Incorporation of Calcium Containing Mesoporous (MCM-41-Type) Particles in Electrospun PCL Fibers by Using Benign Solvents





Abstract
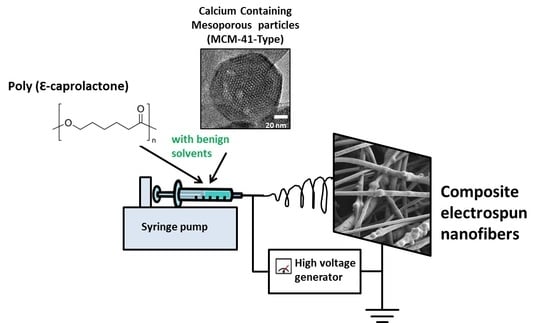
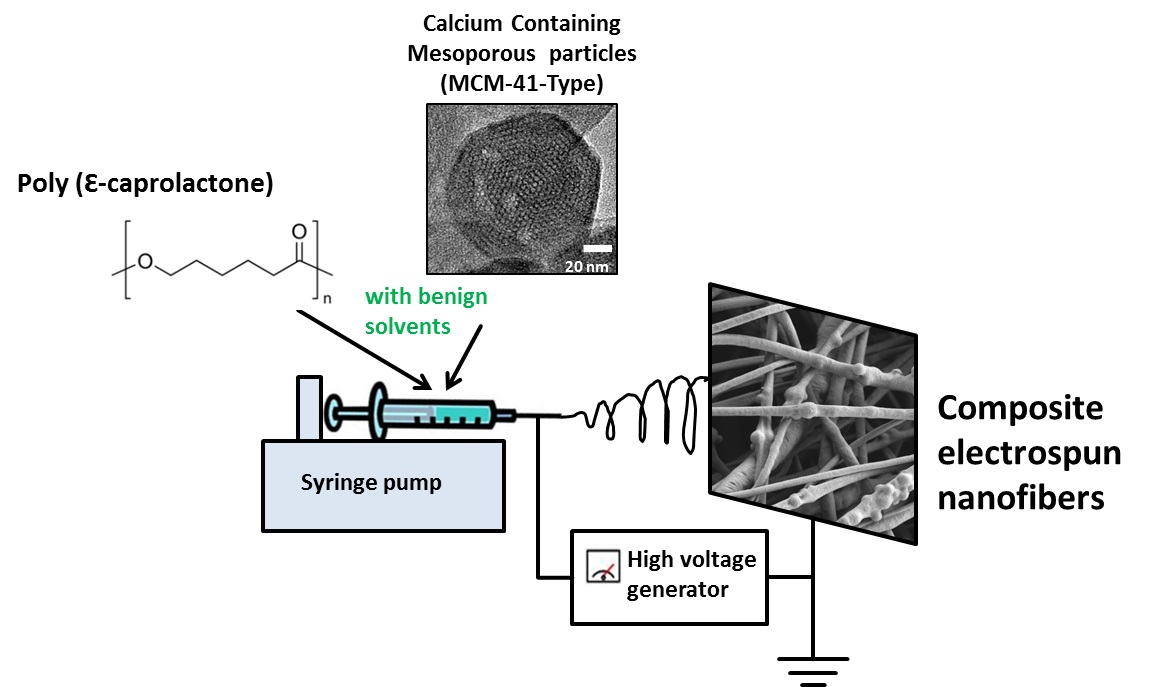
1. Introduction
2. Materials and Methods
2.1. Ca_MCM-41 Particle Synthesis
2.2. Degradation in Simulated Body Fluid (SBF)
2.3. Electrospinning Solution and Process Parameters
2.4. Electrospun Mats Characterization
3. Results
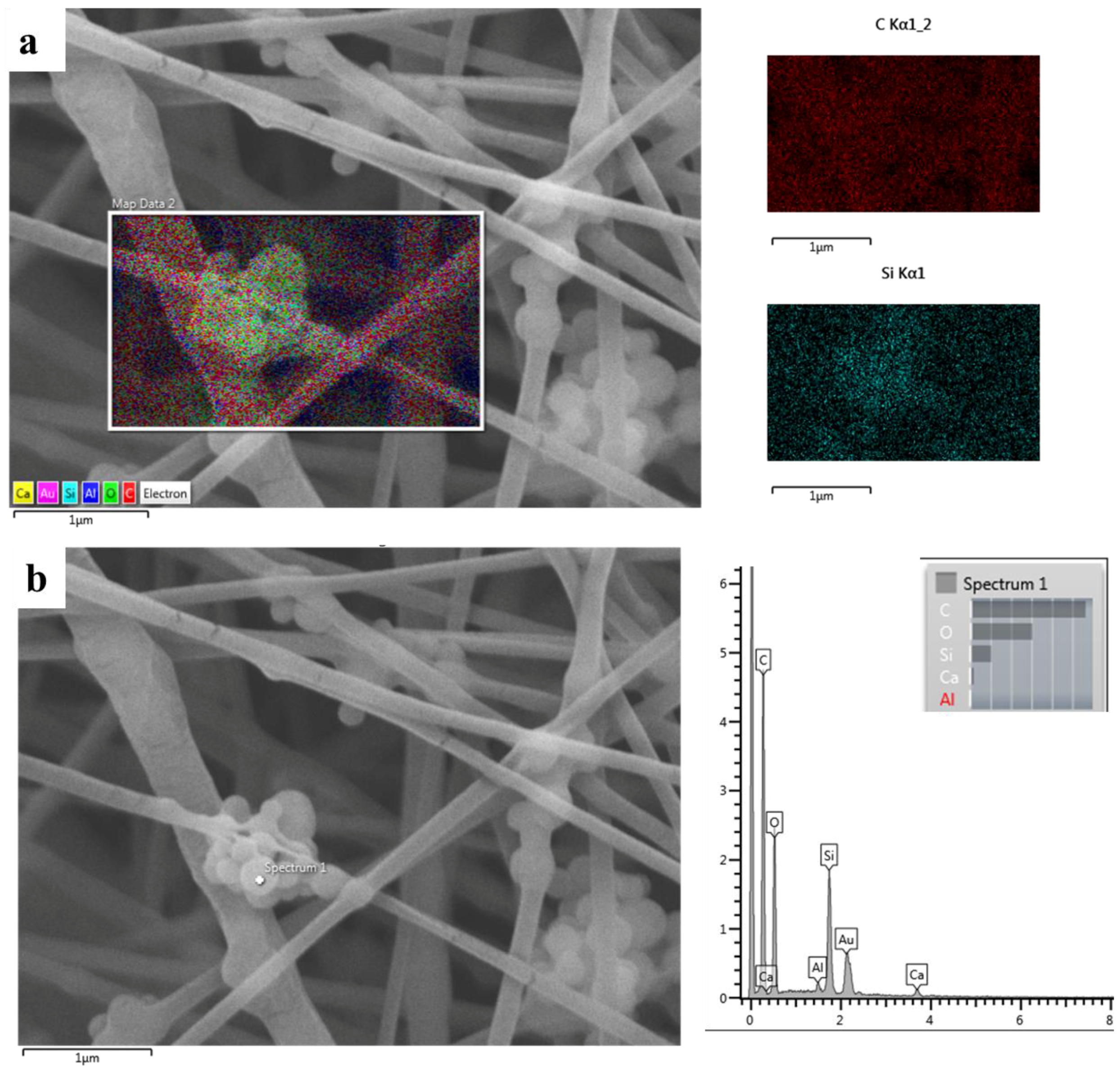
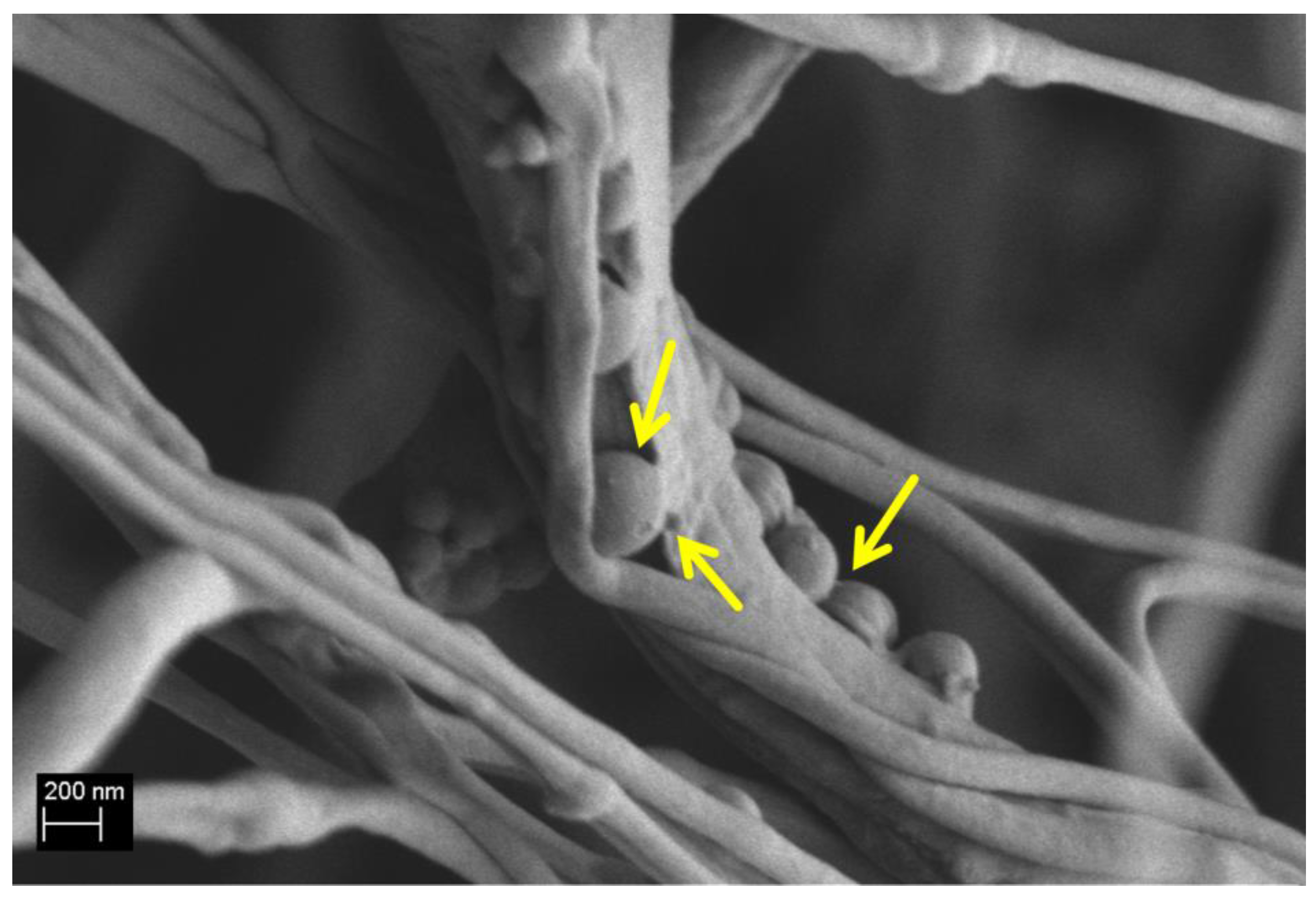
3.1. Ca_MCM-41 Particles Characterization
3.2. Optimization of Electrospinning Parameters
3.3. Mechanical Properties
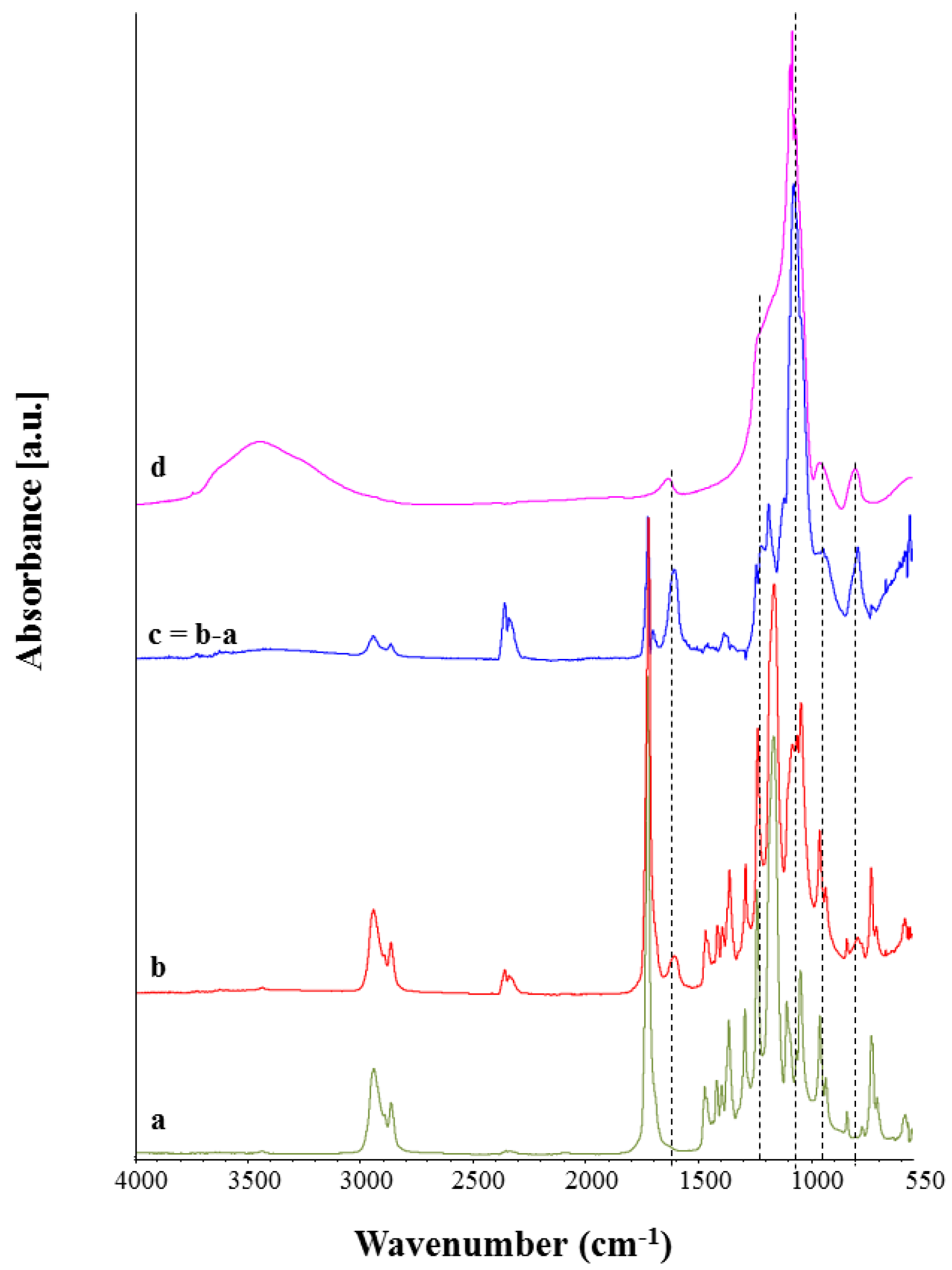
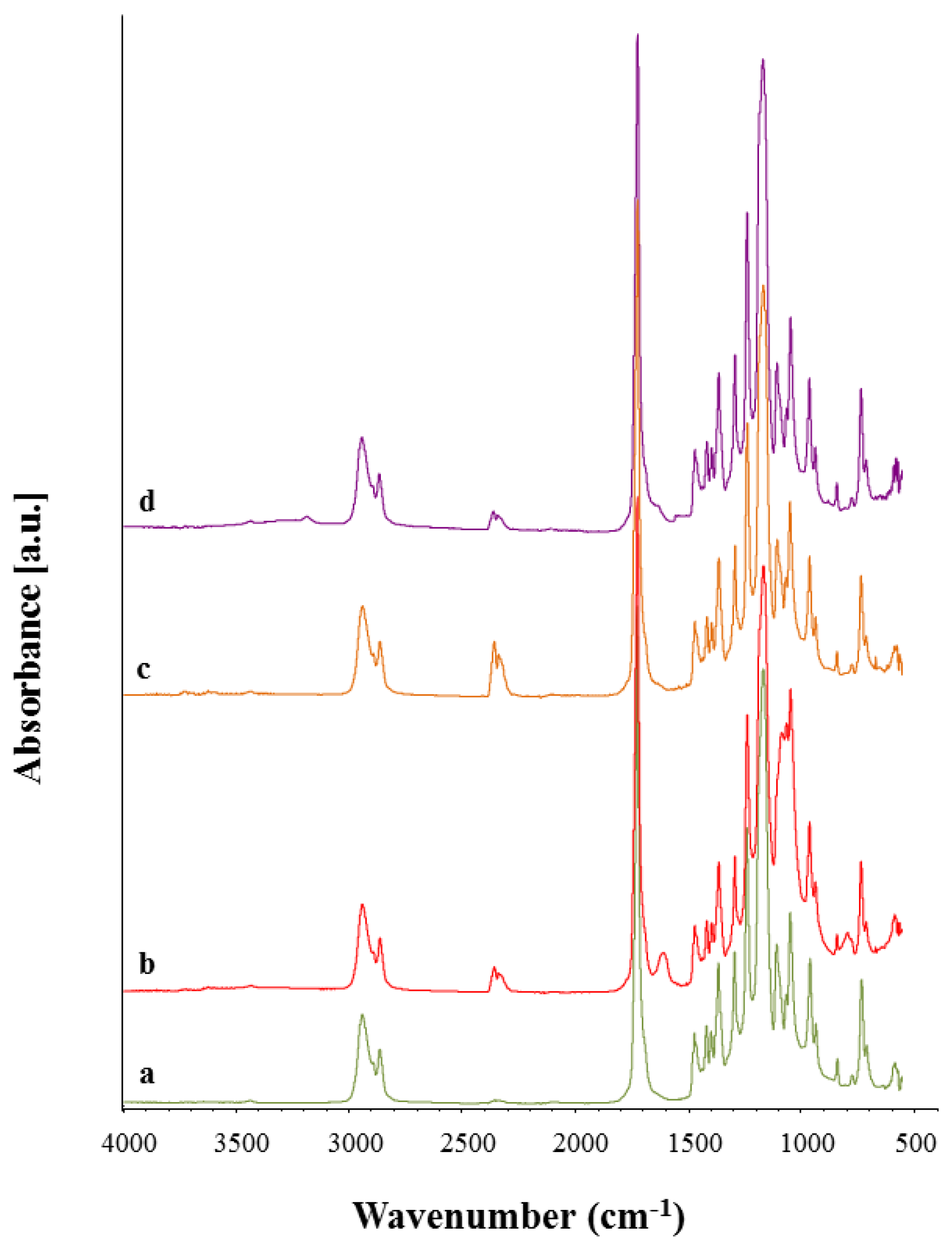
3.4. Chemical Characterization
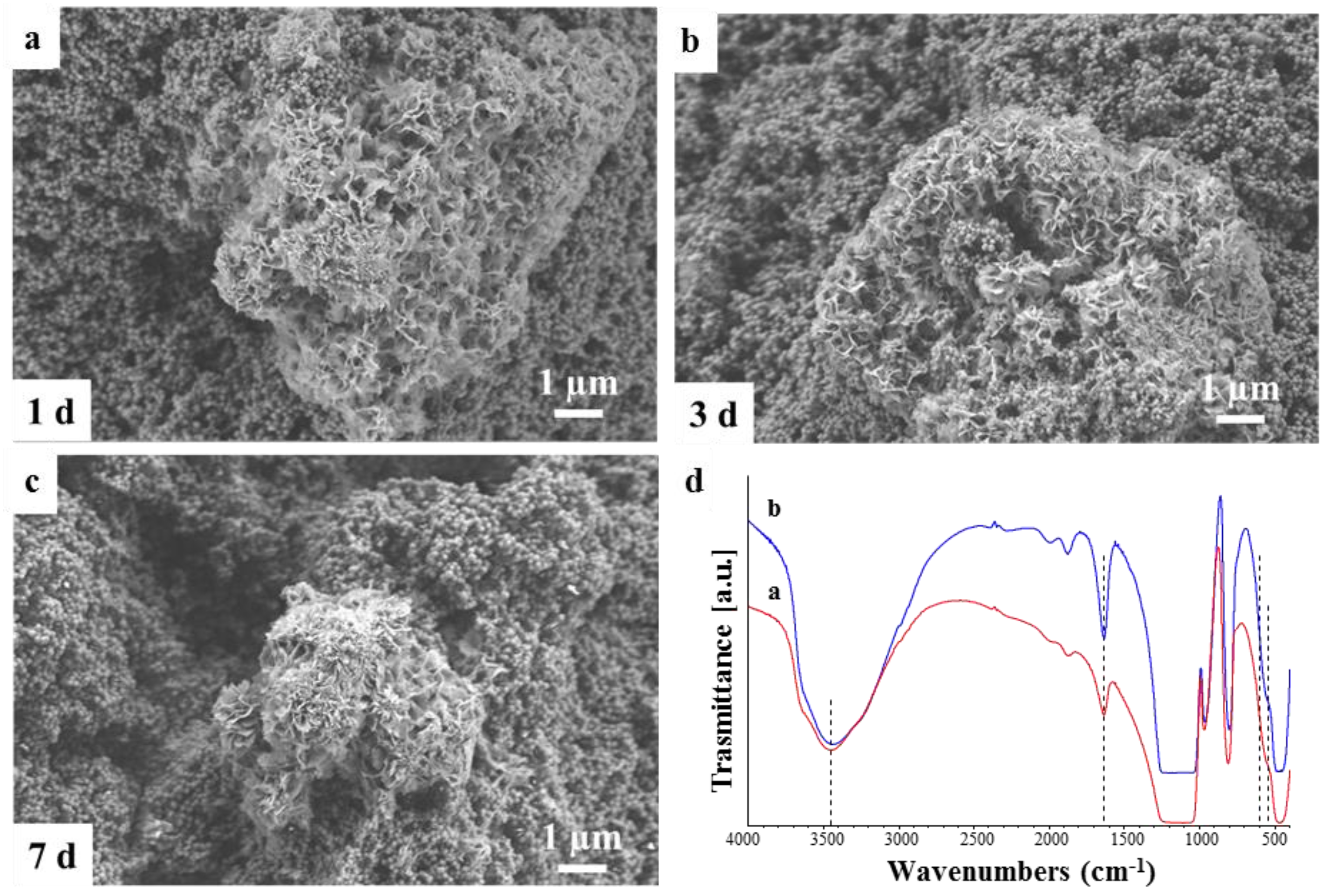
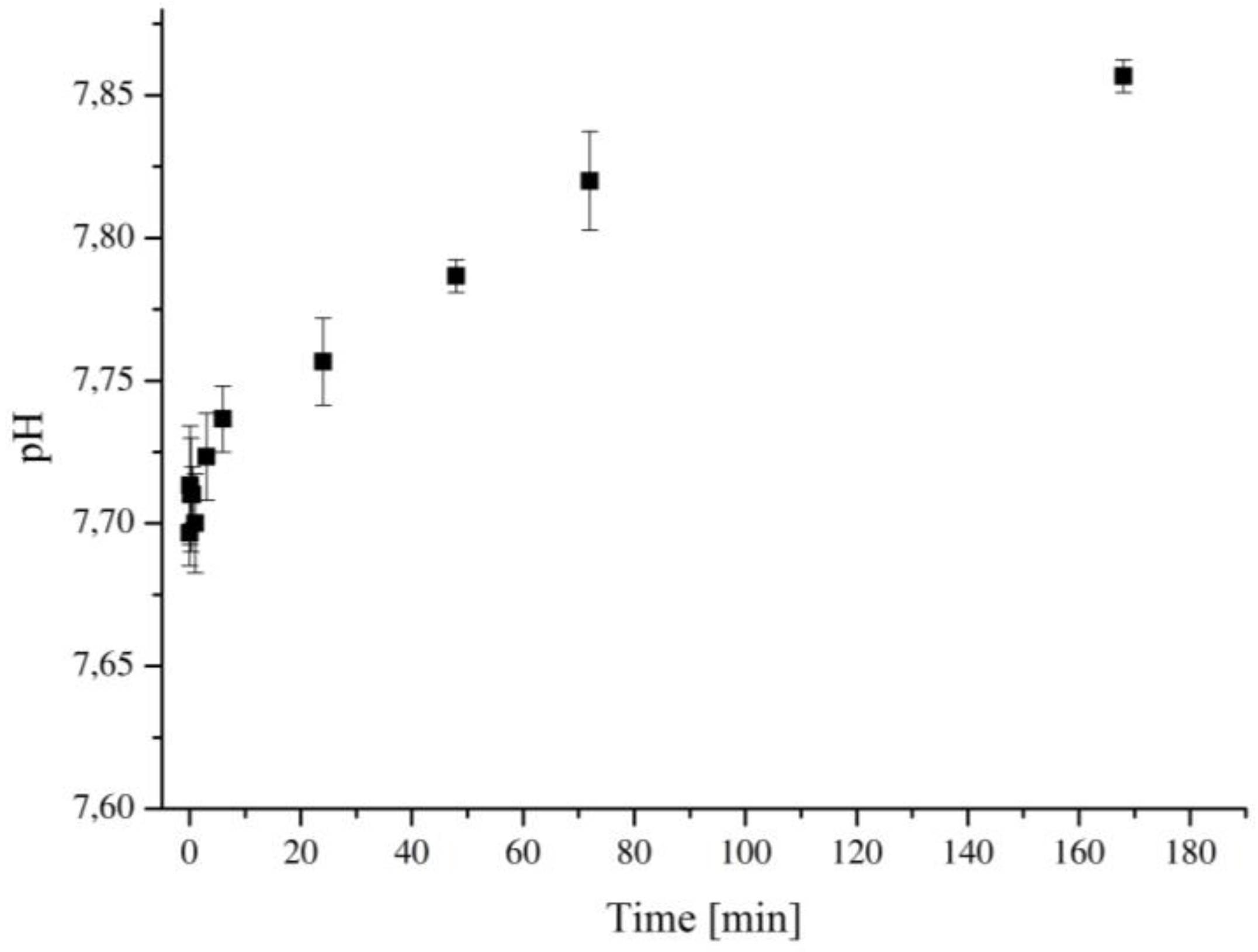
3.5. Bioactivity Assessment
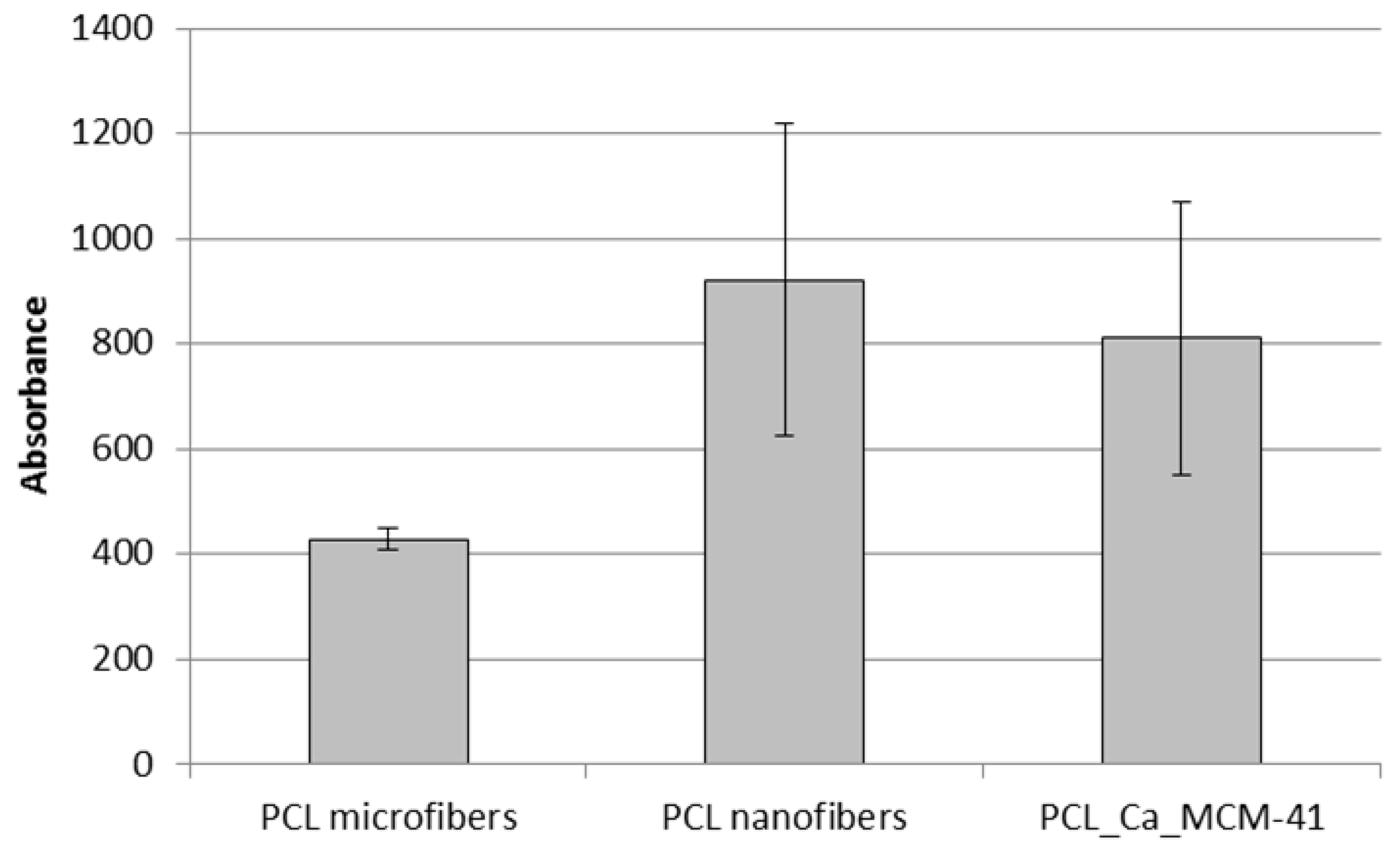
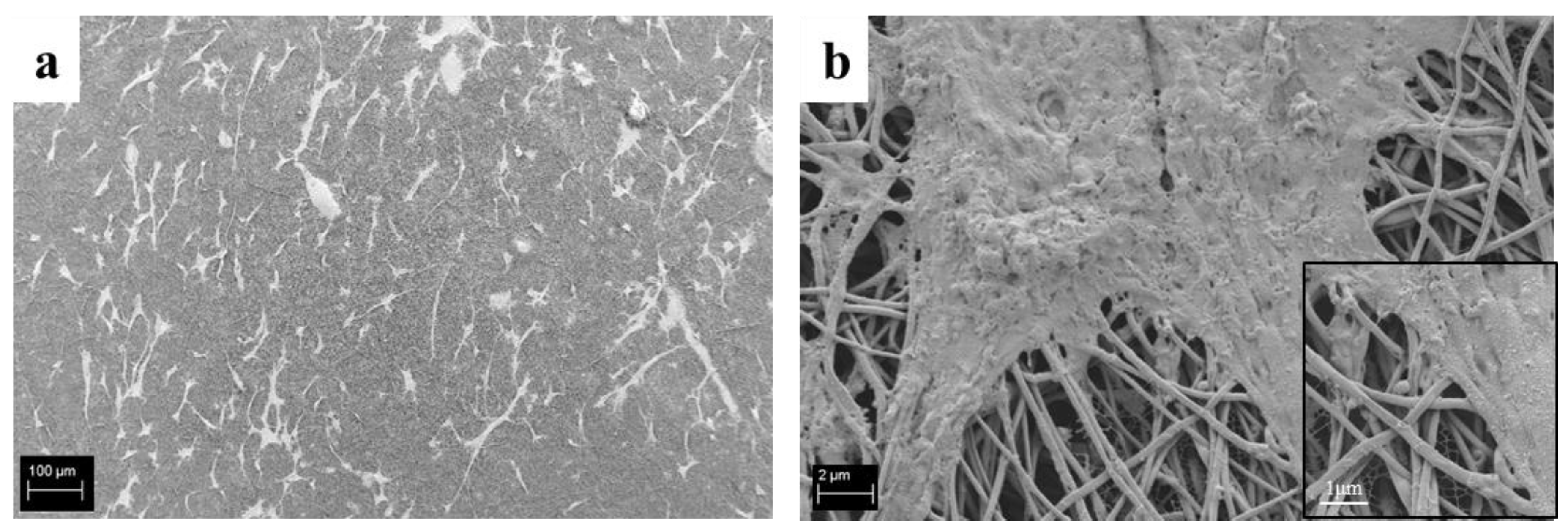
3.6. Cell-Composite Scaffold Interactions: Preliminary Tests
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kai, D.; Liow, S.S.; Loh, X.J. Biodegradable polymers for electrospinning: Towards biomedical applications. Mater. Sci. Eng. C. Mater. Biol. Appl. 2014, 45, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Teo, W.-E.; Inai, R.; Ramakrishna, S. Technological advances in electrospinning of nanofibers. Sci. Technol. Adv. Mater. 2011, 12, 013002. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Duan, X.