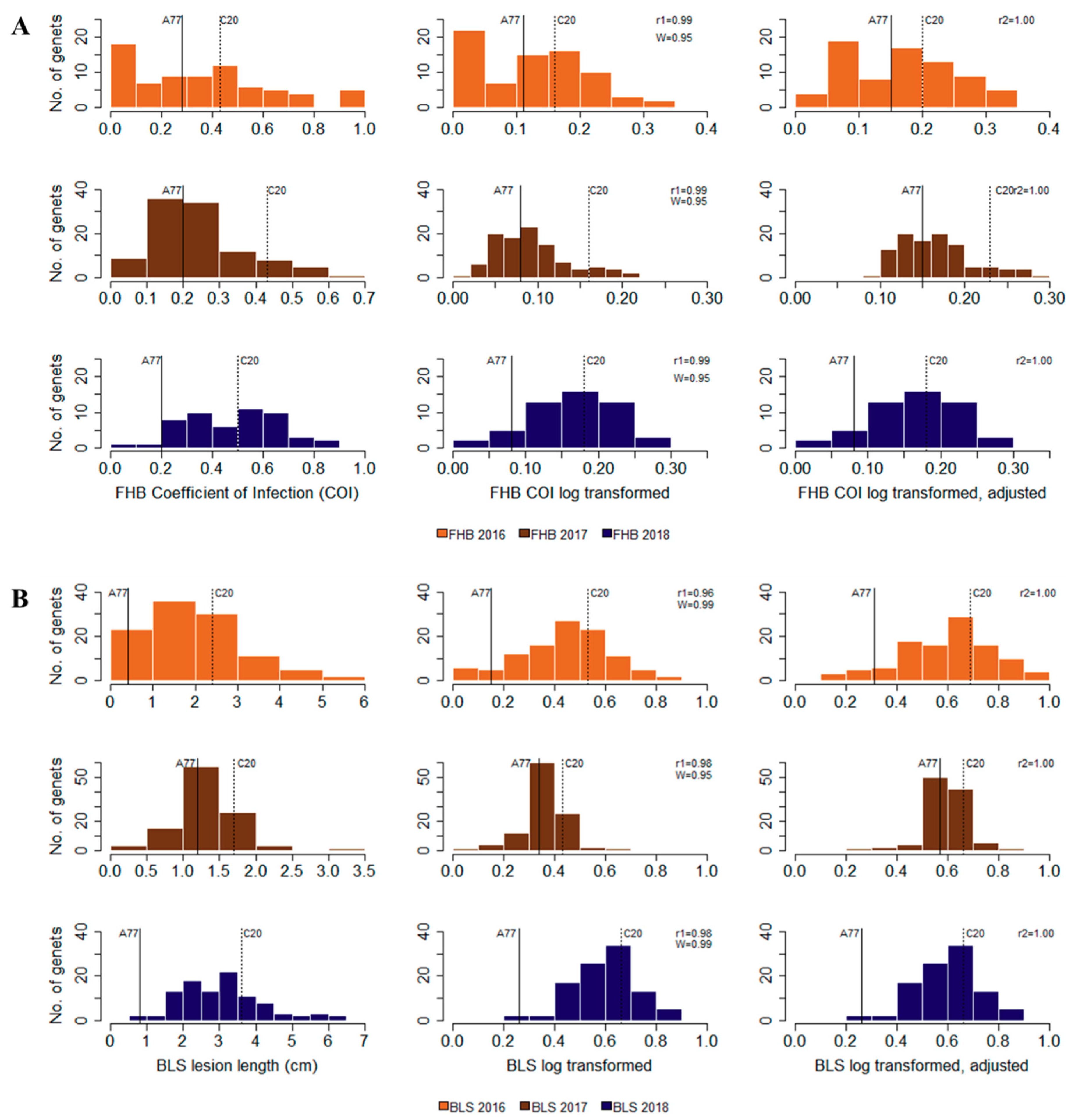
3.1. Disease Evaluation
3.2. Genotyping and Linkage Maps
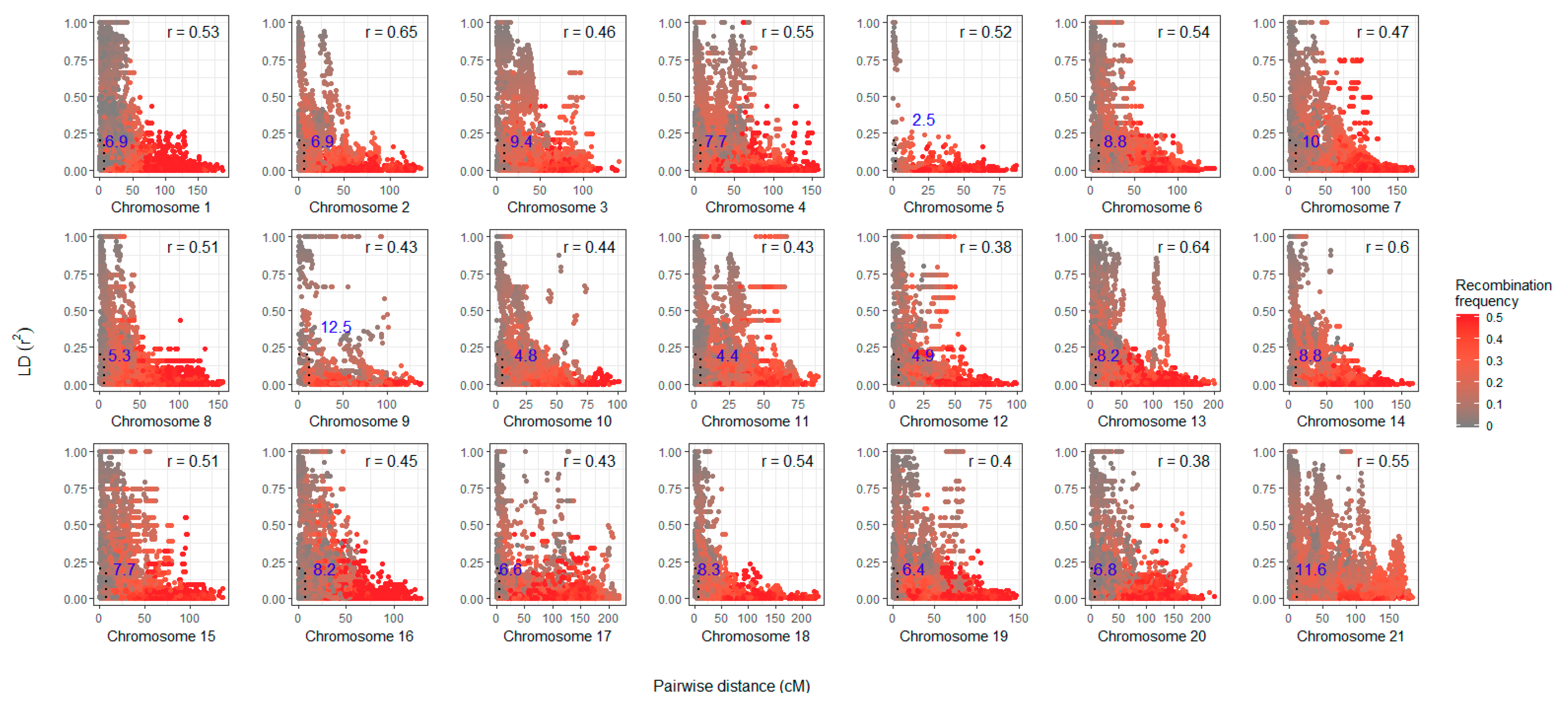
3.3. Linkage Disequilibrium and Recombination Events
3.4. QTL Mapping
- i.
- Two between BLS17 and FHB16 (Ti_Qbls.umn-Chr14.1 and Ti_Qfhb.umn-Chr14.1, and Ti_Qbls.umn-Chr21.2 and Ti_Qfhb.umn-Chr21.2).
- ii.
- One between BLS16 and FHB16 (Ti_Qbls.umn-Chr4.2 and Ti_Qfhb.umn-Chr4.2).
- iii.
- One between BLS16 and FHB17 (Ti_Qbls.umn-Chr4.1 and Ti_Qfhb.umn-Chr4.1).
- iv.
- One between BLS17 and FHB18 (Ti_Qbls.umn-Chr12.3 and Ti_Qfhb.umn-Chr12.3).
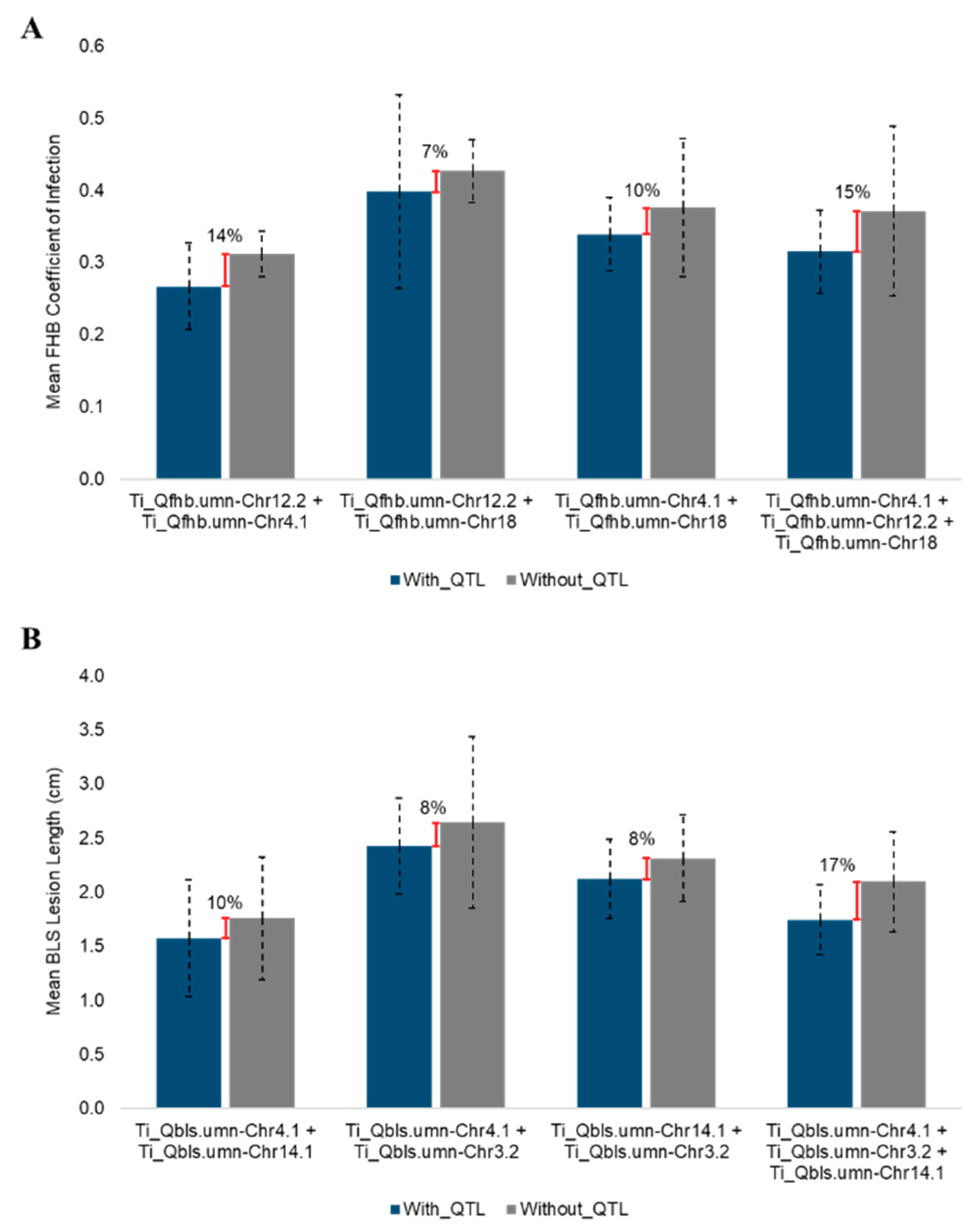
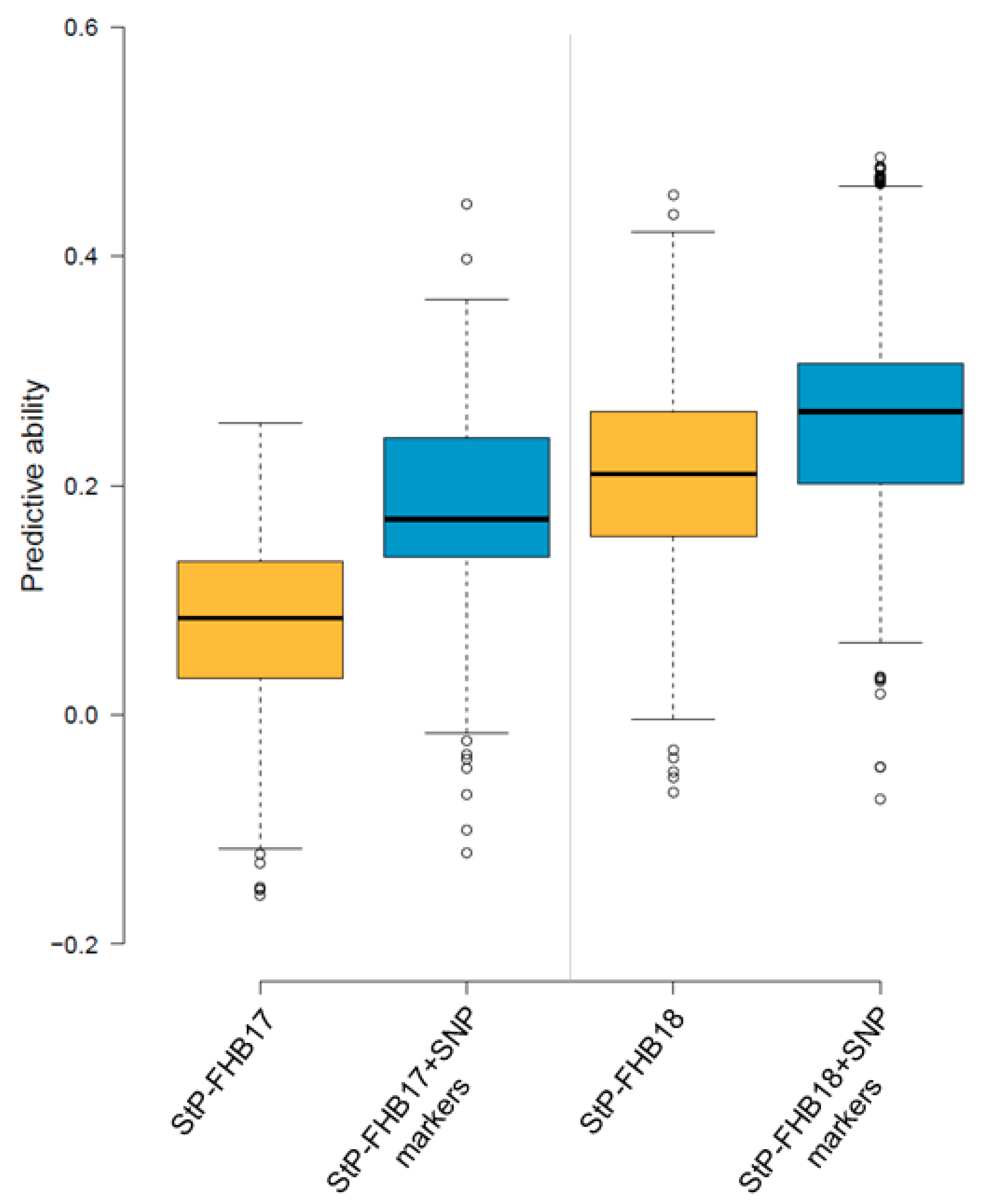
3.5. QTL Models and Genomic Selection for Disease Reduction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oerke, E. Crop losses to pests. J. Agric. Sci. 2006, 144, 31–43. [Google Scholar] [CrossRef]
- Savary, S.; Ficke, A.; Aubertot, J.-N.; Hollier, C. Crop losses due to diseases and their implications for global food production losses and food security. Food Secur. 2012, 4, 519–537. [Google Scholar] [CrossRef]
- Graybosch, R.A.; Peterson, C.J. Genetic improvement in winter wheat yields in the great plains of North America, 1959–2008. Crop Sci. 2010, 50, 1882–1890. [Google Scholar] [CrossRef]
- Garrett, K.A.; Andersen, K.F.; Asche, F.; Bowden, R.L.; Forbes, G.A.; Kulakow, P.A.; Zhou, B. Resistance genes in global crop breeding networks. Phytopathology 2017, 107, 1268–1278. [Google Scholar] [CrossRef]
- Cox, C.M.; Garrett, K.A.; Bockus, W.W. Meeting the challenge of disease management in perennial grain cropping systems. Renew. Agric. Food Syst. 2005, 20, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Tsvelev, N.N. Grasses of the Soviet Union; (Russian Translations Series 8) Hardcover; Springer: Berlin, Germany, 1984. [Google Scholar]
- Friebe, B.; Jiang, J.; Raupp, W.J.; McIntosh, R.A.; Gill, B.S. Characterization of wheat-alien translocations conferring resistance to diseases and pests: Current status. Euphytica 1996, 91, 59–87. [Google Scholar] [CrossRef]
- Luo, P.-G.; Hu, X.-Y.; Chang, Z.-J.; Zhang, M.; Zhang, H.-Q.; Ren, Z.-L. A new stripe rust resistance gene transferred from Thinopyrum intermedium to hexaploid wheat (Triticum aestivum). Phytoprotection 2009, 90, 57–63. [Google Scholar] [CrossRef]
