Structure and Morphology Control in Thin Films of Conjugated Polymers for an Improved Charge Transport

Abstract
:1. Introduction
2. The Morphological and Structural Features Influencing the Charge Transport in Films
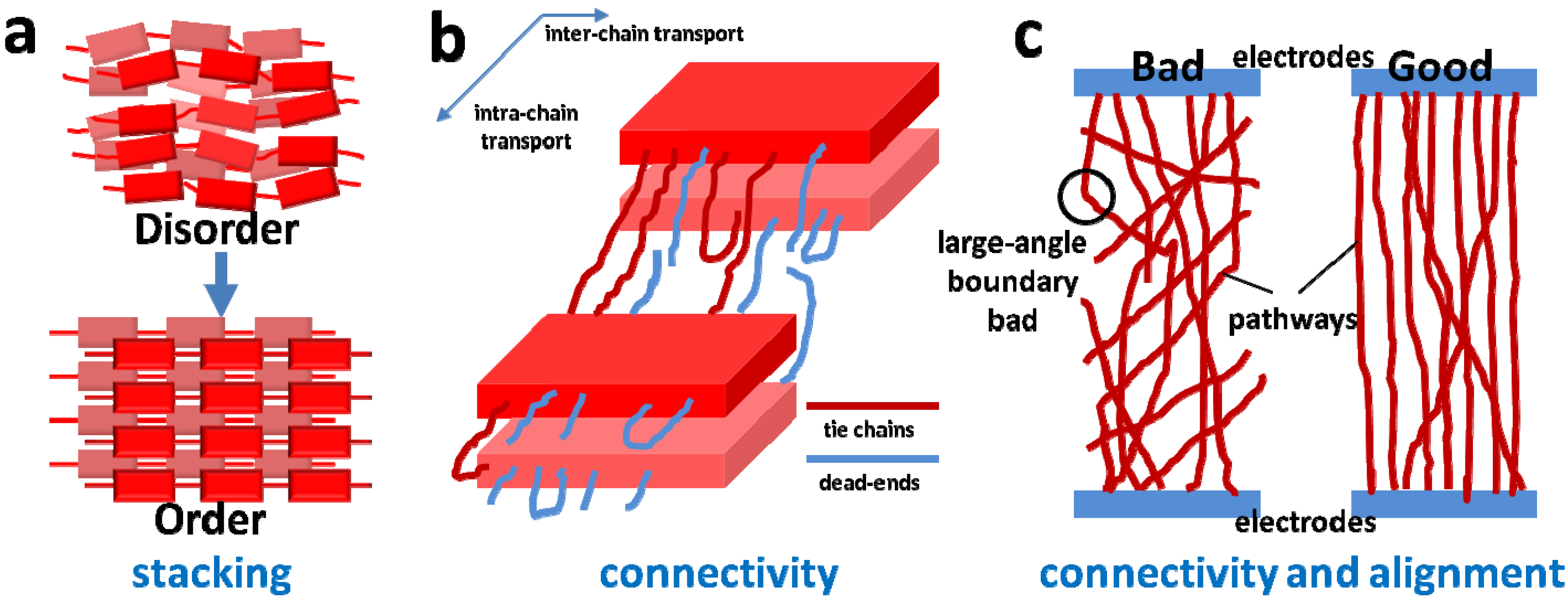
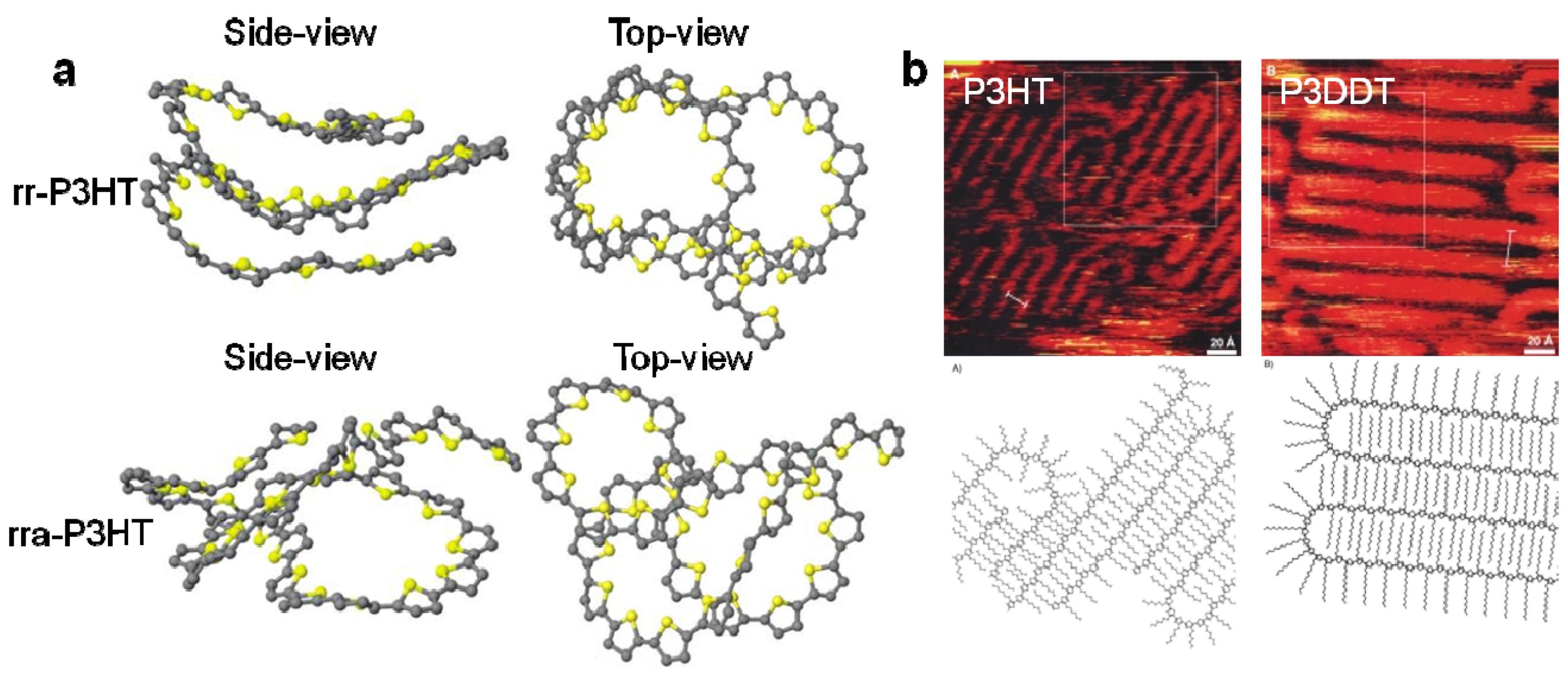
