
Characterization of the NGP4A Gene in Regulating Grain Number Per Panicle of Rice (Oryza sativa L.)

College of Life Sciences, Jiangxi Normal University, Nanchang 330022, China
*
Author to whom correspondence should be addressed.
Agronomy 2022, 12(7), 1549; https://doi.org/10.3390/agronomy12071549
Submission received: 3 May 2022
/
Revised: 18 June 2022
/
Accepted: 26 June 2022
/
Published: 28 June 2022
(This article belongs to the Topic Plant Breeding, Genetics and Genomics)
Abstract
:Grain number per panicle (GNPP) is a major factor influencing rice yield (Oryza sativa L.). However, the molecular mechanisms of GNPP determination are not well understood. A rice GNPP mutant, ngp4a, was isolated from an ethyl methanesulfonate-mutagenized rice library of japonica Nipponbare. ngp4a produced fewer grains than wild-type plants at maturity as the number of secondary branches decreased significantly. The mutant phenotype of ngp4a was controlled by a recessive nuclear gene, which was fine-mapped into a 155.2 kb region on chromosome 4. One GNPP-related gene, Gnp4/LAX2 (LOC_Os04g32510), was found in the mapped region. The deletion of 3-bp nucleotides in the first exon of NGP4A resulted in a threonine residue loss. The mutation in NGP4A was responsible for the mutant phenotype of ngp4a. These results suggest that NGP4A is a new allele for Gnp4 and LAX2, while the mutant phenotype and underlying causation differed. Notably, transcriptome analysis revealed that NGP4A could regulate GNPP determination through the phenylpropanoid biosynthesis and mitogen-activated protein kinase signaling pathways. Our results further elucidated the vital roles of Gnp4/LAX2 in GNPP determination, providing a new genetic resource and theoretical basis to further explore the molecular mechanisms of GNPP in rice.
1. Introduction
Rice (Oryza sativa L.) is one of the major food crops in the world, with nearly half of the world’s population depending on rice as a staple food, particularly Southeast Asia [1]. Owing to the decreasing arable land area and increasing population, breeding rice cultivars with higher yields is crucial for food security [2]. Rice yield is mainly determined by three traits—number of panicles, grain number per panicle (GNPP), and grain weight—all of which are typical quantitative traits and are controlled by numerous genes [3]. Among these three traits, GNPP is a major contributor to rice yield and is crucial for high-yield rice breeding.
In rice breeding practice, different strategies for increasing rice yield have been proposed in China and abroad. For example, the International Rice Research Institute (IRRI) proposed a strategy of “fewer tillers and larger panicles [4]”. Yuan et al. [5] proposed a super-high-yield ideal architectural feature of “high canopy layer, low panicle layer, large panicle, and high lodging resistance”. Yang et al. [6] proposed a strategy of “upright and large panicle” for japonica rice breeding in northern China. Zhou et al. [7] proposed a strategy of “super high-yield breeding of inter-subspecies heavy panicle hybrid rice”. While each of these high-yield breeding strategies has unique characteristics, they are all based on the principles of increasing the number of grains per panicle and increasing the grain weight per panicle, thereby increasing rice yield. Therefore, the development of large panicles with high GNPP has been gaining increasing attention in high-yield breeding programs for rice, and it is of great theoretical significance to clarify the regulatory mechanism of GNPP for the molecular design breeding of rice.
To this date, 238 quantitative trait loci (QTLs) controlling GNPP have been registered for rice on the Gramene website (http://www.gramene.org) (accessed on 2 January 2022). These QTLs are distributed across the 12 chromosomes of rice, and some of them have been well characterized [8,9]. Various mutants are valuable genetic resources for the functional analysis of genes and the elucidation of complex biological processes [10,11,12]. As excellent materials in such studies, GNPP mutants have been widely used to explore the molecular mechanisms of GNPP determination. For example, Zhao et al. [13] constructed a mutant library by mutating a rice variety YIL55 and investigated a mutant pay1 with an upright and compact architecture. The overexpression of PAY1 leads to fewer tillers, taller plant height, more secondary branches, a larger GNPP, and higher grain yield. IPA1 encodes a squamosa promoter binding protein-like protein and positively regulates the number of branches and GNPP [13]. MOC1 encodes a GRAS-family nuclear protein and positively regulates the tiller number, branch number, and GNPP in rice [14]. Gnp4/LAX2 encodes a nuclear protein that regulates the formation of the axillary meristem and positively regulates the number of secondary branches and GNPP [15,16]. Gnp4 and LAX2 share the same locus in the long arm of rice chromosome 4. Moreover, numerous genes, such as LAX1 [17], SP1 [18], DEP1 [19], TAW1 [20], OsNAC2 [21], GNP1 [22], DST [23], and PROG1 [24], have been reported to regulate GNPP. However, the molecular mechanisms by which these genes regulate the GNPP are far from being well understood, and some have not yet been cloned. Therefore, the regulation of GNPP warrants further investigation. It may be possible to increase the GNPP using related genes, thus increasing rice grain yield.
In this study, we identified a GNPP mutant ngp4a in rice. ngp4a was affected by a single recessive nuclear gene, which was named NGP4A. Using map-based cloning, NGP4A was fine mapped into a 155.2 kb region on chromosome 4. The results of the sequence alignment showed that 3 bp nucleotides were deleted in the first exon of LOC_Os04g32510 in ngp4a, which resulted in a threonine residue loss. The complementation analysis confirmed that the genetic mutation of LOC_Os04g32510 was responsible for the ngp4a mutant phenotype. Furthermore, we determined the global transcriptome profile of ngp4a to explore the major molecular function of NGP4A. This study laid a foundation to further illustrate the regulatory mechanism and application potential of NGP4A on GNPP in rice.
2. Materials and Methods
2.1. Plant Materials and Agronomic Evaluation
The ngp4a mutant was obtained from a mutant library, which was generated by subjecting Nipponbare (O. sativa L. ssp. japonica) to ethyl methanesulfonate (EMS). To map the NGP4A gene, an F2 population was developed from the cross between ngp4a and a rice variety Minghui 63 (O. sativa L. ssp. indica). All plants were planted under natural conditions in a paddy field in Nanchang City, Jiangxi Province, China. Agronomic traits, including plant height, tiller number, main panicle length, primary branches of the main panicle, secondary branches of the main panicle, total spikelets of the main panicle, seed setting rate, 1000-grain weight, grain length, and grain width, were measured after maturity. The plant height was measured from the base of the plant to the top of the spike. The main panicle length was measured from the panicle neck to the panicle tip of the main panicle. The seed setting rate was calculated as the percentage of the filled grain number to the total grain number of the main panicle. The grain length and width were measured by an electronic digital vernier caliper, and fully filled grains were used for measuring the 1000-grain weight. The plant height, tiller number, and main panicle length were measured in the field. The remaining agronomic traits were measured in the laboratory after harvest. For each agronomic trait, 10 plants were evaluated as biological replicates. The average and SD values of the data were analyzed using Excel software. Statistical analysis was performed using the Student’s t-test.
2.2. Primer Development and Gene Mapping
Simple sequence repeat (SSR) primers were obtained from the Gramene website (http://www.gramene.org/microsat/) (accessed on 2 January 2019) based on the SSR linkage map constructed by McCouch et al. [25]. Insertion/deletion (InDel) primers were developed based on the different sequences between Nipponbare and 9311 (O. sativa ssp. indica) and designed by the Primer3 (v.0.4.0) software (https://bioinfo.ut.ee/primer3-0.4.0/) (accessed on 2 January 2019). All primers were synthesized by Sangon Biotechnology (Sangon Tech, Shanghai, China) and conserved in the laboratory. To screen linked markers, two DNA pools were constructed by screening the GNPP phenotype in the F2 population after maturity. The first pool was composed of 20 individual plants that showed the GNPP mutant phenotype of ngp4a, while the second pool was composed of 20 individual plants that showed the wild-type GNPP phenotype. In total, 326 SSR markers covering the 12 chromosomes of rice were screened by the two parents, ngp4a and Minghui 63, and the individuals from the F2 population with the mutant GNPP phenotype were used for fine mapping with newly designed InDel markers. Primers used in this study are listed in Table S1.