-P.; Li, Y.-M.; Yang, D.-P.; Long, Y.-Z. Electrospun nanofibers for wound healing. Mater. Sci. Eng. C 2017, 76, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Prabhakaran, M.P.; Venugopal, J.R.; Chyan, T.T.; Hai, L.B.; Chan, C.K.; Lim, A.Y.; Ramakrishna, S. Electrospun biocomposite nanofibrous scaffolds for neural tissue engineering. Tissue Eng. Part A 2008, 14, 1787–1797. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.-H.; Purevdorj, O.; Castano, O.; Planell, J.A.; Kim, H.-W. A short review: Recent advances in electrospinning for bone tissue regeneration. J. Tissue Eng. 2012, 3, 2041731412443530. [Google Scholar] [CrossRef] [PubMed]
- Putti, M.; Simonet, M.; Solberg, R.; Peters, G.W. M. Electrospinning poly(ε-caprolactone) under controlled environmental conditions: Influence on fiber morphology and orientation. Polymer 2015, 63, 189–195. [Google Scholar] [CrossRef]
- Agarwal, S.; Greiner, A. On the way to clean and safe electrospinning-green electrospinning: Emulsion and suspension electrospinning. Polym. Adv. Technol. 2011, 22, 372–378. [Google Scholar] [CrossRef]
- Dong, B.; Arnoult, O.; Smith, M.E.; Wnek, G.E. Electrospinning of collagen nanofiber scaffolds from benign solvents. Macromol. Rapid Commun. 2009, 30, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Van der Schueren, L.; De Schoenmaker, B.; Kalaoglu, Ö.I.; De Clerck, K. An alternative solvent system for the steady state electrospinning of polycaprolactone. Eur. Polym. J. 2011, 47, 1256–1263. [Google Scholar] [CrossRef]