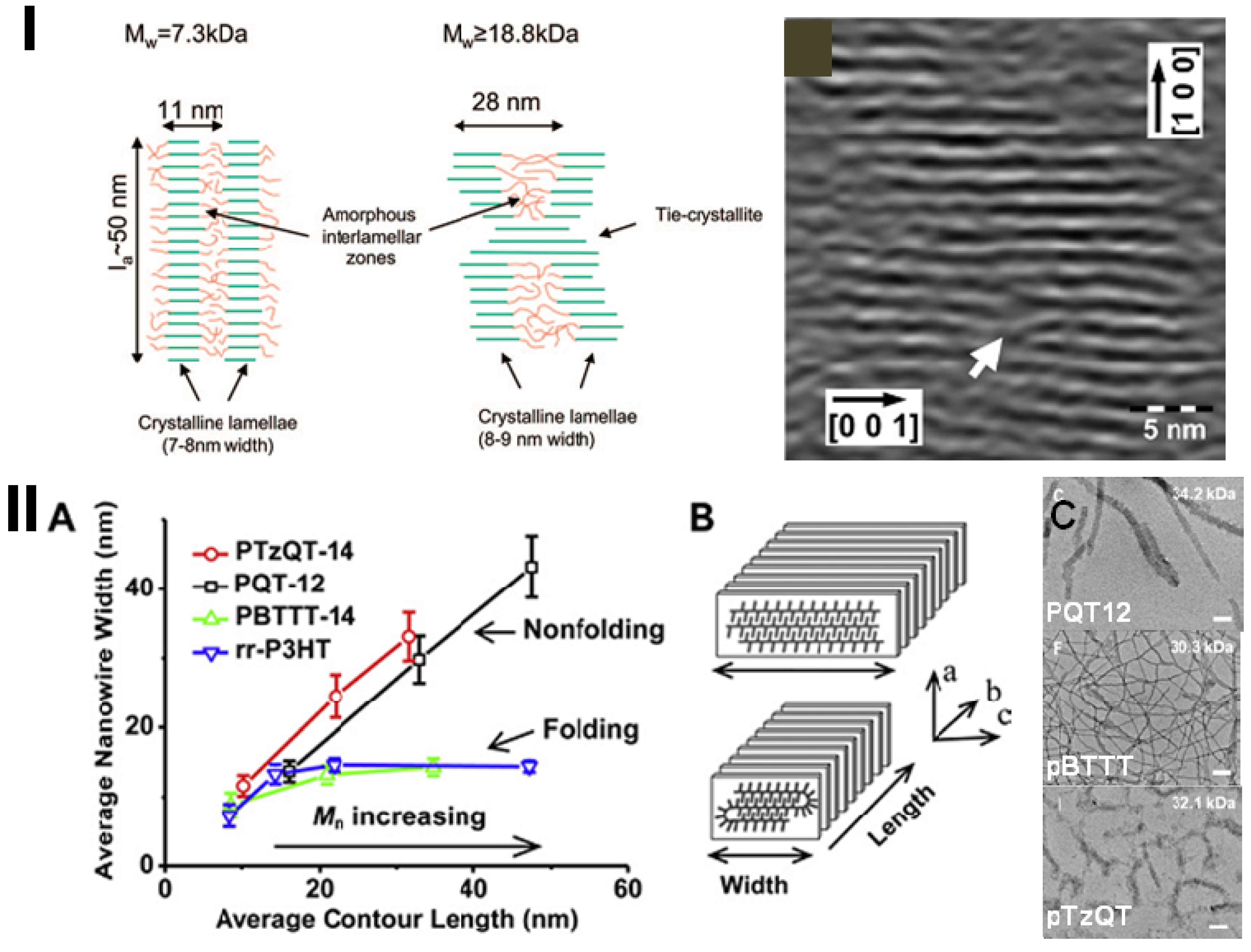
2.1. The Order of Crystalline Regions
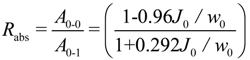
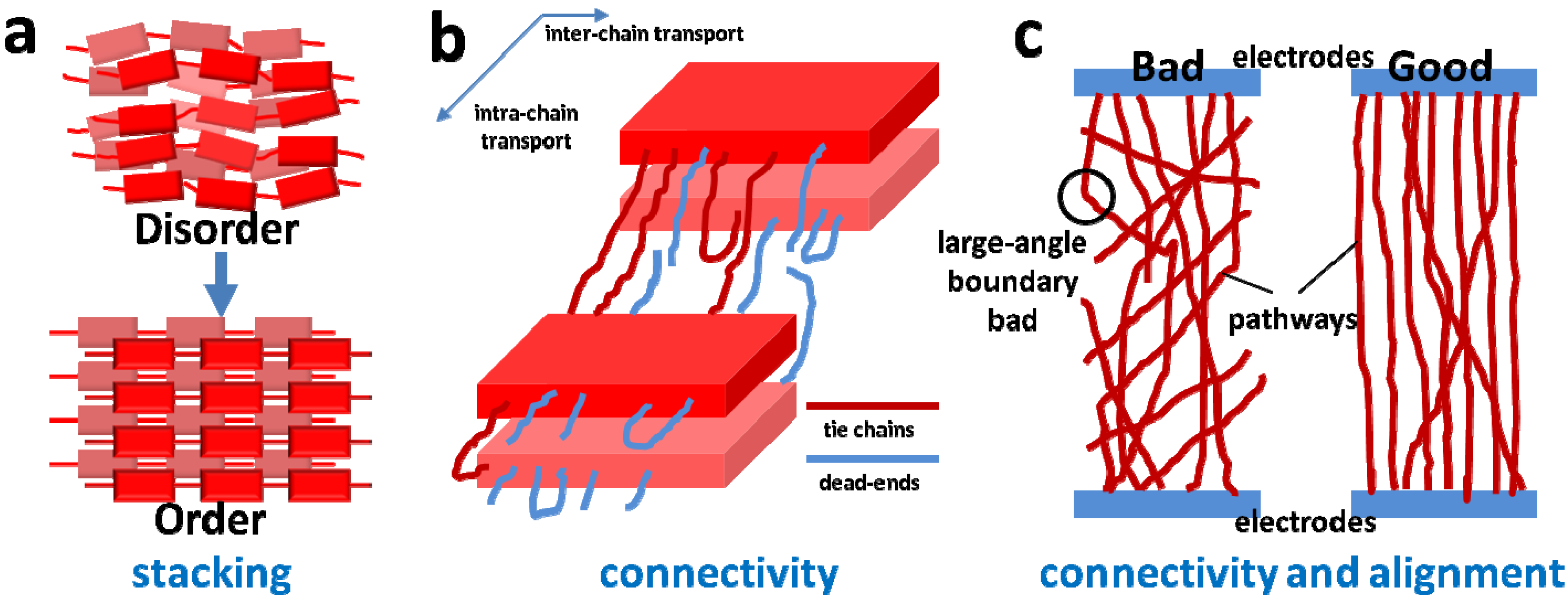
