
The Effect of Molecular Structure and Environment on the Miscibility and Diffusivity in Polythiophene-Methanofullerene Bulk Heterojunctions: Theory and Modeling with the RISM Approach



Abstract
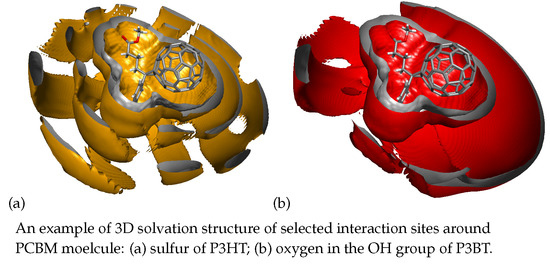
:
1. Introduction
2. A Brief Outline of RISM
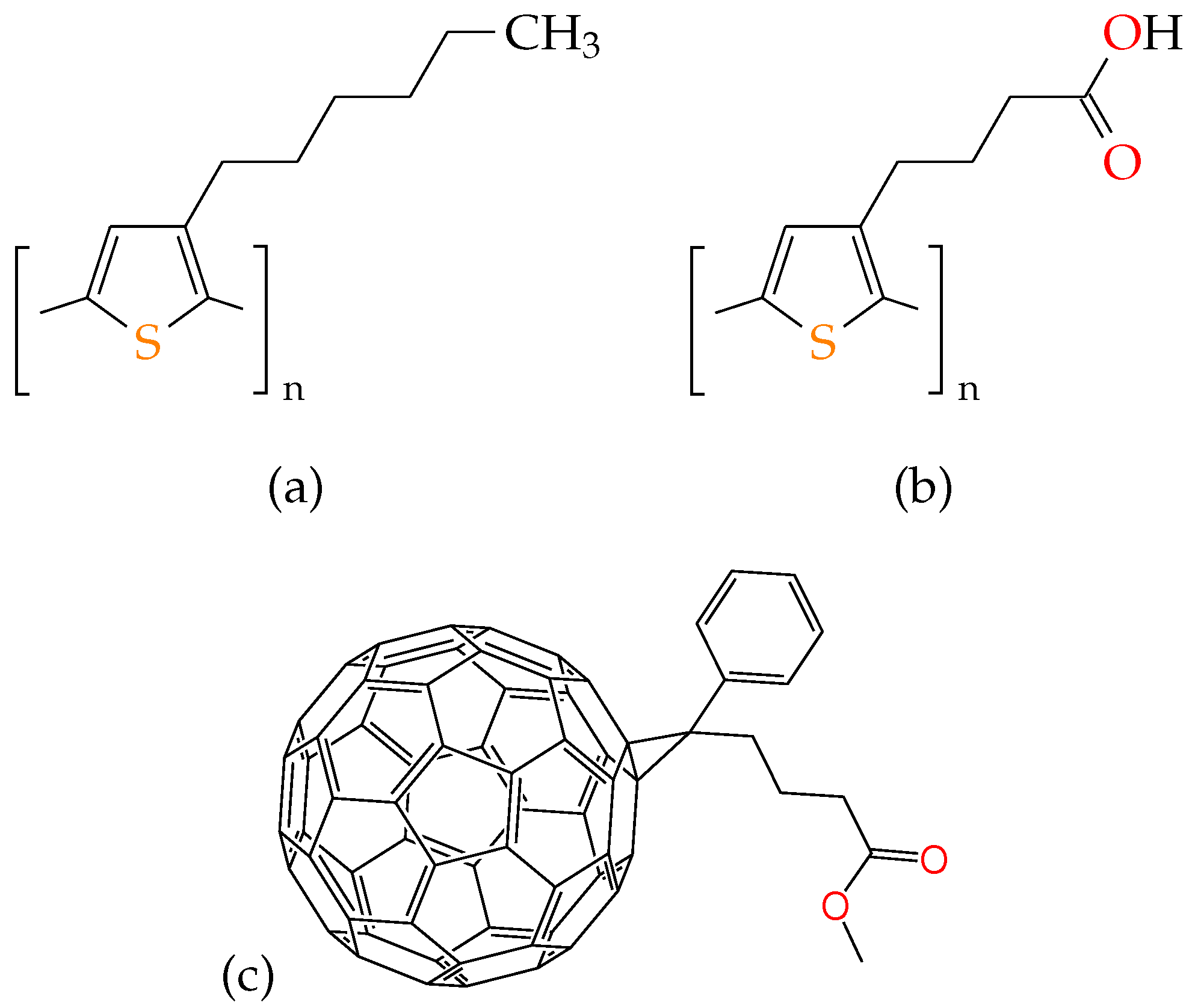
3. Modelling and Simulation Details
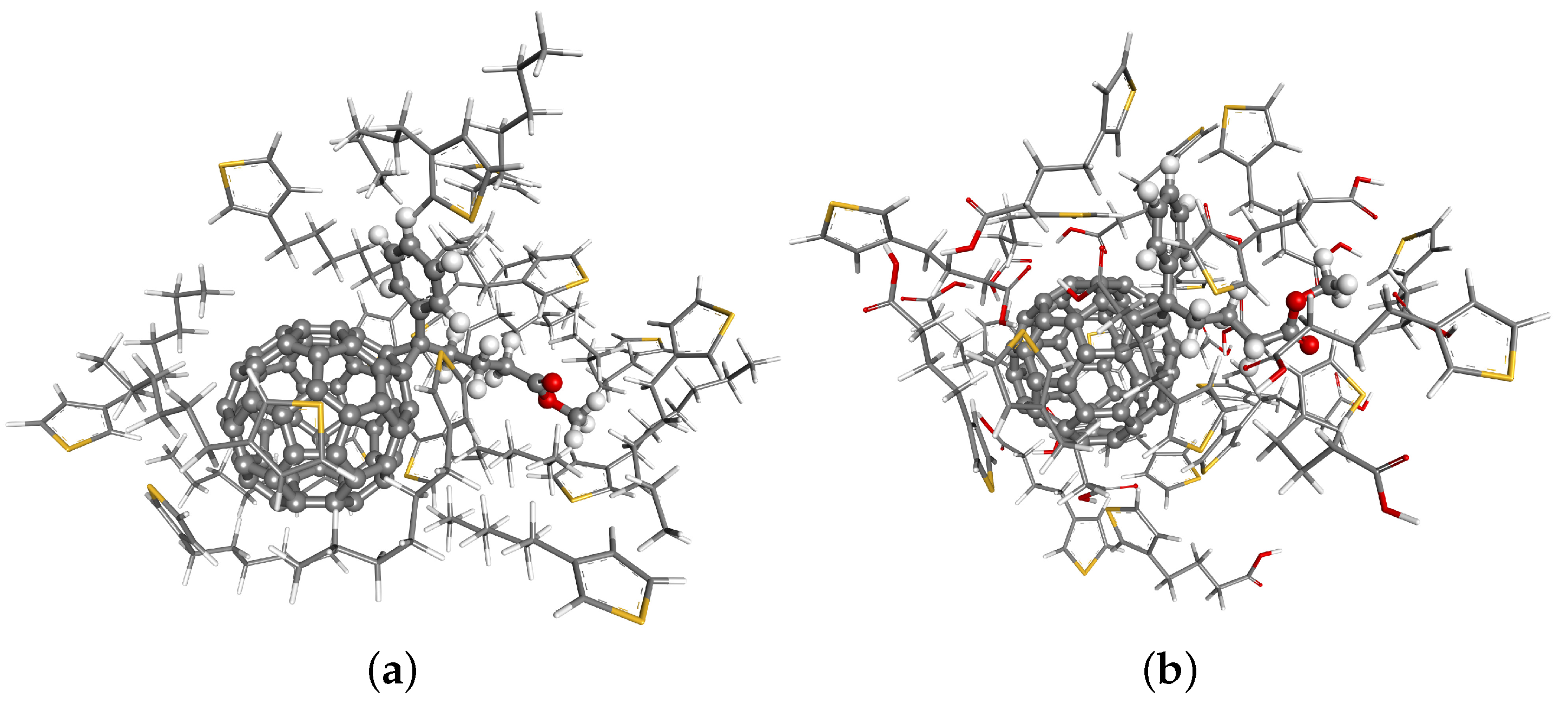
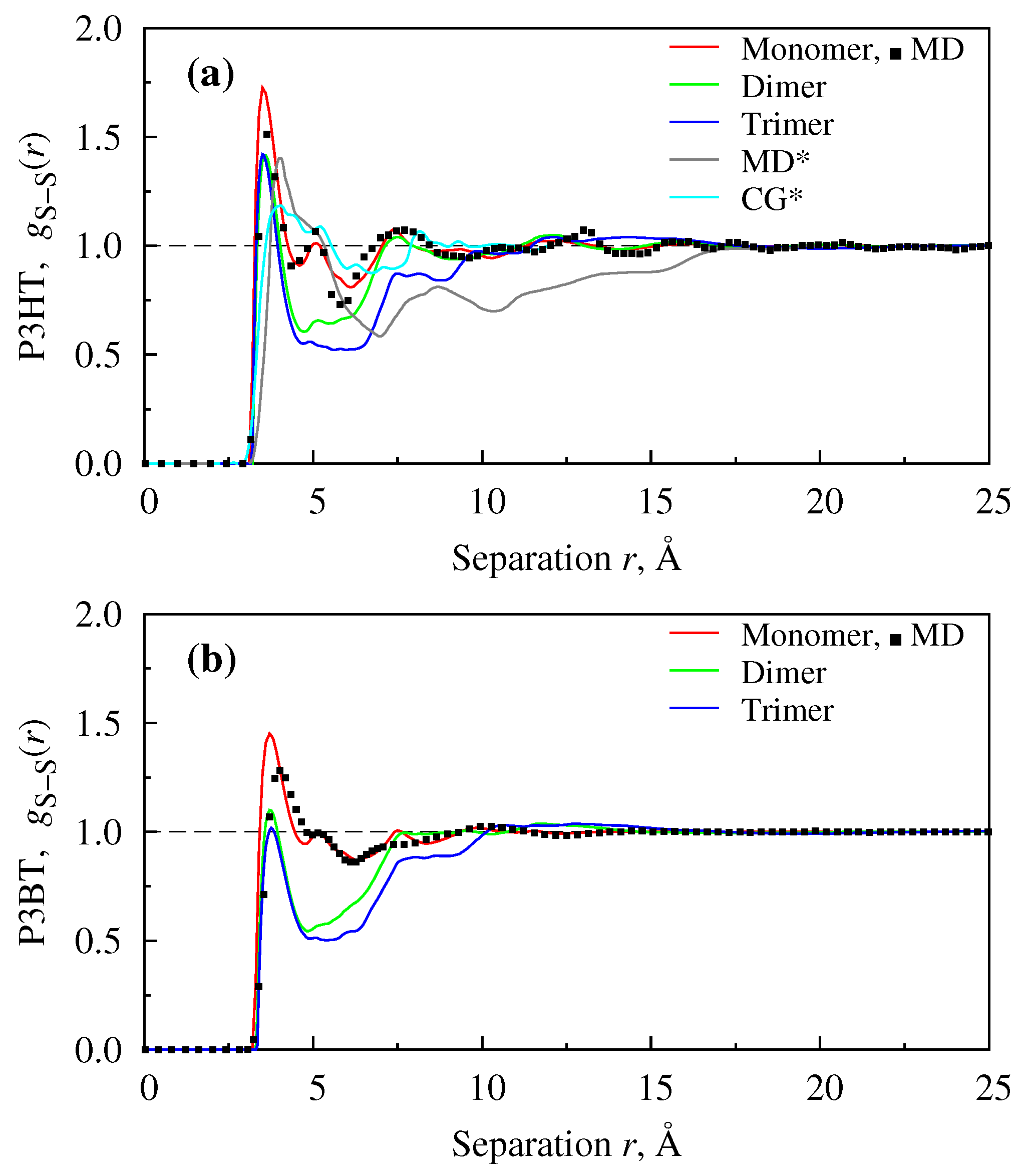
4. Results and Discussions
5. Summary
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Su, Y.W.; Lan, S.C.; Wei, K.H. Organic photovoltaics. Mater. Today 2012, 15, 554–562. [Google Scholar] [CrossRef]
- Mazzio, K.A.; Luscombe, C.K. The future of organic photovoltaics. Chem. Soc. Rev. 2015, 44, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Mazzio, K.A.; Luscombe, C.K. Correction: The future of organic photovoltaics. Chem. Soc. Rev. 2015, 44, 5744–5744. [Google Scholar] [CrossRef] [PubMed]
- Youn, H.; Park, H.J.; Guo, L.J. Organic photovoltaic cells: From performance improvement to manufacturing processes. Small 2015, 11, 2228–2246. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zheng, T.; Wu, Q.; Schneider, A.M.; Zhao, D.; Yu, L. Recent advances in bulk heterojunction polymer solar cells. Chem. Rev. 2015, 115, 12666–12731. [Google Scholar] [CrossRef] [PubMed]
- Müller-Buschbaum, P. The active layer morphology of organic solar cells probed with grazing incidence scattering techniques. Adv. Mater. 2014, 26, 7692–7709. [Google Scholar] [CrossRef] [PubMed]
- Ruderer, M.A.; Müller-Buschbaum, P. Morphology of polymer-based bulk heterojunction films for organic photovoltaics. Soft Matter 2011, 7, 5482–5493. [Google Scholar] [CrossRef]
- Brabec, C.J.; Heeney, M.; McCulloch, I.; Nelson, J. Influence of blend microstructure on bulk heterojunction organic photovoltaic performance. Chem. Soc. Rev. 2011, 40, 1185–1199. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Nakahara, A.; Wei, D.; Nordlund, D.; Russell, T.P. P3HT/PCBM bulk heterojunction organic photovoltaics: Correlating efficiency and morphology. Nano Lett. 2011, 11, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.R.; Jeng, U.S.; Su, C.J.; Wei, K.H.; Su, M.S.; Chiu, M.Y.; Chen, C.Y.; Su, W.B.; Su, C.H.; Su, A.C. Competition between fullerene aggregation and poly(3-hexylthiophene) crystallization upon annealing of bulk heterojunction solar cells. ACS Nano 2011, 5, 6233–6243. [Google Scholar] [CrossRef] [PubMed]
- Giridharagopal, R.; Ginger, D.S. Characterizing morphology in bulk heterojunction organic photovoltaic systems. J. Phys. Chem. Lett. 2010, 1, 1160–1169. [Google Scholar] [CrossRef]
