
Enzymatic Synthesis and Characterization of Hydrophilic Sugar Based Polyesters and Their Modification with Stearic Acid

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Syntheses
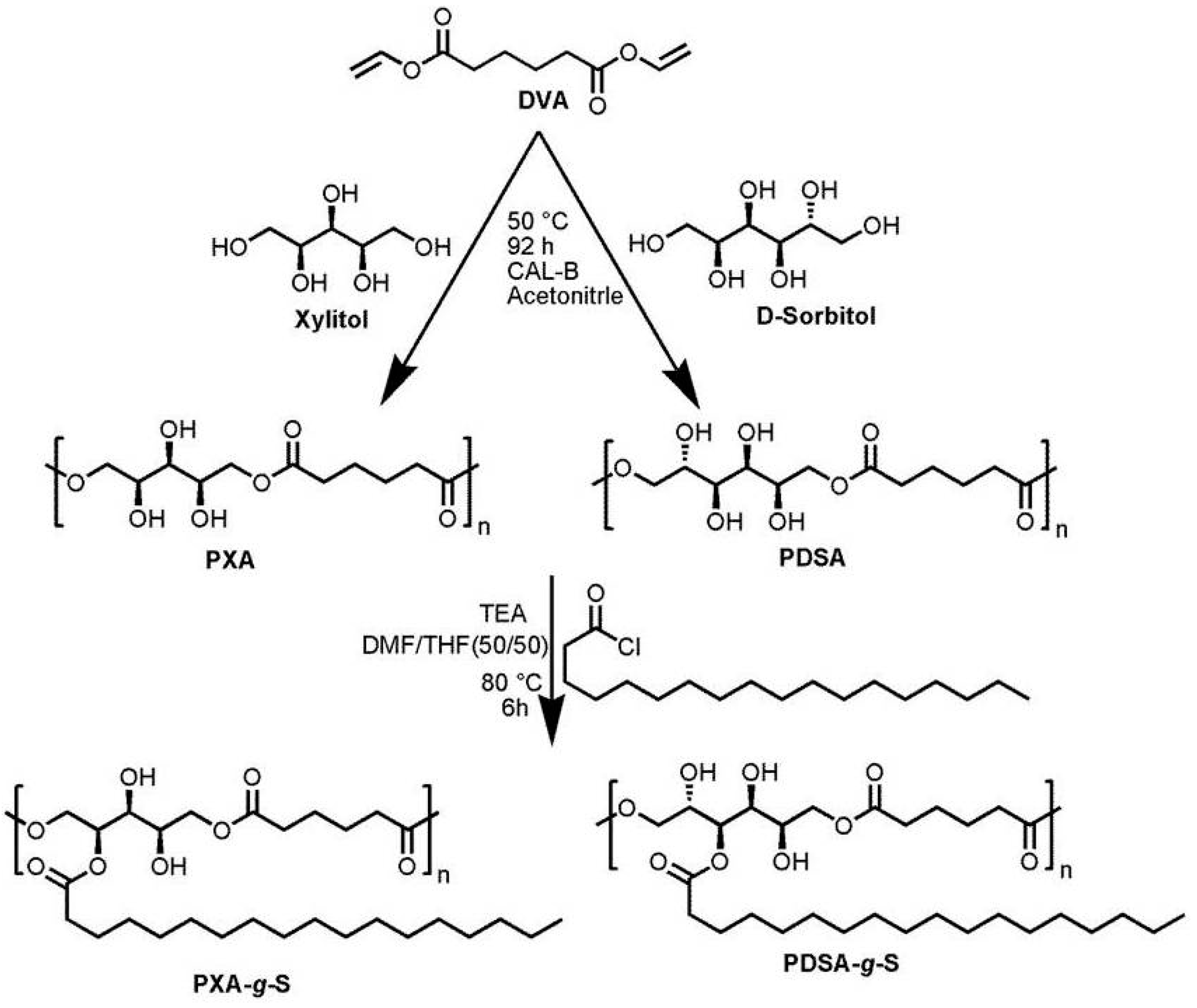
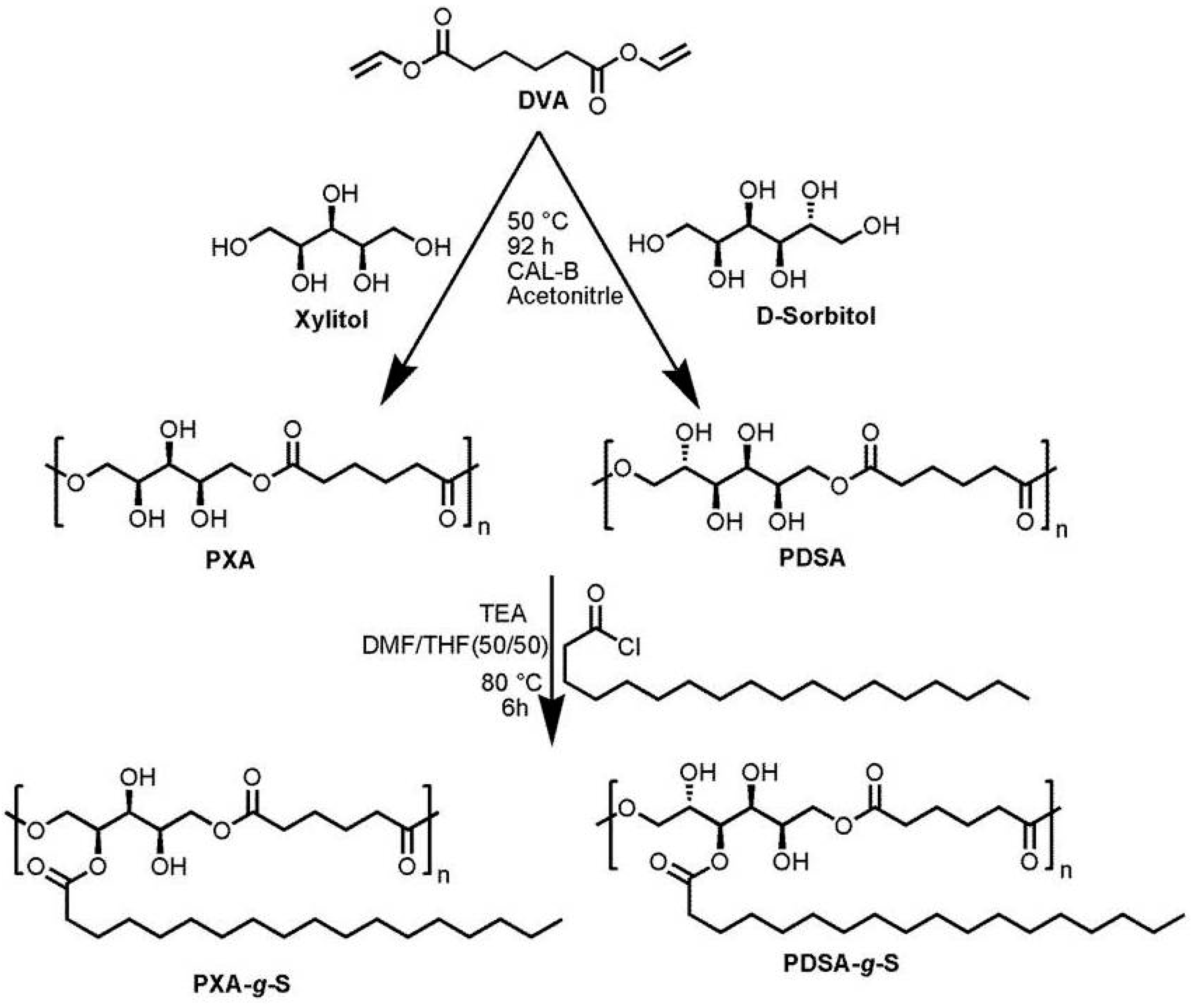
2.2.1. Synthesis of Poly(xylitol adipate)
2.2.2. Synthesis of Poly(d-sorbitol adipate)
2.2.3. Synthesis of Stearoyl Grafted Poly(Xylitol Adipate) and Poly(d-sorbitol adipate)
2.3. Preparation of Nanoparticles
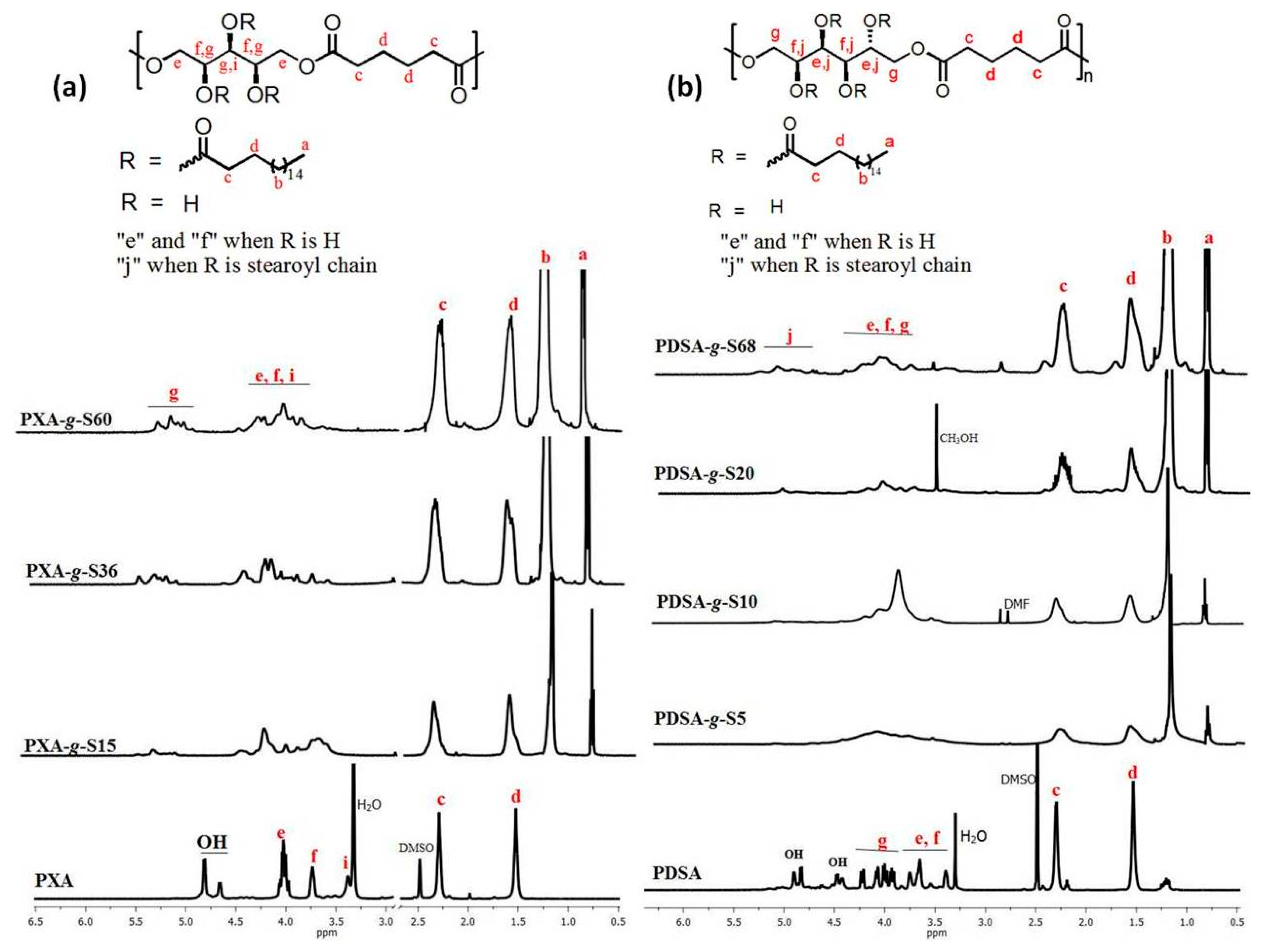
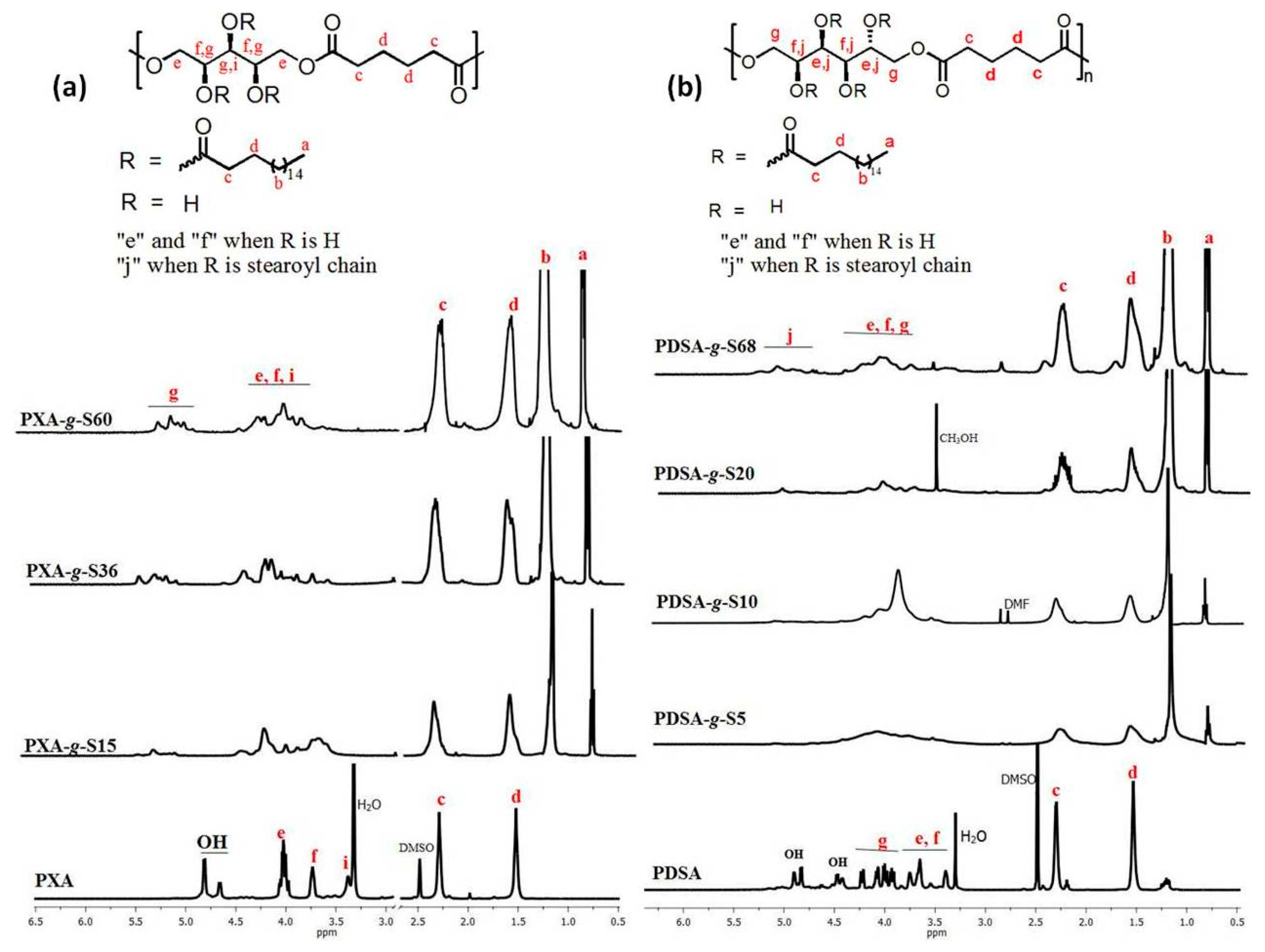
2.4. NMR Spectroscopy
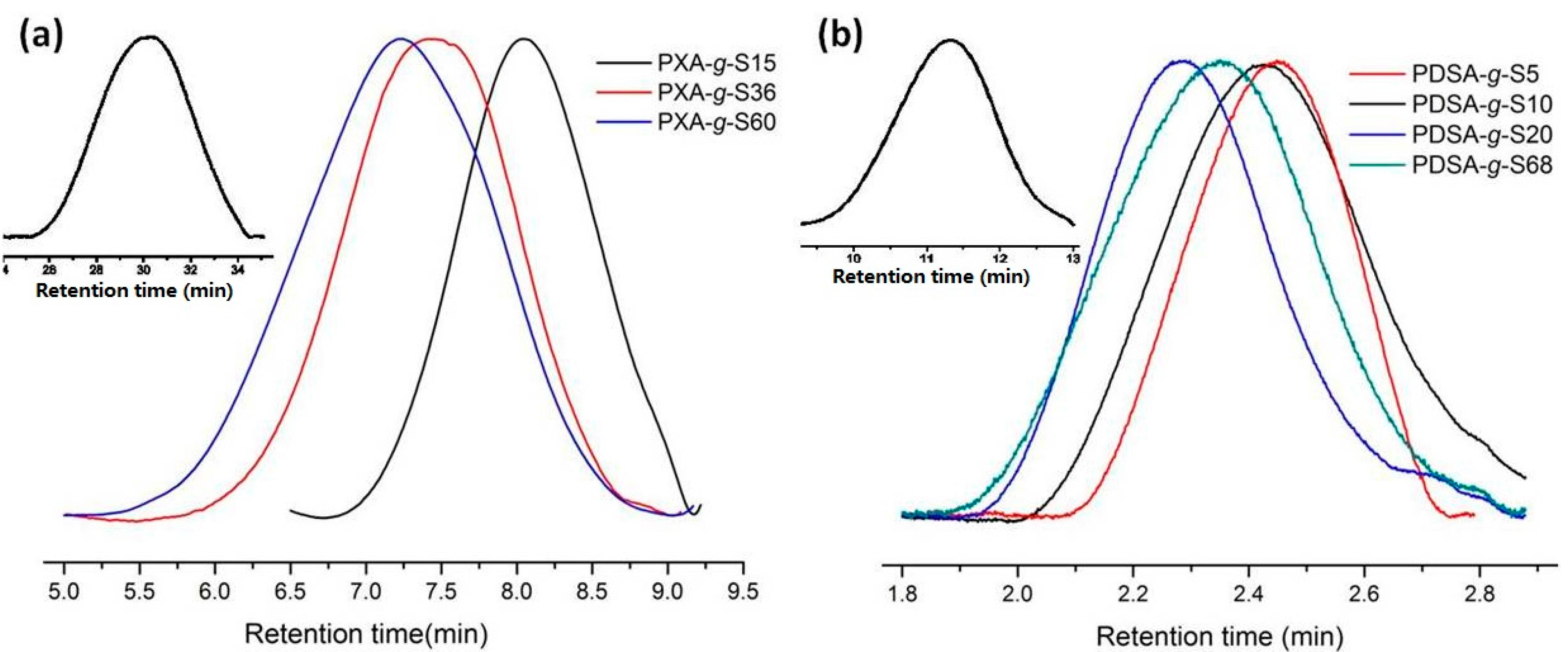
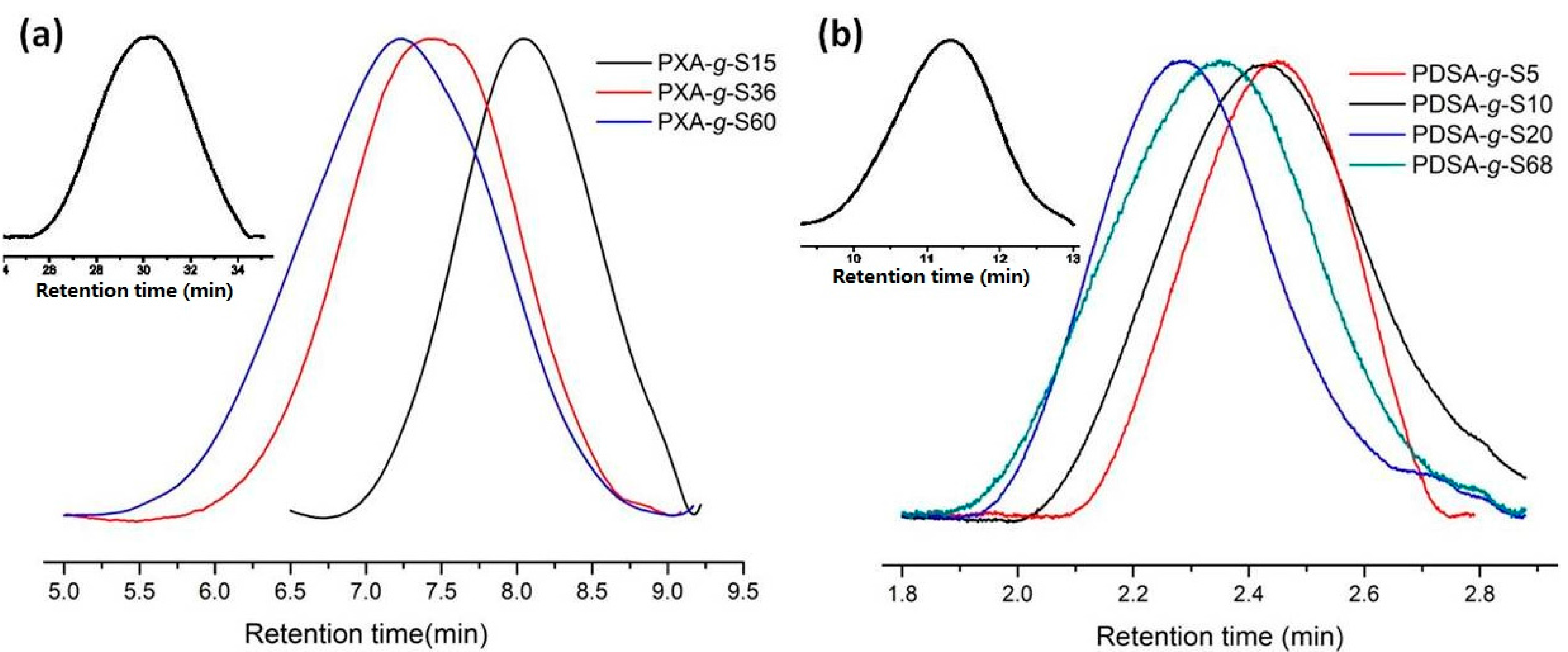
2.5. Size Exclusion Chromatography (SEC)
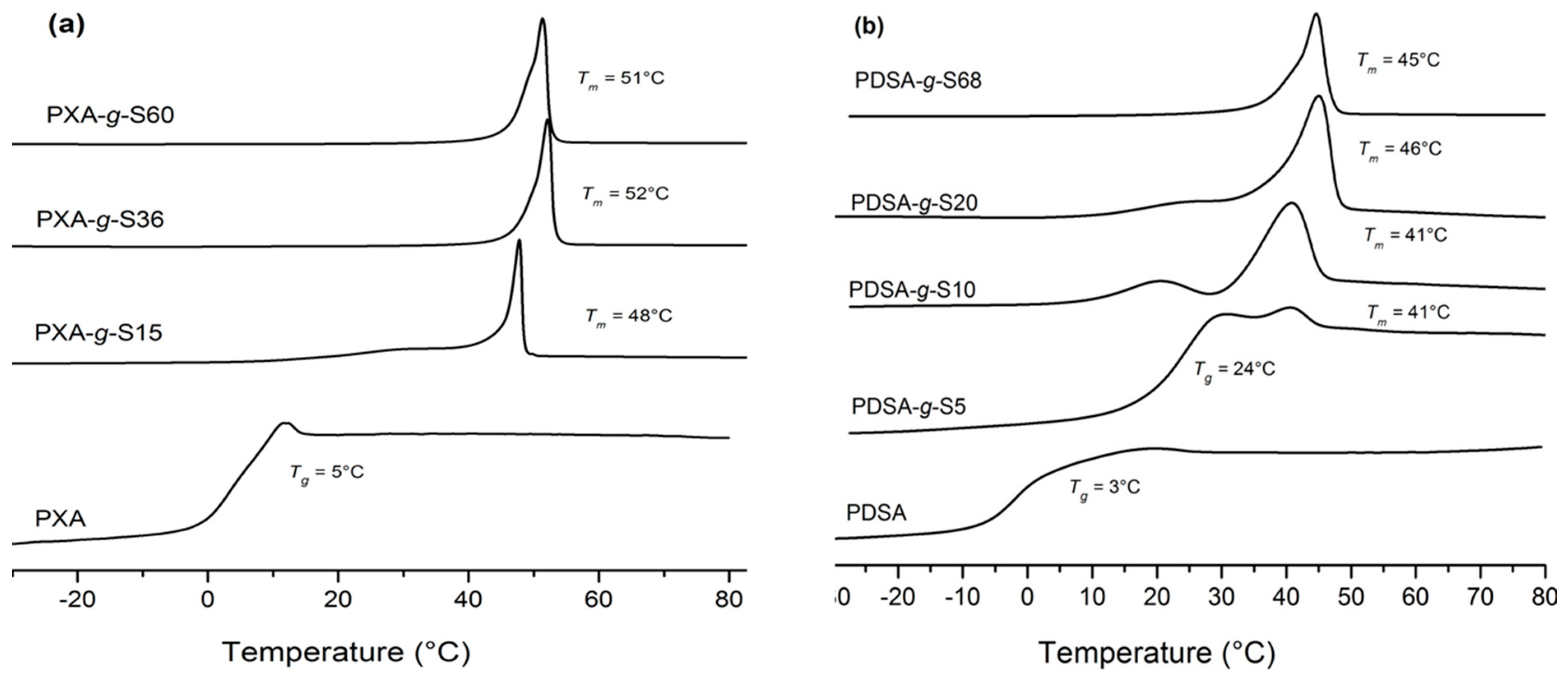
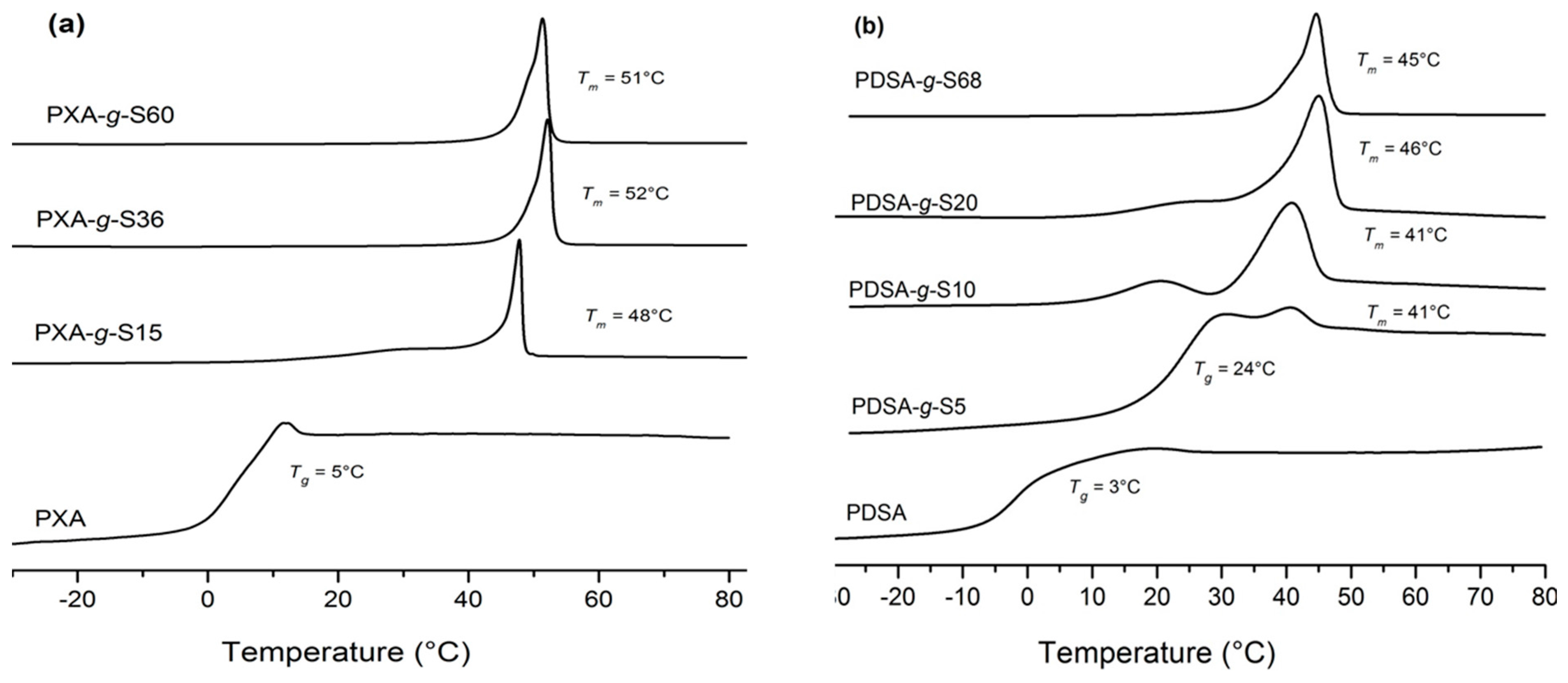
2.6. Differential Scanning Calorimetry (DSC)
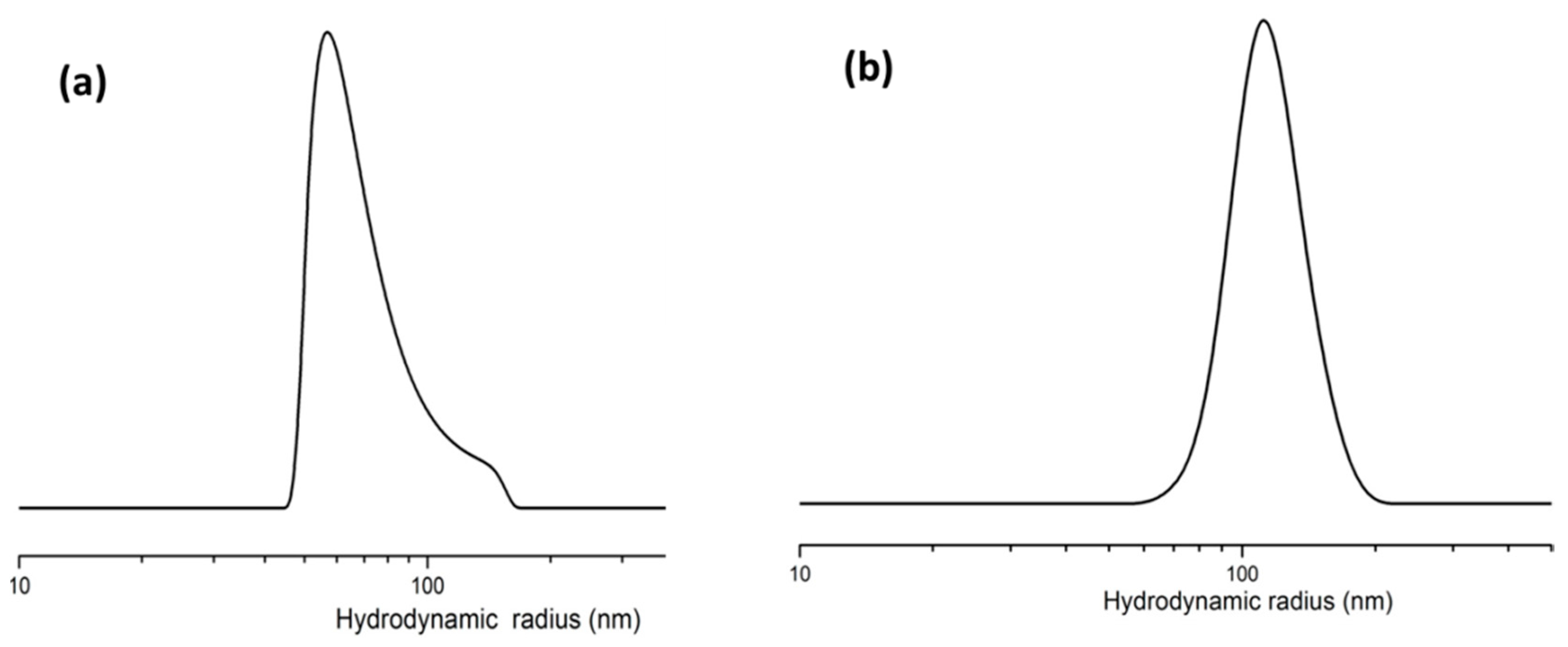
2.7. Dynamic Light Scattering (DLS)
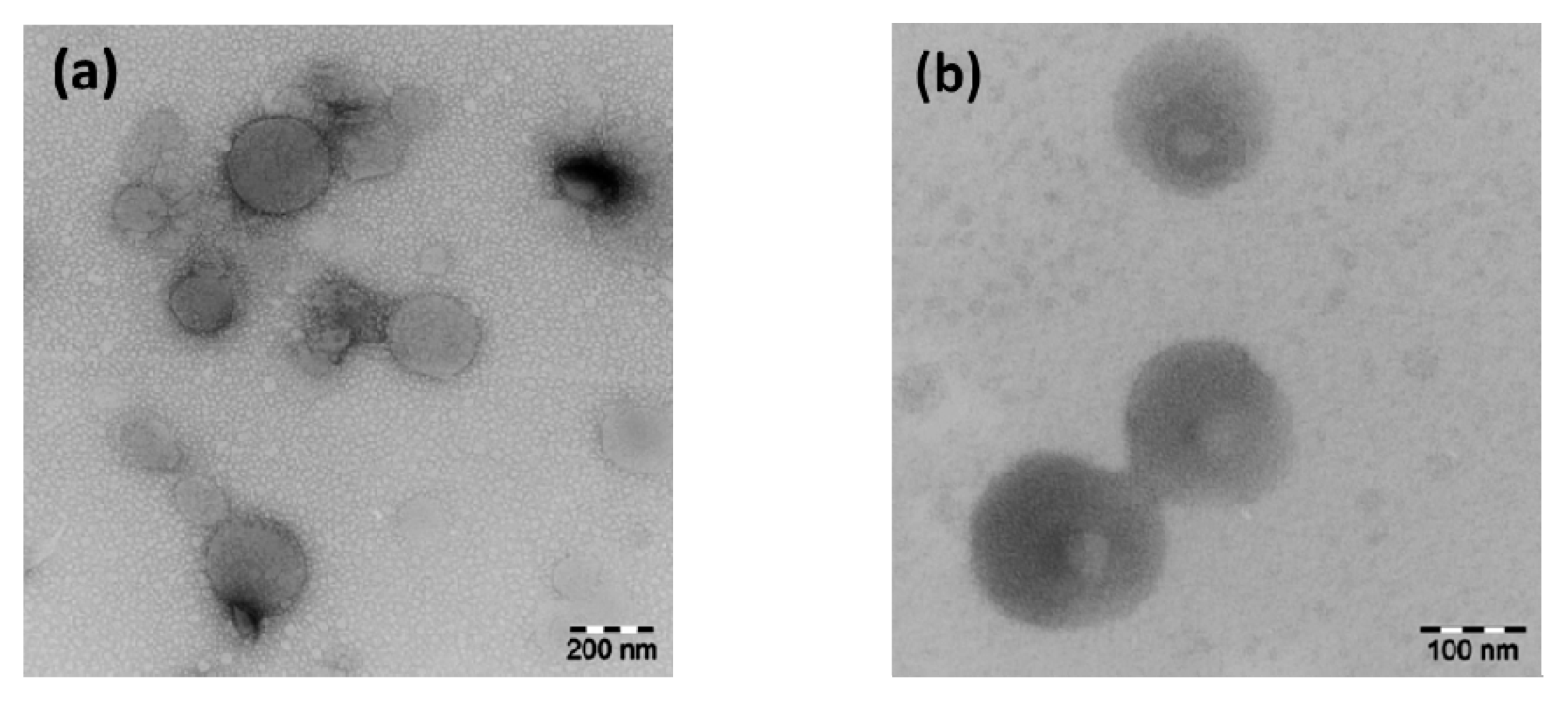
2.8. Negative-Staining TEM
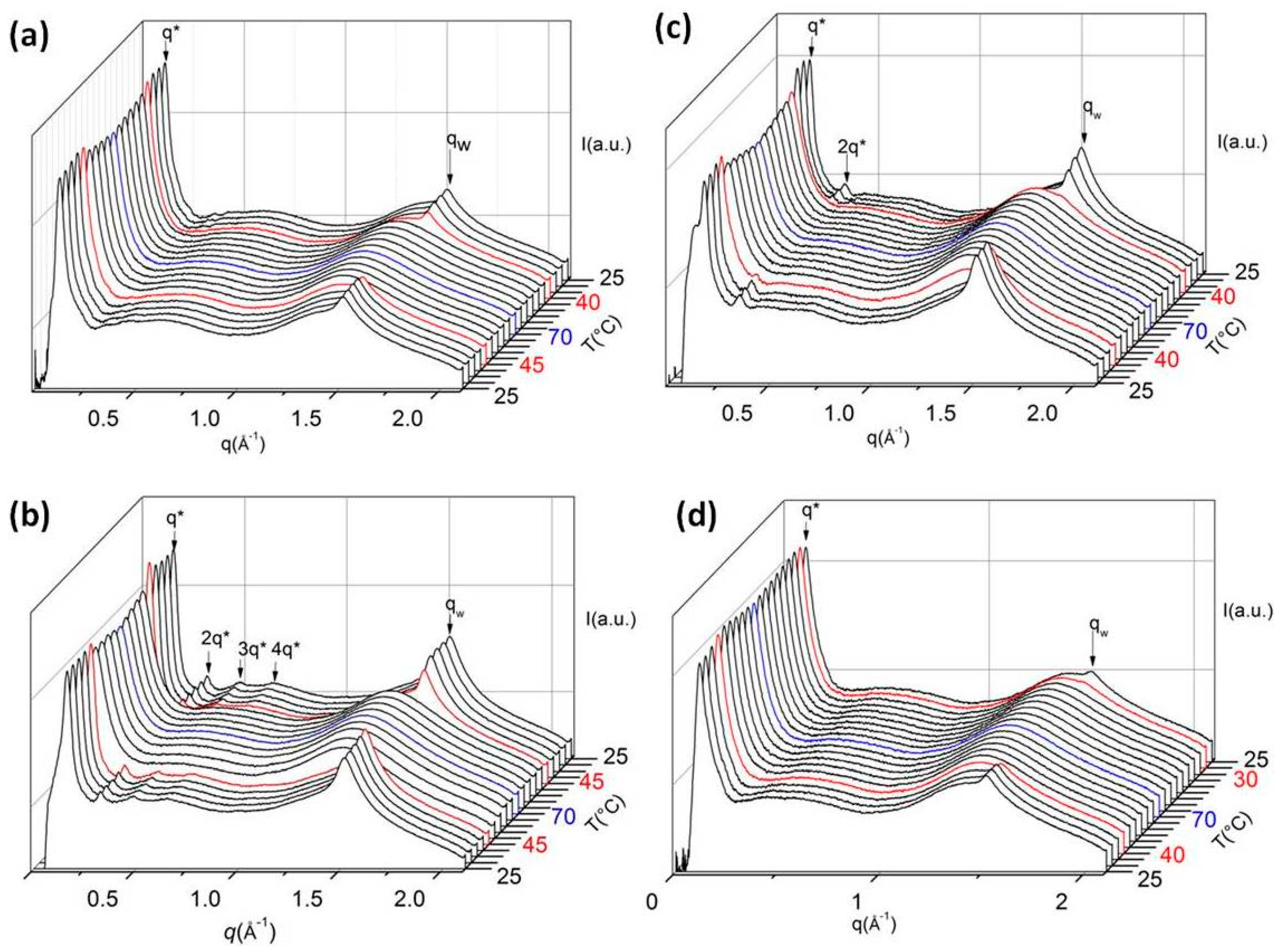
2.9. X-Ray Diffraction (XRD)
3. Results and Discussion