- Liverani, L.; Boccaccini, A.R. Versatile production of poly(Epsilon-caprolactone) fibers by electrospinning using benign solvents. Nanomaterials 2016, 6, 75. [Google Scholar] [CrossRef] [PubMed]
- Liverani, L.; Lacina, J.; Roether, J.A.; Boccardi, E.; Killian, M.S.; Schmuki, P.; Schubert, D.W.; Boccaccini, A.R. Incorporation of bioactive glass nanoparticles in electrospun PCL/chitosan fibers by using benign solvents. Bioact. Mater. 2017. [Google Scholar] [CrossRef]
- Moura, D.; Souza, M.T.; Liverani, L.; Rella, G.; Luz, G.M.; Mano, J.F.; Boccaccini, A.R. Development of a bioactive glass-polymer composite for wound healing applications. Mater. Sci. Eng. C 2017, 76, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Lepry, W.C.; Smith, S.; Liverani, L.; Boccaccini, A.R.; Nazhat, S.N. Acellular Bioactivity of Sol-Gel Derived Borate Glass-Polycaprolactone Electrospun Scaffolds Biomedical Glasses. Biomed. Glas. 2016, 2, 88–98. [Google Scholar]
- Gönen, S.Ö.; Taygun, M.E.; Küçükbayrak, S. Fabrication of Bioactive Glass Containing Nanocomposite Fiber Mats For Bone Tissue Engineering Applications. Compos. Struct. 2016, 138, 96–106. [Google Scholar] [CrossRef]
- Liverani, L.; Abbruzzese, F.; Mozetic, P.; Basoli, F.; Rainer, A.; Trombetta, M. Electrospinning of hydroxyapatite-chitosan nanofibers for tissue engineering applications. Asia-Pac. J. Chem. Eng. 2014, 9, 407–414. [Google Scholar] [CrossRef]