- Li, H.; Wang, X. Thinopyrum ponticum and Th. intermedium: The promising source of resistance to fungal and viral diseases of wheat. J. Genet. Genomics 2009, 36, 557–565. [Google Scholar] [CrossRef]
- Del Ponte, E.M.; Valent, B.; Bergstrom, G.C. A special issue on Fusarium head blight and wheat blast. Trop. Plant Pathol. 2017, 42, 143–145. [Google Scholar] [CrossRef] [Green Version]
- McMullen, M.; Bergstrom, G.; De Wolf, E.; Dill-Macky, R.; Hershman, D.; Shaner, G.; Van Sanford, D. A Unified effort to fight an enemy of wheat and barley: Fusarium head blight. Plant Dis. 2012, 96, 1712–1728. [Google Scholar] [CrossRef]
- Ward, T.J.; Clear, R.M.; Rooney, A.P.; O’Donnell, K.; Gaba, D.; Patrick, S.; Starkey, D.E.; Gilbert, J.; Geiser, D.M.; Nowicki, T.W. An adaptive evolutionary shift in Fusarium head blight pathogen populations is driving the rapid spread of more toxigenic Fusarium graminearum in North America. Fungal Genet. Biol. 2008, 45, 473–484. [Google Scholar] [CrossRef]
- Oliver, R.E.; Cai, X.; Xu, S.; Chen, X.; Stack, R.W. Wheat-alien species derivatives. Crop Sci. 2005, 45, 1353–1360. [Google Scholar]
- Turner, M.K.; DeHaan, L.R.; Jin, Y.; Anderson, J.A. Wheatgrass–wheat partial amphiploids as a novel source of stem rust and fusarium head blight resistance. Crop Sci. 2013, 53, 1994–2005. [Google Scholar] [CrossRef]
- Curland, R.D.; Gao, L.; Bull, C.T.; Vinatzer, B.A.; Dill-Macky, R.; Van Eck, L.; Ishimaru, C.A. Genetic diversity and virulence of wheat and barley strains of Xanthomonas translucens from the Upper Midwestern United States. Phytopathology 2018, 108, 443–453. [Google Scholar] [CrossRef]
- Duveiller, E.; van Ginkel, M.; Thijssen, M. Genetic analysis of resistance to bacterial leaf streak caused by Xanthomonas campestris pv. undulosa in bread wheat. Euphytica 1992, 66, 35–43. [Google Scholar] [CrossRef]
- Duveiller, E. Bacterial leaf streak or black chaff of cereals. EPPO Bull. 1994, 24, 135–157. [Google Scholar] [CrossRef]
- Adhikari, T.B.; Gurung, S.; Hansen, J.M.; Bonman, J.M. Pathogenic and genetic diversity of Xanthomonas translucens pv. undulosa in North Dakota. Phytopathology 2011, 102, 390–402. [Google Scholar] [CrossRef]
- Attari, H.E.; Hayes, P.M.; Rebai, A.; Barrault, G.; Dechamp-Guillaume, G.; Sarrafi, A. Potential of doubled-haploid lines and localization of quantitative trait loci (QTL) for partial resistance to bacterial leaf streak (Xanthomonas campestris pv. hordei) in barley. Theor. Appl. Genet. 1998, 96, 95–100. [Google Scholar] [CrossRef]
