Development of a Biocompatible Layer-by-Layer Film System Using Aptamer Technology for Smart Material Applications


Abstract
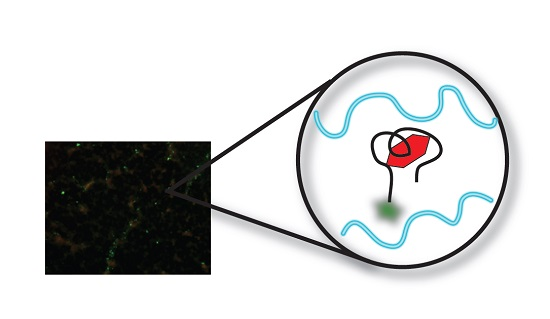
:
1. Introduction
2. Experimental Section
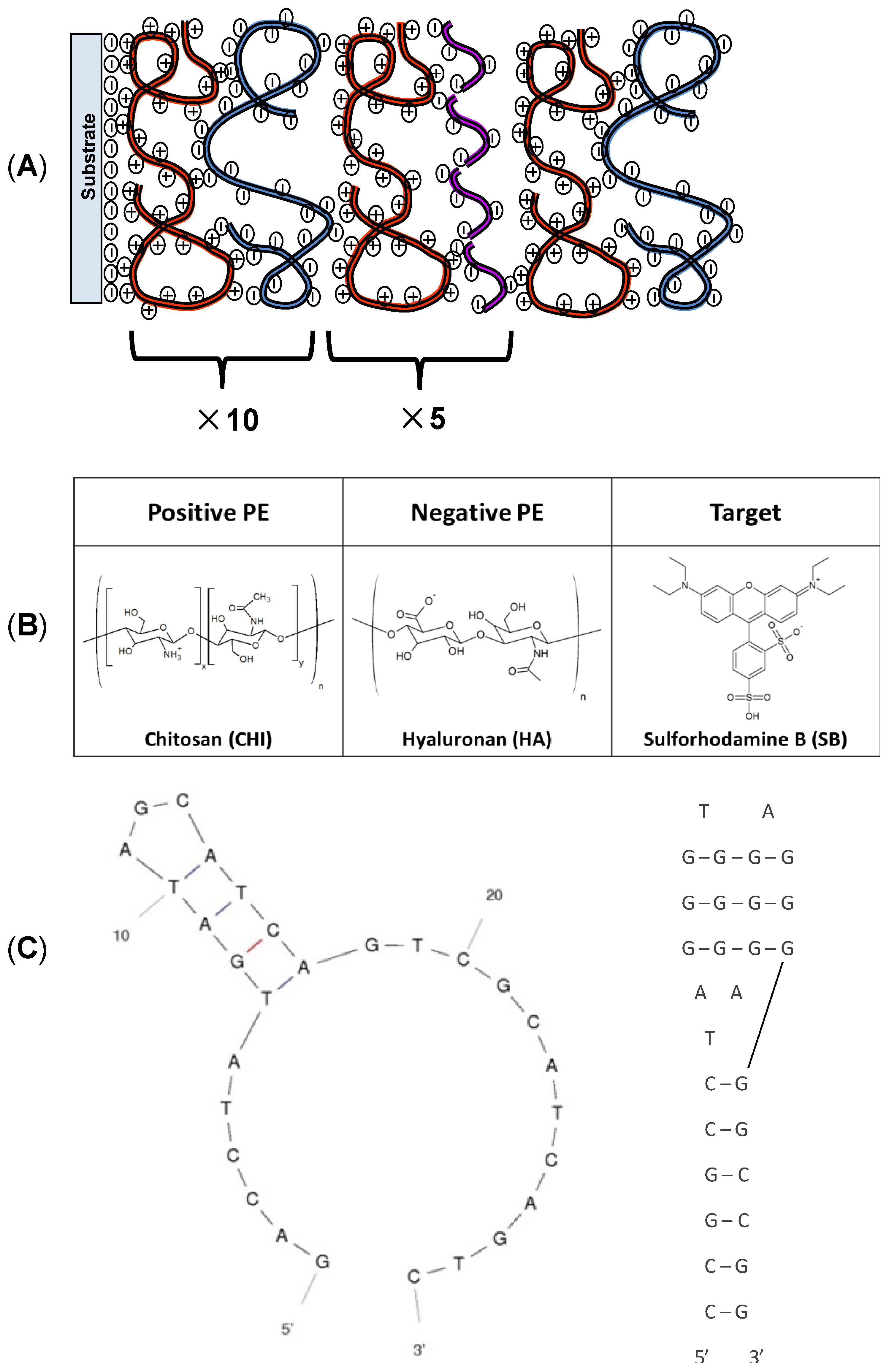
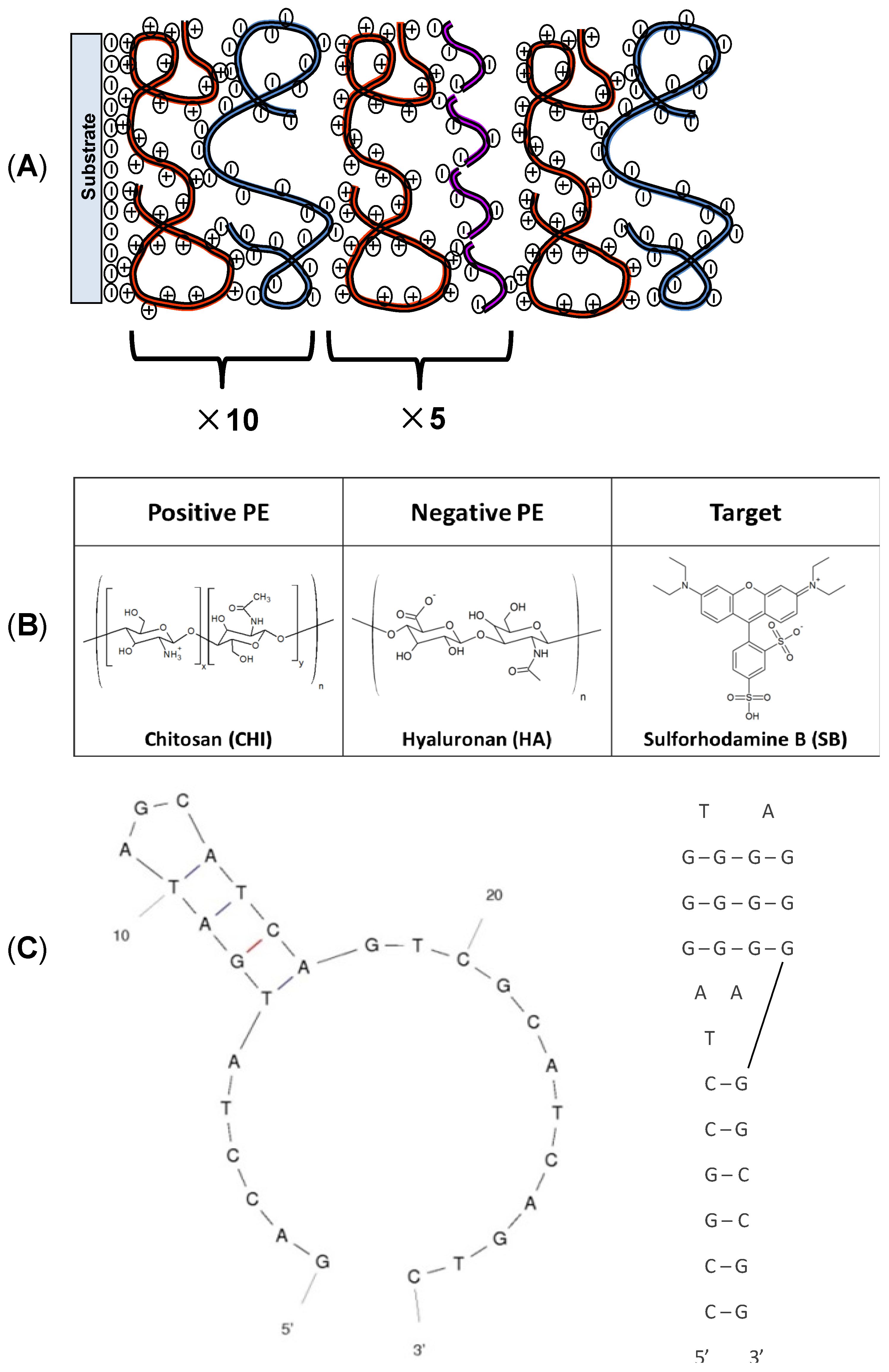
2.1. Materials
2.2. Film Deposition
2.3. Microscopy
2.3.1. Atomic Force Microscopy (AFM)
2.3.2. Scanning Electron Microscopy (SEM)
2.3.3. Fluorescent Microscopy (FM)
3. Results and Discussion
3.1. Physical Characterization of Chitosan (CHI)/Hyaluronan (HA)/DNA Films

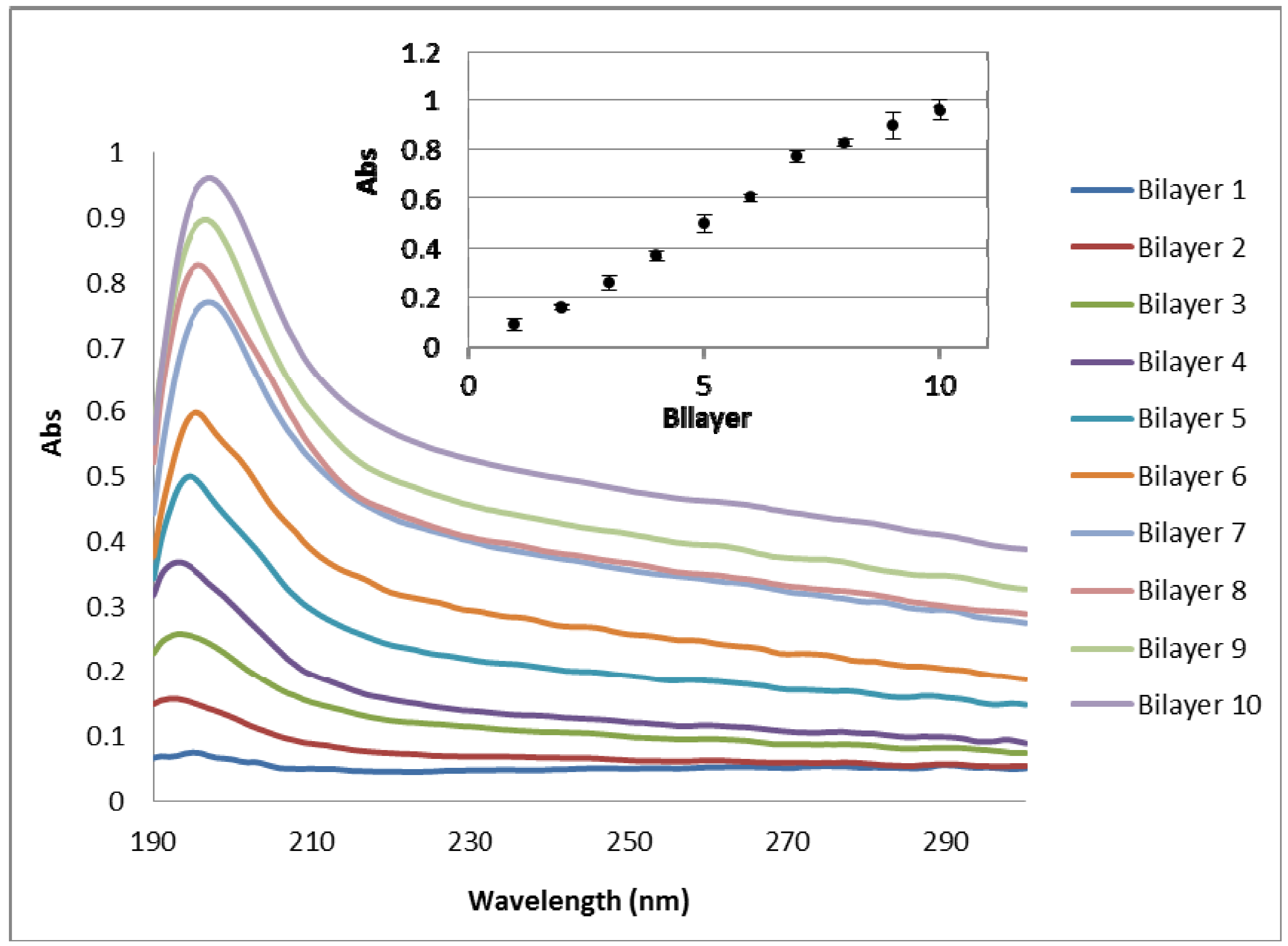
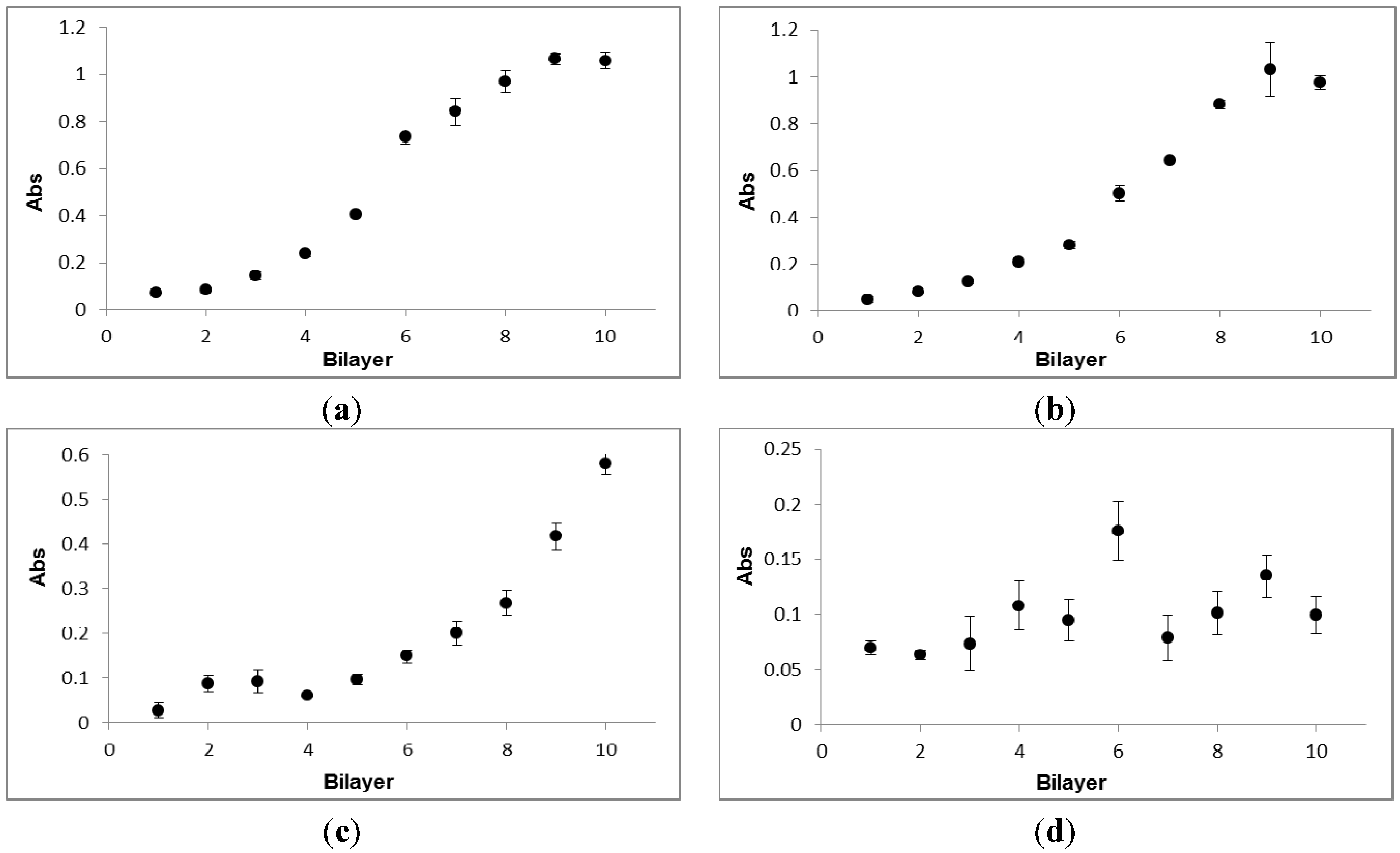
3.2. Investigation of Chitosan/Hyaluronan (CHI/HA) Film Growth by UV-Vis Spectrophotometry

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Technique (s) | Deposition Time (min) | PE Volume (mL) | PE Change Frequency | Rinse Times | Rinse volume (mL) | Rinse Change Frequency | PE Molecular weight (g/mol) |
|---|---|---|---|---|---|---|---|---|
| Foster | • UV-Vis | 15 | 10 | 20 layers | R1 = 10 s | R1 = 10 | 20 layers | HA = 1,580,000 |
| • Hand-dipped | R2 = 5 s | R2 = 10 | CHI = 135,000 | |||||
| Mulligan | • AFM | 15 | Not specified | Not specified | R1 = 1 min | R1 = 350 | 3 layers | HA = 163,000 |
| • Ellipsometry | R2 = 5 min | R2 = 150 | CHI = 50,000 | |||||
| Kujawa * | 20 | 10 (total) | Each use | Not specified | 10 (total) | Each use | HA1 = 360,000 | |
| • SPR | HA2 = 31,000 | |||||||
| • AFM | CHI1 = 160,000 | |||||||
| CHI2 = 30,000 | ||||||||
| Picart | • ATR-FTIR | 15 | 12 | Not specified | R1 = dip R2 = 2.5 min R3 = 2.5 min | R1 = 350 R2 = 40 R3 = 40 | 6 layers | HA = 400,000 CHI1 = 5,000 CHI2 = 100,000 |
| • CLSM | ||||||||
| • QCM | ||||||||
| • Auto-dipped | ||||||||
