
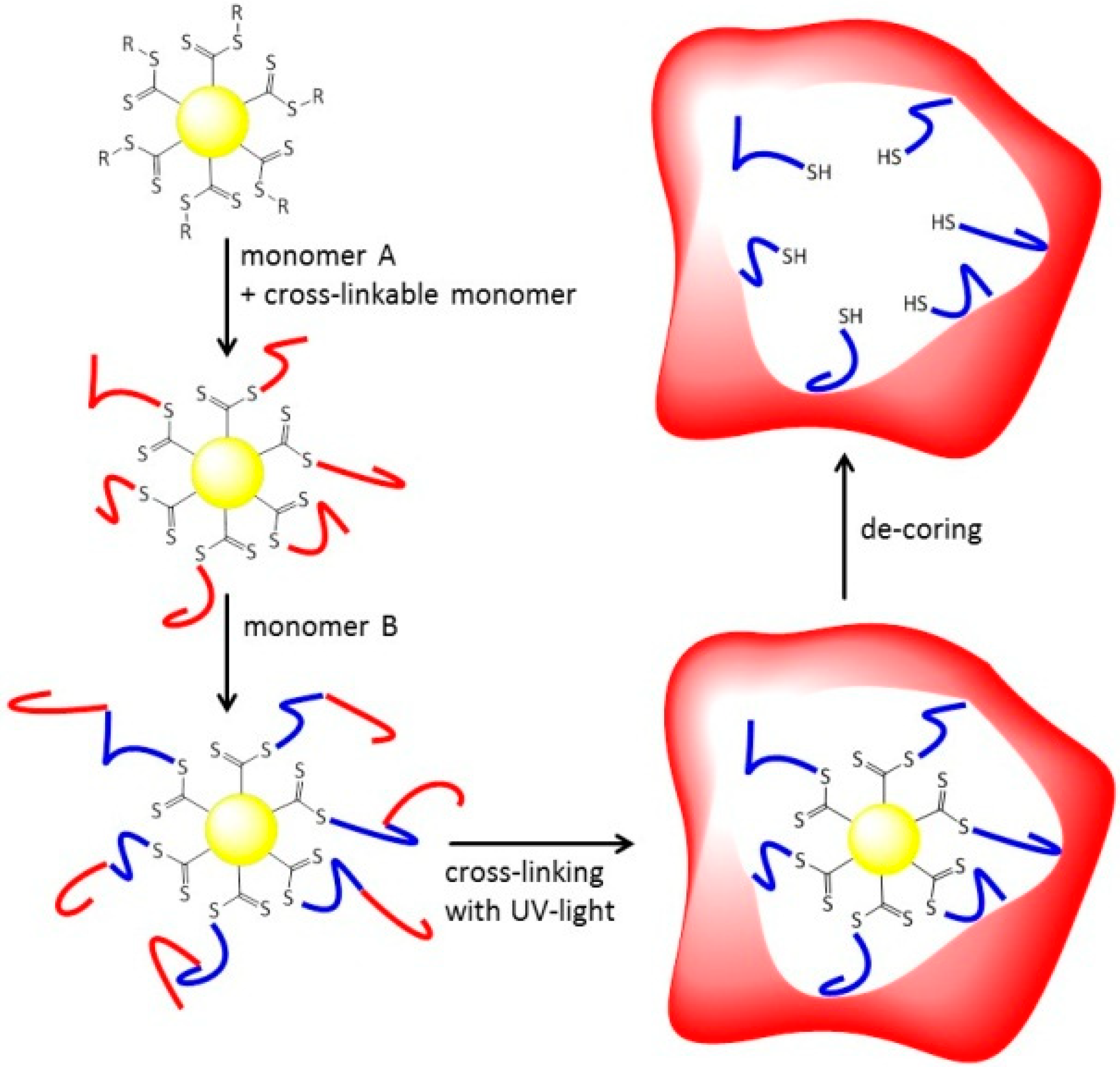
Tailoring Confinement: Nano-Carrier Synthesis via Z-RAFT Star Polymerization


Abstract
:
1. Introduction
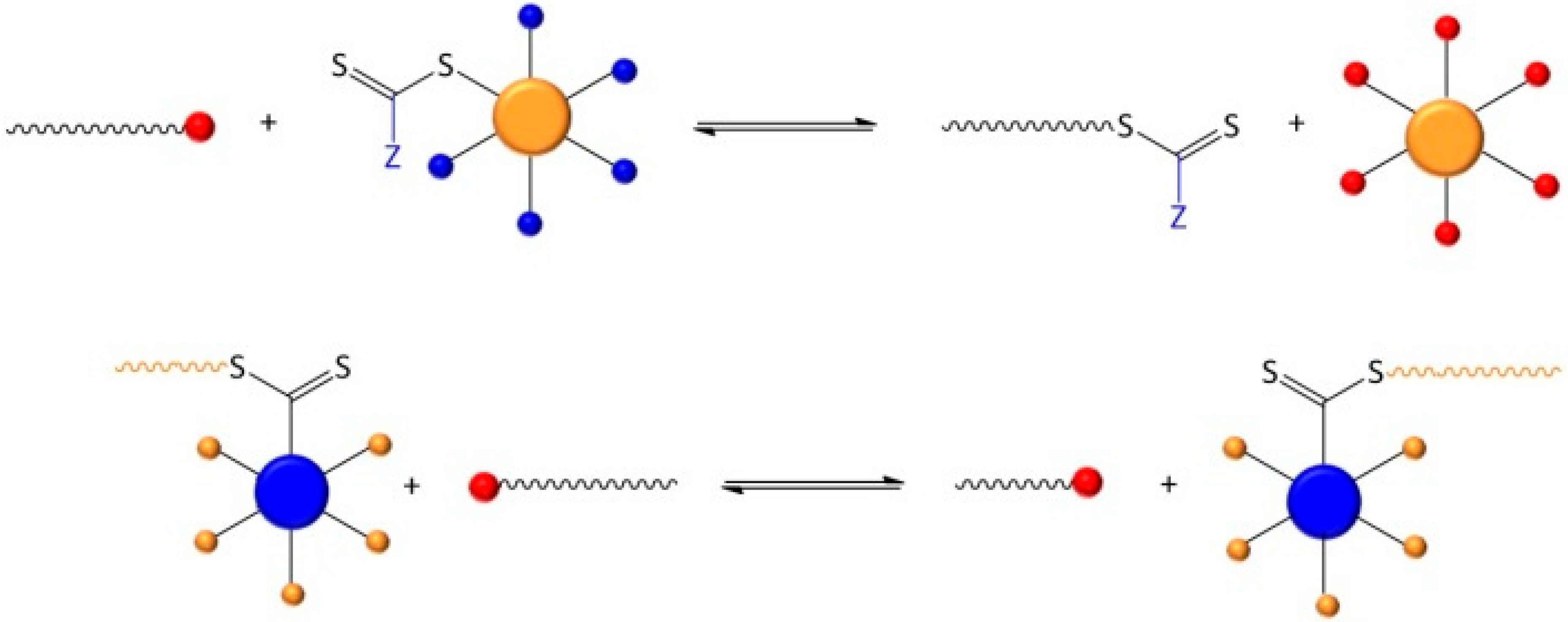
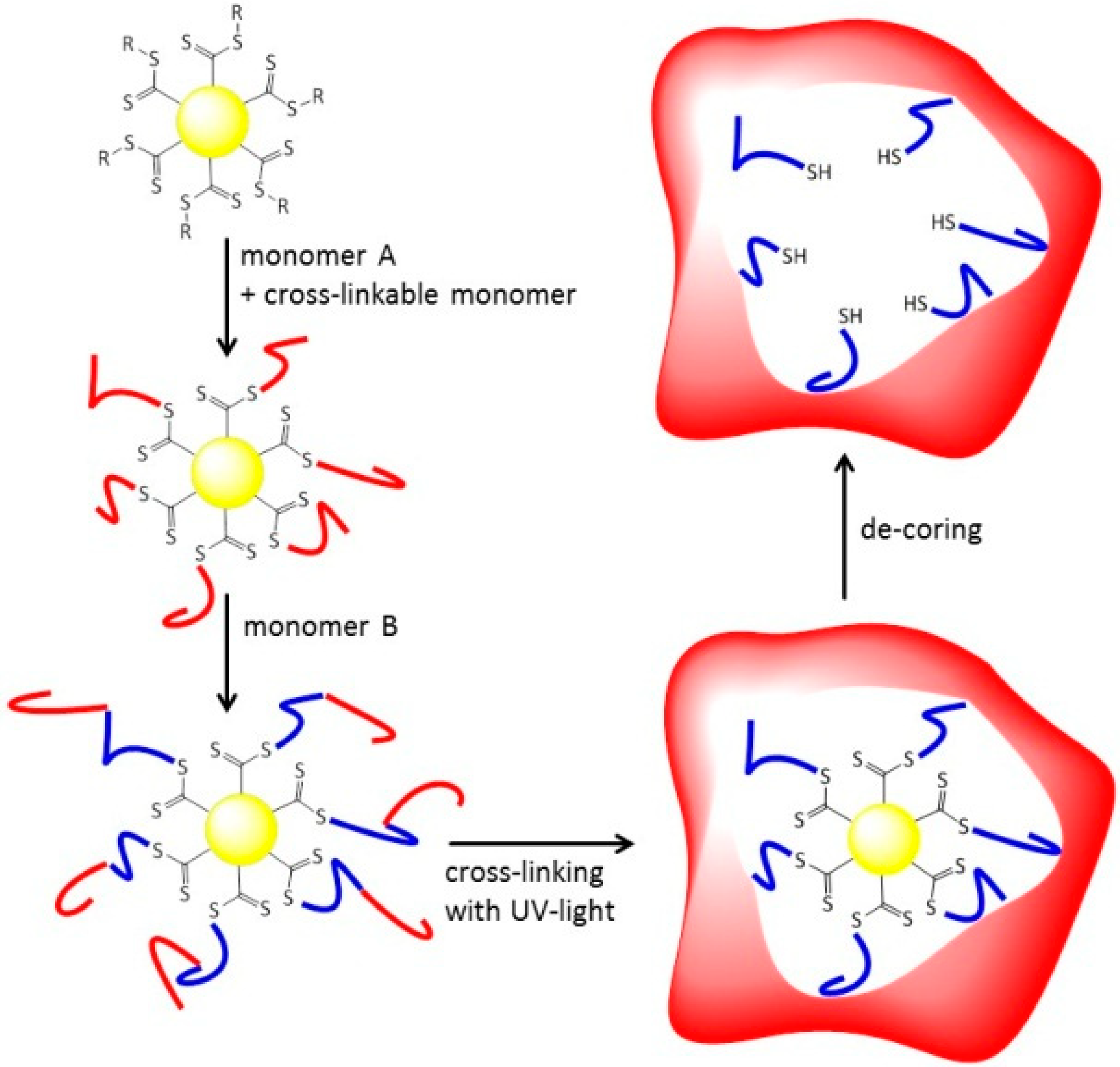
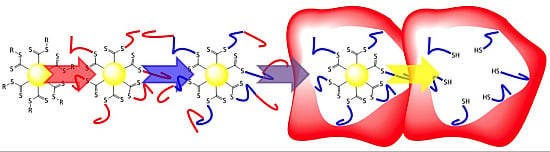
2. Experimental Section
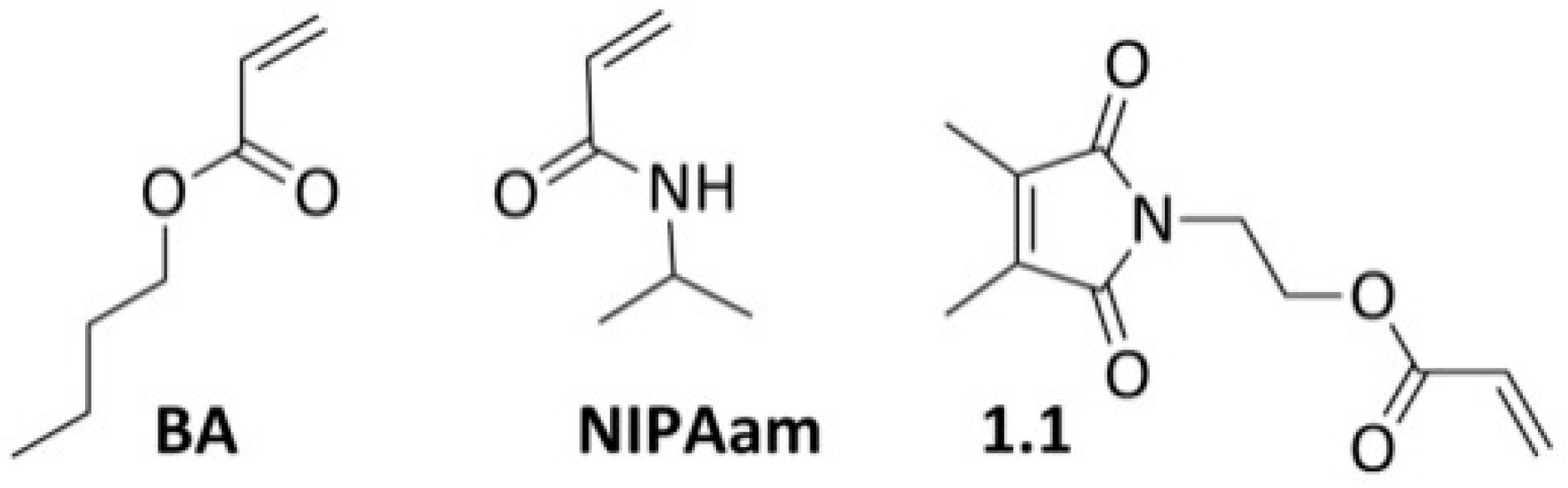
2.1. Materials
2.2. Synthesis
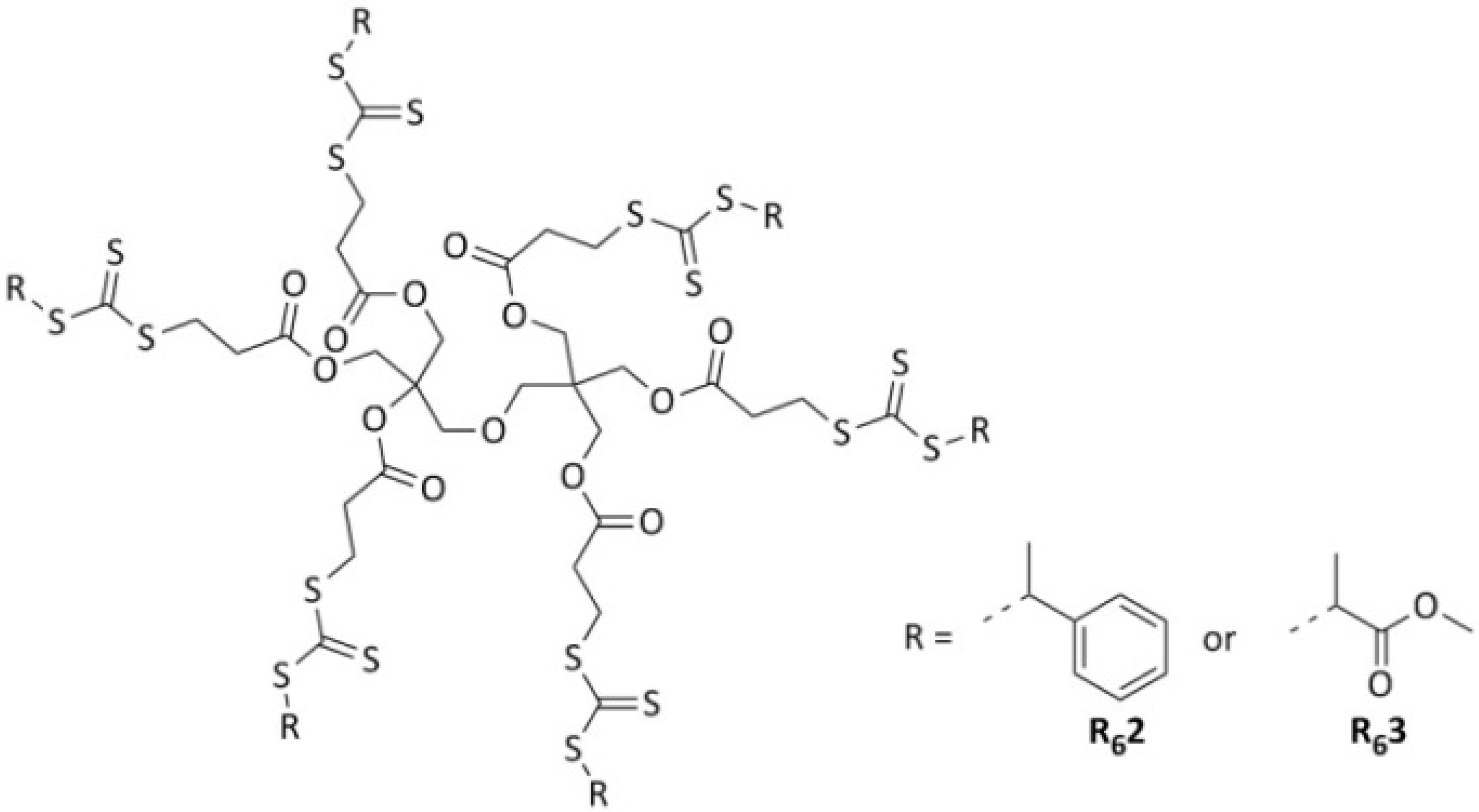
2.3. Polymerizations
2.4. Size Exclusion Chromatography (SEC)