- Adhikari, T.B.; Gurung, S.; Hansen, J.M.; Jackson, E.W.; Bonman, J.M. Association mapping of quantitative trait loci in spring wheat landraces conferring resistance to bacterial leaf streak and spot blotch. Plant Genome 2012, 5, 1–16. [Google Scholar] [CrossRef]
- Gurung, S.; Mamidi, S.; Bonman, J.M.; Xiong, M.; Brown-Guedira, G.; Adhikari, T.B. Genome-wide association study reveals novel quantitative trait loci associated with resistance to multiple leaf spot diseases of spring wheat. PLoS ONE 2014, 9, e108179. [Google Scholar] [CrossRef]
- Mohan, S.K.; Bijman, V.P.; St. John, L. Bacterial leaf stripe caused by Xanthomonas translucens pv. cerealis on intermediate wheatgrass in Idaho. Plant Dis. 2001, 85, 921. [Google Scholar] [CrossRef]
- Rosenthal, J.P.; Dirzo, R. Effects of life history, domestication and agronomic selection on plant defence against insects: Evidence from maizes and wild relatives. Evol. Ecol. 1997, 11, 337–355. [Google Scholar] [CrossRef]
- Soltis, N.E.; Atwell, S.; Shi, G.; Fordyce, R.; Gwinner, R.; Gao, D.; Shafi, A.; Kliebenstein, D.J. Crop domestication and pathogen virulence: Interactions of tomato and Botrytis genetic diversity. bioRxiv 2018, 255992. [Google Scholar] [CrossRef] [Green Version]
- Wulff, B.B.H.; Moscou, M.J. Strategies for transferring resistance into wheat: From wide crosses to GM cassettes. Front. Plant Sci. 2014, 5, 692. [Google Scholar] [CrossRef]
- Ott, J.; Wang, J.; Leal, S.M. Genetic linkage analysis in the age of whole-genome sequencing. Nat. Rev. Genet. 2015, 16, 275–284. [Google Scholar] [CrossRef]
- Kantarski, T.; Larson, S.; Zhang, X.; DeHaan, L.; Borevitz, J.; Anderson, J.; Poland, J. Development of the first consensus genetic map of intermediate wheatgrass (Thinopyrum intermedium) using genotyping-by-sequencing. Theor. Appl. Genet. 2017, 130, 137–150. [Google Scholar] [CrossRef]
- Thinopyrum intermedium Genome Sequencing Consortium Thinopyrum intermedium, v2.1, DOE-JGI. Available online: http://phytozome.jgi.doe.gov/ (accessed on 15 May 2018).
- Bajgain, P.; Zhang, X.; Anderson, J.A. Genome-wide association study of yield component traits in intermediate wheatgrass and implications in genomic selection and breeding. G3 Genes Genomes Genet. 2019. [Google Scholar] [CrossRef]