3.1. Polymerization and Grafting
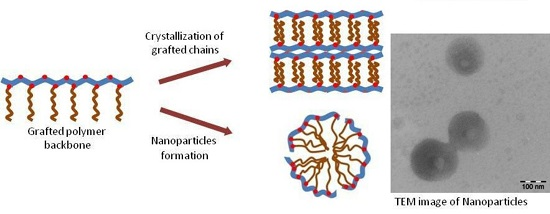
3.2. Nanoparticles
3.2.1. Dynamic Light Scattering
3.2.2. Negative-Staining Transmission Electron Microscopy
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PEO | Poly(ethylene oxide) |
| PGA | Poly(glycerol adipate) |
| PXA | Poly(xylitol adipate) |
| PDSA | Poly(d-sorbitol adipate) |
| PXA-g-Sx | Poly(xylitol adipate)-graft-stearoyl chain (x represents % degree of grafting) |
| PDSA-g-Sx | Poly(d-sorbitol adipate)-graft-stearoyl chain (x represents % degree of grafting) |
| SAXS | Small angle X-ray scattering |
| WAXS | Wide angle X-ray scattering |
| SEC | Size exclusion chromatography |
| DSC | Differential scanning calorimetry |
| DLS | Dynamic light scattering |
References
- Chawla, J.S.; Amiji, M.M. Biodegradable poly(ε-caprolactone) nanoparticles for tumor-targeted delivery of tamoxifen. Int. J. Pharm. 2002, 249, 127–138. [Google Scholar] [CrossRef]