| Schneider | • AFMCLSMAuto-dipped | 15 | 12 | Not specified | R1 = dip | R1 = 350 | 6 layers | HA = 400,000 CHI = 5,000 |
| • AFMCLSMAuto-dipped | R2 = 2.5 min | R2= 40 | ||||||
| • AFMCLSMAuto-dipped | R3 = 2.5 min | R3= 40 | ||||||
| Richert * | • OWLS | 15 | 15 or 0.5 (QCM-D) | Not specified or each use (QCM-D) | R1 = dip R2 = 6 min R3 = 6 min or 10s (QCM-D) | R1 = 350 R2 = 40 R3 = 40 or 0.5 (QCM-D) | 3 layers or each use (QCM-D) | HA = 400,000 |
| • AFM | CHI1 = 110,000 | |||||||
| • QCM-D | CHI2 = 270,000 | |||||||
| • Auto-dipped | CHI3 = 460,000 |
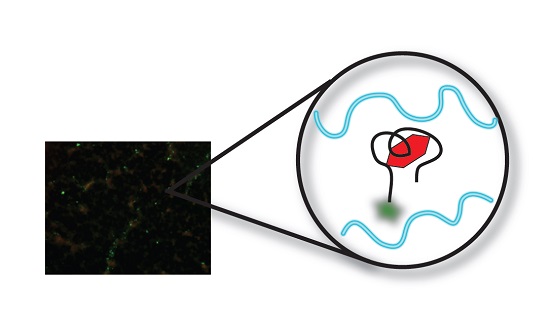
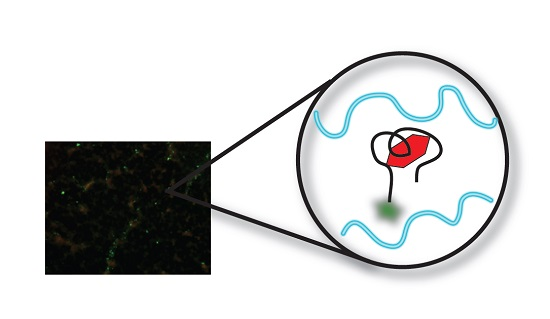
3.3. Fluorescent Microscopy (FM) Analysis of Aptamer-Target Binding
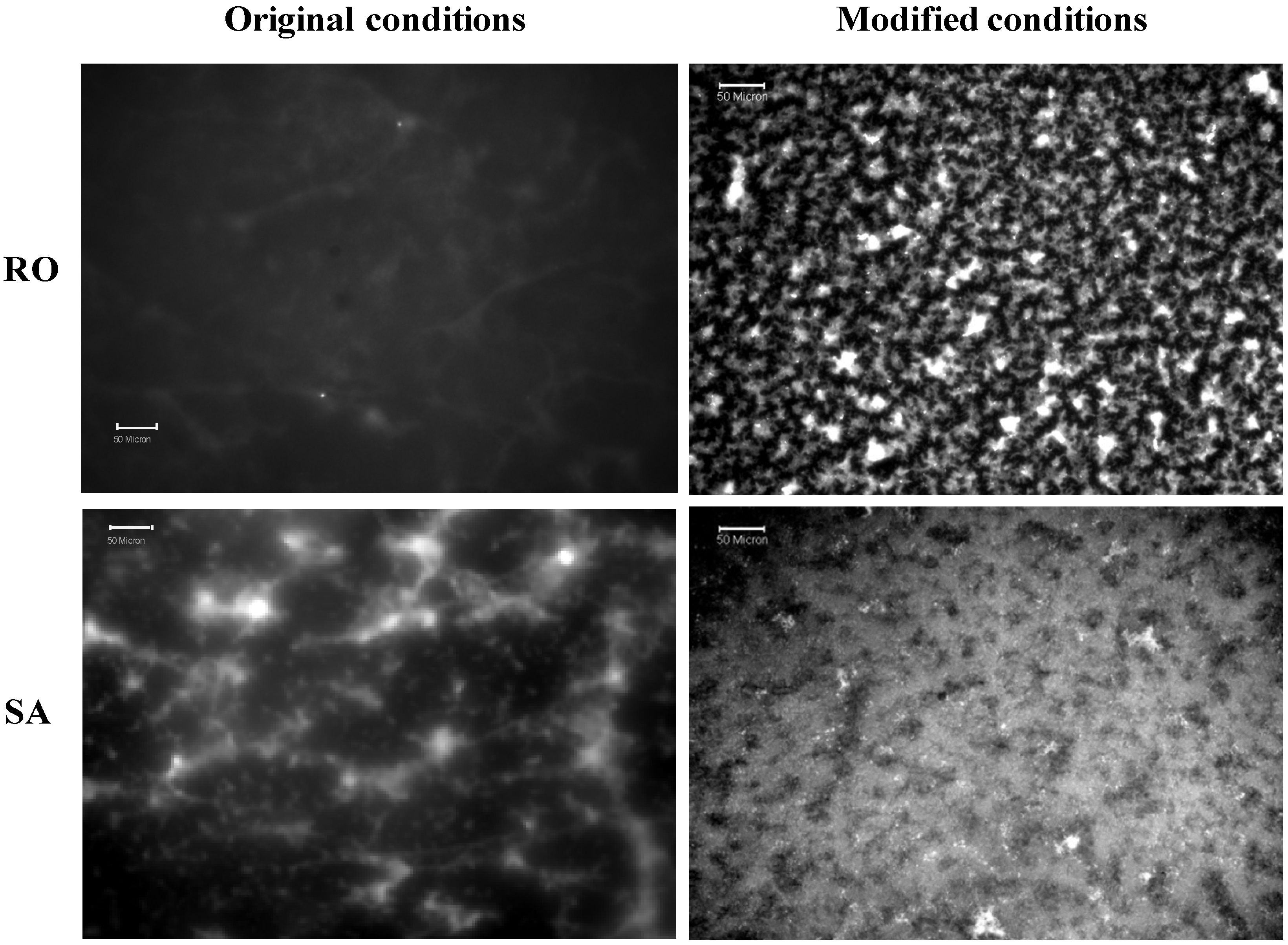
3.3.1. Original Method vs. Modified Method

3.3.2. Target Dye Binding—Original Method
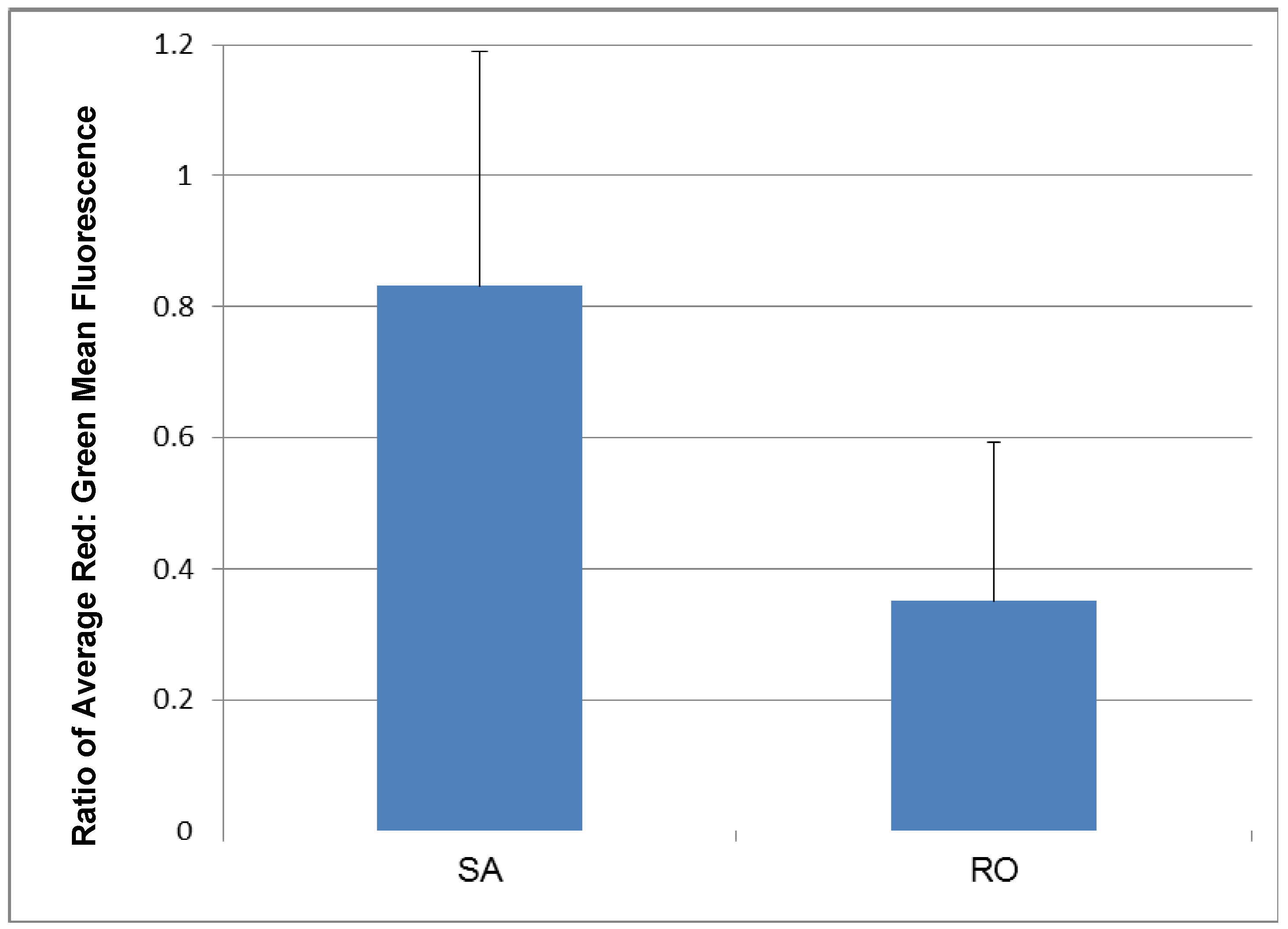
3.3.3. Target Dye Binding—Modified Method
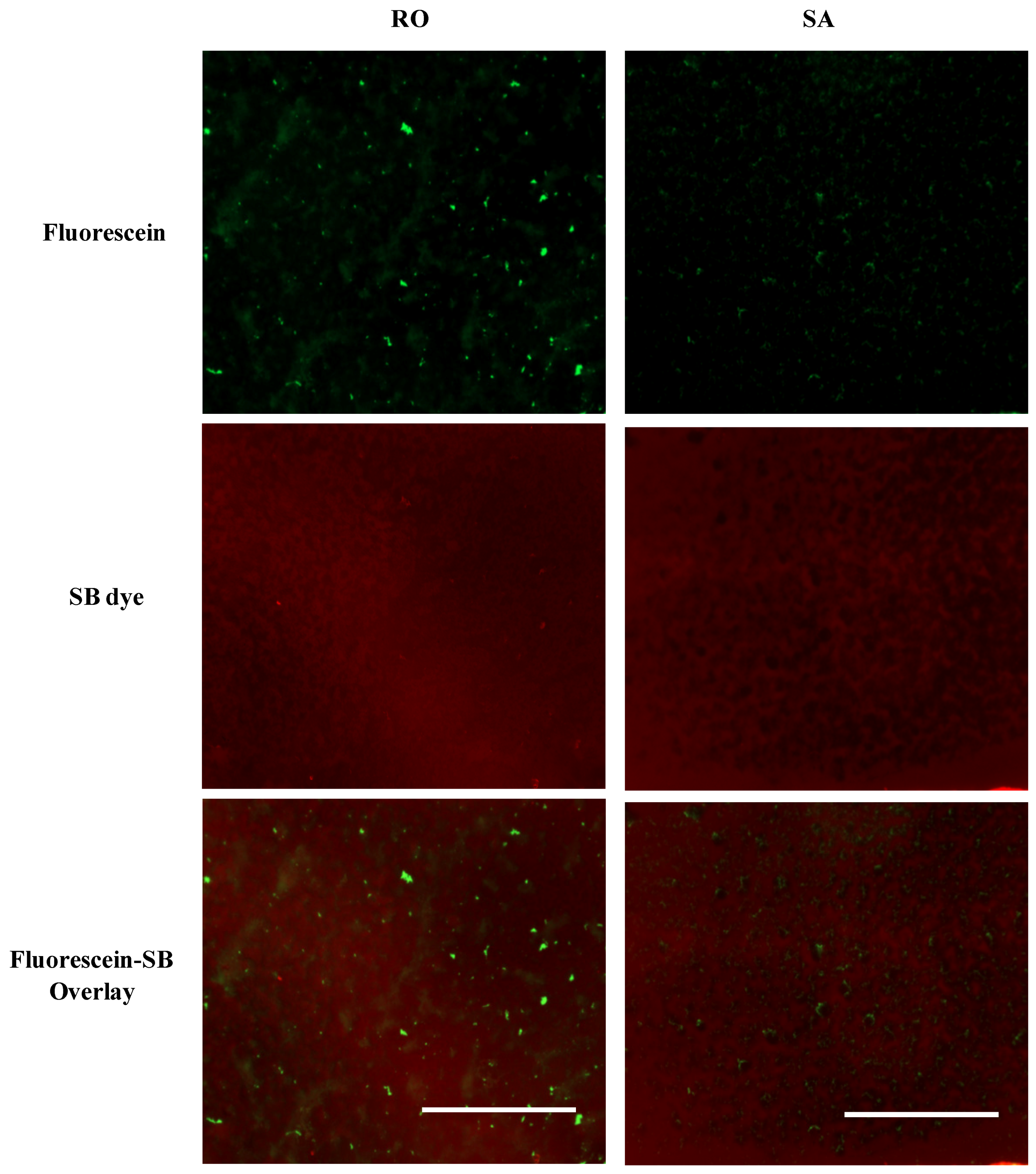

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Decher, G. Fuzzy nanoassemblies: Towards layered polymeric multicomposites. Science 1997, 277, 1232–1237. [Google Scholar] [CrossRef]
- Zasadzinski, J.A.; Viswanathan, R.; Madsen, L.; Garnaes, J.; Schwartz, D.K. Langmuir-blodgett films. Science 1994, 263, 1726–1733. [Google Scholar]
- Crawford, N.F.; Leblanc, R.M. CdSe and CdSe (ZnS) quantum dots in 2D: A Langmuir monolayer approach. Coord. Chem. Rev. 2014, 263, 13–24. [Google Scholar] [CrossRef]
- Ariga, K.; Yamauchi, Y.; Mori, T.; Hill, J.P. 25th anniversary article: What can be done with the langmuir-blodgett method? Recent developments and its critical role in material science. Adv. Mater. 2013, 25, 6477–6512. [Google Scholar]
- Ariga, K.; Yamauchi, Y.; Rydzek, G.; Ji, Q.; Yonamine, Y.; Wu, K.C.W.; Hill, J.P. Layer-by-layer nanoarchitectonics: Invention, innovation, and evolution. Chem. Lett. 2014, 43, 36–68. [Google Scholar] [CrossRef]