- Zhang, X.; Sallam, A.; Gao, L.; Kantarski, T.; Poland, J.; DeHaan, L.R.; Wyse, D.L.; Anderson, J.A. Establishment and optimization of genomic selection to accelerate the domestication and improvement of intermediate wheatgrass. Plant Genome 2016, 9, 1–18. [Google Scholar] [CrossRef]
- Zhang, X.; Larson, S.R.; Gao, L.; Teh, S.L.; DeHaan, L.R.; Fraser, M.; Sallam, A.; Kantarski, T.; Frels, K.; Poland, J.; et al. Uncovering the genetic architecture of seed weight and size in intermediate wheatgrass through linkage and association mapping. Plant Genome 2017, 10, 1–15. [Google Scholar] [CrossRef]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.-L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Van Ooijen, J.W. JoinMap 4; Software for the Calculation of Genetic Linkage Maps in Experimental Populations; Kyazma BV: Wageningen, The Netherlands, 2006. [Google Scholar]
- Hill, W.G.; Weir, B.S. Variances and covariances of squared linkage disequilibria in finite populations. Theor. Popul. Biol. 1988, 33, 54–78. [Google Scholar] [CrossRef]
- R Core Team. R. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Ferdosi, M.H.; Kinghorn, B.P.; van der Werf, J.H.; Lee, S.H.; Gondro, C. hsphase: An R package for pedigree reconstruction, detection of recombination events, phasing and imputation of half-sib family groups. BMC Bioinform. 2014, 15, 172. [Google Scholar] [CrossRef]
- Van Ooijen, J.W. MapQTL 6; Software for the Mapping of Quantitative Trait Loci in Experimental Populations of Diploid Species; Kyazma BV: Wageningen, The Netherlands, 2009. [Google Scholar]
- Sands, O.; Mizrak, G.; Hall, V.; Kim, H.; Bockelman, H.; Golden, M. Seed transmitted bacterial diseases of cereals: Epidemiology and control. Arab J. Plant Prot. 1986, 4, 125–127. [Google Scholar]
- Gautam, P.; Dill-Macky, R. Type I host resistance and trichothecene accumulation in Fusarium-infected wheat heads. Am. J. Agric. Biol. Sci. 2011, 6, 231–241. [Google Scholar] [CrossRef]
- Zadoks, J.C.; Chang, T.T.; Konzak, C.F. A decimal code for the growth stages of cereals. Weed Res. 1974, 14, 415–421. [Google Scholar] [CrossRef]
- Makandar, R.; Essig, J.S.; Schapaugh, M.A.; Trick, H.N.; Shah, J. Genetically engineered resistance to fusarium head blight in wheat by expression of Arabidopsis NPR1. Mol. Plant Microbe Interact. 2006, 19, 123–129. [Google Scholar] [CrossRef]
- Sallam, A.H.; Endelman, J.B.; Jannink, J.-L.; Smith, K.P. Assessing genomic selection prediction accuracy in a dynamic barley breeding population. Plant Genome 2015, 8, 1–15. [Google Scholar] [CrossRef]