2.5. Dynamic Light Scattering
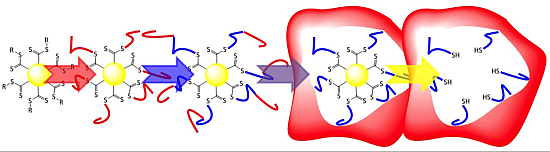
3. Results and Discussion
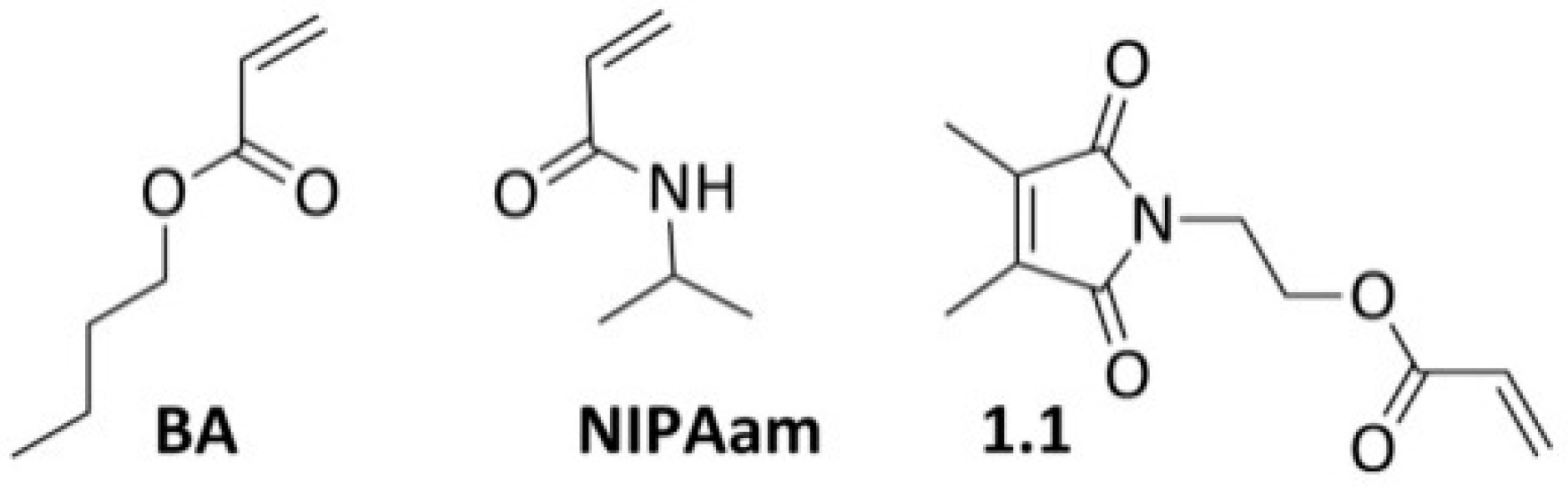
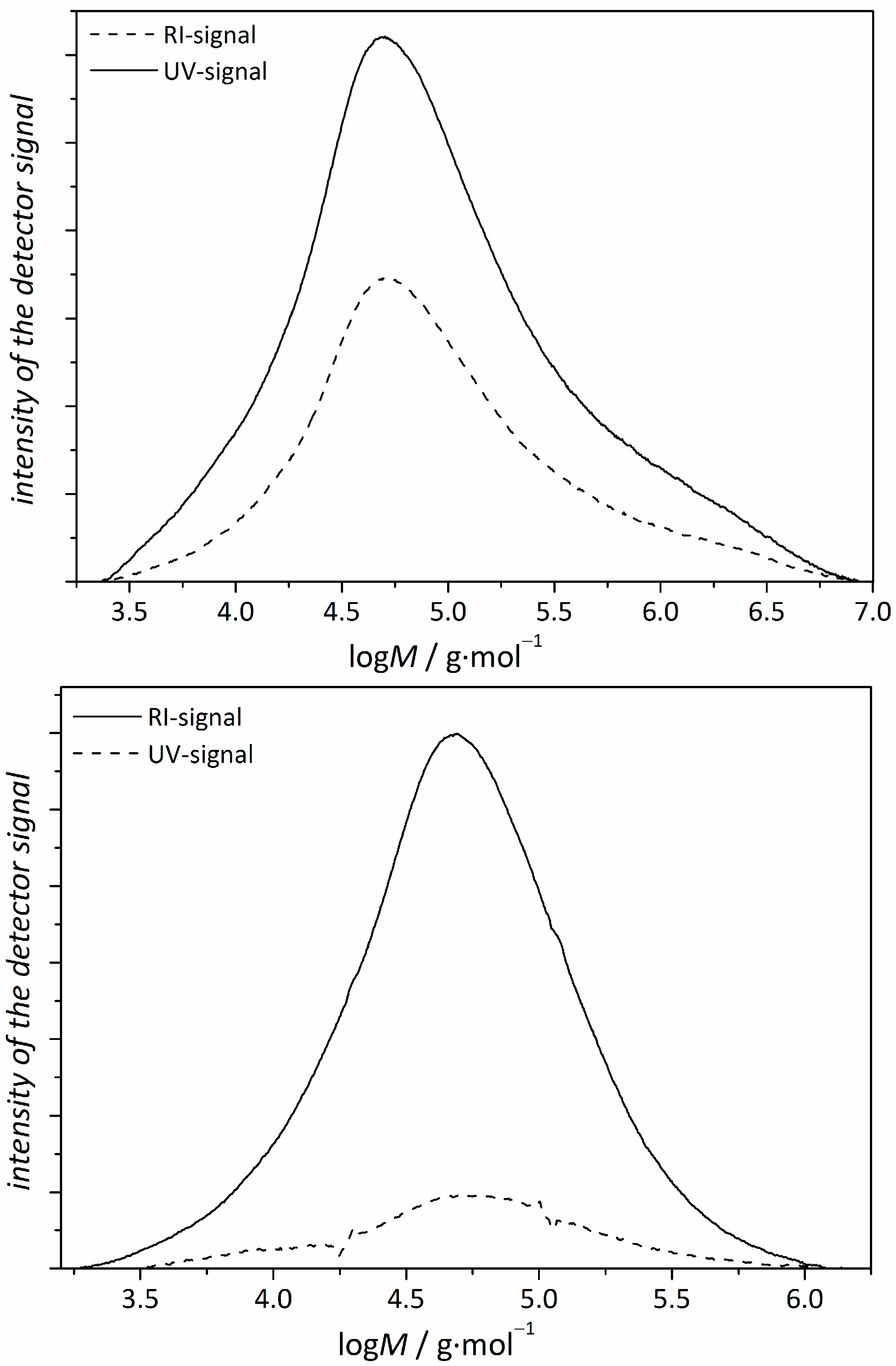
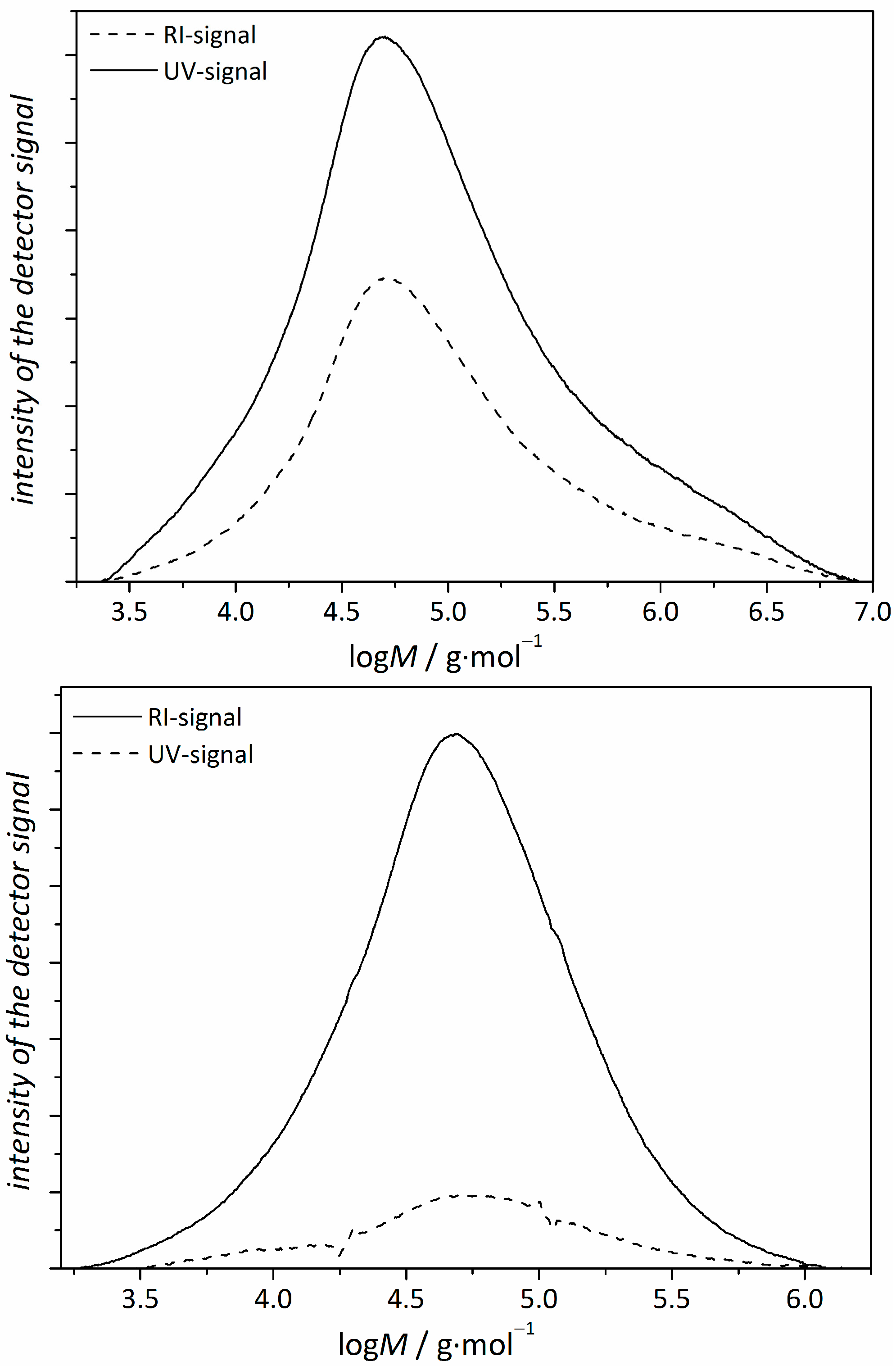
3.1. Copolymerizations with N-Ethylacrylate-3,4-dimethyl-maleimide
- PBA and PNIPAam six-arm star polymers can successfully be synthesized in a well-controlled fashion using the RAFT agents R62 in BA-polymerizations and R63 in NIPAam-polymerizations.