- Liang, Y.; Xu, Z.; Xia, J.; Tsai, S.T.; Wu, Y.; Li, G.; Ray, C.; Yu, L. For the bright future–bulk heterojunction polymer solar cells with power conversion efficiency of 7.4%. Adv. Mater. 2010, 22, E135–E138. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.Y.; Jeng, U.S.; Su, M.S.; Wei, K.H. Morphologies of self-organizing regioregular conjugated polymer/fullerene aggregates in thin film solar cells. Macromolecules 2010, 43, 428–432. [Google Scholar] [CrossRef]
- Moulé, A.J.; Meerholz, K. Morphology control in solution-processed bulk-heterojunction solar cell mixtures. Adv. Funct. Mater. 2009, 19, 3028–3036. [Google Scholar] [CrossRef]
- Chiu, M.Y.; Jeng, U.S.; Su, C.H.; Liang, K.S.; Wei, K.H. Simultaneous use of small- and wide-angle X-ray techniques to analyse nanometerscale phase separation in polymer heterojunction solar cells. Adv. Mater. 2008, 20, 2573–2578. [Google Scholar] [CrossRef]
- Khajeh, A.R.A.; Shankar, K.; Choi, P. Prediction of the active layer nanomorphology in polymer solar cells using molecular dynamics simulation. ACS Appl. Mater. Interfaces 2013, 5, 4617–4624. [Google Scholar] [CrossRef] [PubMed]
- Darling, S.B. Block copolymers for photovoltaics. Energy Environ. Sci. 2009, 2, 1266–1273. [Google Scholar] [CrossRef]
- Yang, C.; Lee, J.K.; Heeger, A.J.; Wudl, F. Well-defined donor-acceptor rod-coil diblock copolymers based on P3HT containing C60: The morphology and role as a surfactant in bulk-heterojunction solar cells. J. Mater. Chem. 2009, 19, 5416–5423. [Google Scholar] [CrossRef] [Green Version]
- Shankar, K.; Mor, G.K.; Prakasam, H.E.; Varghese, O.K.; Grimes, C.A. Self-assembled hybrid polymer-TiO2 nanotube array heterojunction solar cells. Langmuir 2007, 23, 12445–12449. [Google Scholar] [CrossRef] [PubMed]
- Worfolk, B.J.; Rider, D.A.; Elias, A.L.; Thomas, M.; Harris, K.D.; Buriak, J.M. Bulk heterojunction organic photovoltaics based on carboxylated polythiophenes and PCBM on glass and plastic substrates. Adv. Funct. Mater. 2011, 21, 1816–1826. [Google Scholar] [CrossRef]
- Li, W.; Worfolk, B.J.; Li, P.; Hauger, T.C.; Harris, K.D.; Buriak, J.M. Self-assembly of carboxylated polythiophene nanowires for improved bulk heterojunction morphology in polymer solar cells. J. Mater. Chem. 2012, 22, 11354–11363. [Google Scholar] [CrossRef]
- Guo, Z.; Lee, D.; Liu, Y.; Sun, F.; Sliwinski, A.; Gao, H.; Burns, P.C.; Huang, L.; Luo, T. Tuning the thermal conductivity of solar cell polymers through side chain engineering. Phys. Chem. Chem. Phys. 2014, 16, 7764–7771. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; He, G. Molecular dynamics simulation on structural conformation of conjugated polymer functionalised films for optimal fluorescent performance. Molecular Simulation 2015, 41, 1060–1068. [Google Scholar] [CrossRef]