- Endelman, J.B. Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome 2011, 4, 250–255. [Google Scholar] [CrossRef]
- McIntosh, R.; Yamazaki, Y.; Devos, K.; Dubcovsky, J.; Rogers, W.; Appels, R. Catalogue of gene symbols for wheat. In Proceedings of the 10th International Wheat Genetics Symposium, Istituto Sperimentale per la Cerealicoltura, Rome, Italy, 1–3 September 2003; pp. 1–34. [Google Scholar]
- Brettell, R.I.S.; Banks, P.M.; Cauderon, Y.; Chen, X.; Cheng, Z.M.; Larkin, P.J.; Waterhouse, P.M. A single wheatgrass chromosome reduces the concentration of barley yellow dwarf virus in wheat. Ann. Appl. Biol. 1988, 113, 599–603. [Google Scholar] [CrossRef]
- Anderson, J.M.; Bucholtz, D.L.; Greene, A.E.; Francki, M.G.; Gray, S.M.; Sharma, H.; Ohm, H.W.; Perry, K.L. Characterization of wheatgrass-derived barley yellow dwarf virus resistance in a wheat alien chromosome substitution line. Phytopathology 1998, 88, 851–855. [Google Scholar] [CrossRef]
- Dyck, P.L.; Friebe, B. Evaluation of leaf rust resistance from wheat chromosomal translocation lines. Crop Sci. 1993, 33, 687–690. [Google Scholar] [CrossRef]
- Friebe, B.; Jiang, J.; Gill, B.S.; Dyck, P.L. Radiation-induced nonhomoeologous wheat-Agropyron intermedium chromosomal translocations conferring resistance to leaf rust. Theor. Appl. Genet. 1993, 86, 141–149. [Google Scholar] [CrossRef]
- Khan, I.A. Molecular and agronomic characterization of wheat-Agropyron intermedium recombinant chromosomes. Plant Breed. 2000, 119, 25–29. [Google Scholar] [CrossRef]
- Kang, H.-Y.; Tang, L.; Li, D.-Y.; Diao, C.-D.; Zhu, W.; Tang, Y.; Wang, Y.; Fan, X.; Xu, L.-L.; Zeng, J.; et al. Cytogenetic study and stripe rust response of the derivatives from a wheat—Thinopyrum intermedium—Psathyrostachys huashanica trigeneric hybrid. Genome 2016, 60, 393–401. [Google Scholar] [CrossRef]
- Wen, A.; Jayawardana, M.; Fiedler, J.; Sapkota, S.; Shi, G.; Peng, Z.; Liu, S.; White, F.F.; Bogdanove, A.J.; Li, X.; et al. Genetic mapping of a major gene in triticale conferring resistance to bacterial leaf streak. Theor. Appl. Genet. 2018, 131, 649–658. [Google Scholar] [CrossRef]
- Buerstmayr, H.; Ban, T.; Anderson, J.A. QTL mapping and marker-assisted selection for Fusarium head blight resistance in wheat: A review. Plant Breed. 2009, 128, 1–26. [Google Scholar] [CrossRef]