2.2. Connectivity between Crystalline Regions
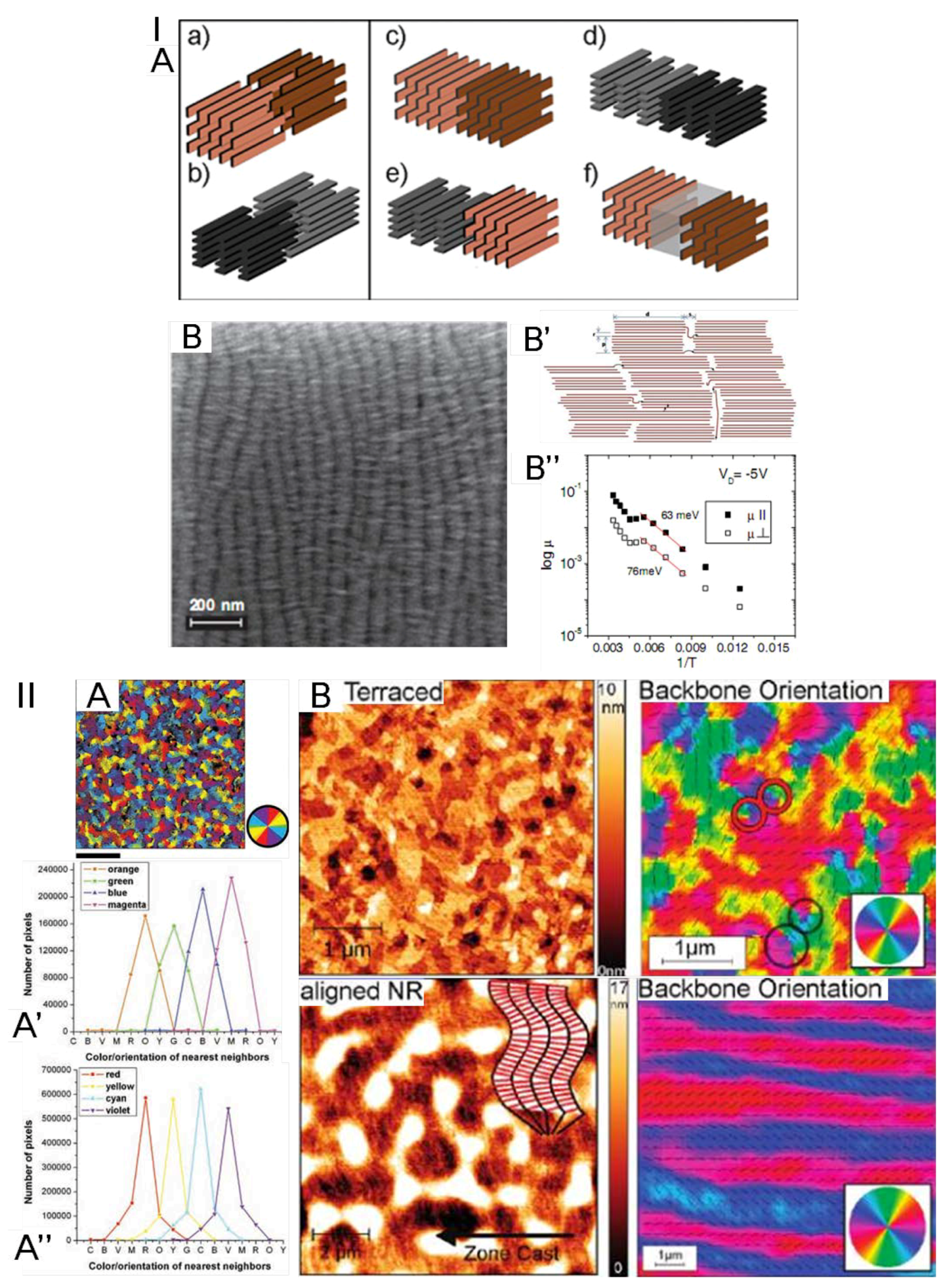
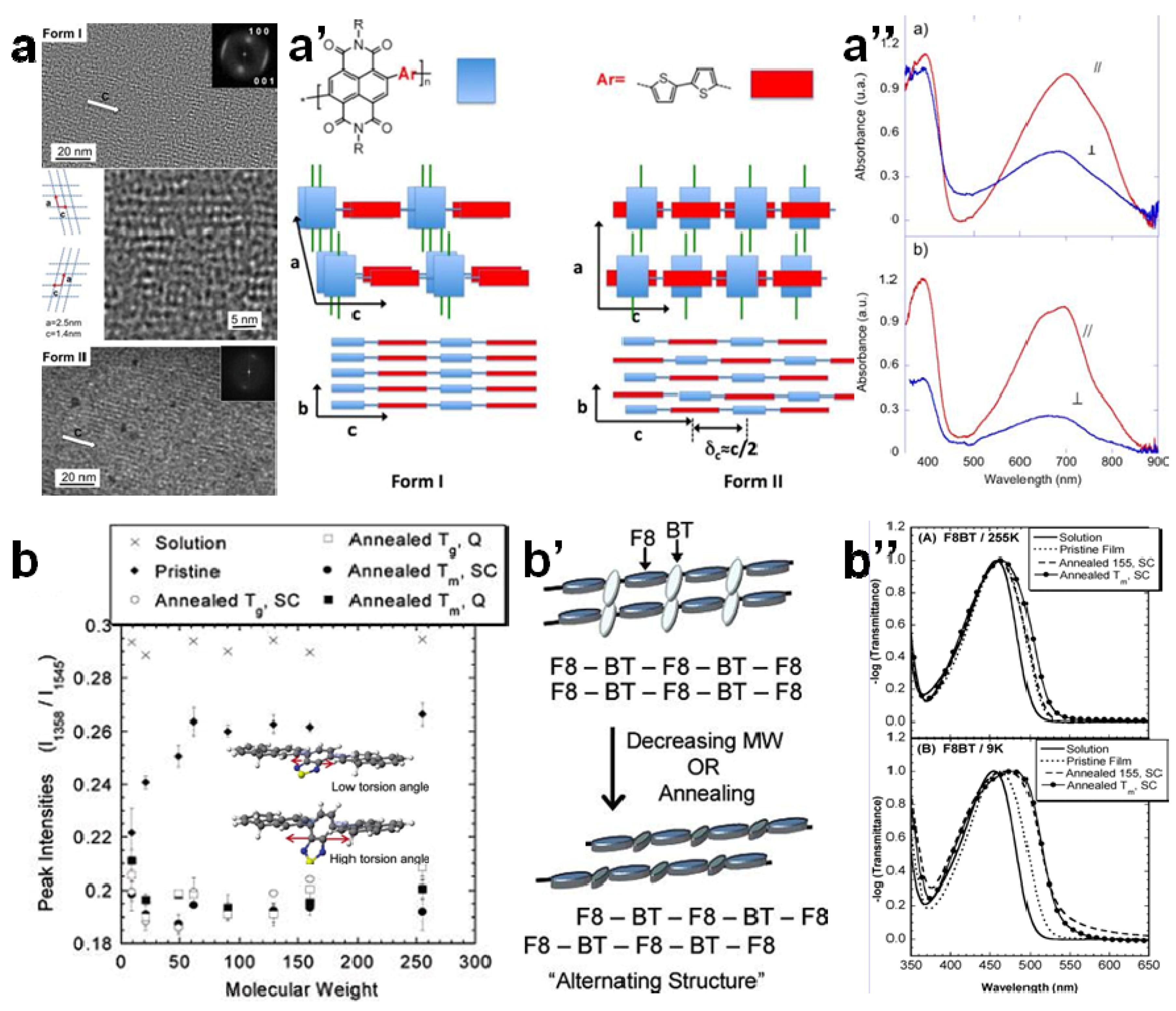