- Lee, C.K.; Pao, C.W.; Chu, C.W. Multiscale molecular simulations of the nanoscale morphologies of P3HT:PCBM blends for bulk heterojunction organic photovoltaic cells. Energy Environ. Sci. 2011, 4, 4124–4132. [Google Scholar] [CrossRef]
- Hirata, F. Chemical processes in solution studied by an integral equation theory of molecular liquids. Bull. Chem. Soc. Jpn. 1998, 71, 1483–1499. [Google Scholar] [CrossRef]
- Hirata, F. Theory of Molecular Liquids. In Molecular Theory of Solvation; Hirata, F., Ed.; Springer Netherlands: Dordrecht, The Netherlands, 2003; Volume 24, pp. 1–60. [Google Scholar]
- Egelstaff, P.A. An Introduction to the Liquid State, 2nd ed.; Oxford Series on Neutron Scattering in Condensed Matter; Clarendon Press: Oxford, UK, 1992; Volume 7. [Google Scholar]
- Gray, C.G.; Gubbins, K.E. Theory of Molecular Fluids. Volume 1: Fundamentals; The International Series of Monographs on Chemistry; Clarendon Press: Oxford, UK, 1984; Volume 9. [Google Scholar]
- Perkyns, J.S.; Pettitt, B.M. A dielectrically consistent interaction site theory for solvent-electrolyte mixtures. Chem. Phys. Lett. 1992, 190, 626–630. [Google Scholar] [CrossRef]
- Perkyns, J.S.; Pettitt, B.M. A site-site theory for finite concentration saline solutions. J. Chem. Phys. 1992, 97, 7656–7666. [Google Scholar] [CrossRef]
- Schweizer, K.S.; Curro, J.G. Integral-equation theory of the structure of polymer melts. Phys. Rev. Lett. 1987, 58, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, K.S.; Curro, J.G. Integral equation theory of the structure and thermodynamics of polymer blends. J. Chem. Phys. 1989, 91, 5059–5081. [Google Scholar] [CrossRef]
- Schweizer, K.S.; Curro, J.G. PRISM Theory of the structure, thermodynamics, and phase transitions of polymer liquids and alloys. In Atomistic Modeling of Physical Properties; Monnerie, L., Suter, U.W., Eds.; Advances in Polymer Science; Springer: Berlin, Germany, 1994; Volume 116, pp. 319–377. [Google Scholar]
- Schweizer, K.S.; Curro, J.G. Integral equation theories of the structure, thermodynamics, and phase transitions of polymer fluids. In Advances in Chemical Physics; Prigogine, I., Rice, S.A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1997; Volume 98, pp. 1–142. [Google Scholar]
- Kovalenko, A. Three-dimensional RISM theory for molecular liquids and solid-liquid interfaces. In Molecular Theory of Solvation; Hirata, F., Ed.; Springer Netherlands: Dordrecht, The Netherlands; Volume 24, pp. 169–275.