- Dweba, C.C.; Figlan, S.; Shimelis, H.A.; Motaung, T.E.; Sydenham, S.; Mwadzingeni, L.; Tsilo, T.J. Fusarium head blight of wheat: Pathogenesis and control strategies. Crop Prot. 2017, 91, 114–122. [Google Scholar] [CrossRef]
- Miedaner, T.; Würschum, T.; Maurer, H.P.; Korzun, V.; Ebmeyer, E.; Reif, J.C. Association mapping for Fusarium head blight resistance in European soft winter wheat. Mol. Breed. 2011, 28, 647–655. [Google Scholar] [CrossRef]
- Arruda, M.P.; Brown, P.; Brown-Guedira, G.; Krill, A.M.; Thurber, C.; Merrill, K.R.; Foresman, B.J.; Kolb, F.L. Genome-wide association mapping of fusarium head blight resistance in wheat using genotyping-by-sequencing. Plant Genome 2016, 9, 1–14. [Google Scholar] [CrossRef]
- Wang, R.; Chen, J.; Anderson, J.A.; Zhang, J.; Zhao, W.; Wheeler, J.; Klassen, N.; See, D.R.; Dong, Y. Genome-wide association mapping of fusarium head blight resistance in spring wheat lines developed in the Pacific Northwest and CIMMYT. Phytopathology 2017, 107, 1486–1495. [Google Scholar] [CrossRef]
- Hao, C.; Wang, Y.; Hou, J.; Feuillet, C.; Balfourier, F.; Zhang, X. Association mapping and haplotype analysis of a 3.1-mb genomic region involved in Fusarium head blight resistance on wheat chromosome 3BS. PLoS ONE 2012, 7, e46444. [Google Scholar] [CrossRef]
- Gaut, B.S.; Long, A.D. The lowdown on linkage disequilibrium. Plant Cell 2003, 15, 1502. [Google Scholar] [CrossRef]
- Meuwissen, T.H.E.; Goddard, M.E. Mapping multiple QTL using linkage disequilibrium and linkage analysis information and multitrait data. Genet. Sel. Evol. GSE 2004, 36, 261–279. [Google Scholar] [CrossRef]
- Bonnett, D.G.; Rebetzke, G.J.; Spielmeyer, W. Strategies for efficient implementation of molecular markers in wheat breeding. Mol. Breed. 2005, 15, 75–85. [Google Scholar] [CrossRef]
- Joshi, R.K.; Nayak, S. Gene pyramiding-A broad spectrum technique for developing durable stress resistance in crops. Biotechnol. Mol. Biol. Rev. 2010, 3, 51–60. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Year | Parent Disease Scores (mean ± SD) | F1 Population Disease Scores | ||
|---|---|---|---|---|---|
| A77-3 | C20-7 | Range | Mean ± SD | ||
| FHB | 2016 | 0.28 ± 0.00 | 0.43 ± 0.18 | 0.00–1.00 | 0.35 ± 0.27 |
| 2017 | 0.20 ± 0.09 | 0.43 ± 0.16 | 0.04–0.70 | 0.24 ± 0.13 | |
| 2018 | 0.20 ± 0.14 | 0.56 ± 0.10 | 0.06–0.90 | 0.48 ± 0.19 | |
| Mean | 0.23 ± 0.08 | 0.47 ± 0.15 | 0.03–0.83 | 0.36 ± 0.20 | |