2.3. Vector Construction and Plant Transformation
Based on the mapping results and literature references [15,16], LOC_Os04g32510 was considered the most likely candidate gene in the gene mapping region. For the genetic complementation test, a DNA fragment containing the entire LOC_Os04g32510 genomic sequence and 3000 bp upstream promoter sequence was amplified from Nipponbare using Q5 high-fidelity DNA polymerase (New England BioLabs, Hitchin, UK). Then, the purified and verified fragment was cloned into a pBWA(V)HII-CCDB-TNOS vector (Biorun biological technology Co., Ltd., Wuhan, China), which was digested by the ECO31I restriction enzyme to generate the resulting binary vector. Subsequently, the resulting binary vector was introduced into the ngp4a mutant using an Agrobacterium tumefaciens-mediated genetic transformation method as previously reported [26]. The PCR products were purified and sequenced by Shanghai Sangon Biological Engineering Technology and Services Co., Ltd. (Shanghai, China).
2.4. Transcriptome Sequencing
For transcriptome sequencing, ngp4a and wild-type plants were grown in hydroponic cultures using an IRRI liquid culture medium recipe (IRRI nutrient solution). At the four-leaf stage, the whole seedlings (all aerial organs plus root tissue) of ngp4a and wild-type were collected and immediately frozen in liquid nitrogen. To minimize the effect of transcriptome unevenness among plants, for RNA extraction, 10 ngp4a mutant and 10 wild-type whole seedlings were collected and mixed separately to form two libraries. Total RNA was then isolated from the samples using the Trizol reagent (Solarbio Science & Technology Co., Ltd., Beijing, China) following the manufacturer’s instructions. RNA quality and quantity were checked using agarose gel electrophoresis and a Nanodrop 2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). Subsequently, the qualified RNA samples were used for transcriptome sequencing. Transcriptome sequencing was performed by BioMarker Technology (Beijing, China) according to the manufacturer’s recommendations.
3. Results
3.1. Characterization of ngp4a Mutant
To characterize the mutant traits of ngp4a, we compared the major agronomic traits between ngp4a and wild-type plants. The morphology of ngp4a from the seedling to the end of the vegetative stage was similar to that of the wild-type plants. However, ngp4a produced fewer grains than the wild-type plants at maturity. The quantitative analysis of the panicles revealed that the number of the primary branches was not affected significantly in the ngp4a mutant plants, while the number of secondary branches decreased significantly (Figure 1). On average, the wild-type plants had approximately 36.7 secondary branches and 166.7 spikelets on the main panicle, while the mutant plants had approximately only 10.0 secondary branches and 106.3 spikelets on the main panicle, which suggests that the GNPP mutant appearance of ngp4a was due to a reduced number of secondary branches and total spikelets of the panicle. The grain size was also affected in the mutant. The statistical analysis revealed that the grain width was not affected in the mutant compared to the wild-type, while the grain length of the mutant increased significantly, which increased the 1000-grain weight. In addition, other major agronomic traits of ngp4a, including the plant height, main panicle length, and tiller number, decreased significantly compared with the wild-type plants (Table 1).
3.2. Fine Mapping of NGP4A
An F2 population was developed from the cross between ngp4a and Minghui 63 for the genetic analysis of the ngp4a mutant. The phenotypic analysis revealed that the F1 plants had the wild-type phenotype; among the 3632 F2 individuals, 2682 plants had the wild-type GNPP phenotype and the rest had the mutant GNPP phenotype. The segregation ratio was approximately equal to 2.82:1 (χ2 = 2.59) and was in accordance with the expected ratio of 3:1, which suggests that the mutant phenotype of ngp4a was controlled by a single nuclear recessive gene, which was named NGP4A. To locate the NGP4A gene, we performed map-based cloning. By screening 326 pairs of SSR markers scattered on the rice 12 chromosomes with proportional spacing, we found that 158 pairs of markers exhibited distinct polymorphisms between the two parents ngp4a and Minghui 63, and these markers were then used for analyzing the relationship with the NGP4A gene. Finally, we found four SSR markers (RM5687, RM6314, RM5424, and RM7563) on chromosome 4 that were linked with NGP4A. Subsequently, using 120 recessive individuals from the F2 population, the region of NGP4A was preliminary framed between markers RM6314 and RM5424 with genetic distances of 6.3 and 5.0 cM, respectively. To further map NGP4A, we used 950 recessive individuals and developed new InDel markers in the preliminary mapping region. Finally, NGP4A was narrowed down to a 0.9 cM interval between InDel4-44 and InDel4-59, and the physical distance was approximately 155.2 kb (Figure 2B,C).
3.3. Sequence and Complementation Analysis
Ten genes were predicted according to the annotation in the Rice Genome Annotation Project (http://rice.uga.edu/) (accessed on 15 February 2019) within the fine mapped region. Among these genes, one (LOC_Os04g32510) was involved in the regulation of the GNPP of rice. To clarify this further, we amplified the entire genomic sequence of LOC_Os04g32510 from the ngp4a mutant and its wild-type parent Nipponbare. Specific primers were designed according to the genome sequence of Nipponbare. The primers for gene cloning and sequence analysis of LOC_Os04g32510 are listed in Table S1. The results of the sequence alignment showed that 3 bp nucleotides were deleted in the first exon of LOC_Os04g32510 in ngp4a, which resulted in the loss of a threonine residue (Figure 2D,E). Therefore, we speculated that the genetic variation of LOC_Os04g32510 could be the underlying reason for the mutant phenotype of ngp4a. To confirm the defect of LOC_Os04g32510 as the causal mutation of ngp4a, a DNA segment including the entire genomic sequence of LOC_Os04g32510 and its promoter was introduced into ngp4a with A. tumefaciens-mediated transformation. Twelve positive transgenic lines in the T2 generation were obtained in total, all of which had a wild-type panicle phenotype (Figure 3). Meanwhile, no significant differences were observed between the wild-type and complementation transgenic plants in terms of other major agronomic traits (Table 2). These results demonstrated that the genetic variation of LOC_Os04g32510 was responsible for the ngp4a mutant phenotype, and LOC_Os04g32510 was indeed the NGP4A gene.
3.4. Transcriptome Analysis
To explore the possible mechanisms underlying the ngp4a mutant phenotype, we performed transcriptome sequencing for ngp4a and wild-type Nipponbare. After filtering, the total numbers of clean reads were 20.48 and 20.59 million for ngp4a and wild-type plants, respectively. At least 93.53% of the clean reads had a quality score of Q30 for both types of samples. The GC contents were 53.10% and 52.83% for ngp4a and wild-type plants, respectively. In total, 39,516,745 (96.47%) and 39,836,670 (96.72%) clean reads from ngp4a and wild-type plants, respectively, were successfully and uniquely mapped to the rice genome using the HISAT2 software (Table 3). These results indicated the high quality of the sequencing data and were competent for subsequent analysis.