- The formed star polymer shows maximum molar masses of 550,000 g∙mol−1 and typical Đ-values between 1.2 and 1.8 (when star-star coupling occurs, see below) for PBA and maximum molar masses of 250,000 g∙mol−1 and typical Đ-values between 1.3 and 1.7 for PNIPAam.
- Star-star coupling occurs with PBA stars, which originates from sporadic macroradical attack to the DMI-moiety, which results in broadened molar mass distributions at high monomer conversions.
- Star-star coupling does not occur in the NIPAam system, which, however, shows a much more prominent occurrence of termination leading to dead polymer material.
- The reactivity ratios both for the BA/1.1-copolymer system and the NIPAm/1.1-system leads to a gradient along the main chain with a higher content of 1.1 at the outer regions of the star.
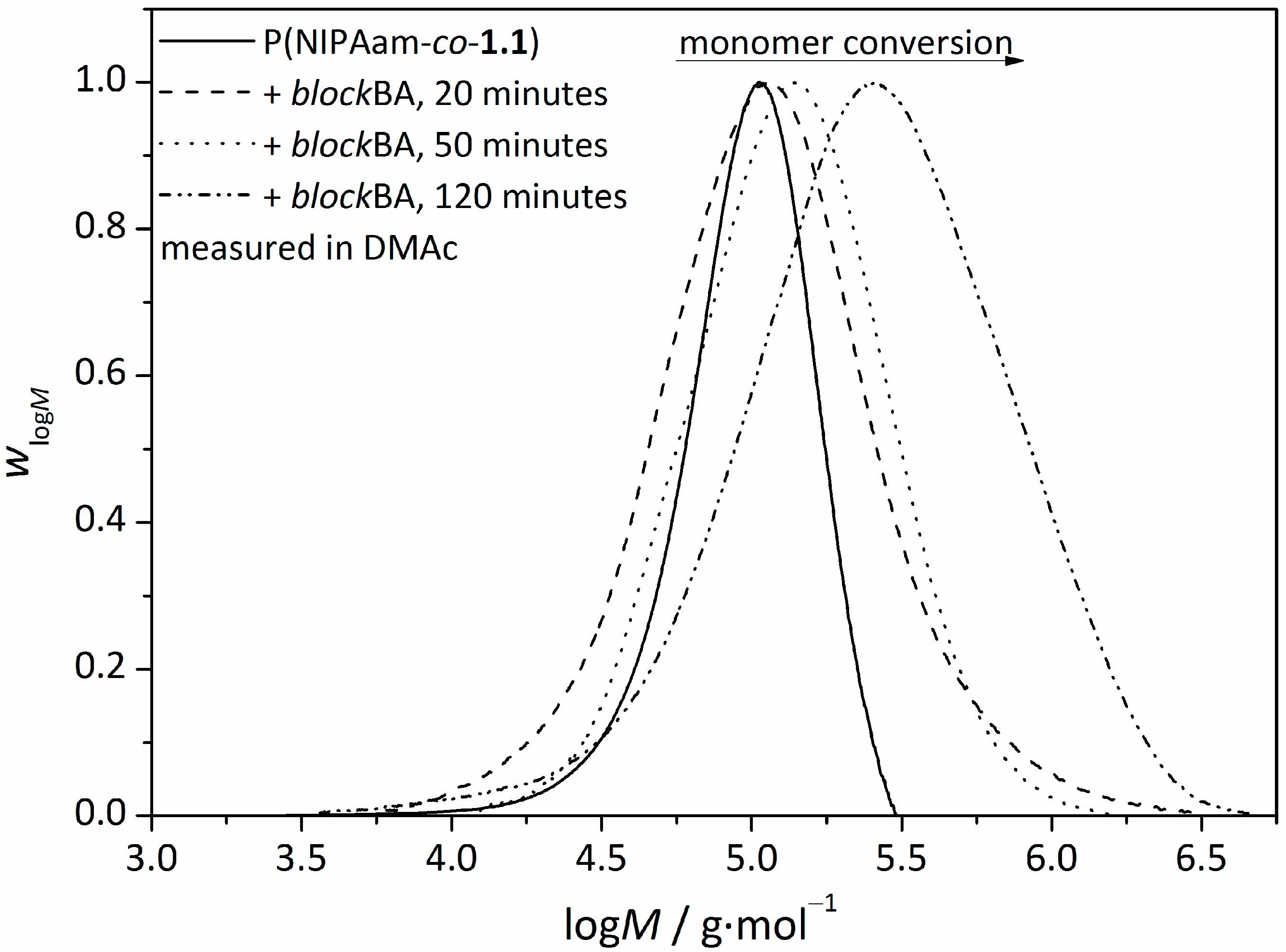
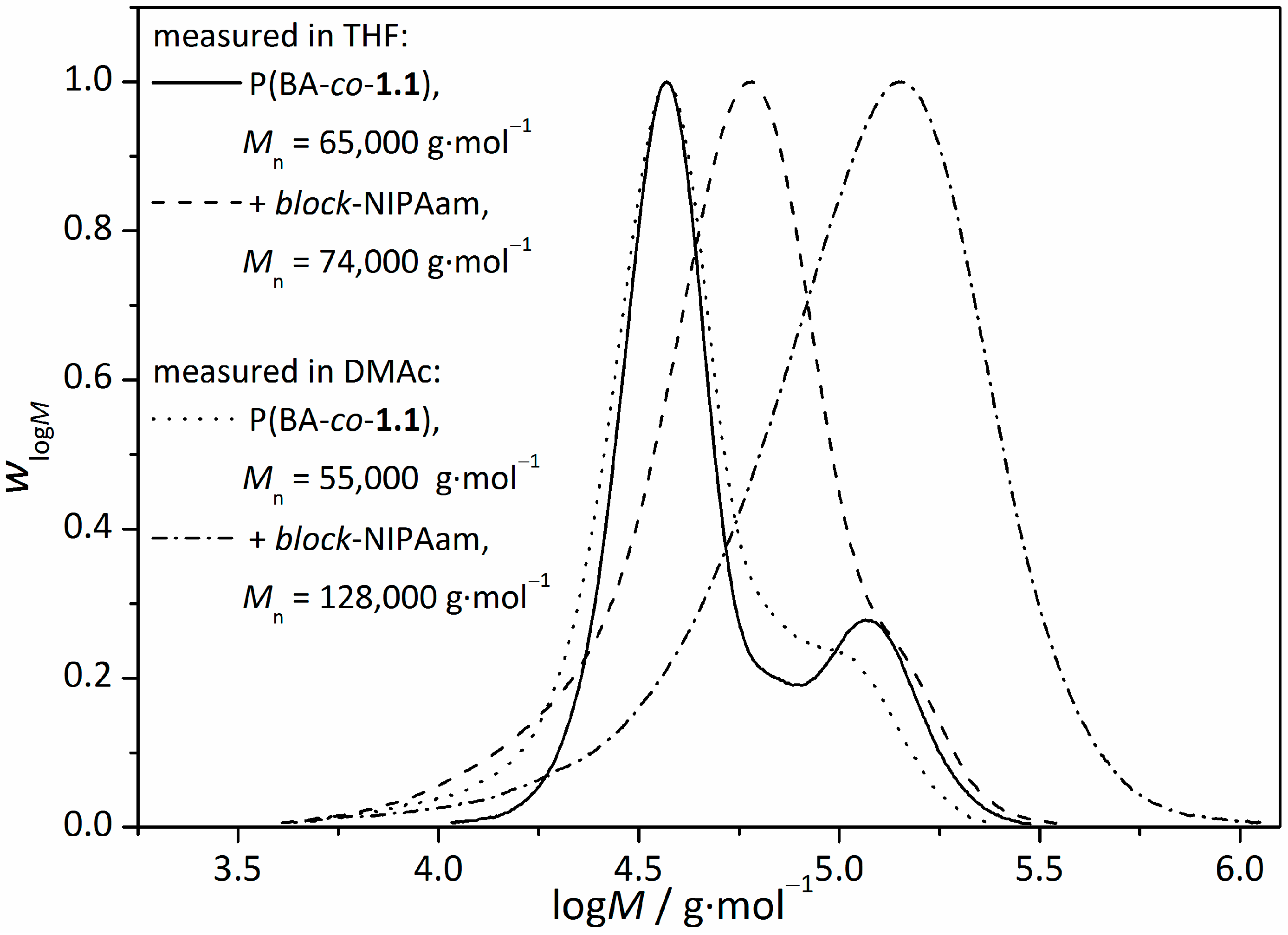
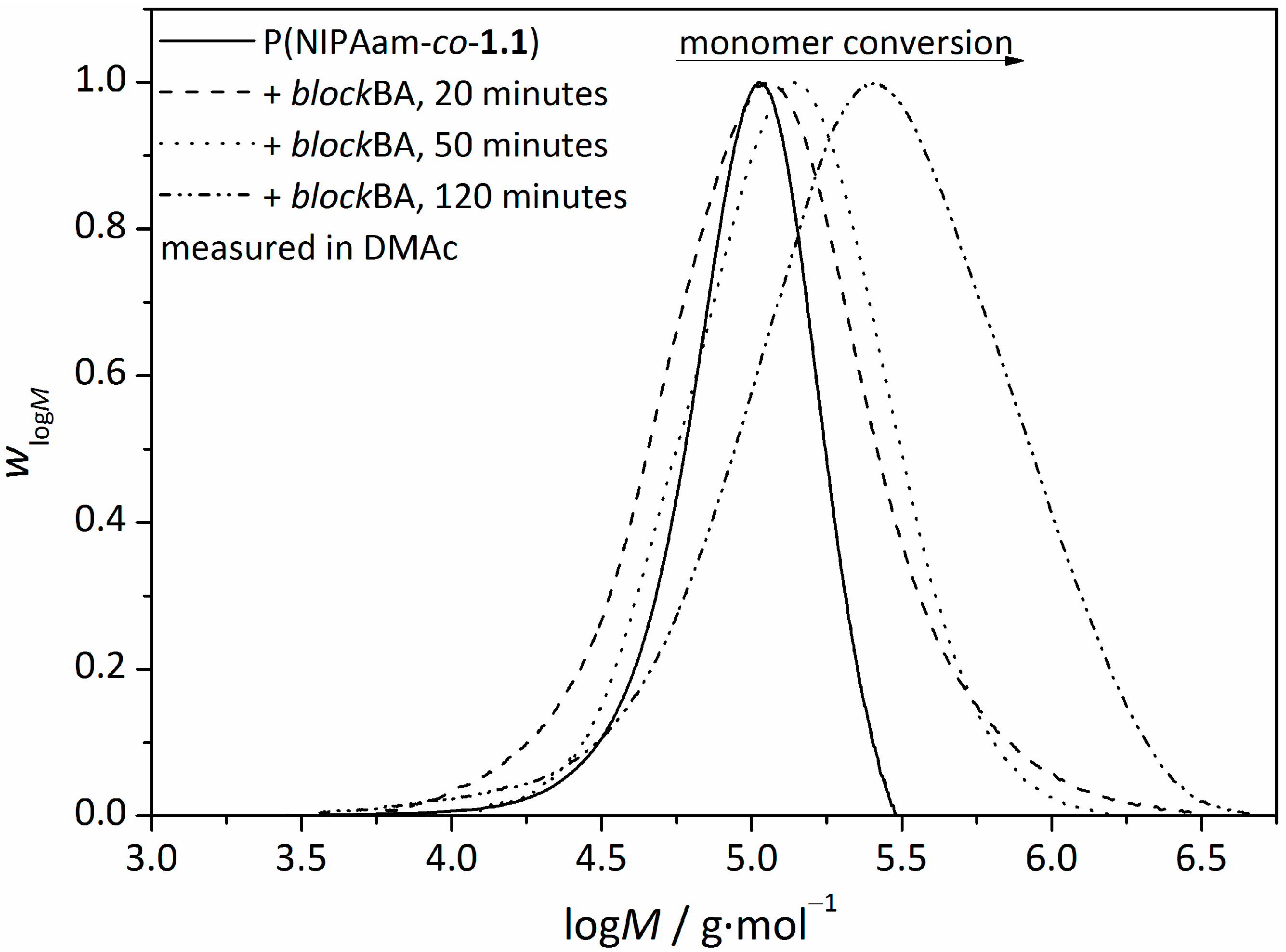
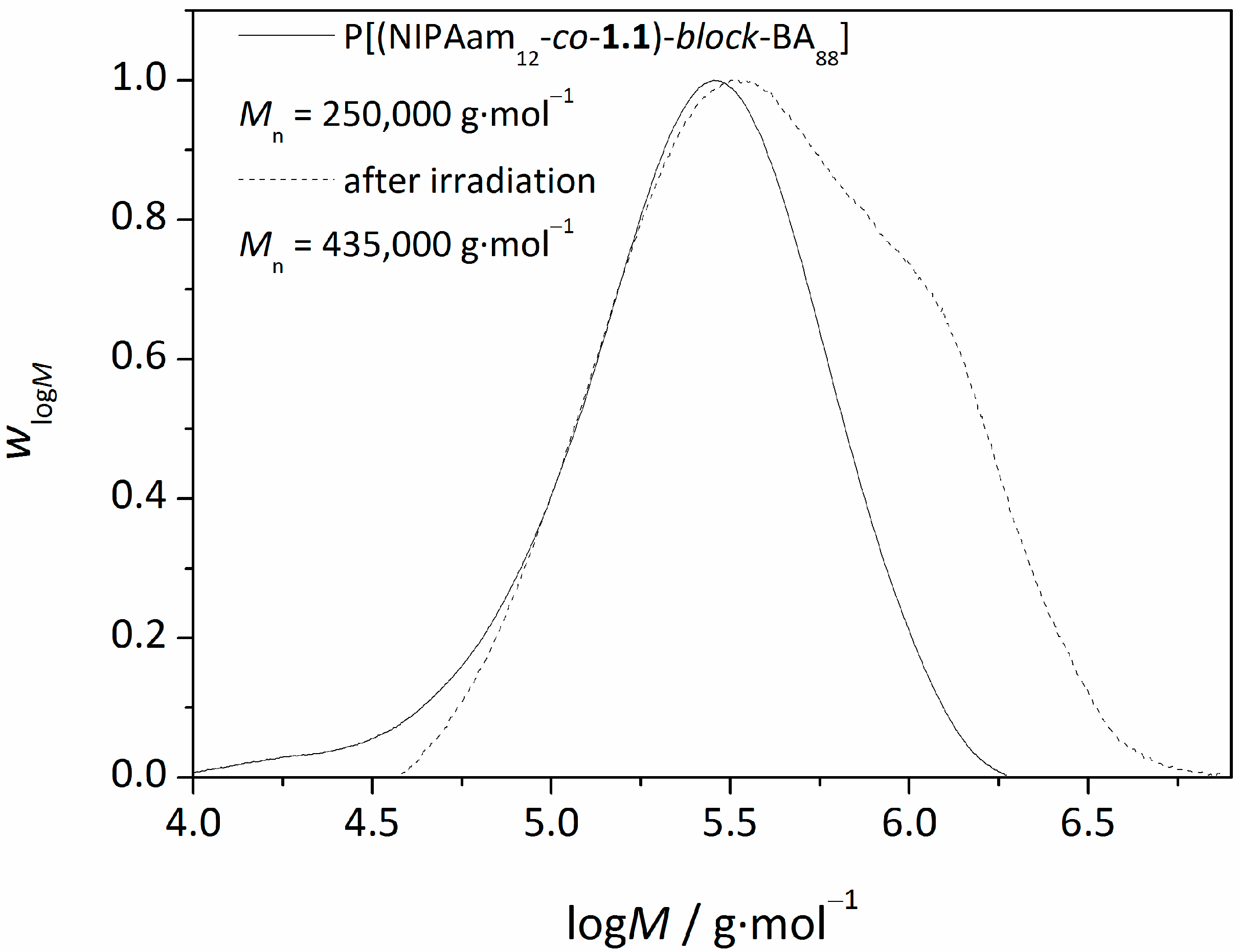
3.2. Synthesis of Amphiphilic Diblock Copolymer Stars


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| t/min | monomer conversion/% | Mn/g∙mol−1 | Ð | FBA/mol% | FNIPAam/mol% |
|---|---|---|---|---|---|
| 20 | 4 | 114,000 | 1.97 | 10.1 | 89.9 |
| 50 | 6 | 150,000 | 1.70 | 38.9 | 61.1 |
| 120 | 7 | 210,000 | 2.67 | 45.9 | 54.1 |
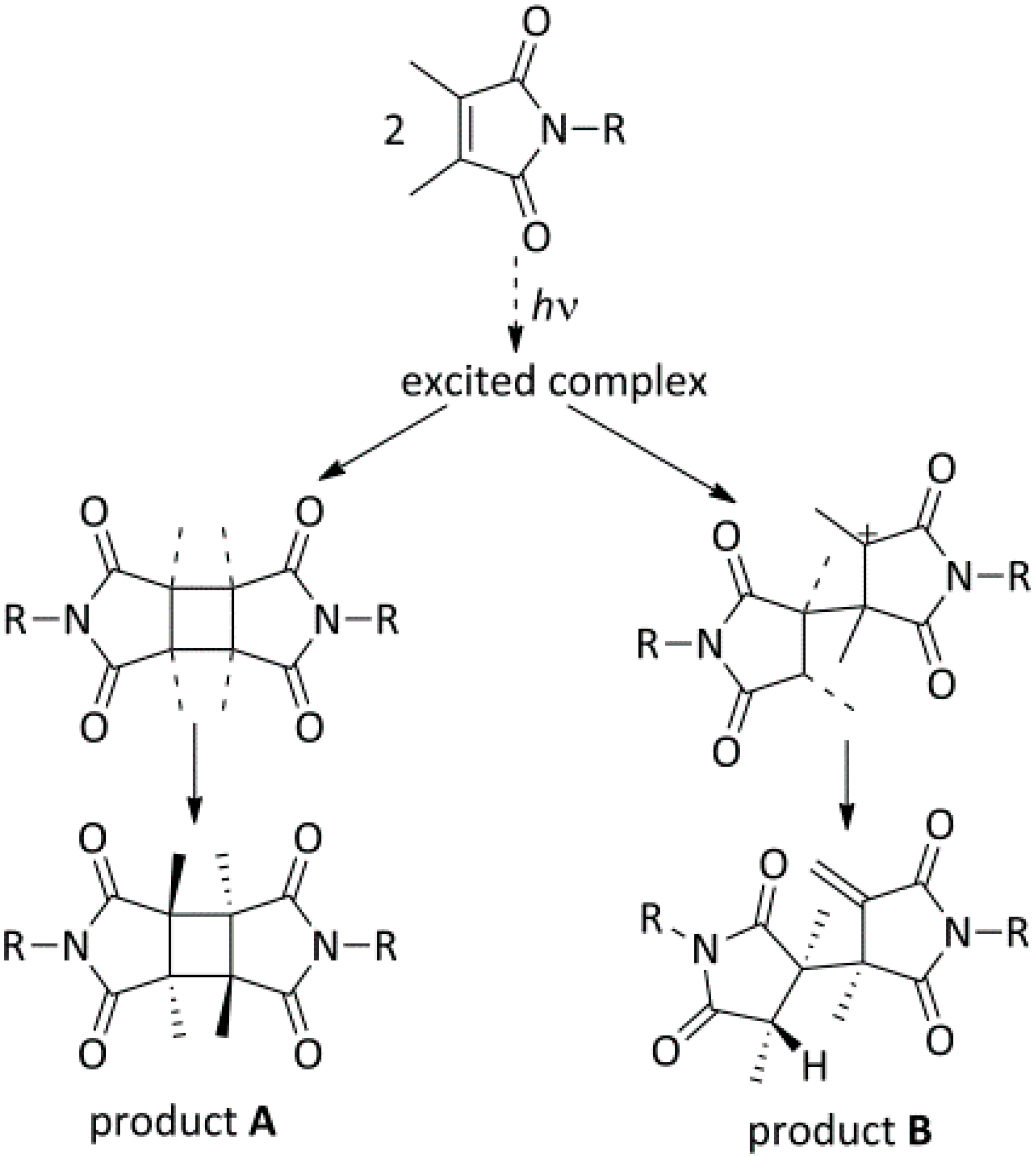
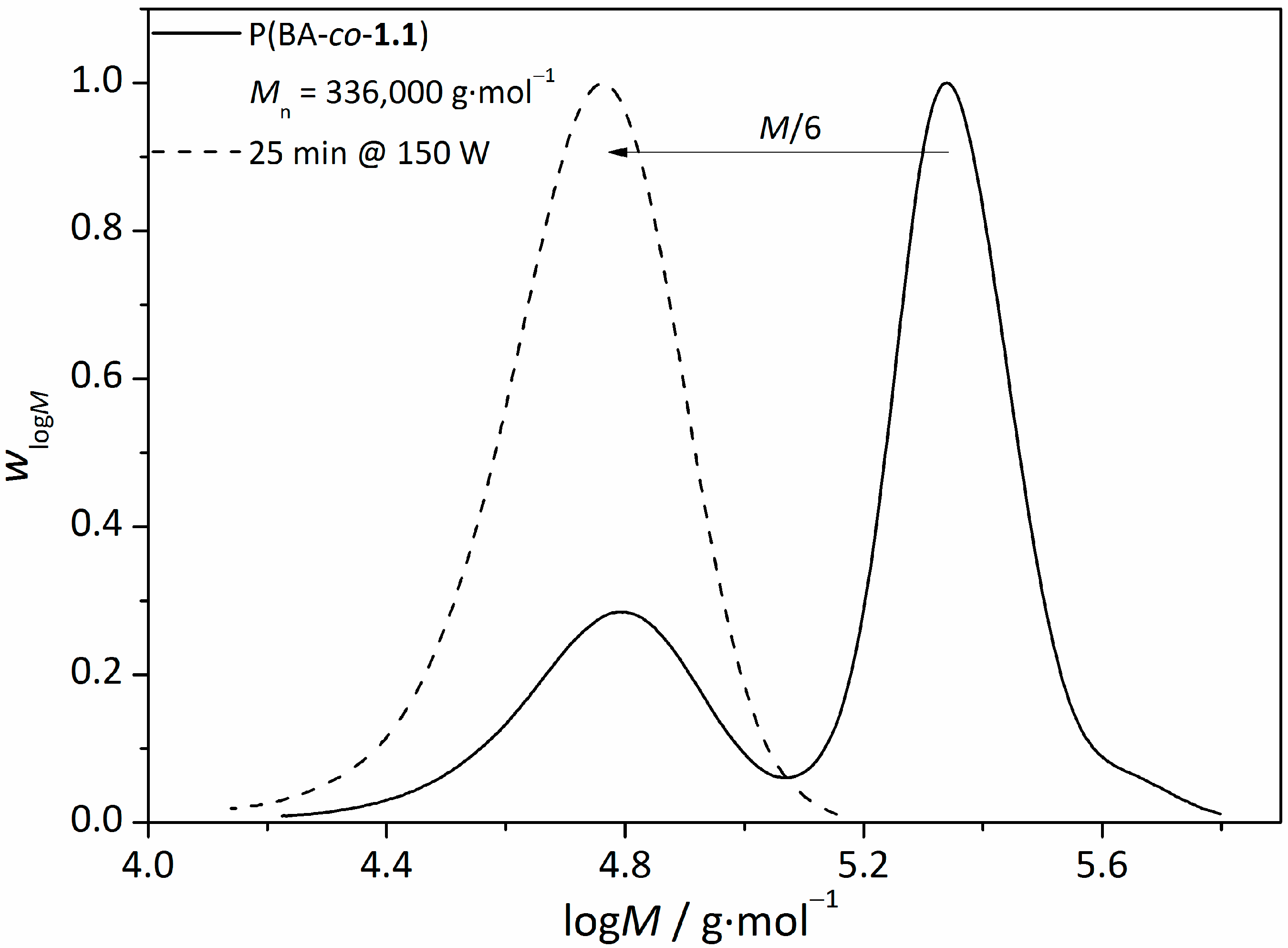
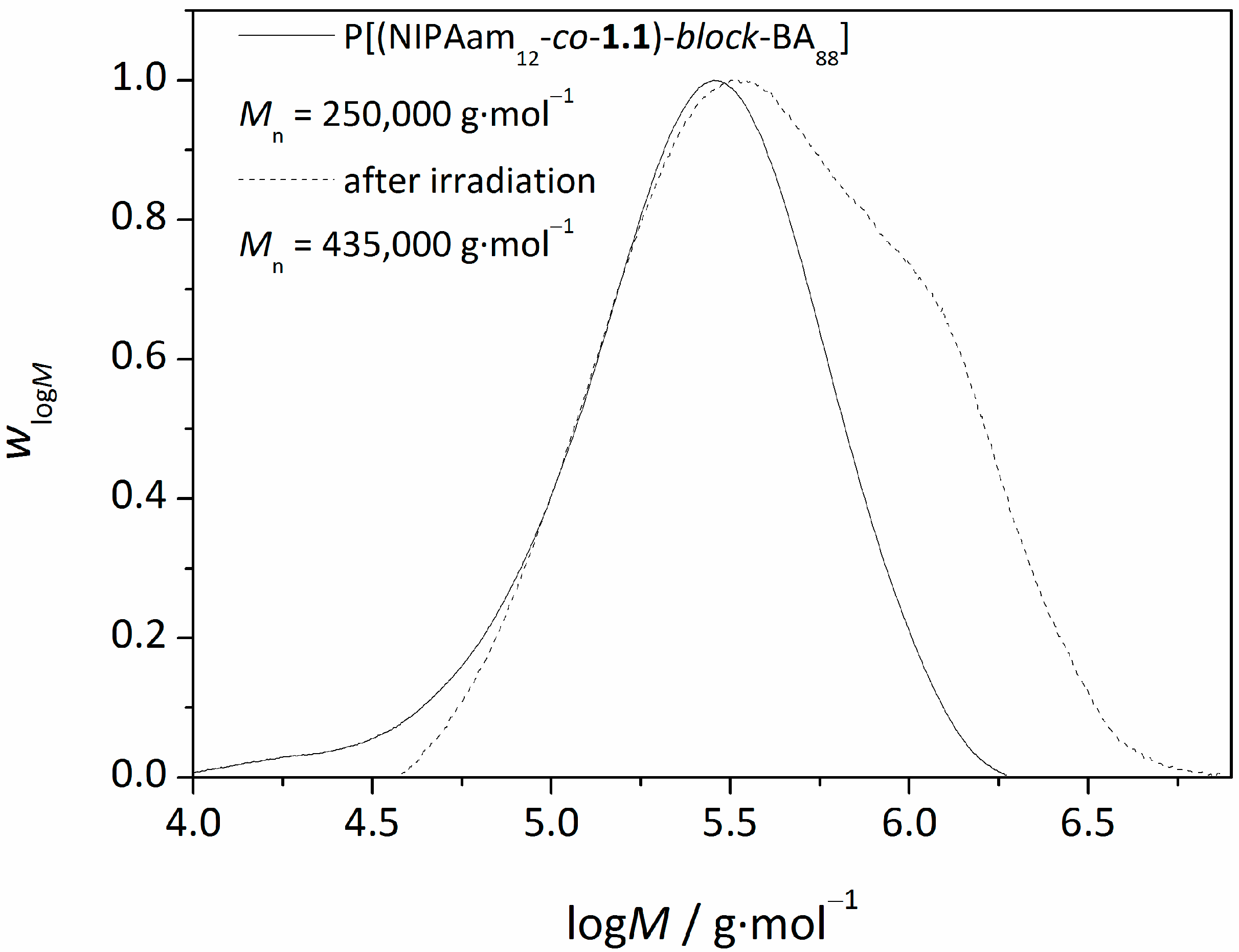
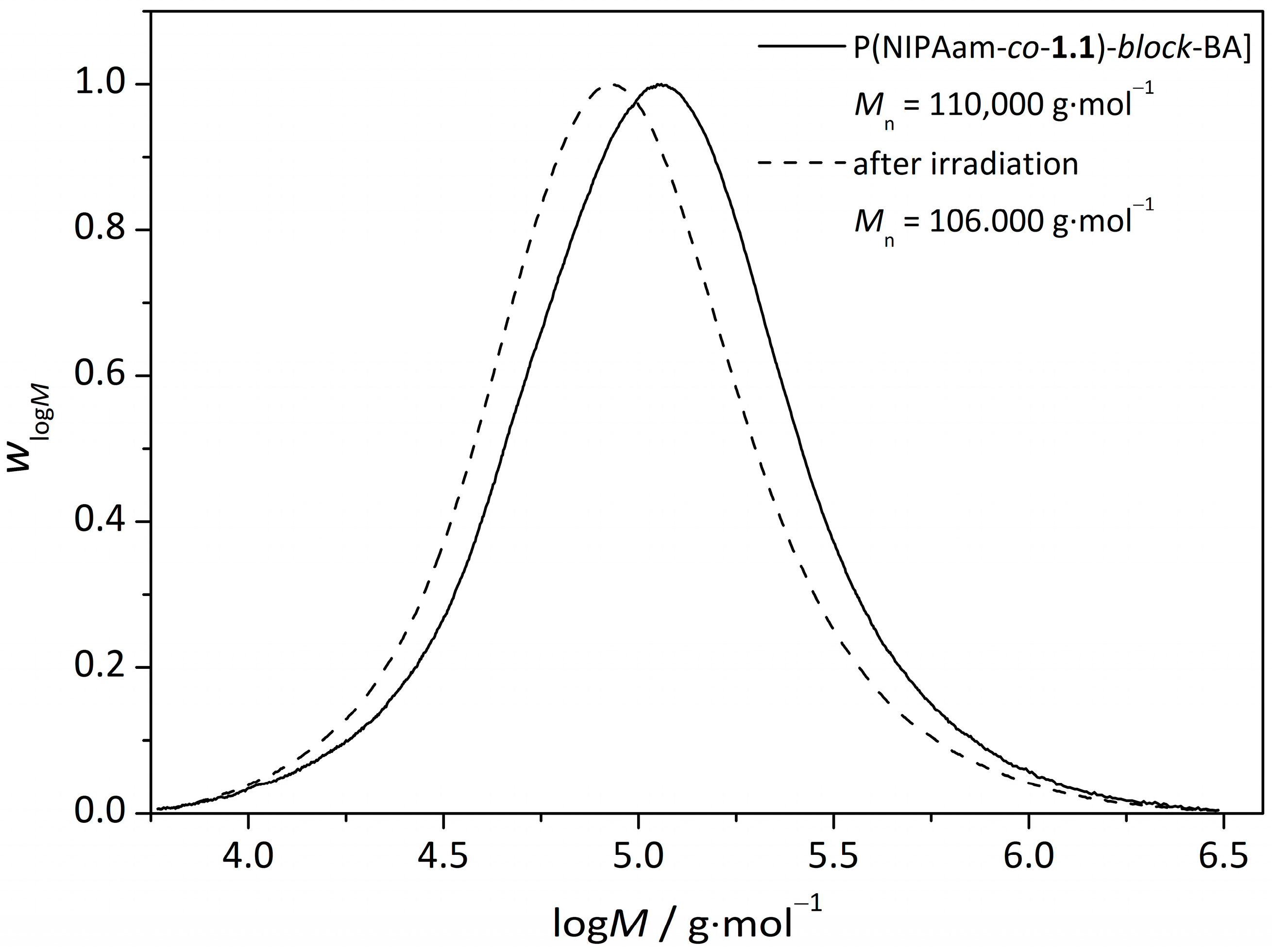
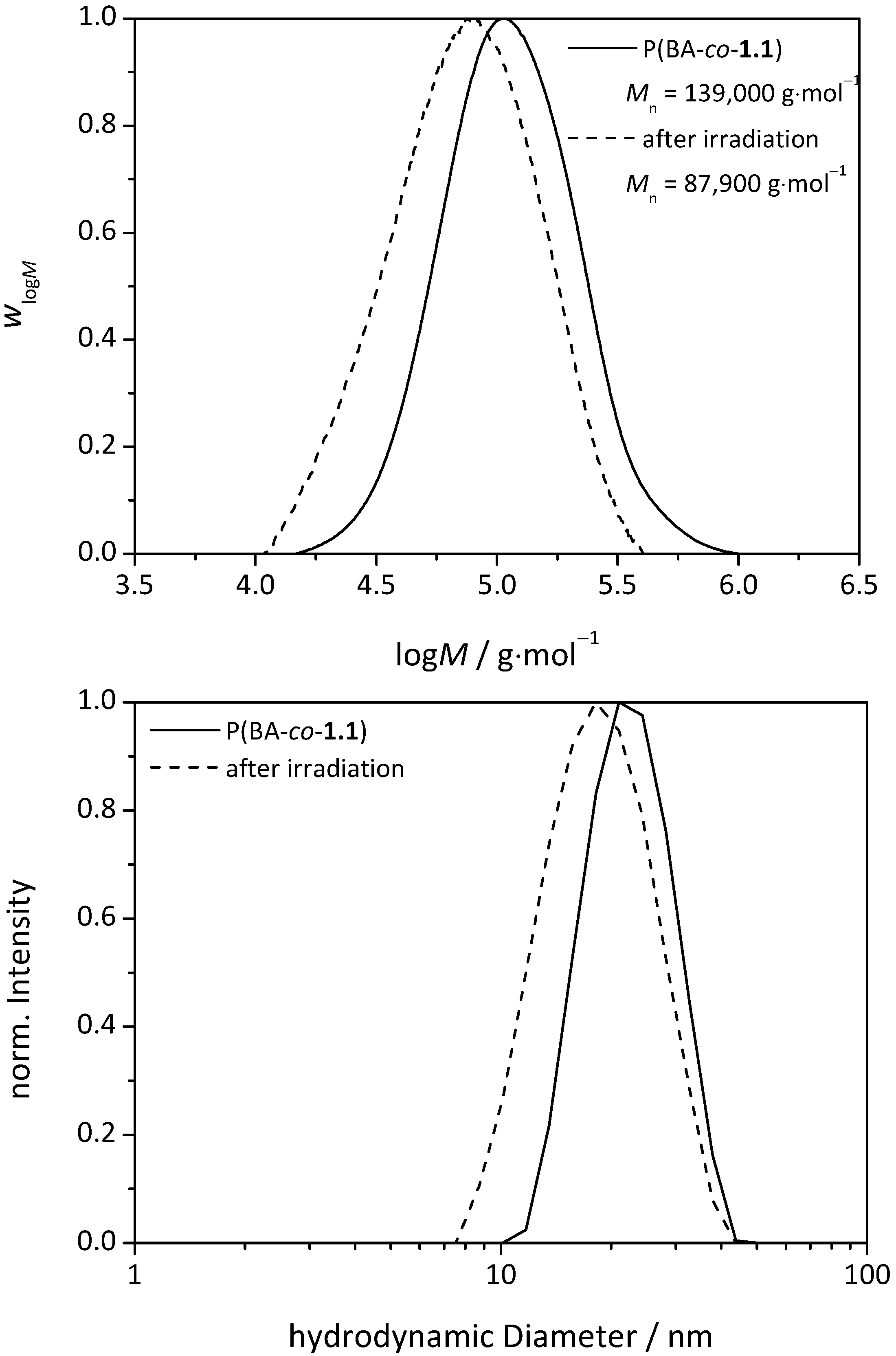
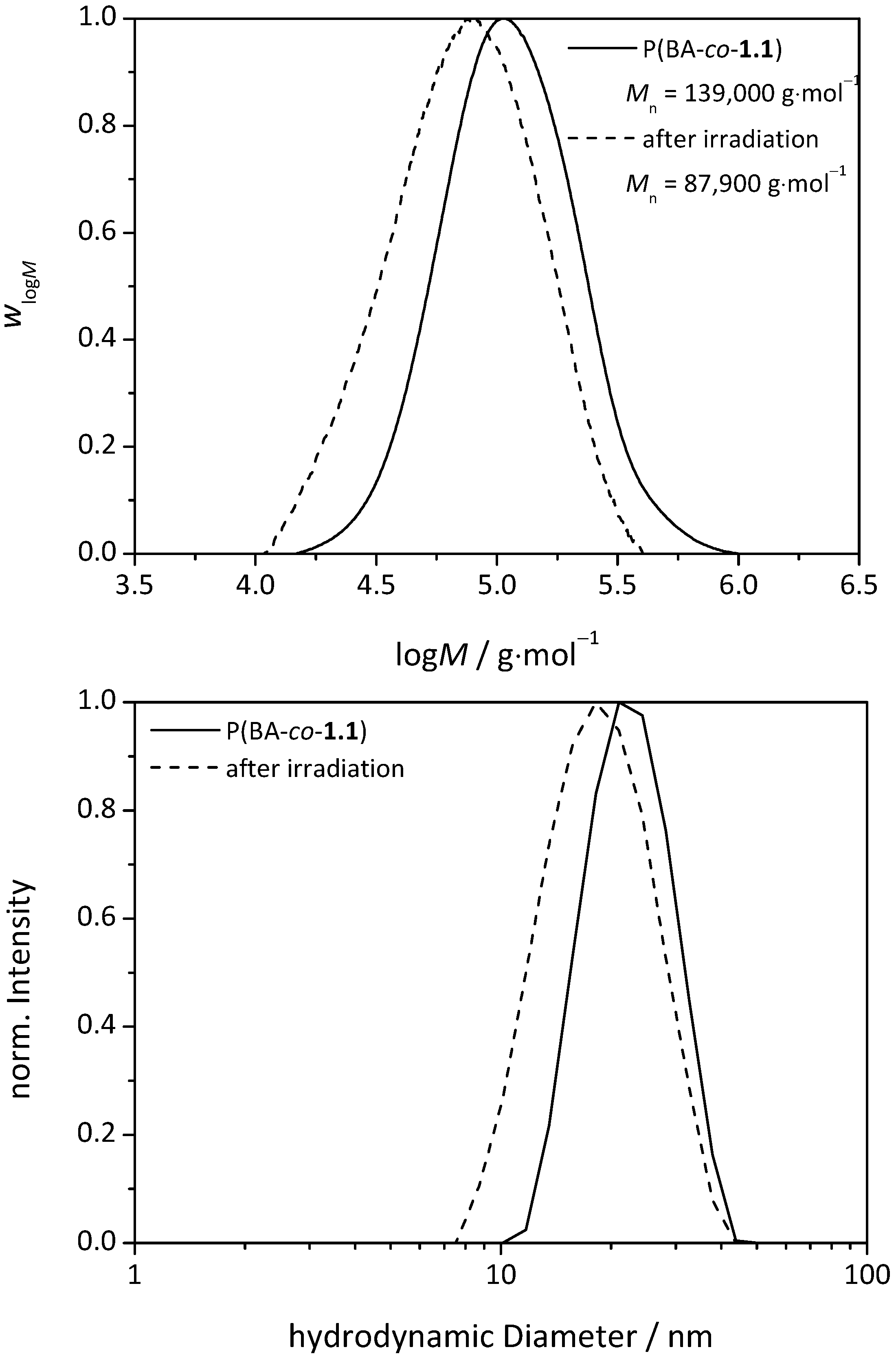
3.3.UV-Induced Cross-Linking of DMI Units in Star Polymers


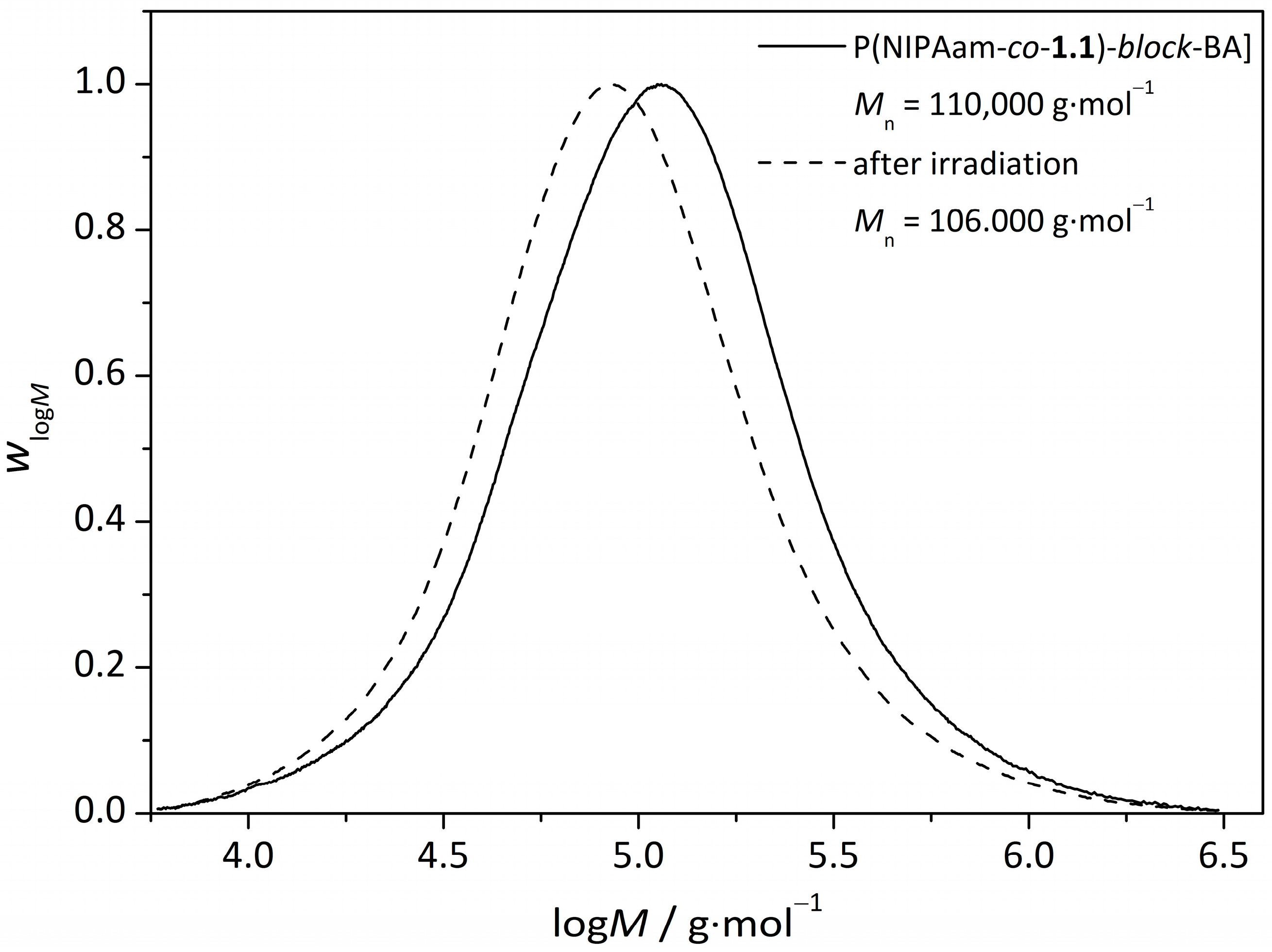
| Sample | Mn (before irradiation) g∙mol−1 | Mn (after irradiation) g∙mol−1 | |
|---|---|---|---|
| star(BA62-co-1.138) | 110,000 | 65,000 | 1.69 |
| star(BA92-co-1.18) | 280,000 | 180,000 | 1.56 |
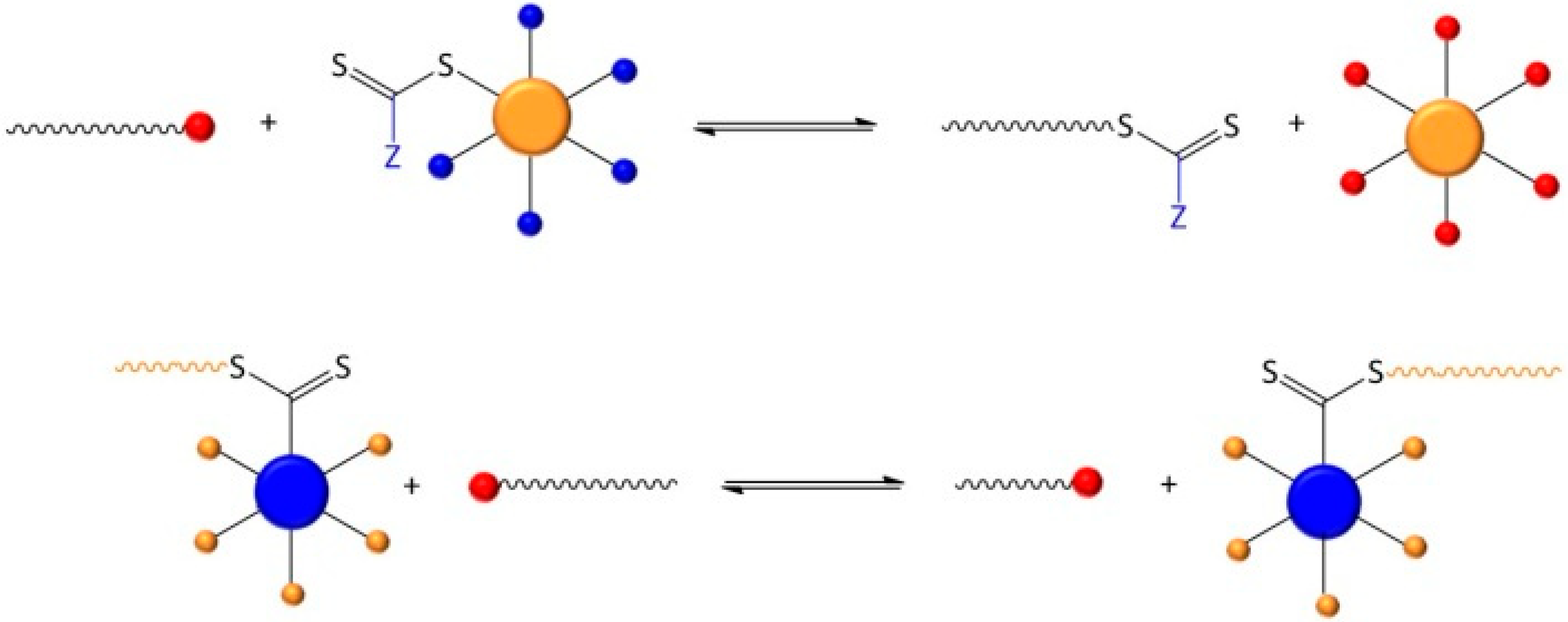
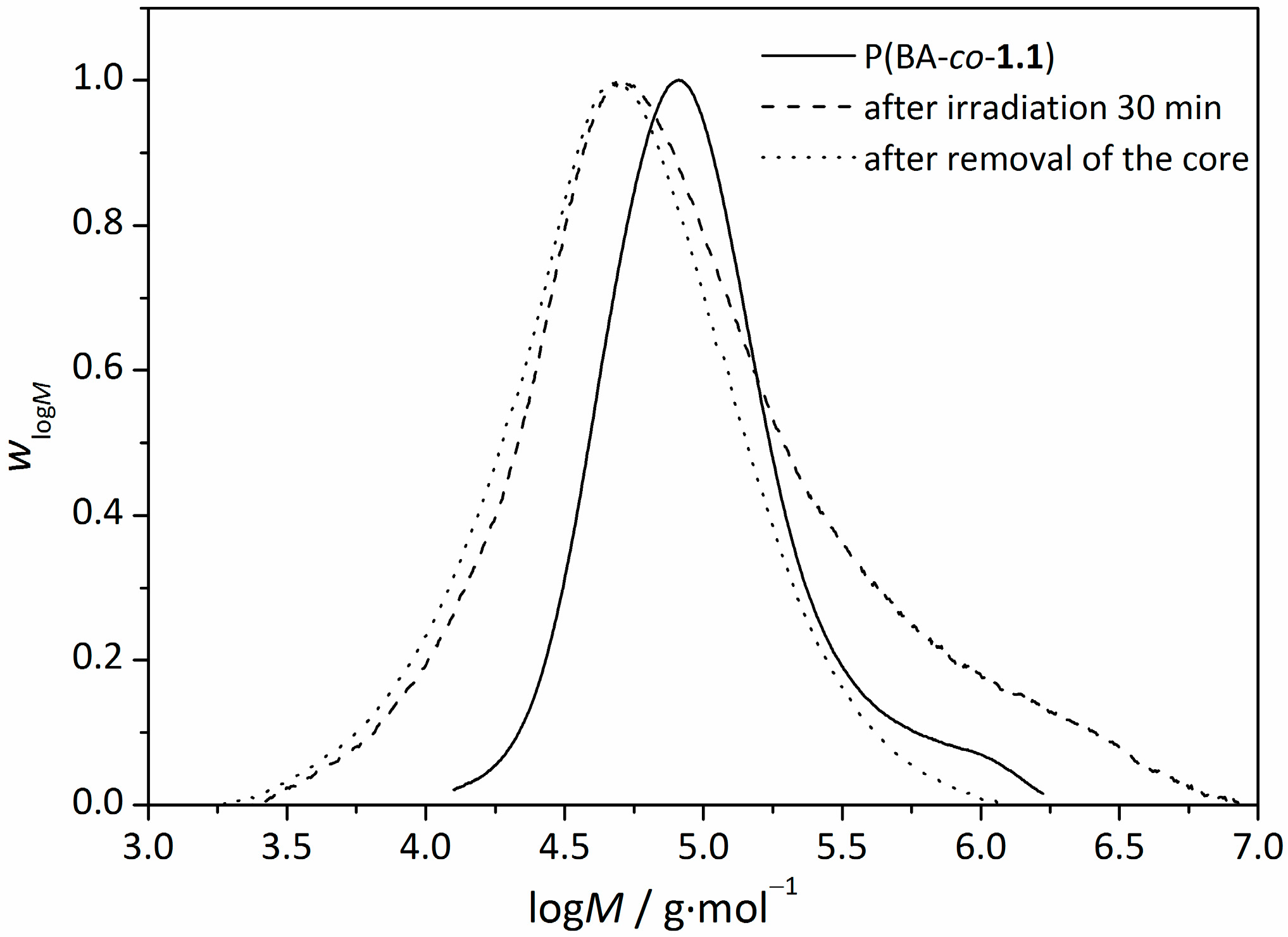
3.4. Removal of the RAFT-Core by Aminolysis
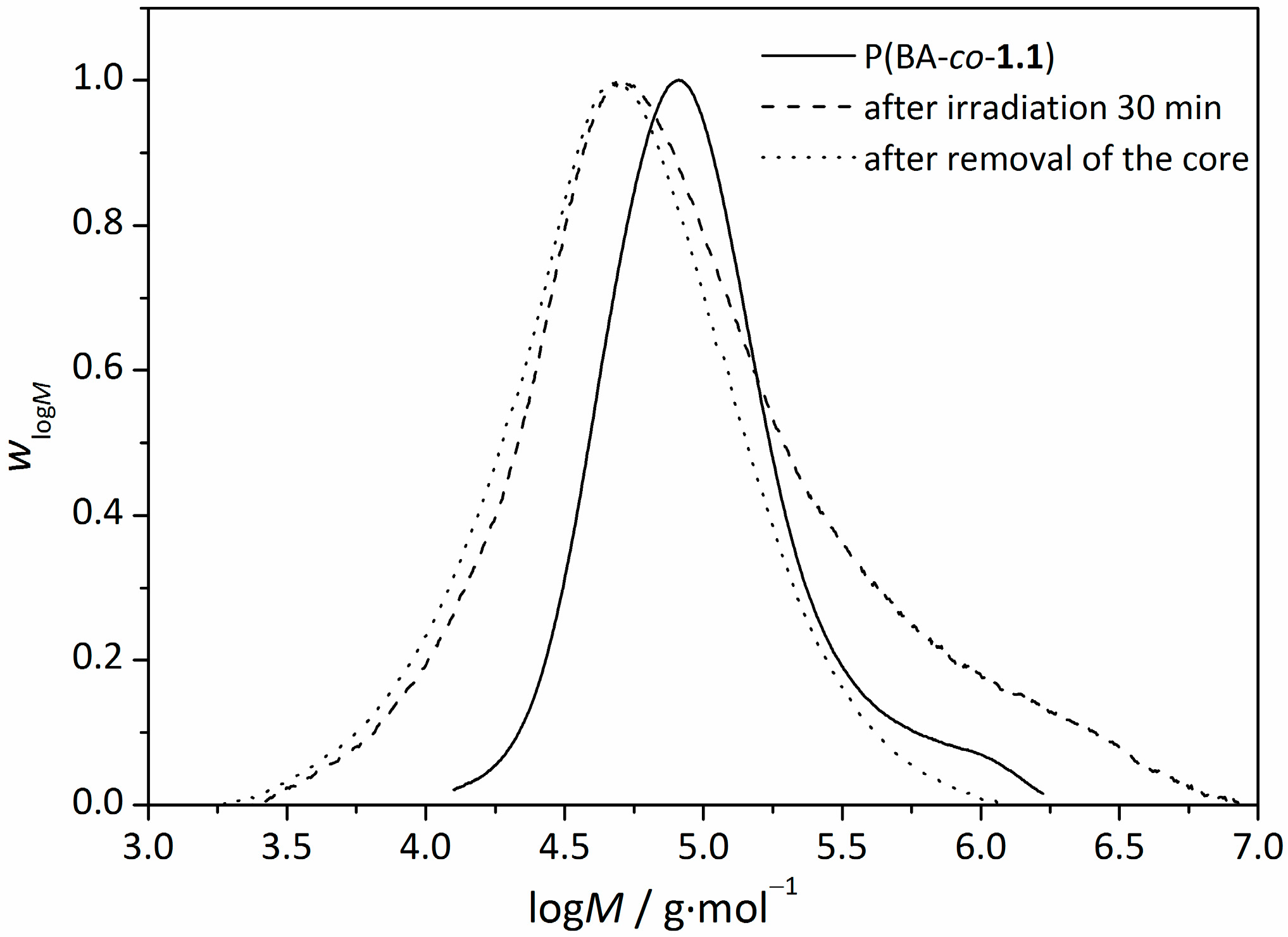
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interests
References
- Moad, G.; Rizzardo, E.; Thang, S.H. Living radical polymerization by the RAFT process. Aust. J. Chem. 2005, 58, 379–410. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living radical polymerization by the RAFT process—A first update. Aust. J. Chem. 2006, 59, 669–692. [Google Scholar]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living radical polymerization by the RAFT process—A second update. Aust. J. Chem. 2009, 62, 1402–1472. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living radical polymerization by the RAFT process—A third update. Aust. J. Chem. 2012, 65, 985–1076. [Google Scholar] [CrossRef]
- Boyer, C.; Stenzel, M.H.; Davis, T.P. Building Nanostructures using RAFT polymerization. J. Polym. Sci. A Polym. Chem. 2011, 49, 551–595. [Google Scholar] [CrossRef]