- Boese, A.D.; Handy, N.C. A new parametrization of exchange-correlation generalized gradient approximation functionals. J. Chem. Phys. 2001, 114, 5497–5503. [Google Scholar] [CrossRef]
- Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Delley, B. Hardness conserving semilocal pseudopotentials. Phys. Rev. B 2002, 66, 155125. [Google Scholar] [CrossRef]
- Klamt, A.; Schuurmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2 1993, 799–805. [Google Scholar] [CrossRef]
- Singh, U.C.; Kollman, P.A. An approach to computing electrostatic charges for molecules. J. Comput. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Materials Studio Modeling Environment, Release 5.5. Accelrys Software Inc.: San Diego, CA, USA, 2010; Available online: http://www.accelrys.com/ (accessed on 4 April 2016).
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Ortmann, F.; Bechstedt, F.; Schmidt, W.G. Semiempirical van der Waals correction to the density functional description of solids and molecular structures. Phys. Rev. B 2006, 73, 205101. [Google Scholar] [CrossRef]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Casewit, C.J.; Colwell, K.S.; Rappe, A.K. Application of a universal force field to organic molecules. J. Am. Chem. Soc. 1992, 114, 10035–10046. [Google Scholar] [CrossRef]
- Casewit, C.J.; Colwell, K.S.; Rappe, A.K. Application of a universal force field to main group compounds. J. Am. Chem. Soc. 1992, 114, 10046–10053. [Google Scholar] [CrossRef]
- Schwarz, K.N.; Kee, T.W.; Huang, D.M. Coarse-grained simulations of the solution-phase self-assembly of poly(3-hexylthiophene) nanostructures. Nanoscale 2013, 5, 2017–2027. [Google Scholar] [CrossRef] [PubMed]
- Thinh, T.T. Multi-Scale Modelling of Organic Photovoltaics System P3HT:PCBM. In Ph.D. Thesis; National University of Singapore: Singapore, 2014; Available online: http://scholarbank.nus.sg/handle/10635/79486 (accessed on 4 April 2016).