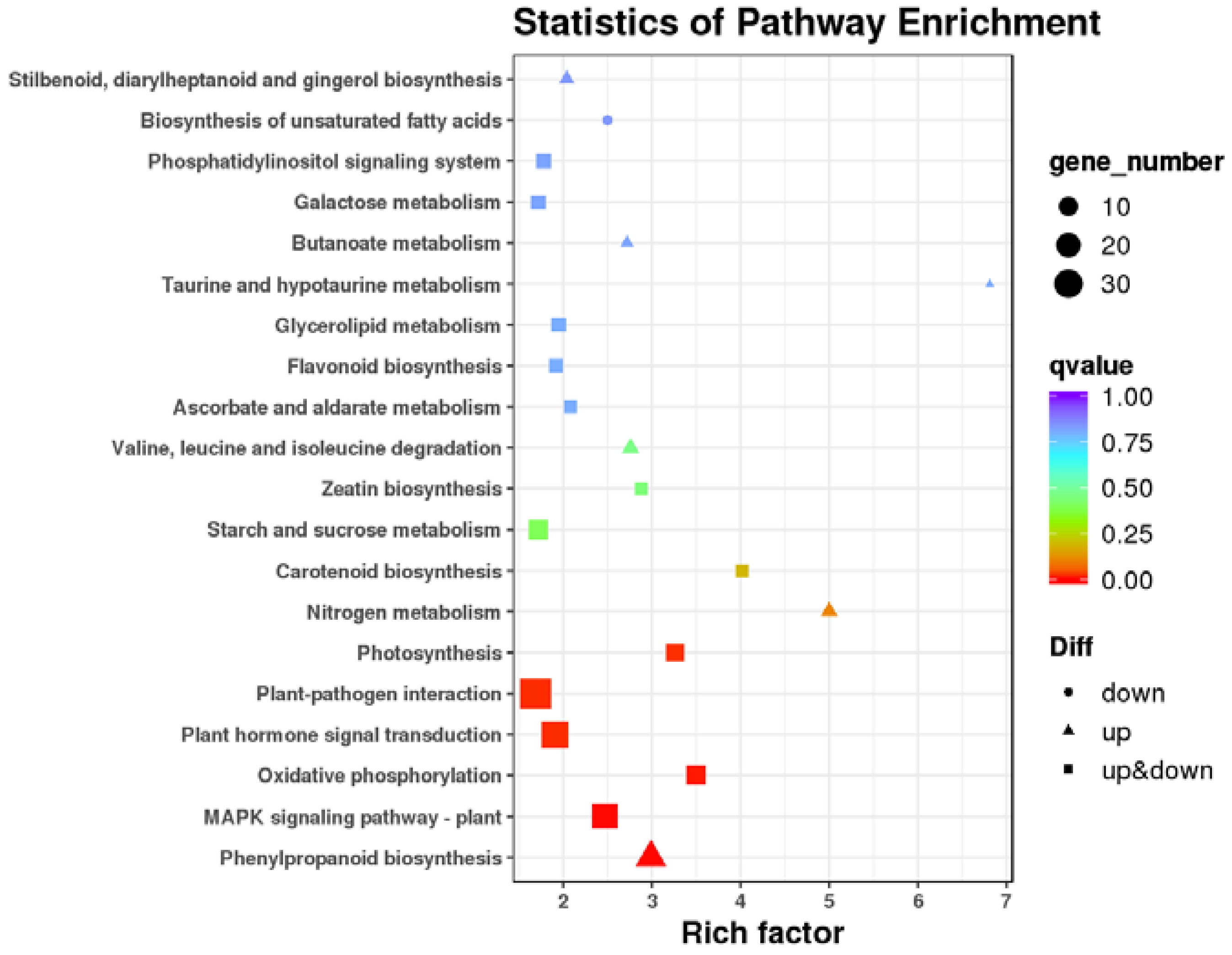
The expression quantities of 30,825 and 30,625 genes were detected in ngp4a and wild-type plants, respectively. Among them, 784 genes were identified as differentially expressed genes (DEGs) with the cut-off for the false discovery rate of <0.01 and fold change of ≥2 (Table S2). Of these DEGs, 581 were up-regulated and the rest were significantly down-regulated. These DEGs were distributed across all chromosomes but most were located on chromosome 1. To characterize the function of DEGs, Gene Ontology (GO) term enrichment analysis was performed to detect the functional pathways of the mutant traits. In total, 538 DEGs were assigned into three GO categories. The statistical results demonstrated that most of the DEGs were related to the cellular component, followed by the biological process and molecular function categories (Figure 4). Many DEGs associated with metabolic process, cellular process, and single-organism process GO terms were significantly enriched, thereby suggesting that extensive biological processes were altered in the mutant plants. Within the cellular component category, cell, cell part, and membrane GO terms were significantly enriched, thereby suggesting that NGP4A affected the membrane and cell parts in the mutant plants. Many DEGs associated with binding, catalytic activity, and transporter activity were also significantly enriched, thereby suggesting that NGP4A influenced the molecular function of the mutants. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was undertaken to predict the biochemical pathways associated with the DEGs (Figure 5). Interestingly, 229 DEGs were assigned to 49 KEGG pathways, of which six pathways were significantly enriched (q-value ≤ 0.05) (Table S3). Among the KEGG pathway-annotated DEGs, 39 were associated with plant–pathogen interaction, 28 were associated with plant hormone signal transduction, and 25 were associated with the mitogen-activated protein kinase (MAPK) signaling pathway-plant. These were the three largest groups of DEGs according to KEGG pathway analysis. However, the NGP4A mutation had the largest impact on the phenylpropanoid biosynthesis (q-value = 0.00115) and MAPK signaling pathways (q-value = 0.00125).
4. Discussion
The development of high-yield cultivars is one of the most important goals of rice breeding. GNPP plays a decisive role in rice yield, and extensive attention has been paid to improving the GNPP trait in various plants [27,28]. Nevertheless, the GNPP mechanism is still poorly understood, especially in rice. Mutant analysis is a powerful approach to illuminate the underlying molecular mechanisms in the complex biological processes of GNPP.
In this study, a GNPP mutant, ngp4a, was found in the mutant library. The GNPP of ngp4a was significantly reduced compared with that of wild-type plants. Meanwhile, the other major agronomic traits, including plant height, tiller number, panicle length, grain length, and 1000-grain weight, were significantly changed in the mutant plants, indicating that NGP4A had pleiotropic effects, thereby corroborating the results of previously published studies. For example, GNS4 encodes a cytochrome P450 protein, and GNS4-overexpressing rice plants have significantly increased grain weight and GNPP [29]. GW2 encodes a ring-type protein with E3 ubiquitin ligase activity, whose loss of function results in a significant increase in 1000-grain weight and a decrease in GNPP [30]. GN2 encodes a peptide of 70 amino acids, and GN2-overexpressing plants exhibit reduced grain numbers, plant height, and heading stage [31]. The results of these studies suggest that a single gene can simultaneously affect several phenotypic traits.