- Yan, Y.; Bjornmalm, M.; Caruso, F. Assembly of layer-by-layer particles and their interactions with biological systems. Chem. Mater. 2014, 26, 452–460. [Google Scholar] [CrossRef]
- Deshmukh, P.K.; Ramani, K.P.; Singh, S.S.; Tekade, A.R.; Chatap, V.K.; Patil, G.B.; Bari, S.B. Stimuli-sensitive layer-by-layer (LBL) self-assembly systems: Targeting and biosensory applications. J. Control. Release 2013, 166, 294–306. [Google Scholar] [CrossRef]
- Ariga, K.; Ji, Q.; Hill, J.P.; Bando, Y.; Aona, M. Forming nanomaterials as layered functional structures toward materials nanoarchitectonics. NPG Asia Mater. 2012, 4. [Google Scholar] [CrossRef]
- Li, C.; Ma, C.; Wang, F.; Xi, Z.; Wang, Z.; Deng, Y.; He, N. Preparation and biomedical applications of core-shell silica/magnetic nanoparticle composites. J. Nanosci. Nanotechnol. 2012, 12, 2964–2972. [Google Scholar]
- Joseph, N.; Ahmadiannamini, P.; Hoogenboom, R.; Vankelecom, I.F.J. Layer-by-layer preparation of polyelectrolyte multilayer membranes for separation. Polym. Chem. 2013, 5, 1817–1831. [Google Scholar]
- Kharlampieva, E.; Sukhishvili, S.A. Hydrogen-bonded layer-by-layer polymer films. J. Macromol. Sci. CPolym. 2006, 46, 377–395. [Google Scholar]
- Sievers, T.K.; Vergin, A.; Mohwald, H.; Kurth, D.G. Thin film of cross-linked metallo-supramolecular coordination polyelectrolytes. Langmuir 2007, 23, 12179–12184. [Google Scholar] [CrossRef]
- Kotov, N.A. Layer-by-layer self-assembly: The contribution of hydrophobic interactions. Nanostruct. Mater. 1999, 12, 789–796. [Google Scholar] [CrossRef]
- Schoeler, B.; Kumaraswamy, G.; Caruso, F. Investigation of the influence of polyelectrolyte charge density on the growth of multilayer thin films prepared by the layer-by-layer technique. Macromolecules 2002, 35, 889–897. [Google Scholar] [CrossRef]