- Trollsås, M.; Lee, V.Y.; Mecerreyes, D.; Löwenhielm, P.; Möller, M.; Miller, R.D.; Hedrick, J.L. Hydrophilic aliphatic polyesters: Design, synthesis, and ring-opening polymerization of functional cyclic esters. Macromolecules 2000, 33, 4619–4627. [Google Scholar] [CrossRef]
- Mateo, C.; Palomo, J.M.; Fernandez-Lorente, G.; Guisan, J.M.; Fernandez-Lafuente, R. Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzym. Microb. Technol. 2007, 40, 1451–1463. [Google Scholar] [CrossRef]
- Kim, D.-Y.; Dordick, J.S. Combinatorial array-based enzymatic polyester synthesis. Biotechnol. Bioeng. 2001, 76, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Kulshrestha, A.S.; Sahoo, B.; Gao, W.; Fu, H.; Gross, R.A. Lipase catalysis. A direct route to linear aliphatic copolyesters of bis(hydroxymethyl)butyric acid with pendant carboxylic acid groups. Macromolecules 2005, 38, 3205–3213. [Google Scholar] [CrossRef]
- Kallinteri, P.; Higgins, S.; Hutcheon, G.A.; St. Pourçain, C.B.; Garnett, M.C. Novel functionalized biodegradable polymers for nanoparticle drug delivery systems. Biomacromolecules 2005, 6, 1885–1894. [Google Scholar] [CrossRef] [PubMed]