Using fine mapping, NGP4A was finally delimited into a 155.2 kb region on chromosome 4. Based on the annotation results of the Rice Genome Annotation Project (http://rice.uga.edu/) (accessed on 15 February 2019), ten genes were predicted in the fine-mapped region. Among these genes, one (LOC_Os04g32510) was named Gnp4 and LAX2 and is involved in the regulation of the GNPP trait of rice [15,16]. To this end, LOC_Os04g32510 was considered the most likely candidate gene in the gene mapping region. Using sequence and complementation analysis, we confirmed that NGP4A was a novel allele for Gnp4 and LAX2. However, ngp4a was different from gnp4 and lax2 in the mutant phenotype and underlying causation. For gnp4, there was no significant difference in the primary branch number and grain width, while the number of secondary branches and grains decreased significantly, and the grain length and 1000-grain weight increased significantly; these results were similar to those of ngp4a. However, no significant difference was observed in the tillering number of gnp4, while that of ngp4a decreased significantly. Meanwhile, the number of secondary branches, grains, and tillers of lax2 decreased significantly; these results were similar to those for ngp4a. The number of primary branches of lax2 increased; however, no significant difference was obtained in the number of primary branches of ngp4a. These results showed that different mutant phenotypes occurred in the three mutants, ngp4a, gnp4, and lax2. Furthermore, 3 bp nucleotides were deleted in the first exon of LOC_Os04g32510 in ngp4a, which resulted in the loss of a threonine residue. However, no nucleotide differences occurred in gnp4 and the phenotypic mutation could have been caused by the changes in DNA methylation levels in the promoter of LOC_Os04g32510. Tabuchi et al. [16] isolated three independent mutant alleles for LAX2 (LAX2-1, LAX2-2, and LAX2-3). They found that lax2-1 and lax2-3 deleted a nucleotide in the first and third exons of LOC_Os04g32510, respectively, while a nucleotide was inserted into the first exon of LOC_Os04g32510 in lax2-2. These results suggested that the mutation pattern of NGP4A was different from those of Gnp4 and LAX2.
The molecular mechanisms through which Gnp4 and LAX2 affect GNPP are poorly understood, warranting further investigation. MOC1 [14] and LAX1 [16] are two important genes in axillary meristem formation. Tabuchi et al. [16] proposed a regulation model for LAX2 and suggested that there were multiple pathways in which LAX1, LAX2, and MOC1 function for branching. LAX2 acts as a cofactor of LAX1, and it is unlikely that LAX2 regulates LAX1 expression. Meanwhile, LAX2 may or may not form dimers with MOC1. Moreover, Zhang et al. [15] reported that Gnp4 and LAX1 indirectly regulate tiller formation and directly or indirectly regulate secondary branch formation in multiple genetic pathways. Similar results were obtained in our transcriptome analysis as the expression levels of LAX1 and MOC1 did not change significantly in ngp4a mutant plants.
In recent years, the rapid development of high-throughput sequencing technology has resulted in the emergence of transcriptome analysis by RNA sequencing as a powerful strategy to explore molecular mechanisms underlying complex biological processes [32,33]. In this study, we found that the NGP4A mutation caused global changes in the rice transcriptome using RNA sequencing. Six significantly enriched pathways were observed in ngp4a, including phenylpropanoid biosynthesis, MAPK signaling, oxidative phosphorylation, plant hormone signal transduction, plant–pathogen interaction, and photosynthesis. The NGP4A mutation had the largest impact on the phenylpropanoid biosynthesis and MAPK signaling pathways. The phenylpropanoid biosynthesis pathway, identified to synthesize lignin, anthocyanins, and flavonoids in plants, is important for plant growth and development and contributes to plant responses to abiotic and biotic stresses [34,35,36,37]. Recent studies have suggested that the seed size of plants is correlated with lignin content. For example, the overexpression of SlMBP3 results in a 60% increase in lignin content and larger seeds in tomato [38]. GS3.1 regulates grain size via metabolic flux allocation between two branches of phenylpropanoid metabolism in rice [39]. In the present study, the grain length of ngp4a was significantly affected, which implies that the phenylpropanoid biosynthesis pathway may play multiple roles in the phenotypic mutation of grain number and seed size. MAPK is a serine/threonine protein kinase, whose cascades are highly conserved and is associated with ubiquitous signaling pathways in eukaryotes [40,41]. Plant MAPK signaling modules play fundamental roles in many aspects of biological processes, including immunity, stress responses, and developmental programs [42,43]. To date, several genes involved in the MAPK signaling pathway associated with grain number have been identified and characterized in rice. Guo et al. [44] reported that GSN1 can act as a negative regulator of a MAPK cascade that determines panicle architecture. Compared with wild-type plants, the grain length, grain width, and 1000-grain weight of the gsn1 mutant increased significantly, while the GNPP and seed setting rate decreased significantly. Other examples include an MAPK gene OsMPK15, which results in a marked decrease in the GNPP and seed setting rate and an increase in the 1000-grain weight in mutant plants [45]. OsER1 can act upstream of a MAPK cascade to control the spikelet number of panicles [44]. Mutant oser1 results in a markedly increased number of spikelets per panicle compared with the wild-type plants. Therefore, the up-regulation and down-regulation of core components of the MAPK signaling pathway in ngp4a suggest its flexibility in regulating the GNPP of rice, although the exact mechanisms remain unclear.
Furthermore, plant hormones, including cytokinin (CK), auxin, gibberellin (GA), abscisic acid (ABA), and brassinolactone (BR), involved in regulating the GNPP of rice, are becoming increasingly well understood [46,47,48,49]. A complex network of multiple hormonal pathways collide and communicate to regulate GNPP in rice. For example, GNP1 is involved in GA biosynthesis and affects the GNPP in rice [22]. Gn1a is a gene for CK oxidase/dehydrogenase and reduces the expression of Gn1a that causes CK accumulation and increases the number of grains, thereby resulting in enhanced grain yield [23,50]. OsRLCK57 negatively regulates the BR signaling pathway and reduces the expression of OsRLCK57, thereby resulting in significantly fewer tillers and panicle secondary branching [51]. Based on our transcriptome sequencing data, we found 28 DEGs that were significantly enriched in the pathways related to plant hormone signal transduction processes in ngp4a mutant plants. These findings suggest that the knockout of NGP4A altered plant hormone homeostasis, which may be the main cause of the GNPP mutant phenotype.
5. Conclusions
In conclusion, we characterized a novel GNPP mutant ngp4a. The genetic analysis and map-based cloning revealed that the causal gene NGP4A was a novel mutant allele gene for Gnp4 and LAX2. However, ngp4a was different from gnp4 and lax2 in the mutant phenotype and underlying causation. The transcriptome analysis showed significant changes in the expression level of genes involved in phenylpropanoid biosynthesis, MAPK signaling, oxidative phosphorylation, plant hormone signal transduction, plant–pathogen interaction, and photosynthesis in ngp4a. These findings provide a theoretical basis for further molecular studies of NGP4A involved in the GNPP of rice and subsequent high-yield rice breeding programs.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/agronomy12071549/s1, Table S1. Primers for map-based cloning and genetic complementation of NGP4A. Table S2. Differentially expressed genes between ngp4a and wild-type plants. Table S3. KEGG pathways of the differentially expressed genes.