| BLS | 2016 | 0.48 ± 0.69 | 2.37 ± 0.49 | 0.00–5.90 | 1.99 ± 1.21 |
| 2017 | 1.22 ± 0.43 | 1.63 ± 0.30 | 0.16–3.50 | 1.33 ± 0.43 | |
| 2018 | 0.77 ± 0.43 | 3.68 ± 0.72 | 0.88–6.08 | 3.07 ± 1.11 | |
| Mean | 0.82 ± 0.52 | 2.56 ± 0.50 | 0.35–5.12 | 2.13 ± 0.92 | |
| Chromosome | No. of SNPs | Length (cM) | Average Inter-SNP Distance (cM) | Correlation with Physical Distance |
|---|---|---|---|---|
| 1 | 352 | 207.8 | 0.59 | 0.59 |
| 2 | 224 | 134.2 | 0.60 | 0.74 |
| 3 | 164 | 141.3 | 0.87 | 0.73 |
| 4 | 270 | 168.1 | 0.62 | 0.77 |
| 5 | 73 | 91.5 | 1.27 | −0.47 |
| 6 | 286 | 159.6 | 0.56 | 0.76 |
| 7 | 233 | 180.6 | 0.79 | 0.91 |
| 8 | 263 | 170.0 | 0.65 | 0.88 |
| 9 | 94 | 137.7 | 1.48 | 0.69 |
| 10 | 163 | 109.2 | 0.67 | −0.83 |
| 11 | 230 | 97.3 | 0.42 | 0.80 |
| 12 | 172 | 99.2 | 0.58 | 0.73 |
| 13 | 378 | 224.1 | 0.59 | 0.77 |
| 14 | 193 | 169.5 | 0.88 | 0.77 |
| 15 | 237 | 153.3 | 0.65 | 0.79 |
| 16 | 198 | 141.8 | 0.72 | 0.83 |
| 17 | 179 | 218.4 | 1.23 | 0.69 |
| 18 | 201 | 251.6 | 1.26 | 0.83 |
| 19 | 258 | 155.3 | 0.60 | 0.78 |
| 20 | 200 | 233.8 | 1.17 | −0.72 |
| 21 | 254 | 194.6 | 0.77 | 0.75 |
| Sum | 4622 | 3438.9 | - | - |
| Average | 220 | 163.8 | 0.81 | 0.75 |
| Trait | QTL Name | Peak SNP Marker | Chr | Pos (cM) | LOD | R2 (%) | Contributing Parent |
|---|---|---|---|---|---|---|---|
| FHB16 | Ti_Qfhb.umn-Chr4.2 | S04_93002901 | 4 | 64.83 | 3.90 | 4.3 | C20-7 |
| Ti_Qfhb.umn-Chr7 | S07_145440056 | 7 | 54.14 | 4.95 | 11.9 | A77-3 | |
| Ti_Qfhb.umn-Chr9 | S09_325459398 | 9 | 73.43 | 7.69 | 11.3 | A77-3 | |
| Ti_Qfhb.umn-Chr12.1 | S12_224745368 | 12 | 17.03 | 6.54 | 10.4 | A77-3 | |
| Ti_Qfhb.umn-Chr12.2 | S12_297702937 | 12 | 35.84 | 8.74 | 12.0 | A77-3 | |
| Ti_Qfhb.umn-Chr14.1 | S14_1567568 | 14 | 25.64 | 8.46 | 10.1 | C20-7 | |
| Ti_Qfhb.umn-Chr18 | S18_472824840 | 18 | 11.41 | 7.57 | 10.8 | A77-3 | |
| Ti_Qfhb.umn-Chr21.2 | S21_325649929 | 21 | 91.58 | 4.19 | 9.1 | A77-3 | |
| FHB17 | Ti_Qfhb.umn-Chr3.1 | S03_57465451 | 3 | 16.31 | 3.63 | 8.1 | C20-7 |
| Ti_Qfhb.umn-Chr4.1 | S04_44702081 | 4 | 25.16 | 8.79 | 15.3 | A77-3 | |
| Ti_Qfhb.umn-Chr7 | S07_145440056 | 7 | 54.14 | 5.73 | 11.0 | A77-3 | |
| Ti_Qfhb.umn-Chr12.1 | S12_224745368 | 12 | 17.03 | 6.80 | 11.4 | A77-3 | |
| Ti_Qfhb.umn-Chr12.2 | S12_297702937 | 12 | 35.84 | 10.36 | 17.6 | A77-3 | |
| Ti_Qfhb.umn-Chr13 | S13_257779646 | 13 | 207.45 | 3.73 | 3.1 | C20-7 | |