- Kline, B.J.; Beckman, E.J.; Russell, A.J. One-step biocatalytic synthesis of linear polyesters with pendant hydroxyl groups. J. Am. Chem. Soc. 1998, 120, 9475–9480. [Google Scholar] [CrossRef]
- Korupp, C.; Weberskirch, R.; Müller, J.J.; Liese, A.; Hilterhaus, L. Scaleup of lipase-catalyzed polyester synthesis. Org. Process Res. Dev. 2010, 14, 1118–1124. [Google Scholar] [CrossRef]
- Shafioul, A.S.M.; Pyo, J.I.; Kim, K.S.; Cheong, C.S. Synthesis of poly(glycerol-co-dioate-co-butanedioate -co-xanthorrhizol) ester and a study of chain length effect on pendant group loading. J. Mol. Catal. B Enzym. 2012, 84, 198–204. [Google Scholar] [CrossRef]
- Naolou, T.; Conrad, D.; Busse, K.; Mäder, K.; Kressler, J. Fatty acid modified poly(glycerol adipate)-polymeric analogues of glycerides. In Tailored Polymer Architectures for Pharmaceutical and Biomedical Applications; Scholz, C., Kressler, J., Eds.; American Chemical Society: Washington, DC, USA, 2013; Volume 1135, pp. 39–52. [Google Scholar]