Author Contributions
Performed the field experiments and map-based cloning, Y.C. and W.Y.; performed the vector construction and plant transformation experiments, M.Z. and Y.Z.; developed new molecular markers: G.D.; performed statistical analyses, J.X.; drafted the manuscript, Y.C. and F.Z.; contributed to the experimental design and edition of the manuscript, F.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was partially supported by the National Natural Science Foundation of China (31960370, 32070374), the Natural Science Foundation of Jiangxi Province, China (20202ACB205002), and the Key Project of Natural Science of Jiangxi Province (20202ACBL205002).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lu, Z.; Guo, X.; Huang, Z.; Xia, J.; Li, X.; Wu, J.; Yu, H.; Shahid, M.Q.; Liu, X. Transcriptome and gene editing analyses reveal MOF1a defect alters the expression of genes associated with tapetum development and chromosome behavior at meiosis stage resulting in low pollen fertility of tetraploid rice. Int. J. Mol. Sci. 2020, 21, 7489. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Xu, K.; Li, Z.; Hu, Y.; Hu, Z.; Zheng, X.; Song, S.; Tang, Z.; Li, L. Genome-wide association study and Mendelian randomization analysis provide insights for improving rice yield potential. Sci. Rep. 2021, 11, 6894. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Feng, F.; Zhang, Y.; Elesawi, I.E.; Xu, K.; Li, T.; Mei, H.; Liu, H.; Gao, N.; Chen, C.; et al. A novel rice grain size gene OsSNB was identified by genome-wide association study in natural population. PLoS Genet. 2019, 15, e1008191. [Google Scholar] [CrossRef]
- Kushibuchi, K. Historical changes in rice cultivars. In Science of the Rice Plant—Volume 3, Genetics; Matsuo, T., Futsuhara, Y., Kikuchi, F., Yamaguchi, H., Eds.; Food and Agriculture Policy Research Center: Tokyo, Japan, 1997; pp. 837–875. [Google Scholar]
- Yuan, L.P. Hybrid rice breeding for super high yield. Hybrid Rice 1997, 12, 1–6. (In Chinese) [Google Scholar]
- Yang, S.R.; Zhang, L.B.; Chen, W.F.; Xu, Z.J.; Wang, J.M. Theories and methods of rice breeding for maximum yield. Acta Agromomic Sin. 1995, 22, 295–304. (In Chinese) [Google Scholar]
- Zhou, K.D.; Wang, X.D.; Li, S.G.; Li, P.; Li, H.Y.; Huang, G.S.; Liu, T.Q.; Shen, M.S. The study on heavy panicle type of inter-subspecific hybrid rice (Oryza sativa L.). Sci. Agric. Sin. 1997, 30, 91–93. (In Chinese) [Google Scholar]
- Gouda, G.; Gupta, M.K.; Donde, R.; Mohapatra, T.; Vadde, R.; Behera, L. Marker-assisted selection for grain number and yield-related traits of rice (Oryza sativa L.). Physiol. Mol. Biol. Plants 2020, 26, 885–898. [Google Scholar] [CrossRef]
- Yin, C.; Zhu, Y.; Li, X.; Lin, Y. Molecular and genetic aspects of grain number determination in rice (Oryza sativa L.). Int. J. Mol. Sci. 2021, 22, 728. [Google Scholar] [CrossRef]
- Xie, J.; Li, F.; Khan, N.U.; Zhu, X.; Wang, X.; Zhang, Z.; Ma, X.; Zhao, Y.; Zhang, Q.; Zhang, S.; et al. Identifying natural genotypes of grain number per panicle in rice (Oryza sativa L.) by association mapping. Genes Genom. 2019, 41, 283–295. [Google Scholar] [CrossRef]
- Singh, V.K.; Ellur, R.K.; Singh, A.K.; Nagarajan, M.; Singh, B.D.; Singh, N.K. Effect of qGN4.1 QTL for grain number per panicle in genetic backgrounds of twelve different mega varieties of rice. Rice 2018, 11, 8. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Cao, L.; Sun, X.; Zhu, Y.; Zhang, T.; Jiang, L.; Liu, Y.; Dong, S.; Sun, D.; Yang, J.; et al. Fine mapping of a major quantitative trait locus, qgnp7(t), controlling grain number per panicle in African rice (Oryza glaberrima S.). Breed. Sci. 2018, 68, 606–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Tan, L.; Zhu, Z.; Xiao, L.; Xie, D.; Sun, C. PAY1 improves plant architecture and enhances grain yield in rice. Plant J. 2015, 83, 528–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Qian, Q.; Fu, Z.; Wang, Y.; Xiong, G.; Zeng, D.; Wang, X.; Liu, X.; Teng, S.; Hiroshi, F.; et al. Control of tillering in rice. Nature 2003, 422, 618–621. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Li, J.J.; Yao, G.X.; Zhang, H.L.; Dou, H.J.; Shi, H.L.; Sun, X.M.; Li, Z.C. Fine mapping and cloning of the grain number per-panicle gene (Gnp4) on chromosome 4 in rice (Oryza sativa L.). J. Integr. Agric. 2011, 10, 1825–1833. [Google Scholar] [CrossRef]
- Tabuchi, H.; Zhang, Y.; Hattori, S.; Omae, M.; Shimizu-Sato, S.; Oikawa, T.; Qian, Q.; Nishimura, M.; Kitano, H.; Xie, H.; et al. LAX PANICLE2 of rice encodes a novel nuclear protein and regulates the formation of axillary meristems. Plant Cell 2011, 23, 3276–3287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Maekawa, M.; Shimamoto, K.; Kyozuka, J. The LAX1 and FRIZZY PANICLE 2 genes determine the inflorescence architecture of rice by controlling rachis-branch and spikelet development. Dev. Biol. 2001, 231, 364–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Qian, Q.; Fu, Z.; Zeng, D.; Meng, X.; Kyozuka, J.; Maekawa, M.; Zhu, X.; Zhang, J.; Li, J.; et al. Short panicle1 encodes a putative PTR family transporter and determines rice panicle size. Plant J. 2009, 58, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Qian, Q.; Liu, Z.; Sun, H.; He, S.; Luo, D.; Xia, G.; Chu, C.; Li, J.; Fu, X. Natural variation at the DEP1 locus enhances grain yield in rice. Nat. Genet. 2009, 41, 494–497. [Google Scholar] [CrossRef]
- Yoshida, A.; Sasao, M.; Yasuno, N.; Takagi, K.; Daimon, Y.; Chen, R.; Yamazaki, R.; Tokunaga, H.; Kitaguchi, Y.; Sato, Y.; et al. TAWAWA1, a regulator of rice inflorescence architecture, functions through the suppression of meristem phase transition. Proc. Natl. Acad. Sci. USA 2013, 110, 767–772. [Google Scholar] [CrossRef] [Green Version]
- Mao, C.; Ding, W.; Wu, Y.; Yu, J.; He, X.; Shou, H.; Wu, P. Overexpression of a NAC-domain protein promotes shoot branching in rice. New Phytol. 2007, 176, 288–298. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, Y.; Mi, X.F.; Shan, J.X.; Li, X.M.; Xu, J.L.; Lin, H.X. The QTL GNP1 encodes GA20ox1, which increases grain number and yield by increasing cytokinin activity in rice panicle meristems. PLoS Genet. 2016, 12, e1006386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Zhao, B.; Yuan, D.; Duan, M.; Qian, Q.; Tang, L.; Wang, B.; Liu, X.; Zhang, J.; Wang, J.; et al. Rice zinc finger protein DST enhances grain production through controlling Gn1a/OsCKX2 expression. Proc. Natl. Acad. Sci. USA 2013, 110, 3167–3172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, L.; Li, X.; Liu, F.; Sun, X.; Li, C.; Zhu, Z.; Fu, Y.; Cai, H.; Wang, X.; Xie, D.; et al. Control of a key transition from prostrate to erect growth in rice domestication. Nat. Genet. 2008, 40, 1360–1364. [Google Scholar] [CrossRef] [PubMed]