- Lavalle, P.; Picart, C.; Mutterer, J.; Gergely, C.; Reiss, H.; Voegel, J.C.; Senger, B.; Scaaf, P. Modeling the buildup of polyelectrolyte multilayer films having exponential growth. J. Phys. Chem. 2004, 108, 635–648. [Google Scholar]
- Lvov, Y.; Decher, G.; Mohwald, H. Assembly, structural characterization, and thermal behavior of layer-by-layer deposited ultrathin films of poly(vinyl sulfate) and poly(allylamine). Langmuir 1993, 9, 481–486. [Google Scholar] [CrossRef]
- Mesquita, J.P.; Donnici, C.L.; Pereira, F.V. Biobased nanocomposites from layer-by-layer assembly of cellulose nanowhiskers with chitosan. Biomacromolecules 2010, 11, 473–480. [Google Scholar]
- Richert, L.; Lavalle, P.; Payan, E.; Shu, X.Z.; Prestwich, G.D.; Stoltz, J.F.; Schaaf, P.; Voegel, J.C.; Picart, C. Layer by layer buildup of polysaccharide films: physical chemistry and cellular adhesion aspects. Langmuir 2004, 20, 448–458. [Google Scholar] [CrossRef]
- Elbert, D.L.; Herbert, C.B.; Hubbell, J.A. Thin polymer layers formed by polyelectrolyte multilayer techniques on biological surfaces. Langmuir 1999, 15, 5355–5362. [Google Scholar] [CrossRef]
- Picart, C.; Lavalle, P.; Hubert, P.; Cusinier, F.J.G.; Decher, G.; Schaaf, P.; Voegel, J.C. Buildup mechanism for poly(l-lysine)/hyaluronic acid films onto a solid surface. Langmuir 2001, 17, 7414–7424. [Google Scholar] [CrossRef]
- Porcel, C.; Lavalle, P.; Decher, G.; Senger, B.; Voegel, J.C.; Schaaf, P. Influence of the polyelectrolyte molecular weight on exponentially growing multilayer films in the linear regime. Langmuir 2007, 23, 1898–1904. [Google Scholar]