- Weiss, V.M.; Naolou, T.; Hause, G.; Kuntsche, J.; Kressler, J.; Mäder, K. Poly(glycerol adipate)-fatty acid esters as versatile nanocarriers: From nanocubes over ellipsoids to nanospheres. J. Control. Release 2012, 158, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Weiss, V.M.; Naolou, T.; Amado, E.; Busse, K.; Mäder, K.; Kressler, J. Formation of structured polygonal nanoparticles by phase-separated comb-like polymers. Macromol. Rapid Commun. 2012, 33, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Gao, W.; Kulshrestha, A.; Gross, R.A. “Sweet Polyesters”: Lipase-catalyzed condensation—Polymerizations of alditols. Macromolecules 2006, 39, 6789–6792. [Google Scholar] [CrossRef]
- Naolou, T.; Busse, K.; Kressler, J. Synthesis of well-defined graft copolymers by combination of enzymatic polycondensation and “click” chemistry. Biomacromolecules 2010, 11, 3660–3667. [Google Scholar] [CrossRef] [PubMed]
- Naolou, T.; Meister, A.; Schöps, R.; Pietzsch, M.; Kressler, J. Synthesis and characterization of graft copolymers able to form polymersomes and worm-like aggregates. Soft Matter 2013, 9, 10364. [Google Scholar] [CrossRef]