- McCouch, S.R.; Teytelman, L.; Xu, Y.; Lobos, K.B.; Clare, K.; Walton, M.; Fu, B.; Maghirang, R.; Li, Z.; Xing, Y.; et al. Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.). DNA Res. 2002, 9, 199–207. [Google Scholar] [CrossRef]
- Liu, X.Q.; Bai, X.Q.; Wang, X.J.; Chu, C.C. OsWRKY71, a rice transcription factor, is involved in rice defense response. J. Plant Physiol. 2007, 164, 969–979. [Google Scholar] [CrossRef]
- Maignan, V.; Géliot, P.; Avice, J.C. Glutacetine® biostimulant applied on wheat under contrasting field conditions improves grain number leading to better yield, upgrades N-related traits and changes grain ionome. Plants 2021, 10, 456. [Google Scholar] [CrossRef]
- Dampanaboina, L.; Jiao, Y.; Chen, J.; Gladman, N.; Chopra, R.; Burow, G.; Hayes, C.; Christensen, S.A.; Burke, J.; Ware, D.; et al. Sorghum MSD3 encodes an ω-3 fatty acid desaturase that increases grain number by reducing jasmonic acid levels. Int. J. Mol. Sci. 2019, 20, 5359. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Tao, Y.; Zhu, J.; Miao, J.; Liu, J.; Liu, Y.; Yi, C.; Yang, Z.; Gong, Z.; Liang, G. GNS4, a novel allele of DWARF11, regulates grain number and grain size in a high-yield rice variety. Rice 2017, 10, 34. [Google Scholar] [CrossRef] [Green Version]
- Hao, J.; Wang, D.; Wu, Y.; Huang, K.; Duan, P.; Li, N.; Xu, R.; Zeng, D.; Dong, G.; Zhang, B.; et al. The GW2-WG1-OsbZIP47 pathway controls grain size and weight in rice. Mol. Plant 2021, 14, 1266–1280. [Google Scholar] [CrossRef]
- Chen, H.; Tang, Y.; Liu, J.; Tan, L.; Jiang, J.; Wang, M.; Zhu, Z.; Sun, X.; Sun, C. Emergence of a novel chimeric gene underlying grain number in rice. Genetics 2017, 205, 993–1002. [Google Scholar] [CrossRef]
- Nguyen, K.L.; Grondin, A.; Courtois, B.; Gantet, P. Next-generation sequencing accelerates crop gene discovery. Trends Plant Sci. 2019, 24, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Dueñas, C.J.; Slamet-Loedin, I.; Macovei, A. Transcriptomics view over the germination landscape in biofortified rice. Genes 2021, 12, 2013. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, A.B.; Bottcher, A.; Kiyota, E.; Mayer, J.L.; Vicentini, R.; Brito Mdos, S.; Creste, S.; Landell, M.G.; Mazzafera, P. Water stress alters lignin content and related gene expression in two sugarcane genotypes. J. Agric. Food Chem. 2015, 63, 4708–4720. [Google Scholar] [CrossRef] [PubMed]
- Ksouri, N.; Jiménez, S.; Wells, C.E.; Contreras-Moreira, B.; Gogorcena, Y. Transcriptional responses in root and leaf of Prunus persica under drought stress using RNA sequencing. Front. Plant Sci. 2016, 7, 1715. [Google Scholar] [CrossRef]
- Li, J.; Fan, F.; Wang, L.; Zhan, Q.; Wu, P.; Du, J.; Yang, X.; Liu, Y. Cloning and expression analysis of cinnamoyl-CoA reductase (CCR) genes in sorghum. PeerJ 2016, 4, e2005. [Google Scholar] [CrossRef]
- Jardim-Messeder, D.; Felix-Cordeiro, T.; Barzilai, L.; de Souza-Vieira, Y.; Galhego, V.; Bastos, G.A.; Valente-Almeida, G.; Aiube, Y.R.A.; Faria-Reis, A.; Corrêa, R.L.; et al. Genome-wide analysis of general phenylpropanoid and monolignol-specific metabolism genes in sugarcane. Funct. Integr. Genom. 2021, 21, 73–99. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Naeem, M.; Zhu, M.; Li, J.; Yu, X.; Hu, Z.; Chen, G. An AGAMOUS MADS-box protein, SlMBP3, regulates the speed of placenta liquefaction and controls seed formation in tomato. J. Exp. Bot. 2019, 70, 909–924. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Yu, H.X.; Ye, W.W.; Shan, J.X.; Dong, N.Q.; Guo, T.; Kan, Y.; Xiang, Y.H.; Zhang, H.; Yang, Y.B.; et al. A rice QTL GS3.1 regulates grain size through metabolic-flux distribution between flavonoid and lignin metabolons without affecting stress tolerance. Commun. Biol. 2021, 4, 1171. [Google Scholar] [CrossRef]
- Li, S.; Han, X.; Lu, Z.; Qiu, W.; Yu, M.; Li, H.; He, Z.; Zhuo, R. MAPK cascades and transcriptional factors: Regulation of heavy metal tolerance in plants. Int. J. Mol. Sci. 2022, 23, 4463. [Google Scholar] [CrossRef]
- Wen, X.; Jiao, L.; Tan, H. MAPK/ERK pathway as a central regulator in vertebrate organ regeneration. Int. J. Mol. Sci. 2022, 23, 1464. [Google Scholar] [CrossRef]
- Wang, N.; Liu, Y.; Dong, C.; Zhang, Y.; Bai, S. MdMAPKKK1 regulates apple resistance to Botryosphaeria dothidea by interacting with MdBSK1. Int. J. Mol. Sci. 2022, 23, 4415. [Google Scholar] [CrossRef]
- Ma, H.; Gao, Y.; Wang, Y.; Dai, Y.; Ma, H. Regulatory mechanisms of mitogen-activated protein kinase cascades in plants: More than sequential phosphorylation. Int. J. Mol. Sci. 2022, 23, 3572. [Google Scholar] [CrossRef]
- Guo, T.; Chen, K.; Dong, N.Q.; Shi, C.L.; Ye, W.W.; Gao, J.P.; Shan, J.X.; Lin, H.X. GRAIN SIZE AND NUMBER1 negatively regulates the OsMKKK10-OsMKK4-OsMPK6 cascade to coordinate the trade-off between grain number per panicle and grain size in rice. Plant Cell 2018, 30, 871–888. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.; Liu, Q.; Cao, Y.; Zhang, Y.; Chen, D.; Lou, X.; Cheng, S.; Cao, L. The OsMPK15 negatively regulates Magnaporthe oryza and Xoo disease resistance via SA and JA signaling pathway in rice. Front. Plant Sci. 2019, 10, 752. [Google Scholar] [CrossRef]
- Nadolska-Orczyk, A.; Rajchel, I.K.; Orczyk, W.; Gasparis, S. Major genes determining yield-related traits in wheat and barley. Theor. Appl. Genet. 2017, 130, 1081–1098. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, S.; Kudoyarova, G.R.; Veselov, D.S.; Arkhipova, T.N.; Davies, W.J. Plant hormone interactions: Innovative targets for crop breeding and management. J. Exp. Bot. 2012, 63, 3499–3509. [Google Scholar] [CrossRef]
- Deveshwar, P.; Prusty, A.; Sharma, S.; Tyagi, A.K. Phytohormone-mediated molecular mechanisms involving multiple genes and QTL govern grain number in rice. Front. Genet. 2020, 11, 586462. [Google Scholar] [CrossRef]
- Shirley, N.J.; Aubert, M.K.; Wilkinson, L.G.; Bird, D.C.; Lora, J.; Yang, X.; Tucker, M.R. Translating auxin responses into ovules, seeds and yield: Insight from Arabidopsis and the cereals. J. Integr. Plant Biol. 2019, 61, 310–336. [Google Scholar] [CrossRef] [Green Version]
- Tu, B.; Tao, Z.; Wang, S.; Zhou, L.; Zheng, L.; Zhang, C.; Li, X.; Zhang, X.; Yin, J.; Zhu, X.; et al. Loss of Gn1a/OsCKX2 confers heavy-panicle rice with excellent lodging resistance. J. Integr. Plant Biol. 2022, 64, 23–38. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, J.; Peng, C.; Zhu, X.; Yin, J.; Li, W.; He, M.; Wang, J.; Chern, M.; Yuan, C.; et al. Four receptor-like cytoplasmic kinases regulate development and immunity in rice. Plant Cell Environ. 2016, 39, 1381–1392. [Google Scholar] [CrossRef]
Figure 1.