- Picart, C.; Gergely, C.; Arntz, Y.; Voegel, J.C.; Schaaf, P.; Cuisinier, F.J.G.; Senger, B. Measurement of film thickness up to several hundreds of nanometers using optical waveguide lightmode spectroscopy. Biosens. Bioelectron. 2004, 20, 553–561. [Google Scholar] [CrossRef]
- Shiratori, S.S.; Rubner, M.F. pH-dependent thickness behavior of sequentially adsorbed layers of weak polyelectrolytes. Macromolecules 2000, 33, 4213–4219. [Google Scholar] [CrossRef]
- Bieker, P.; Schonhoff, M. Linear and exponential growth regimes of multilayers of weak polyelectrolytes in dependence on pH. Macromolecules 2010, 43, 5052–5059. [Google Scholar] [CrossRef]
- Salomaki, M.; Vinokurov, I.A.; Kankare, J. Effect of temperature on the buildup of polyelectrolyte multilayers. Langmuir 2005, 21, 11232–11240. [Google Scholar] [CrossRef]
- Wilson, C.; Szostak, J.W. Isolation of a fluorophore-specific DNA aptamer with weak redox activity. Chem. Biol. 1998, 5, 609–617. [Google Scholar] [CrossRef]
- Schneider, A.; Richert, L.; Francius, G.; Voegel, J.C.; Picart, C. Elasticity, biodegradability and cell adhesive properties of chitosan/hyaluronan multilayer films. Biomed. Mater. 2007, 2, 45–51. [Google Scholar] [CrossRef]
- Kujawa, P.; Moraille, P.; Sanchez, J.; Badia, A.; Winnik, F.M. Effect of molecular weight on the exponential growth and morphology of hyaluronan/chitosan multilayers: A surface plasmon resonance spectroscopy and atomic force microscopy investigation. J. Am. Chem. Soc. 2005, 127, 9224–9234. [Google Scholar] [CrossRef]
- Picart, C.; Schneider, A.; Etienne, O.; Mutterer, J.; Schaaf, P.; Egles, C.; Jessel, N.; Voegel, J.C. Controlled degradability of polysaccharide multilayer films in vitro and in vivo. Adv. Funct. Mater. 2005, 15, 1771–1780. [Google Scholar] [CrossRef]
- Zhu, Z.; Wu, C.; Liu, H.; Zou, Y.; Zhang, X.; Kang, H.; Yang, C.J.; Tan, W. An aptamer cross-linked hydrogel as a colorimetric platform for visual detection. Angew. Chem. 2010, 122, 1070–1074. [Google Scholar]