- Kobryn, A.E.; Yamaguchi, T.; Hirata, F. Site-site memory equation approach in study of density/pressure dependence of translational diffusion coefficient and rotational relaxation time of polar molecular solutions: Acetonitrile in water, methanol in water, and methanol in acetonitrile. J. Chem. Phys. 2005, 122, 184511. [Google Scholar] [CrossRef] [PubMed]
- Kobryn, A.E.; Kovalenko, A. Molecular theory of hydrodynamic boundary conditions in nanofluidics. J. Chem. Phys. 2008, 129, 134701. [Google Scholar] [CrossRef] [PubMed]
- Kobryn, A.E.; Ichiki, K.; Kovalenko, A. Thermodynamic dependences of slip length for nanofluidic flows over crystalline surfaces: predictions of molecular theory of solvation. Int. J. Quant. Chem. 2009, 109, 1666–1671. [Google Scholar] [CrossRef]
- Kobryn, A.E.; Kovalenko, A. Slip boundary conditions in nanofluidics from the molecular theory of solvation. Mol. Simulat. 2011, 37, 733–737. [Google Scholar] [CrossRef]
- Lyubimova, O.; Liu, X.; Gusarov, S.; Kobryn, A.E.; Kovalenko, A. Solvation structure and gelation ability of polyelectrolytes: predictions by quantum chemistry methods and integral equation theory of molecular liquids. Procedia Comput. Sci. 2011, 4, 1186–1192. [Google Scholar] [CrossRef]
- Liu, X.; Lyubimova, O.; Kobryn, A.E.; Gusarov, S.; Kovalenko, A. Mesoscopic study of dynamics and gelation ability of oligomeric electrolyte gelator with dissipative particle dynamics. Procedia Comput. Sci. 2011, 4, 1031–1038. [Google Scholar] [CrossRef]
- Kovalenko, A.; Kobryn, A.E.; Gusarov, S.; Lyubimova, O.; Liu, X.; Blinov, N.; Yoshida, M. Molecular theory of solvation for supramolecules and soft matter structures: Application to ligand binding, ion channels, and oligomeric polyelectrolyte gelators. Soft Matter 2012, 8, 1508–1520. [Google Scholar] [CrossRef]