| Ti_Qfhb.umn-Chr14.2 | S14_282002781 | 14 | 62.27 | 3.15 | 4.8 | C20-7 | |
| FHB18 | Ti_Qfhb.umn-Chr10 | S10_32674865 | 10 | 91.40 | 3.23 | 5.3 | C20-7 |
| Ti_Qfhb.umn-Chr12.3 | S12_335131053 | 12 | 46.83 | 6.93 | 15.3 | A77-3 | |
| Ti_Qfhb.umn-Chr18 | S18_472824840 | 18 | 11.41 | 11.81 | 15.6 | A77-3 | |
| Ti_Qfhb.umn-Chr21.1 | S21_73121994 | 21 | 31.77 | 3.19 | 5.1 | A77-3 | |
| BLS16 | Ti_Qbls.umn-Chr1.2 | S01_371412974 | 1 | 192.78 | 4.60 | 10.5 | A77-3 |
| Ti_Qbls.umn-Chr3.2 | S03_146509486 | 3 | 68.56 | 4.47 | 10.0 | A77-3 | |
| Ti_Qbls.umn-Chr4.1 | S04_44702081 | 4 | 25.55 | 5.81 | 11.3 | A77-3 | |
| Ti_Qbls.umn-Chr4.2 | S04_93002901 | 4 | 64.83 | 3.66 | 6.8 | C20-7 | |
| Ti_Qbls.umn-Chr4.2 | S04_158111261 | 4 | 93.69 | 3.12 | 10.2 | C20-7 | |
| Ti_Qbls.umn-Chr15 | S15_73155829 | 15 | 103.01 | 6.69 | 10.8 | A77-3 | |
| BLS17 | Ti_Qbls.umn-Chr1.1 | S01_501973344 | 1 | 132.18 | 3.39 | 3.7 | C20-7 |
| Ti_Qbls.umn-Chr8 | S08_387846008 | 8 | 132.01 | 3.53 | 3.2 | C20-7 | |
| Ti_Qbls.umn-Chr12.3 | S12_335131053 | 12 | 46.83 | 7.75 | 11.6 | A77-3 | |
| Ti_Qbls.umn-Chr14.1 | S14_1567568 | 14 | 25.64 | 9.23 | 12.9 | A77-3 | |
| Ti_Qbls.umn-Chr21.2 | S21_325649929 | 21 | 91.58 | 3.79 | 7.3 | A77-3 | |
| BLS18 | Ti_Qbls.umn-Chr3.2 | S03_146509486 | 3 | 68.56 | 4.77 | 11.6 | A77-3 |
| Ti_Qbls.umn-Chr20 | S20_201616698 | 20 | 9.66 | 3.32 | 5.6 | C20-7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bajgain, P.; Zhang, X.; Turner, M.K.; Curland, R.D.; Heim, B.; Dill-Macky, R.; Ishimaru, C.A.; Anderson, J.A. Characterization of Genetic Resistance to Fusarium Head Blight and Bacterial Leaf Streak in Intermediate Wheatgrass (Thinopyrum intermedium). Agronomy 2019, 9, 429. https://doi.org/10.3390/agronomy9080429
Bajgain P, Zhang X, Turner MK, Curland RD, Heim B, Dill-Macky R, Ishimaru CA, Anderson JA. Characterization of Genetic Resistance to Fusarium Head Blight and Bacterial Leaf Streak in Intermediate Wheatgrass (Thinopyrum intermedium). Agronomy. 2019; 9(8):429. https://doi.org/10.3390/agronomy9080429
Chicago/Turabian StyleBajgain, Prabin, Xiaofei Zhang, M. Kathryn Turner, Rebecca D. Curland, Brett Heim, Ruth Dill-Macky, Carol A. Ishimaru, and James A. Anderson. 2019. "Characterization of Genetic Resistance to Fusarium Head Blight and Bacterial Leaf Streak in Intermediate Wheatgrass (Thinopyrum intermedium)" Agronomy 9, no. 8: 429. https://doi.org/10.3390/agronomy9080429