- Farokhzad, O.C.; Cheng, J.; Teply, B.A.; Sherifi, I.; Jon, S.; Kantoff, P.W.; Richie, J.P.; Langer, R. Targeted nanoparticle-aptamer bioconjugates for cancer therapy in vivo. PNAS 2006, 103, 6315–6320. [Google Scholar] [CrossRef]
- Cao, Z.; Tong, R.; Mishra, A.; Xu, W.; Wong, G.C.L.; Cheng, J.; Lu, Y. Reversible cell-specific drug delivery with aptamer-functionalized liposomes. Angew. Chem. 2009, 48, 6494–6498. [Google Scholar] [CrossRef]
- Wu, Y.; Sefah, K.; Liu, H.; Wang, R.; Tan, W. DNA aptamer-micelle as an efficient detection/delivery vehicle toward cancer cells. PNAS 2010, 107, 5–10. [Google Scholar] [CrossRef]
- So, H.; Won, K.; Kim, Y.H.; Kim, B.; Ryu, B.H.; Na, P.S.; Kim, H.; Lee, J.O. Single-walled carbon nanotube biosensors using aptamers as molecular recognition elements. J. Am. Chem. Soc. 2005, 127, 11906–11907. [Google Scholar]
- Mastronardi, E.; Foster, A.; Zhang, X.; DeRosa, M.C. Smart materials based on DNA aptamers: Taking aptasensing to the next level. Sensors 2014, 14, 3156–3171. [Google Scholar] [CrossRef]
- Sultan, Y.; Walsh, R.; Monreal, C.; DeRosa, M. Preparation of functional aptamers films using layer-by-layer self-assembly. Biomacromolecules 2009, 10, 1149–1154. [Google Scholar] [CrossRef]
- Sultan, Y.; DeRosa, M. Target binding influences permeability in aptamer-polyelectrolyte microcapsules. Small 2011, 7, 1219–1226. [Google Scholar] [CrossRef]
- Malile, B.; Chen, J.I.L. Morphology-based plasmonic nanoparticle sensors: Controlling etching kinetics with target-responsive permeability gate. J. Am. Chem. Soc. 2013, 135, 16042–16045. [Google Scholar] [CrossRef]
- Du, Y.; Chen, C.; Li, B.; Zhou, M.; Wang, E.; Dong, S. Layer-by-layer electrochemical biosensor with aptamer-appended active polyelectrolyte multilayer for sensitive protein determination. Biosens. Bioelectron. 2010, 25, 1902–1907. [Google Scholar] [CrossRef]