- Nikolić, D.; Moffat, K.A.; Farrugia, V.M.; Kobryn, A.E.; Gusarov, S.; Wosnick, J.H.; Kovalenko, A. Multi-scale modelling and synthesis of polyester ionomers. Phys. Chem. Chem. Phys. 2013, 15, 6128–6138. [Google Scholar] [CrossRef] [PubMed]
- Kobryn, A.E.; Nikolić, D.; Lyubimova, O.; Gusarov, S.; Kovalenko, A. Dissipative particle dynamics with an effective pair potential from integral equation theory of molecular liquids. J. Phys. Chem. B 2014, 118, 12034–12049. [Google Scholar] [CrossRef] [PubMed]
- Kovalenko, A.; Hirata, F. First-principles realization of a van der Waals-Maxwell theory for water. Chem. Phys. Lett. 2001, 349, 496–502. [Google Scholar] [CrossRef]
- Watts, B.; Belcher, W.J.; Thomsen, L.; Ade, H.; Dastoor, P.C. A quantitative study of PCBM diffusion during annealing of P3HT:PCBM blend films. Macromolecules 2009, 42, 8392–8397. [Google Scholar] [CrossRef]
- Shang, Y.; Kazmer, D. Modeling and Simulation. In Characterization of Polymer Blends: Miscibility, Morphology and Interfaces; Thomas, S., Grohens, Y., Jyotishkumar, P., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015; pp. 457–522. [Google Scholar]
- Kim, J.Y.; Frisbie, C.D. Correlation of phase behaviour and charge transport in conjugated polymer/fullerene blends. J. Phys. Chem. C 2008, 112, 17726–17736. [Google Scholar] [CrossRef]
- Collins, B.A.; Gann, E.; Guignard, L.; He, X.; McNeill, C.R.; Ade, H. Molecular miscibility of polymer-fullerene blends. J. Phys. Chem. Lett. 2010, 1, 3160–3166. [Google Scholar] [CrossRef]
- Kozub, D.R.; Vakhshouri, K.; Orme, L.M.; Wang, C.; Hexemer, A.; Gomez, E.D. Polymer crystallization of partially miscible polythiophene/fullerene mixtures controls morphology. Macromolecules 2011, 44, 5722–5726. [Google Scholar] [CrossRef]
- Ruderer, M.A.; Meier, R.; Porcar, L.; Cubitt, R.; Müller-Buschbaum, P. Phase separation and molecular intermixing in polymer-fullerene bulk heterojunction thin films. J. Phys. Chem. Lett. 2012, 3, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Kohn, P.; Rong, Z.; Scherer, K.H.; Sepe, A.; Sommer, M.; Müller-Buschbaum, P.; Friend, R.H.; Steiner, U.; Hüttner, S. Crystallization-induced 10-nm structure formation in P3HT/PCBM blends. Macromolecules 2013, 46, 4002–4013. [Google Scholar] [CrossRef]