- Landfester, K.; Musyanovych, A.; Maila, V. From polymeric particles to multifunctional nanocapsules for biomedical applications using the miniemulsion process. J. Polym. Sci. A Polym. Chem. 2010, 48, 493–515. [Google Scholar] [CrossRef]
- Wang, Y.; Angelatos, A.S.; Caruso, F. Template synthesis of nanostructured materials via layer-by-layer. Chem. Mater. 2008, 20, 848–858. [Google Scholar] [CrossRef]
- Boyer, C.; Whittaker, M.R.; Nouvel, C.; Davis, T.P. Synthesis of hollow polymer nanocapsules exploiting gold nanoparticles as sacrificial templates. Macromolecules 2010, 43, 1792–1799. [Google Scholar] [CrossRef]
- McCormick, C.L.; Sumerlin, B.S.; Lokitz, B.S.; Stempka, J.E. RAFT-synthesized diblock and triblock copolymers: Thermally-induced supramolecular assembly in aqueous media. Soft Matter 2008, 4, 1760. [Google Scholar] [CrossRef]
- Xu, J.; Tao, L.; Boyer, C.; Lowe, A.B.; Davis, T.P. Facile Access to polymeric vesicular nanostructures: Remarkable ω-end group effects in cholesterol and pyrene functional (co)polymers. Macromolecules 2011, 44, 299–312. [Google Scholar] [CrossRef]
- Xu, J.; Liu, S. Polymeric nanocarriers possessing thermoresponsive coronas. Soft Matter 2008, 4, 1745. [Google Scholar] [CrossRef]
- Elsabahy, M.; Wooley, K.L. Strategies toward well-defined polymer nanoparticles inspired by nature: Chemistry versus versatility. J. Polym. Sci. A Polym. Chem. 2012, 50, 1869–1880. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Luehmann, H.P.; Sun, G.; Samarajeewa, S.; Zou, J.; Zhang, S.; Zhang, F.; Welch, M.J.; Liu, Y.; Wooley, K.L. Synthesis and in vivo pharmacokinetic evaluation of degradable shell cross-linked polymer nanoparticles with poly(carboxybetaine) versus poly(ethylene glycol) surface-grafted coatings. ACS Nano 2012, 6, 8970–8982. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Wei, H.; Wu, D.-Q.; Yang, B.; Chen, N.; Cheng, S.-X.; Zhang, X.-Z.; Zhuo, R.-X. Thermo-responsive shell cross-linked PMMA-b-P(NIPAAm-co-NAS) micelles for drug delivery. Int. J. Pharm. 2011, 420, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Flores, J.D.; Mccormick, C.L. Reversible imine shell cross-linked micelles from aqueous raft-synthesized thermoresponsive triblock copolymers as potential nanocarriers for “PH-triggered” drug release. Macromolecules 2011, 44, 1327–1334. [Google Scholar] [CrossRef]
- Gao, H. Development of star polymers as unimolecular containers for nanomaterials. Macromol. Rapid Commun. 2012, 33, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Liu, L.; Luo, Y.; Wang, X.; Liu, Y. Biotinylated thermoresponsive core cross-linked nanoparticles via RAFT polymerization and “click” chemistry. J. Colloid Interface Sci. 2011, 356, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Kafouris, D.; Patrickios, C.S. Synthesis and characterization of shell-cross-linked polymer networks and large-core star polymers: Effect of the volume of the cross-linking mixture. Eur. Polym. J. 2009, 45, 10–18. [Google Scholar] [CrossRef]