Phenotypic traits of ngp4a and wild-type plants. (A): Plant phenotypes of the ngp4a mutant at maturity. (B): Main panicle. (C): Primary and secondary branches. PB: primary branch. SB: secondary branch. ngp4a: mutant plants; WT: wild-type plants.
Figure 1.
Phenotypic traits of ngp4a and wild-type plants. (A): Plant phenotypes of the ngp4a mutant at maturity. (B): Main panicle. (C): Primary and secondary branches. PB: primary branch. SB: secondary branch. ngp4a: mutant plants; WT: wild-type plants.

Figure 2.
Genetic and physical map of NGP4A. (A): NGP4A locus mapped in the long arm of chromosome 4. (B,C): Fine mapping of NGP4A. The numbers in the parentheses indicate the number of recombinants. (D): Structure of NGP4A. The black boxes indicate the coding sequence and the lines between the boxes represent introns. (E): Mutant site of NGP4A. The 3 bp nucleotide deletion of NGP4A is framed by the red dotted box. WT: wild-type plants.
Figure 2.
Genetic and physical map of NGP4A. (A): NGP4A locus mapped in the long arm of chromosome 4. (B,C): Fine mapping of NGP4A. The numbers in the parentheses indicate the number of recombinants. (D): Structure of NGP4A. The black boxes indicate the coding sequence and the lines between the boxes represent introns. (E): Mutant site of NGP4A. The 3 bp nucleotide deletion of NGP4A is framed by the red dotted box. WT: wild-type plants.
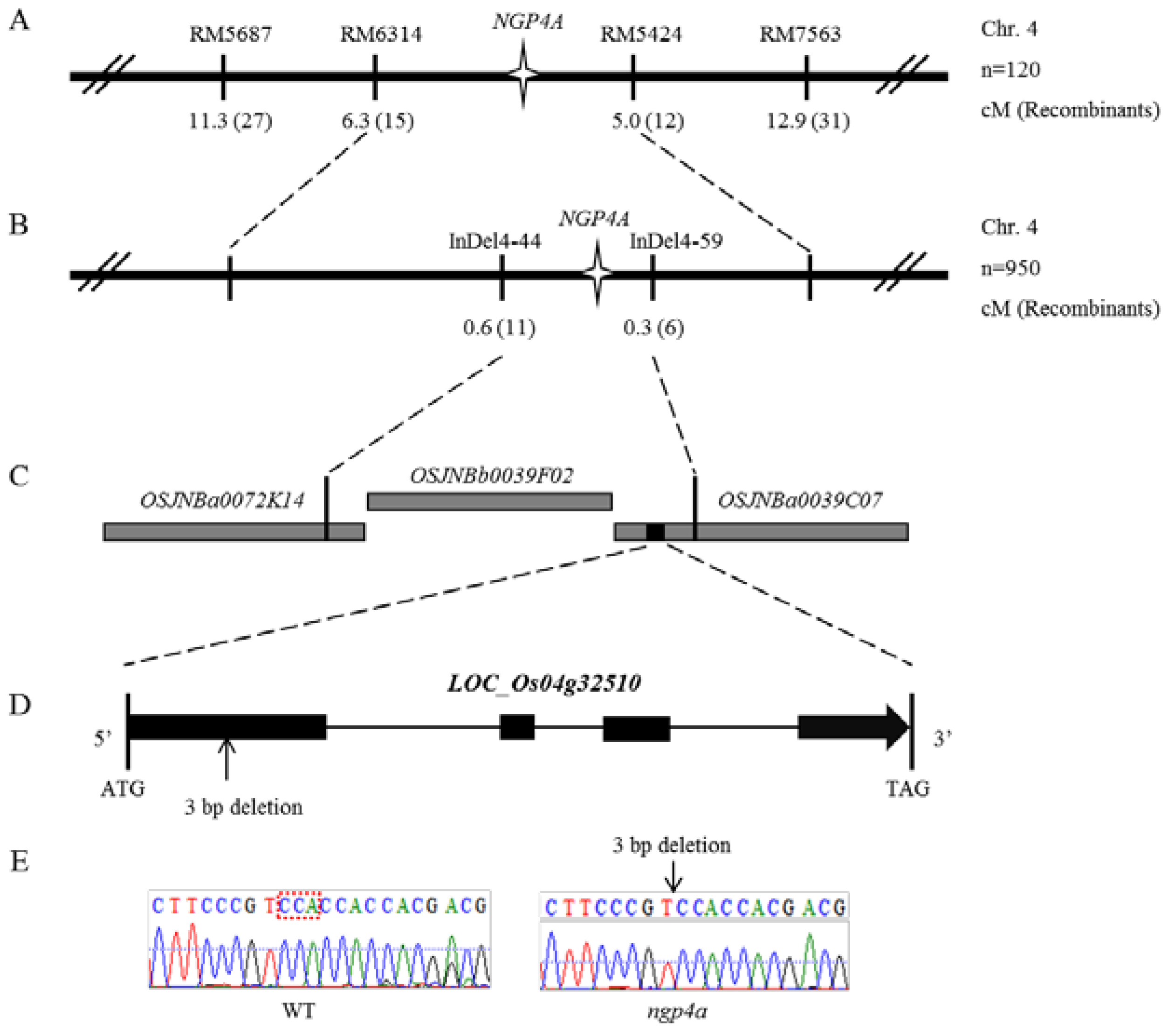
Figure 3.
Panicle phenotype of complementation transgenic plants. While ngp4a exhibited the grain number per panicle mutant phenotype, the complementation transgenic plants exhibited the wild-type phenotype. (A): Main panicle. (B): Primary and secondary branches. WT: Wild type; ngp4a: mutant; ngp4a/NGP4A: complementation transgenic plants. SB: Secondary branch.
Figure 3.
Panicle phenotype of complementation transgenic plants. While ngp4a exhibited the grain number per panicle mutant phenotype, the complementation transgenic plants exhibited the wild-type phenotype. (A): Main panicle. (B): Primary and secondary branches. WT: Wild type; ngp4a: mutant; ngp4a/NGP4A: complementation transgenic plants. SB: Secondary branch.

Figure 4.
Gene ontology (GO) classification of the differentially expressed genes.