- Jbeily, M.; Naolou, T.; Bilal, M.; Amado, E.; Kressler, J. Enzymatically synthesized polyesters with pendent OH groups as macroinitiators for the preparation of well-defined graft copolymers by atom transfer radical polymerization. Polym. Int. 2014, 63, 894–901. [Google Scholar] [CrossRef]
- Naolou, T.; Jbeily, M.; Scholtysek, P.; Kressler, J. Synthesis and characterization of stearoyl modified poly(glycerol adipate) containing ATRP initiator on its backbone. Adv. Mater. Res. 2013, 812, 1–11. [Google Scholar] [CrossRef]
- Uyama, H.; Klegraf, E.; Wada, S.; Kobayashi, S. Regioselective polymerization of sorbitol and divinyl sebacate using lipase catalyst. Chem. Lett. 2000, 29, 800–801. [Google Scholar] [CrossRef]
- Foster, D.P.; Jasnow, D.; Balazs, A.C. Macrophase and microphase separation in random comb copolymers. Macromolecules 1995, 28, 3450–3462. [Google Scholar] [CrossRef]
- Beiner, M.; Huth, H. Nanophase separation and hindered glass transition in side-chain polymers. Nat. Mater. 2003, 2, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Ungar, G. Structure of rotator phases in n-alkanes. J. Phys. Chem. 1983, 87, 689–695. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Decuzzi, P.; Pasqualini, R.; Arap, W.; Ferrari, M. Intravascular delivery of particulate systems: Does geometry really matter? Pharm. Res. 2009, 26, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cui, W.; Bei, J.; Wang, S. Preparation of poly(lactide-co-glycolide-co-caprolactone) nanoparticles and their degradation behaviour in aqueous solution. Polym. Degrad. Stab. 2006, 91, 1929–1936. [Google Scholar] [CrossRef]
- Fessi, H.; Puisieux, F.; Devissaguet, J.P.; Ammoury, N.; Benita, S. Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int. J. Pharm. 1989, 55, 1–4. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Volume fraction of stearoyl chains 5 | Mn (g·mol−1) | PDI | Tm (°C) 4 | Tg (°C) 4 | ΔHm (J·g−1) 4 |
|---|---|---|---|---|---|---|
| PXA | - | 5,000 1 | 2.4 1 | - | 5 | |
| PXA-g-S15 | 0.37 | 7,300 2 | 2.3 3 | 48 | 20 | 60 |
| PXA-g-S36 | 0.58 | 10,600 2 | 2.6 3 | 52 | - | 100 |
| PXA-g-S60 | 0.69 | 14,600 2 | 1.8 3 | 51 | - | 85 |
| PDSA | - | 3,500 1 | 1.9 1 | - | 3 | - |