- Stenzel-Rosenbaum, M.; Davis, T.P.; Chen, V.; Fane, A.G. Star-polymer synthesis via radical reversible addition—Fragmentation chain-transfer polymerization. J. Polym. Sci. A Polym. Chem. 2001, 39, 2777–2783. [Google Scholar] [CrossRef]
- Stenzel, M.H.; Davis, T.P. Star polymer synthesis using trithiocarbonate functional β-cyclodextrin cores (reversible addition-fragmentation chain-transfer polymerization). J. Polym. Sci. A Polym. Chem. 2002, 40, 4498–4512. [Google Scholar] [CrossRef]
- Boschmann, D.; Mänz, M.; Pöppler, A.-C.; Sörensen, N.; Vana, P. Tracing arm-growth initiation in z-raft star polymerization by NMR: The impact of the leaving r-group on star topology. J. Polym. Sci. A Polym. Chem. 2008, 46, 7280–7286. [Google Scholar] [CrossRef]
- Mayadunne, R.T.A.; Jeffery, J.; Moad, G.; Rizzardo, E. Living free radical polymerization with reversible addition–fragmentation chain transfer (RAFT polymerization): Approaches to star polymers. Macromolecules 2003, 36, 1505–1513. [Google Scholar] [CrossRef]
- Boschmann, D.; Vana, P. Z-RAFT star polymerizations of acrylates: Star coupling via intermolecular chain transfer to polymer. Macromolecules 2007, 40, 2683–2693. [Google Scholar] [CrossRef]
- Boschmann, D.; Vana, P. Poly(vinyl acetate) and poly(vinyl propionate) star polymers via reversible addition fragmentation chain transfer (RAFT) polymerization. Polym. Bull. 2005, 53, 231–242. [Google Scholar] [CrossRef]
- Fröhlich, M.G.; Vana, P.; Zifferer, G. Shielding effects in polymer-polymer reactions. II. Reactions between linear and star-branched chains with up to six arms. J. Chem. Phys. 2007, 127, 164906. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, M.G.; Vana, P.; Zifferer, G. Shielding effects in polymer–polymer reactions, 1. Z-RAFt star polymerization of four-arm stars. Macromol. Theory Simul. 2007, 16, 610–618. [Google Scholar] [CrossRef]
- Fröhlich, M.G.; Nardai, M.M.; Förster, N.; Vana, P.; Zifferer, G. Shielding effects in polymer–polymer reactions, 3. Z-RAFT star polymerization under various solvent conditions. Polymer 2010, 51, 5122–5134. [Google Scholar] [CrossRef]
- Shen, W.; Qiu, Q.; Wang, Y.; Miao, M.; Li, B.; Zhang, T.; Cao, A.; An, Z. Hydrazine as a nucleophile and antioxidant for fast aminolysis of raft polymers in air. Macromol. Rapid Commun. 2010, 31, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Corten, C.; Görner, H.; Wolff, T.; Kuckling, D. Photodimers of N-alkyl-3,4-dimethylmaleimides—Product ratios and reaction mechanism. J. Photochem. Photobiol. A Chem. 2008, 198, 34–44. [Google Scholar] [CrossRef]
- Bouas-Laurent, H.; Desvergne, J.-P.; Castellan, A.; Lapouyade, R. Photodimerization of anthracenes in fluid solutions: (part 2) Mechanistic aspects of the photocycloaddition and of the photochemical and thermal cleavage. Chem. Soc. Rev. 2001, 30, 248–263. [Google Scholar] [CrossRef]