- Huang, D.M. Computational study of P3HT/C60-fullerene miscibility. Aust. J. Chem. 2014, 67, 585–591. [Google Scholar] [CrossRef]
- Huang, W.Y.; Lee, C.C.; Wang, S.G.; Han, Y.K.; Chang, M.Y. Side chain effects of poly(3-alkylthiophene) on the morphology and performance of polymer solar cells. J. Electrochem. Soc. 2010, 157, B1336–B1342. [Google Scholar] [CrossRef]
- Kline, R.J.; McGehee, M.D.; Kadnikova, E.N.; Liu, J.; Fréchet, J.M.J.; Toney, M.F. Dependence of regioregular poly(3-hexylthiophene) film morphology and field-effect mobility on molecular weight. Macromolecules 2005, 38, 3312–3319. [Google Scholar] [CrossRef]
- He, M.; Ge, J.; Fang, M.; Qiu, F.; Yang, Y. Fabricating polythiophene into highly aligned microwire film by fast evaporation of its whisker solution. Polymer 2010, 51, 2236–2243. [Google Scholar] [CrossRef]
- Alexiadis, O.; Mavrantzas, V.G. All-atom molecular dynamics simulation of temperature effects on the structural, thermodynamic, and packing properties of the pure amorphous and pure crystalline phases of regioregular P3HT. Macromolecules 2013, 46, 2450–2467. [Google Scholar] [CrossRef]
- Williams, T.; Kelley, C. gnuplot 4.6.5 for Linux x86 64, 2014. Available online: http://www.gnuplot.info/ (accessed on 4 April 2016).
- Friedman, H.L. A Course in Statistical Mechanics; Prentice-Hall, Inc.: Englewood Cliffs, NJ, USA, 1985. [Google Scholar]
- Cussler, E.L. Diffusion: Mass Transfer in Fluid Systems, 3rd ed.; Cambridge Series in Chemical Engineering; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Treat, N.D.; Brady, M.A.; Smith, G.; Toney, M.F.; Kramer, E.J.; Hawker, C.J.; Chabinyc, M.L. Interdiffusion of PCBM and P3HT reveals miscibility in a photovoltaically active blend. Adv. Energy Mater. 2011, 1, 82–89. [Google Scholar] [CrossRef]