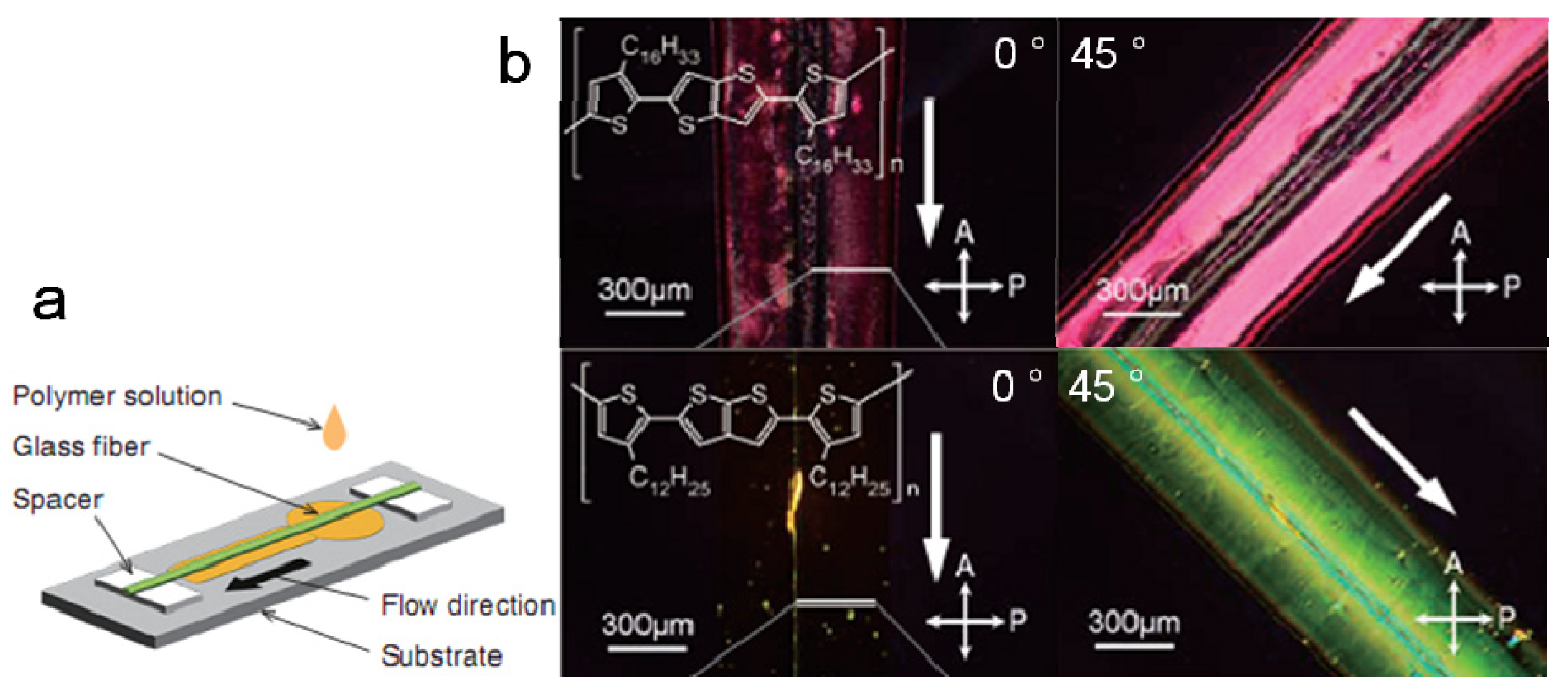
2.3. Crystallinity and Crystallite Alignment

3. The Effects of Conjugation Structures of Semiconducting Polymers on