- Schütz, A.; Wolff, T. Regioselectivity in the photodimerization of 9-hydroxy-methylanthracene and 9-anthracene carboxylic acid esters in surfactant systems. J. Photochem. Photobiol. A Chem. 1997, 109, 251–258. [Google Scholar] [CrossRef]
- Menon, S.; Thekkayil, R.; Varghese, S.; Das, S. Photoresponsive soft materials: Synthesis and photophysical studies of a stilbene-based diblock copolymer. J. Polym. Sci. A Polym. Chem. 2011, 49, 5063–5073. [Google Scholar] [CrossRef]
- Atta, A.M.; El-Ghazawy, R.A.; Farag, R.K.; El-Kafrawy, A.F.; Abdel-Azim, A.-A.A. Crosslinked cinnamoyloxyethyl methacrylate and isooctyl acrylate copolymers as oil sorbers. Polym. Int. 2005, 54, 1088–1096. [Google Scholar] [CrossRef]
- Lendlein, A.; Jiang, H.; Jünger, O.; Langer, R. Light-induced shape-memory polymers. Nature 2005, 434, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, M.; Konishi, K.; Aida, T. A photocrosslinkable dendrimer consisting of a nucleobase. Chem. Lett. 2000, 29, 374–375. [Google Scholar] [CrossRef]
- Jiang, J.; Qi, B.; Lepage, M. Polymer micelles stabilization on demand through reversible photo-cross-linking. Macromolecules 2007, 40, 790–792. [Google Scholar] [CrossRef]
- Tietz, K.; Finkhäuser, S.; Samwer, K.; Vana, P. Stabilizing the microphase separation of block copolymers by controlled photo-crosslinking. Macromol. Chem. Phys. 2014, 215, 1563–1572. [Google Scholar] [CrossRef]
- Xu, J.; Boyer, C. Visible light photocatalytic thiol—Ene reaction: An elegant approach for fast polymer postfunctionalization and step-growth polymerization. Macromolecules 2015, 48, 520–529. [Google Scholar] [CrossRef]
- Singh, D.; Kuckling, D.; Choudhary, V.; Adler, H.-J.; Koul, V. Synthesis and characterization of poly(N-isopropylacrylamide) films by photopolymerization. Polym. Adv. Technol. 2006, 17, 186–192. [Google Scholar] [CrossRef]
- Vo, C.D.; Kuckling, D.; Adler, H.-P.; Schönhoff, M. Preparation of thermosensitive nanogels by photo-cross-linking. Colloid Polym. Sci. 2002, 409, 400–409. [Google Scholar] [CrossRef]
- Gupta, S.; Kuckling, D.; Kretschmer, K.; Choudhary, V.; Adler, H.-J. Synthesis and characterization of stimuli-sensitive micro- and nanohydrogels based on photocrosslinkable poly(dimethylaminoethyl methacrylate). J. Polym. Sci. A Polym. Chem. 2007, 45, 669–679. [Google Scholar] [CrossRef]
- Förster, N.; Pöppler, A.-C.; Stalke, D.; Vana, P. Photocrosslinkable star polymers via RAFT-copolymerizations with N-ethylacrylate-3,4-dimethylmaleimide. Polymers 2013, 5, 706–729. [Google Scholar] [CrossRef]