| PDSA-g-S5 | 0.15 | 4,300 2 | 1.4 3 | 41 | 24 | 1.15 |
| PDSA-g-S10 | 0.30 | 4,800 2 | 1.9 3 | 41 | - | 36 |
| PDSA-g-S20 | 0.43 | 5,800 2 | 1.5 3 | 46 | - | 136 |
| PDSA-g-S68 | 0.71 | 12,200 2 | 1.7 3 | 45 | - | 73 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bilal, M.H.; Prehm, M.; Njau, A.E.; Samiullah, M.H.; Meister, A.; Kressler, J. Enzymatic Synthesis and Characterization of Hydrophilic Sugar Based Polyesters and Their Modification with Stearic Acid. Polymers 2016, 8, 80. https://doi.org/10.3390/polym8030080
Bilal MH, Prehm M, Njau AE, Samiullah MH, Meister A, Kressler J. Enzymatic Synthesis and Characterization of Hydrophilic Sugar Based Polyesters and Their Modification with Stearic Acid. Polymers. 2016; 8(3):80. https://doi.org/10.3390/polym8030080
Chicago/Turabian StyleBilal, Muhammad Humayun, Marko Prehm, Andrew Efraim Njau, Muhammad Haris Samiullah, Annette Meister, and Jörg Kressler. 2016. "Enzymatic Synthesis and Characterization of Hydrophilic Sugar Based Polyesters and Their Modification with Stearic Acid" Polymers 8, no. 3: 80. https://doi.org/10.3390/polym8030080