- Ding, Y.; Yao, Q.; Li, W.; Schubert, D.W.; Boccaccini, A.R.; Roether, J.A. The evaluation of physical properties and in vitro cell behavior of PHB/PCL/sol–gel derived silica hybrid scaffolds and PHB/PCL/fumed silica composite scaffolds. Colloids Surf. B Biointerfaces 2015, 136, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Wan, Y.; Zhou, W. Ordered Mesoporous Materials; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013. [Google Scholar]
- Vallet-Regí, M.; García, M.M.; Colilla, M. Biomedical Applications of Mesoporous Ceramics: Drug Delivery, Smart Materials and Bone Tissue Engineering; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Grün, M.; Unger, K.K.; Matsumoto, A.; Tsutsumib, K. Novel pathways for the preparation of mesoporous MCM-41 materials: Control of porosity and morphology. Microporous Mesoporous Mater. 1999, 27, 207–216. [Google Scholar] [CrossRef]
- Stöber, W.; Fink, A.; Bohn, E. Controlled growth of monodisperse silica spheres in the micron size range. J. Colloid. Interface Sci. 1968, 26, 62–69. [Google Scholar] [CrossRef]
- Vallet-Regi, M.; Rámila, A.; del Real, R.P.; Pérez-Pariente, J. A New Property of MCM-41: Drug Delivery System. Chem. Mater. 2000, 13, 308–311. [Google Scholar] [CrossRef]
- Vallet-Regí, M. Ordered Mesoporous Materials in the Context of Drug Delivery Systems and Bone Tissue Engineering. Chem. A Eur. J. 2006, 12, 5934–5943. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-Barba, I.; Colilla, M.; Manzano, M.; Vallet-Regí, M. In vitro stability of SBA-15 under physiological conditions. Microporous Mesoporous Mater. 2010, 132, 442–452. [Google Scholar] [CrossRef]