- Stenzel, M.H.; Barner-Kowollik, C.; Davis, T.P.; Dalton, H.M. Amphiphilic block copolymers based on poly(2-acryloyloxyethyl phosphorylcholine) prepared via RAFT polymerisation as biocompatible nanocontainers. Macromol. Biosci. 2004, 4, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Garay-Jimenez, J.C.; Gergeres, D.; Young, A.; Lim, D.V.; Turos, E. Physical properties and biological activity of poly(butyl acrylate-styrene) nanoparticle emulsions prepared with conventional and polymerizable surfactants. Nanomedicine 2009, 5, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Garay-Jimenez, J.C.; Young, A.; Gergeres, D.; Greenhalgh, K.; Turos, E. Methods for purifying and detoxifying sodium dodecyl sulfate-stabilized polyacrylate nanoparticles. Nanomedicine 2008, 4, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Cui, Y.; Levenson, R.M.; Chung, L.W.K.; Nie, S. In vivo cancer targeting and imaging with semiconductor quantum dots. Nat. Biotechnol. 2004, 22, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Asua, J.M.; Beuermann, S.; Buback, M.; Castignolles, P.; Charleux, B.; Gilbert, R.G.; Hutchinson, R.A.; Leiza, J.R.; Nikitin, A.N.; Vairon, J.-P.; et al. Critically evaluated rate coefficients for free-radical polymerization, 5. Macromol. Chem. Phys. 2004, 205, 2151–2160. [Google Scholar]
- Boschmann, D.; Drache, M.; Fröhlich, M.; Zifferer, G. Mechanism of z-raft star polymerization. Polym. Repr. 2008, 49, 189–190. [Google Scholar]
- Boschmann, D.; Edam, R.; Schoenmakers, P.J.; Vana, P. Z-RAFT star polymerization of styrene: Comprehensive characterization using size-exclusion chromatography. Polymer 2008, 49, 5199–5208. [Google Scholar] [CrossRef]
- Boschmann, D. Sternpolymere mittels RAFT-Polymerisation. Ph.D. Thesis, Georg-August-Universität Göttingen, Göttingen, Germany, May 2008. [Google Scholar]
- Yohannes, G.; Shan, J.; Jussila, M.; Nuopponen, M.; Tenhu, H.; Riekkola, M.-L. Characterisation of poly(N-isopropylacrylamide) by asymmetrical flow field-flow fractionation, dynamic light scattering, and size exclusion chromatography. J. Sep. Sci. 2005, 28, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Ebeling, B.; Eggers, S.; Hendrich, M.; Nitschke, A.; Vana, P. Flipping the pressure- and temperature-dependent cloud-point behavior in the cononsolvency system of poly(N-isopropylacrylamide) in water and ethanol. Macromolecules 2014, 47, 1462–1469. [Google Scholar] [CrossRef]
- Seifert, D.; Kipping, M.; Adler, H.-J.P.; Kuckling, D. A study of simple RAFT transfer agents for the polymerization of (meth-)acrylates and acrylamides. Macromol. Symp. 2007, 254, 386–391. [Google Scholar] [CrossRef]
- Vega-Rios, A.; Licea-Claveríe, A. Controlled synthesis of block copolymers containing N-isopropylacrylamide by reversible addition-fragmentation chain-transfer (RAFT) polymerization. J. Mex. Chem. Soc. 2011, 55, 21–32. [Google Scholar]
- Neises, B.; Steglich, W. Einfaches Verfahren zur veresterung von carbonsäuren. Angew. Chem. 1978, 90, 556–557. [Google Scholar] [CrossRef]