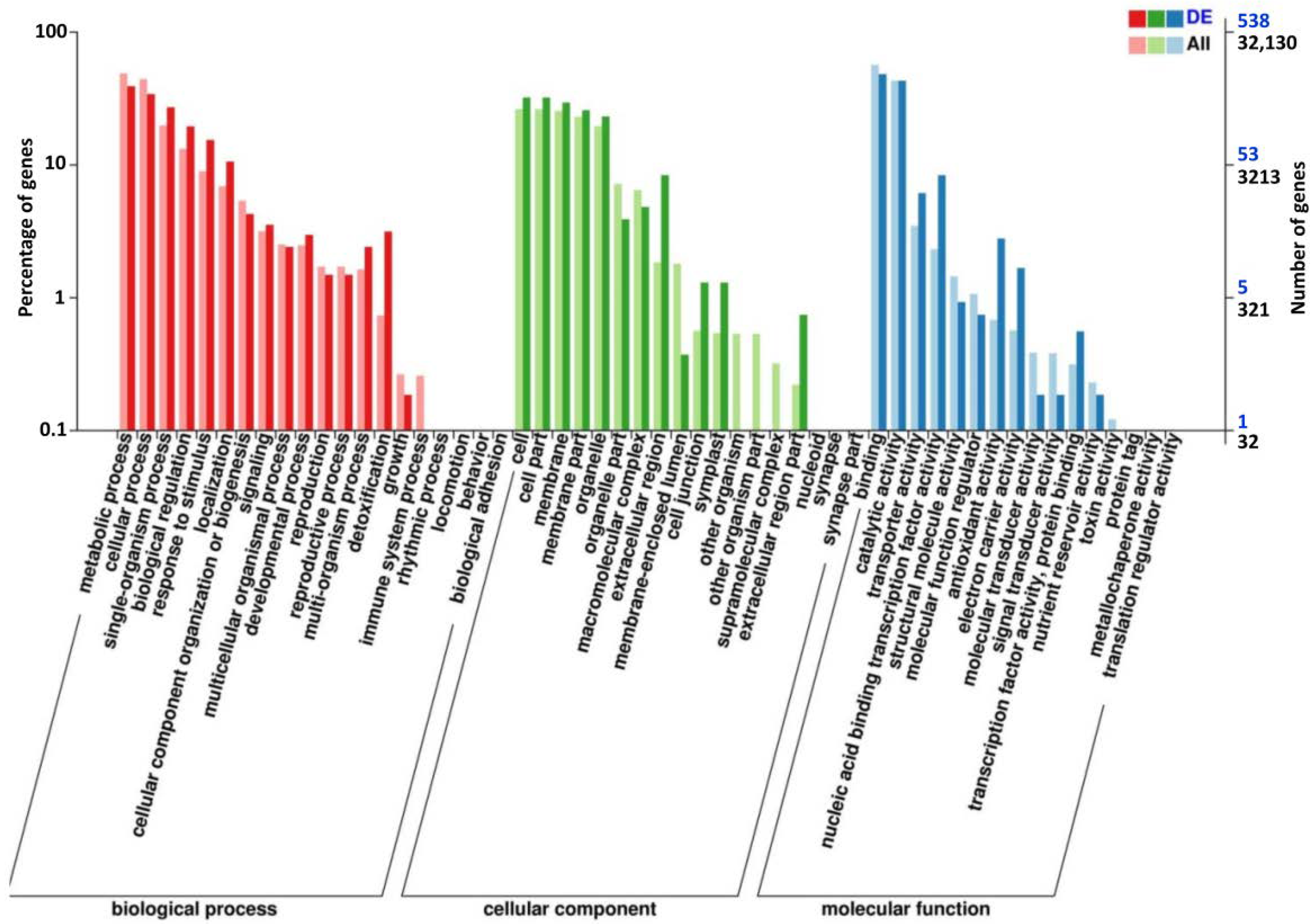
Figure 5.
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway assignments for the differentially expressed genes.
Figure 5.
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway assignments for the differentially expressed genes.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Comparison of major agronomic traits between ngp4a and wild-type plants. ngp4a: mutant plants; WT: wild-type plants.
Table 1.
Comparison of major agronomic traits between ngp4a and wild-type plants. ngp4a: mutant plants; WT: wild-type plants.
| Traits | Wild Type (WT) | ngp4a | Compared with WT (%) |
|---|---|---|---|
| Plant height (cm) | 95.7 ± 1.5 | 84.0 ± 0.8 | −12.2 ** |
| Tiller number | 23.7 ± 1.5 | 16.0 ± 0.5 | −32.5 ** |
| Main panicle length (cm) | 29.3 ± 1.1 | 26.9 ± 1.0 | −8.2 * |
| Primary branches of main panicle | 10.7 ± 0.6 | 10.0 ± 0.3 | −6.5 |
| Secondary branches of main panicle | 36.7 ± 2.1 | 10.0 ± 0.5 | −72.8 ** |
| Total spikelets of main panicle | 166.7 ± 4.7 | 106.3 ± 2.5 | −36.2 ** |
| Seed setting rate (%) | 85.0 ± 1.4 | 88.01 ± 1.8 | +3.5 |
| 1000-grain weight (g) | 22.8 ± 0.4 | 26.09 ± 0.7 | +14.4 * |
| Grain length (mm) | 7.3 ± 0.1 | 8.0 ± 0.1 | +9.6 * |
| Grain width (mm) | 3.8 ± 0.1 | 3.9 ± 0.1 | +2.6 |
Note: * represents a significant difference at the 0.05 level. ** represents a significant difference at the 0.01 level. For each agronomic trait, 10 plants as biological replicates were evaluated. All data are represented as mean ± SD. Statistical analysis was performed with the Student’s t-test.
Table 2.
Comparison of major agronomic traits between complementation transgenic plants ngp4a/NGP4A and wild-type plants.
Table 2.
Comparison of major agronomic traits between complementation transgenic plants ngp4a/NGP4A and wild-type plants.
| Traits | Wild Type (WT) | ngp4a/ NGP4A | Compared with WT (%) |
|---|---|---|---|
| Plant height (cm) | 95.6 ± 0.8 | 93.0 ± 1.1 | −2.7 |
| Tiller number | 22.7 ± 1.5 | 23.3 ± 0.9 | +2.8 |
| Main panicle length (cm) | 30.0 ± 0.4 | 28.8 ± 0.7 | −4.0 |
| Primary branches of main panicle | 10.7 ± 0.6 | 10.3 ± 0.6 | −3.1 |
| Secondary branches of main panicle | 35.7 ± 1.7 | 33.8 ± 2.5 | −5.2 |
| Total spikelets of main panicle | 172.0 ± 5.3 | 162.7 ± 3.7 | −5.4 |
| Seed setting rate (%) | 85.5 ± 0.7 | 87.2 ± 0.5 | +2.0 |
| 1000-grain weight (g) | 22.8 ± 0.9 | 23.4 ± 0.8 | +2.8 |
| Grain length (mm) | 7.3 ± 0.1 | 7.4 ± 0.1 | +0.5 |
| Grain width (mm) | 3.8 ± 0.1 | 3.9 ± 0.1 | +0.9 |
Table 3.
RNA sequencing data of two samples.
| Sample | Wild Type (WT) | ngp4a |
|---|---|---|
| Clean reads | 20,593,555 | 20,481,813 |
| GC content | 52.83% | 53.10% |
| Clean bases | 6,150,165,034 | 6,126,925,284 |
| Total mapped reads (%) | 39,836,670 (96.72%) | 39,516,745 (96.47%) |
| Multiple mapped reads (%) | 1,184,323 (2.88%) | 1,130,576 (2.76%) |
| Unique mapped reads (%) | 38,652,348 (93.85%) | 38,386,169 (93.71%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chen, Y.; Yang, W.; Zhao, M.; Ding, G.; Zhou, Y.; Xie, J.; Zhang, F. Characterization of the NGP4A Gene in Regulating Grain Number Per Panicle of Rice (Oryza sativa L.). Agronomy 2022, 12, 1549. https://doi.org/10.3390/agronomy12071549
AMA Style
Chen Y, Yang W, Zhao M, Ding G, Zhou Y, Xie J, Zhang F. Characterization of the NGP4A Gene in Regulating Grain Number Per Panicle of Rice (Oryza sativa L.). Agronomy. 2022; 12(7):1549. https://doi.org/10.3390/agronomy12071549
Chicago/Turabian StyleChen, Yanhong, Wanling Yang, Minmin Zhao, Gumu Ding, Yi Zhou, Jiankun Xie, and Fantao Zhang. 2022. "Characterization of the NGP4A Gene in Regulating Grain Number Per Panicle of Rice (Oryza sativa L.)" Agronomy 12, no. 7: 1549. https://doi.org/10.3390/agronomy12071549
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.