- Qin, X.; Wu, D. Effect of different solvents on poly(caprolactone) (PCL) electrospun nonwoven membranes. J. Therm. Anal. Calorim. 2012, 107, 1007–1013. [Google Scholar] [CrossRef]
- Soliman, S.; Pagliari, S.; Rinaldi, A.; Forte, G.; Fiaccavento, R.; Pagliari, F.; Franzese, O.; Minieri, M.; Di Nardo, P.; Licoccia, S.; et al. Multiscale three-dimensional scaffolds for soft tissue engineering via multimodal electrospinning. Acta Biomater. 2010, 6, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.L.; Gomes, S.; Henriques, C.; Borges, J.P.; Silva, J.C. Electrospinning polycaprolactone dissolved in glacial acetic acid: Fiber production, nonwoven characterization, and In Vitro evaluation. J. Appl. Polym. Sci. 2014, 131, 37–39. [Google Scholar] [CrossRef]
- Song, B.; Wu, C.; Chang, J. Controllable delivery of hydrophilic and hydrophobic drugs from electrospun poly(lactic-co-glycolic acid)/mesoporous silica nanoparticles composite mats. J. Biomed. Mater. Res. Part B Appl. Biomater. 2012, 100B, 2178–2186. [Google Scholar] [CrossRef] [PubMed]
- Mehrasa, M.; Asadollahi, M.A.; Ghaedi, K.; Salehi, H.; Arpanaei, A. Electrospun aligned PLGA and PLGA/gelatin nanofibers embedded with silica nanoparticles for tissue engineering. Int. J. Biol. Macromol. 