- Buback, M.; Junkers, T.; Vana, P. Laser single pulse initiated RAFT polymerization for assessing chain-length dependent radical termination kinetics. Macromol. Rapid Commun. 2005, 26, 796–802. [Google Scholar] [CrossRef]
- Beuermann, S.; Paquet, D.A.; McMinn, J.H.; Hutchinson, R.A. Determination of free-radical propagation rate coefficients of butyl, 2-ethylhexyl, and dodecyl acrylates by pulsed-laser polymerization. Macromolecules 1996, 29, 4206–4215. [Google Scholar] [CrossRef]
- Mckee, J.R.; Ladmiral, V.; Niskanen, J.; Tenhu, H.; Armes, S.P. Synthesis of sterically-stabilized polystyrene latexes using well-defined thermoresponsive poly(N-isopropylacrylamide) macromonomers. Macromolecules 2011, 44, 7692–7703. [Google Scholar]
- Sinkel, C.; Greiner, A.; Agarwal, S. A polymeric drug depot based on 7-(2'-methacryloyloxyethoxy)-4-methylcoumarin copolymers for photoinduced release of 5-fluorouracil designed for the treatment of secondary cataracts. Macromol. Chem. Phys. 2010, 211, 1857–1867. [Google Scholar] [CrossRef]
- Vana, P.; Davis, T.P.; Barner-Kowollik, C. Kinetic analysis of reversible addition fragmentation chain transfer (RAFT) polymerizations: Conditions for inhibition, retardation, and optimum living polymerization. Macromol. Theory Simulations 2002, 11, 823–835. [Google Scholar] [CrossRef]
- De Schryver, F.C.; Boens, N.; Smets, G. Photochemistry of nonconjugated bichromophoric systems. Photopolymerization of N,N-alkylenebis(dimethylmaleimides). J. Am. Chem. Soc. 1974, 96, 6463–6469. [Google Scholar] [CrossRef]
- Mänz, M. Wege zu makromolekularen Nanotransportern aus Sternpolymeren mittels RAFT-Polymerisation; Cuvillier Vlag: Göttingen, Germany, 2010. [Google Scholar]
- Quinn, J.F.; Barner, L.; Barner-kowollik, C.; Rizzardo, E.; Davis, T.P. Reversible addition-fragmentation chain transfer polymerization initiated with ultraviolet radiation. Macromolecules 2002, 35, 7620–7627. [Google Scholar] [CrossRef]
- Štěpánek, M.; Uchman, M.; Procházka, K. Self-assemblies formed by four-arm star copolymers with amphiphilic diblock arms in aqueous solutions. Polymer 2009, 50, 3638–3644. [Google Scholar] [CrossRef]
- Qiu, X.-P.; Winnik, F.M. Facile and efficient one-pot transformation of RAFT polymer end groups via a mild aminolysis/michael addition sequence. Macromol. Rapid Commun. 2006, 27, 1648–1653. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Förster, N.; Schmidt, S.; Vana, P. Tailoring Confinement: Nano-Carrier Synthesis via Z-RAFT Star Polymerization. Polymers 2015, 7, 695-716. https://doi.org/10.3390/polym7040695
Förster N, Schmidt S, Vana P. Tailoring Confinement: Nano-Carrier Synthesis via Z-RAFT Star Polymerization. Polymers. 2015; 7(4):695-716. https://doi.org/10.3390/polym7040695
Chicago/Turabian StyleFörster, Nadja, Sonja Schmidt, and Philipp Vana. 2015. "Tailoring Confinement: Nano-Carrier Synthesis via Z-RAFT Star Polymerization" Polymers 7, no. 4: 695-716. https://doi.org/10.3390/polym7040695