- Ren, K.; Ji, J.; Shen, J. Construction and enzymatic degradation of multilayered poly-l-lysine/DNA films. Biomaterials 2006, 27, 1152–1159. [Google Scholar] [CrossRef]
- Gu, B.X.; Xu, C.X.; Zhu, G.P.; Liu, S.Q.; Chen, L.Y.; Wang, M.L.; Zhu, J.J. Layer by layer immobilized horseradish peroxidase on zinc oxide nanorods for biosensing. J. Phys. Chem. B 2009, 113, 6553–6557. [Google Scholar] [CrossRef]
- He, P.; Hu, N.; Zhou, G. Assembly of electroactive layer-by-layer films of hemoglobin and polycationic poly(diallyldimethylammonium). Biomacromolecules 2002, 3, 139–146. [Google Scholar] [CrossRef]
- Mulligan, K.; Jakubek, Z.J.; Johnston, L.J. Supported lipid bilayers on biocompatible polysaccharide multilayers. Langmuir 2011, 27, 14352–14359. [Google Scholar] [CrossRef]
- Fujii, N.; Fujimoto, K.; Michinobu, T.; Akada, M.; Hill, J.P.; Shiratori, S.; Argia, K.; Shigehara, K. The simplest layer-by-layer assembly structure: Best paired polymer electrolytes with one charge per main chain carbon atom for multilayered thin films. Macromolecules 2010, 43, 3947–3955. [Google Scholar] [CrossRef]
- Crouzier, T.; Boudou, T.; Picart, C. Polysaccharide-based polyelectrolyte multilayers. Curr. Opin. Colloid Interface Sci. 2010, 15, 417–426. [Google Scholar] [CrossRef]
- Wu, T.; Zivanovic, S. Determination of the degree of acetylation (DA) of chitin and chitosan by an improved first derivative UV method. Carbohydr. Polym. 2008, 73, 248–253. [Google Scholar] [CrossRef]
- Liu, D.; Wei, Y.; Yao, P.; Jiang, L. Determination of the degree of acetylation of chitosan by UV spectrophotometry using dual standards. Carbohydr. Res. 2006, 341, 782–785. [Google Scholar] [CrossRef]