2015, 79, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, L.; Dong, X.; Liang, J.; Shi, J. Preparation of mesoporous calcium doped silica spheres with narrow size dispersion and their drug loading and degradation behavior. Microporous Mesoporous Mater. 2007, 102, 151–158. [Google Scholar] [CrossRef]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef] [PubMed]
- Cerruti, M.; Greenspan, D.; Powers, K. Effect of pH and ionic strength on the reactivity of Bioglass 45S5. Biomaterials 2005, 26, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Maçon, A.L.B.; Kim, T.B.; Valliant, E.M.; Goetschius, K.; Brow, R.K.; Day, D.E.; Hoppe, A.; Boccaccini, A.R.; Kim, I.Y.; Ohtsuki, C.; et al. A unified in vitro evaluation for apatite-forming ability of bioactive glasses and their variants. J. Mater. Sci. Mater. Med. 2015, 26, 115. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Hotaling, N.A.; Bharti, K.; Kriel, H.; Simon, C.G. DiameterJ: A validated open source nanofiber diameter measurement tool. Biomaterials 2015, 61, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, H.; Serra, J.; González, P.; León, B. Structural study of sol–gel silicate glasses by IR and Raman spectroscopies. J. Non. Cryst. Solids 2009, 355, 475–480. [Google Scholar] [CrossRef]
- Zheng, K.; Solodovnyk, A.; Li, W.; Goudouri, O.-M.; Stähli, C.; Nazhat, S.N.; Boccaccini, A.R. Aging Time and Temperature Effects on the Structure and Bioactivity of Gel-Derived 45S5 Glass-Ceramics. J. Am. Ceram. Soc. 2015, 98, 30–38. [Google Scholar] [CrossRef]