- Treat, N.D.; Mates, T.E.; Hawker, C.J.; Kramer, E.J.; Chabinyc, M.L. Temperature dependence of the diffusion coefficient of PCBM in poly(3-hexylthiophene). Macromolecules 2013, 46, 1002–1007. [Google Scholar] [CrossRef]
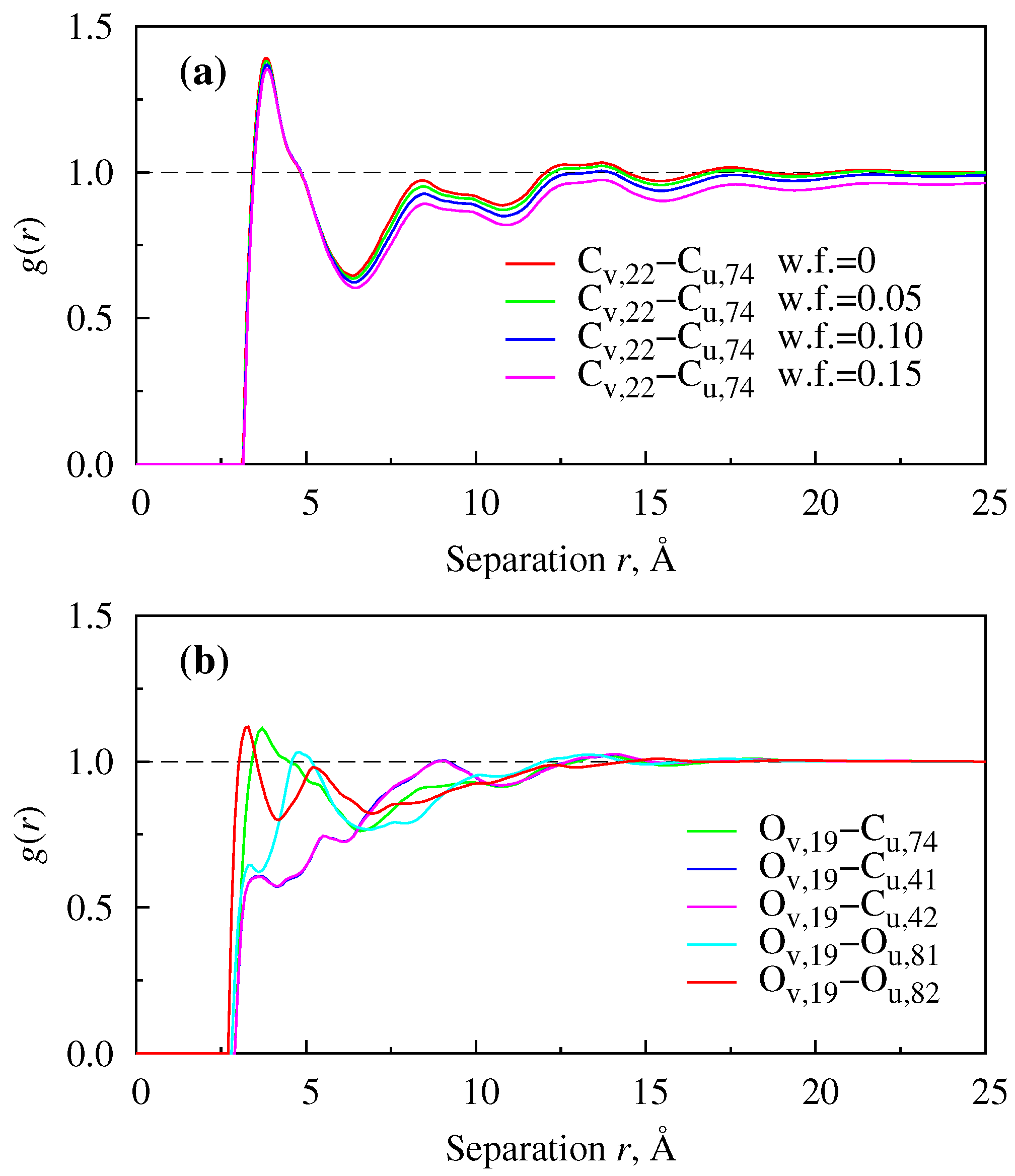


{kind=link}
{kind=link}
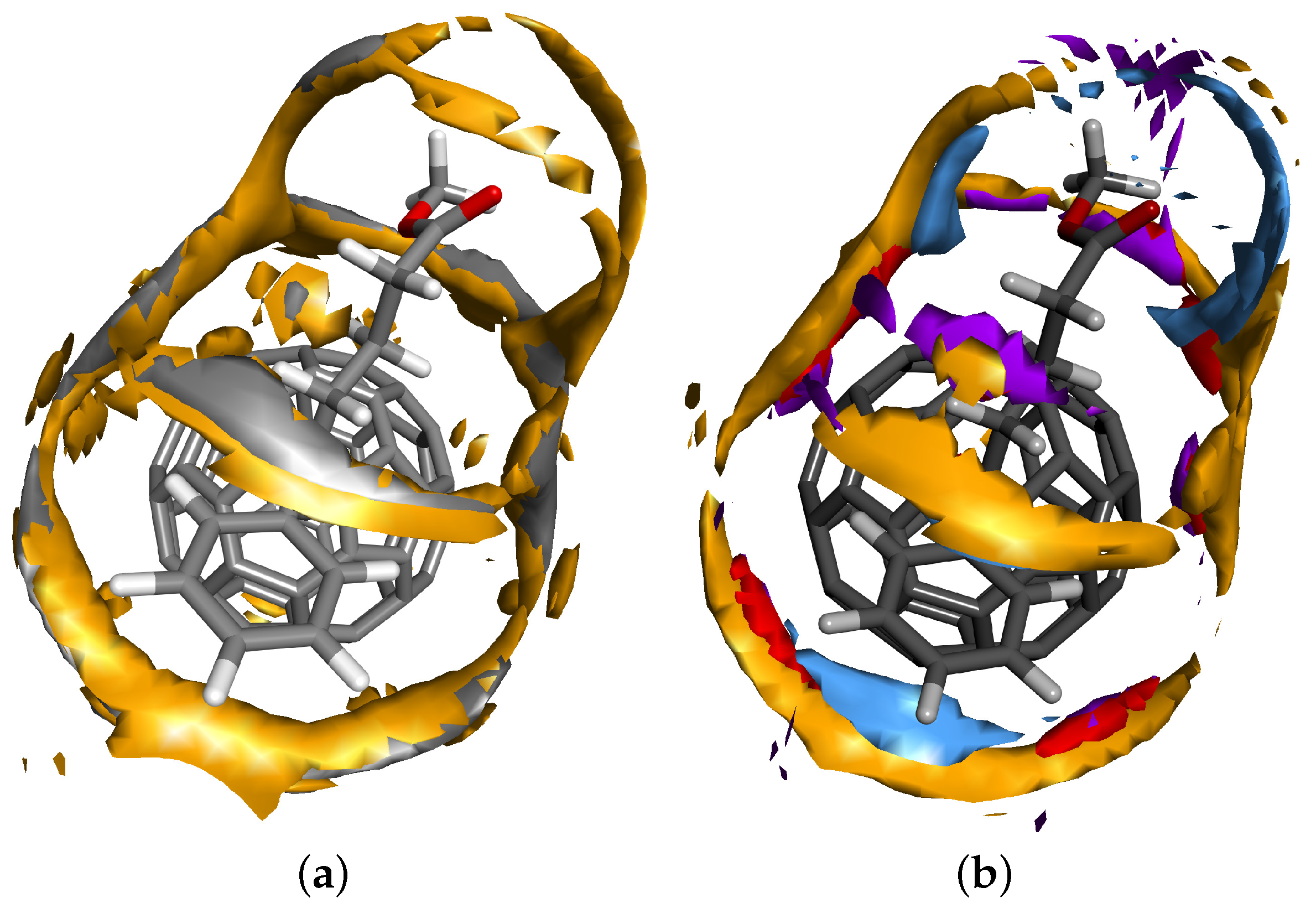
{kind=link}
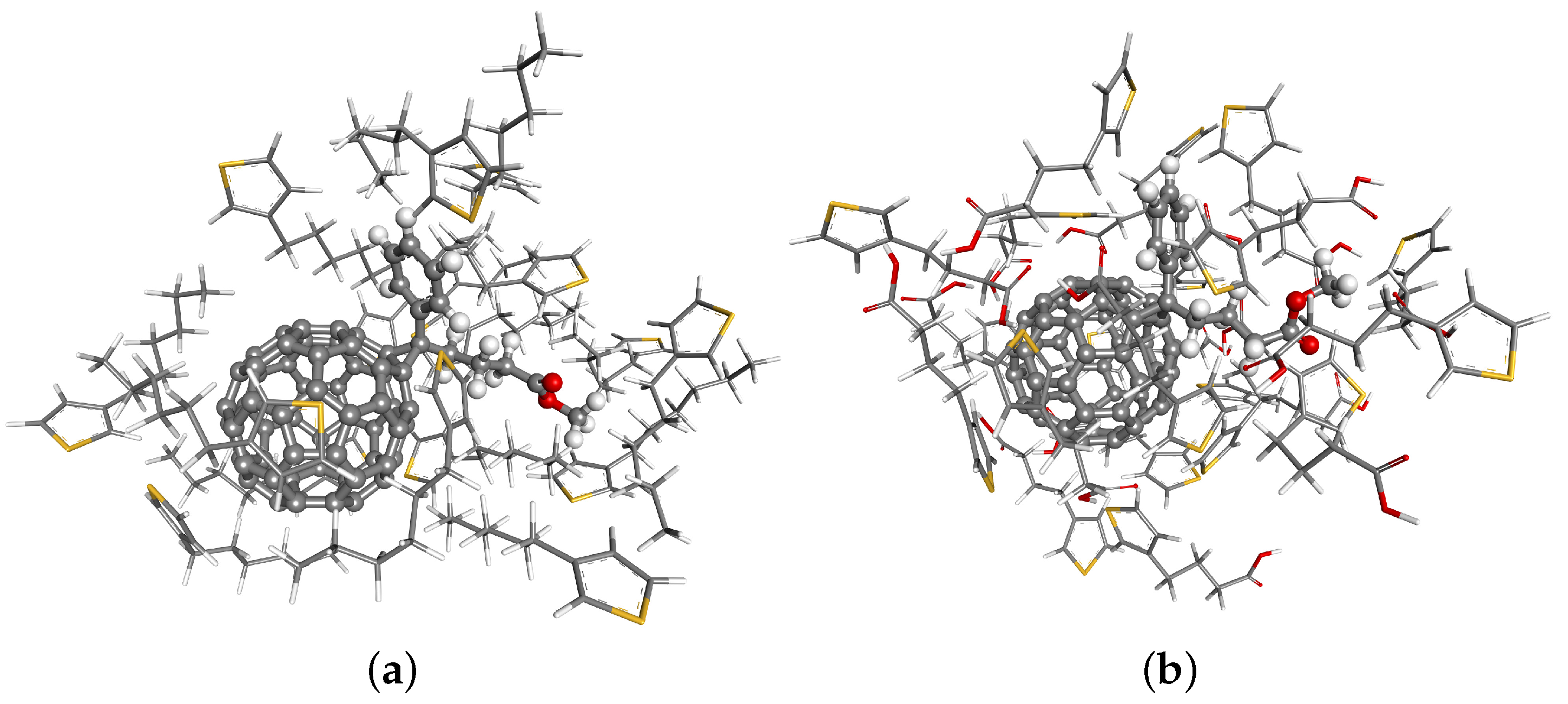
{kind=link}
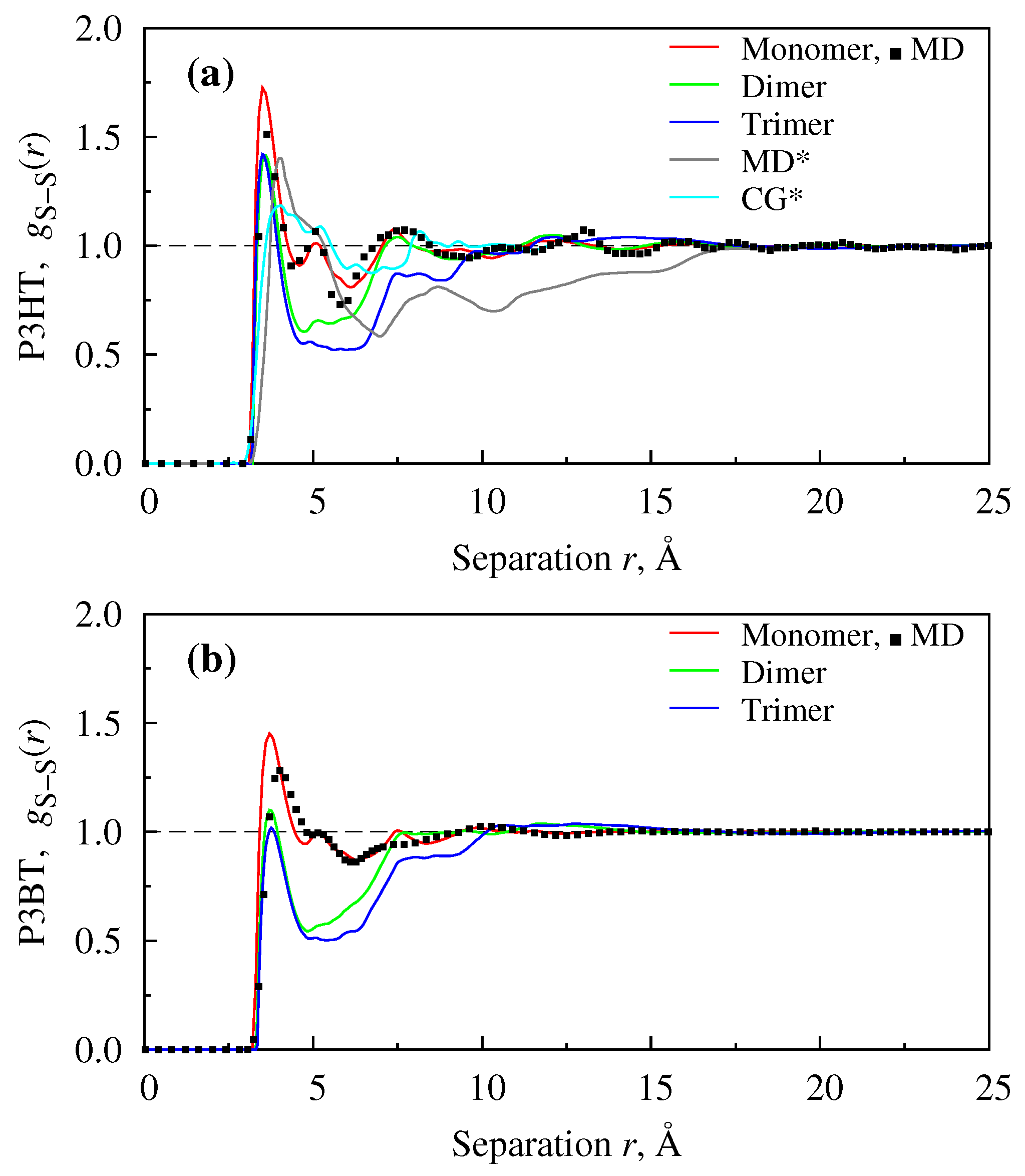
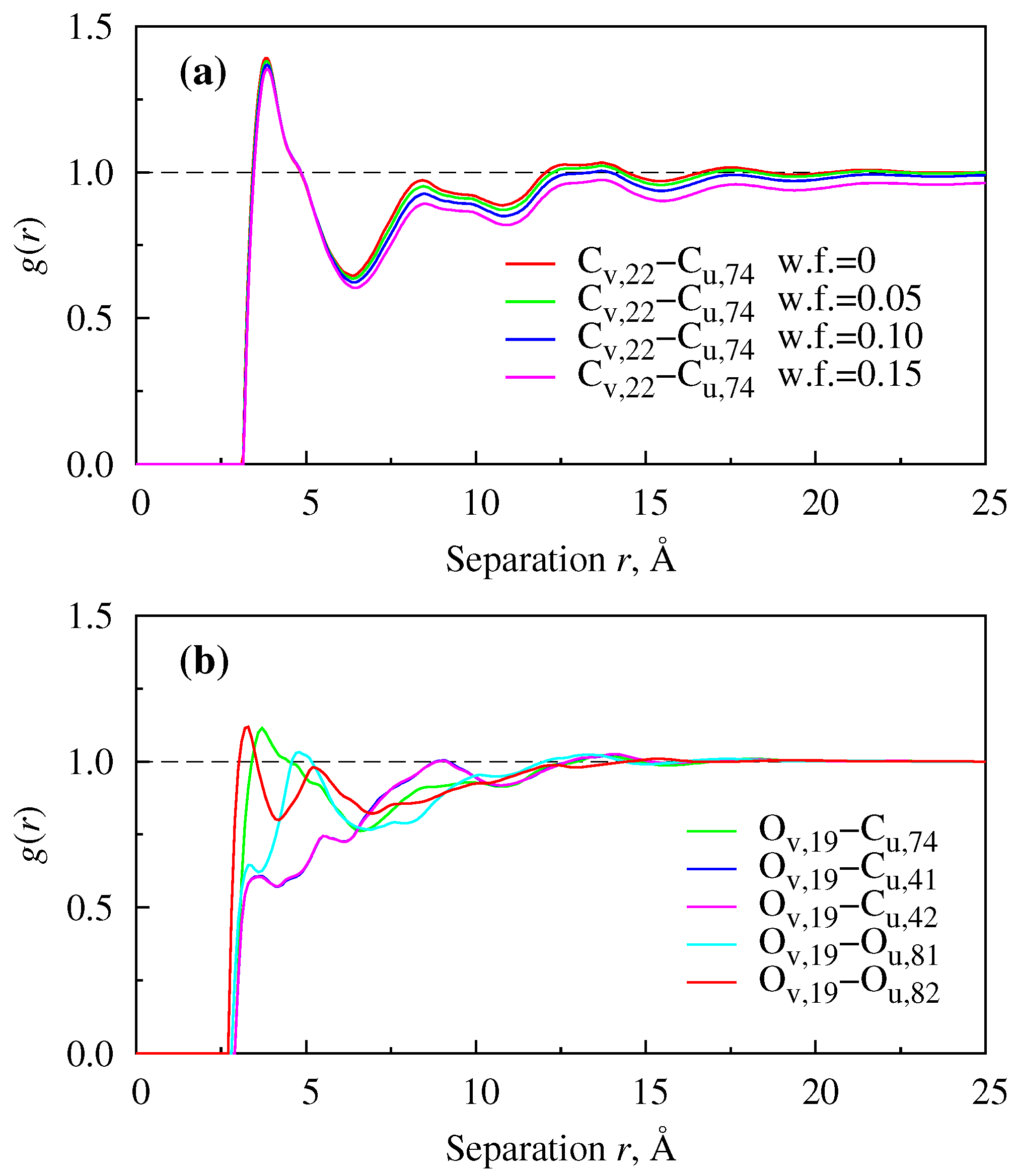{kind=link}
{kind=link}
{kind=link}
| System | T, K | D, cm/s | Ref. | |
|---|---|---|---|---|
| PCBM in P3HT | 423 | 40% w.f | [74] | |
| 413 | 19% v/v | [59] | ||
| 383 | 0.001% v/v | [75] | ||
| 383 | 0.01% v/v | [75] | ||
| 423 | 0.01% v/v | [75] | ||
| 383 | 0.05% v/v | [75] | ||
| 400 | 0 a | MD | ||
| 400 | 0 b | MD | ||
| PCBM in P3BT | 400 | 0 a | MD | |
| 400 | 0 b | MD |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobryn, A.E.; Gusarov, S.; Shankar, K. The Effect of Molecular Structure and Environment on the Miscibility and Diffusivity in Polythiophene-Methanofullerene Bulk Heterojunctions: Theory and Modeling with the RISM Approach. Polymers 2016, 8, 136. https://doi.org/10.3390/polym8040136
Kobryn AE, Gusarov S, Shankar K. The Effect of Molecular Structure and Environment on the Miscibility and Diffusivity in Polythiophene-Methanofullerene Bulk Heterojunctions: Theory and Modeling with the RISM Approach. Polymers. 2016; 8(4):136. https://doi.org/10.3390/polym8040136
Chicago/Turabian StyleKobryn, Alexander E., Sergey Gusarov, and Karthik Shankar. 2016. "The Effect of Molecular Structure and Environment on the Miscibility and Diffusivity in Polythiophene-Methanofullerene Bulk Heterojunctions: Theory and Modeling with the RISM Approach" Polymers 8, no. 4: 136. https://doi.org/10.3390/polym8040136