- Hubsch, E.; Ball, V.; Senger, B.; Decher, G.; Voegel, J.C.; Schaaf, P. Controlling the growth regime of polyelectrolyte multilayer films: Changing from exponential to linear growth by adjusting the composition of polyelectrolyte mixtures. Langmuir 2004, 20, 1980–1985. [Google Scholar] [CrossRef]
- Porcel, C.; Lavalle, P.; Ball, V.; Decher, G.; Senger, B.; Voegel, J.C.; Schaaf, P. From exponential to linear growth in polyelectrolyte multilayers. Langmuir 2006, 22, 4376–4383. [Google Scholar] [CrossRef]
- Michel, M.; Izquierdo, A.; Decher, G.; Voegel, J.C.; Schaaf, P.; Ball, V. Layer by layer self-assembled polyelectrolyte multilayers with embedded phospholipid vesicles obtained by spraying: Integrity of the vesicles. Langmuir 2005, 21, 7854–7859. [Google Scholar] [CrossRef]
- Ghostine, R.A.; Markarian, M.Z.; Schlenoff, J.B. Asymmetric growth in polyelectrolyte multilayers. J. Am. Chem. Soc. 2013, 135, 7636–7646. [Google Scholar]
- White, R.J.; Rowe, A.A.; Plaxco, K.W. Re-engineering aptamers to support reagentless, self-reporting electrochemical sensors. Analyst 2010, 135, 589–594. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Foster, A.; DeRosa, M.C. Development of a Biocompatible Layer-by-Layer Film System Using Aptamer Technology for Smart Material Applications. Polymers 2014, 6, 1631-1654. https://doi.org/10.3390/polym6051631
Foster A, DeRosa MC. Development of a Biocompatible Layer-by-Layer Film System Using Aptamer Technology for Smart Material Applications. Polymers. 2014; 6(5):1631-1654. https://doi.org/10.3390/polym6051631
Chicago/Turabian StyleFoster, Amanda, and Maria C. DeRosa. 2014. "Development of a Biocompatible Layer-by-Layer Film System Using Aptamer Technology for Smart Material Applications" Polymers 6, no. 5: 1631-1654. https://doi.org/10.3390/polym6051631