- Izquierdo-Barba, I.; Arcos, D.; Sakamoto, Y.; Terasaki, O.; López-Noriega, A.; Vallet-Regí, M. High-Performance Mesoporous Bioceramics Mimicking Bone Mineralization. Chem. Mater. 2008, 20, 3191–3198. [Google Scholar] [CrossRef]
- Liverani, L.; Roether, J.A.; Boccaccini, A.R. Nanofiber composites in bone tissue engineering. In Nanofiber Composite Materials for Biomedical Applications; Ramalingam, M., Ramakrishna, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Zhou, P.; Cheng, X.; Xia, Y.; Wang, P.; Zou, K.; Xu, S.; Du, J. Organic/Inorganic Composite Membranes Based on Poly(l-lactic-co-glycolic acid) and Mesoporous Silica for Effective Bone Tissue Engineering. ACS Appl. Mater. Interfaces 2014, 6, 20895–20903. [Google Scholar] [CrossRef] [PubMed]
- Paşcu, E.I.; Stokes, J.; McGuinness, G.B. Electrospun composites of PHBV, silk fibroin and nano-hydroxyapatite for bone tissue engineering. Mater. Sci. Eng. C 2013, 33, 4905–4916. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Lv, F.; Zhang, Y.; Yi, Z.; Ke, Q.; Wu, C.; Liu, M.; Chang, J. Hierarchically micro-patterned nanofibrous scaffolds with a nanosized bio-glass surface for accelerating wound healing. Nanoscale 2015, 7, 18446–18452. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.H.; Lee, E.J.; Shin, D.S.; Kim, H.E.; Kim, H.W.; Koh, Y.H.; Jang, J.H. In vitro/in vivo biocompatibility and mechanical properties of bioactive glass nanofiber and poly(ε-caprolactone) composite materials. J. Biomed. Mater. Res. Part B Appl. Biomater. 2009, 91, 213–220. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Young’s Modulus (MPa) | UTS (MPa) | Tensile Strain (%) |
|---|---|---|---|
| Neat PCL1 | 11.0 ± 0.8 | 6.2 ± 0.9 | 115 ± 2 |
| PCL_Ca_MCM41 | 13 ± 4 | 3.3 ± 0.9 | 49 ± 10 |
| PCL_Ca_MCM-41 after SBF | 10 ± 2 | 2.4 ± 0.4 | 55 ± 3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liverani, L.; Boccardi, E.; Beltrán, A.M.; Boccaccini, A.R. Incorporation of Calcium Containing Mesoporous (MCM-41-Type) Particles in Electrospun PCL Fibers by Using Benign Solvents. Polymers 2017, 9, 487. https://doi.org/10.3390/polym9100487
Liverani L, Boccardi E, Beltrán AM, Boccaccini AR. Incorporation of Calcium Containing Mesoporous (MCM-41-Type) Particles in Electrospun PCL Fibers by Using Benign Solvents. Polymers. 2017; 9(10):487. https://doi.org/10.3390/polym9100487
Chicago/Turabian StyleLiverani, Liliana, Elena Boccardi, Ana Maria Beltrán, and Aldo R. Boccaccini. 2017. "Incorporation of Calcium Containing Mesoporous (MCM-41-Type) Particles in Electrospun PCL Fibers by Using Benign Solvents" Polymers 9, no. 10: 487. https://doi.org/10.3390/polym9100487
APA StyleLiverani, L., Boccardi, E., Beltrán, A. M., & Boccaccini, A. R. (2017). Incorporation of Calcium Containing Mesoporous (MCM-41-Type) Particles in Electrospun PCL Fibers by Using Benign Solvents. Polymers, 9(10), 487. https://doi.org/10.3390/polym9100487