Their Electronic Properties, Structures and Morphology
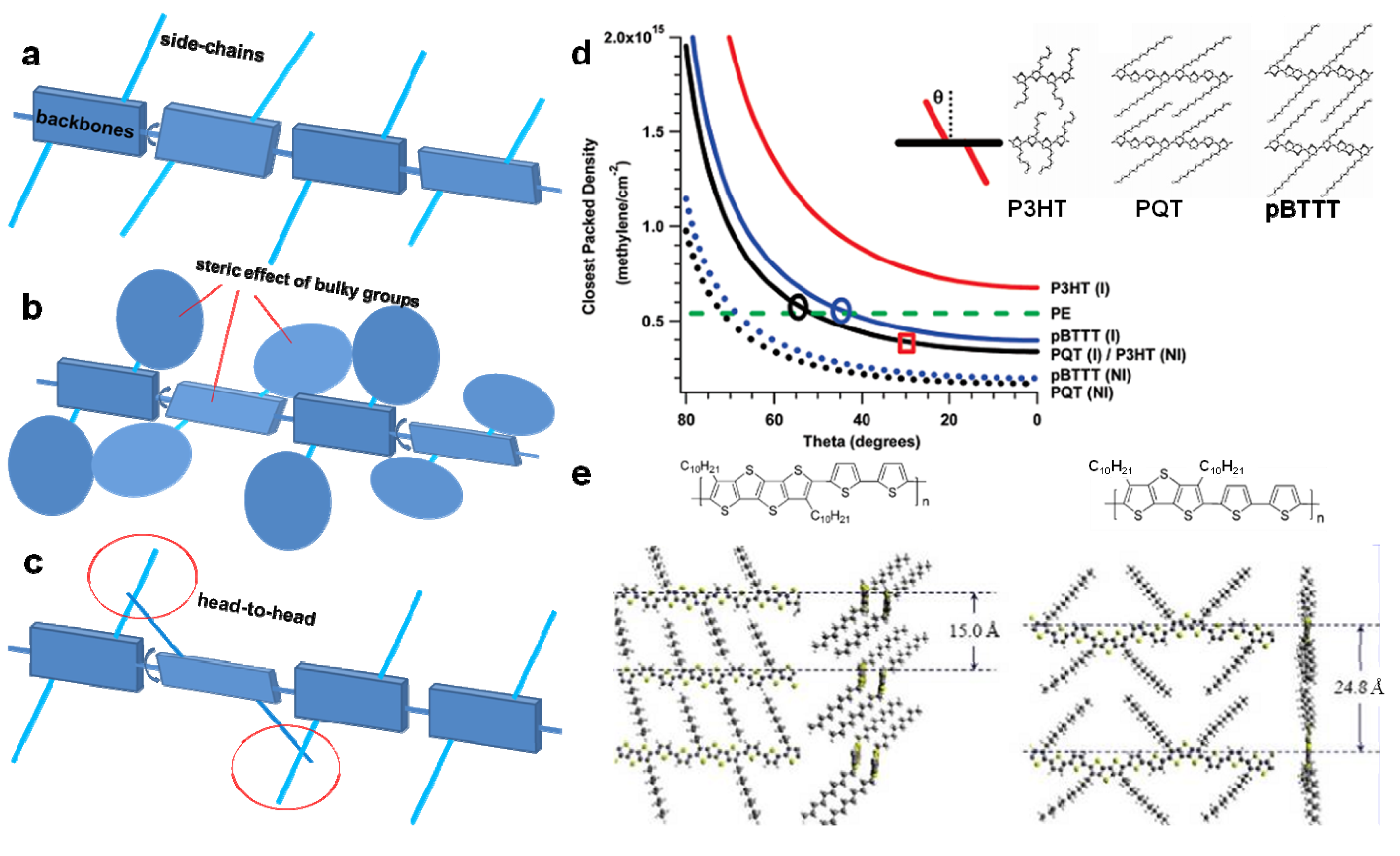
3.1. Structural Differences between Conjugated Polymers and Flexible Polymers and the Corresponding Effects on Morphology
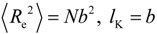
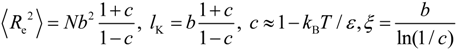
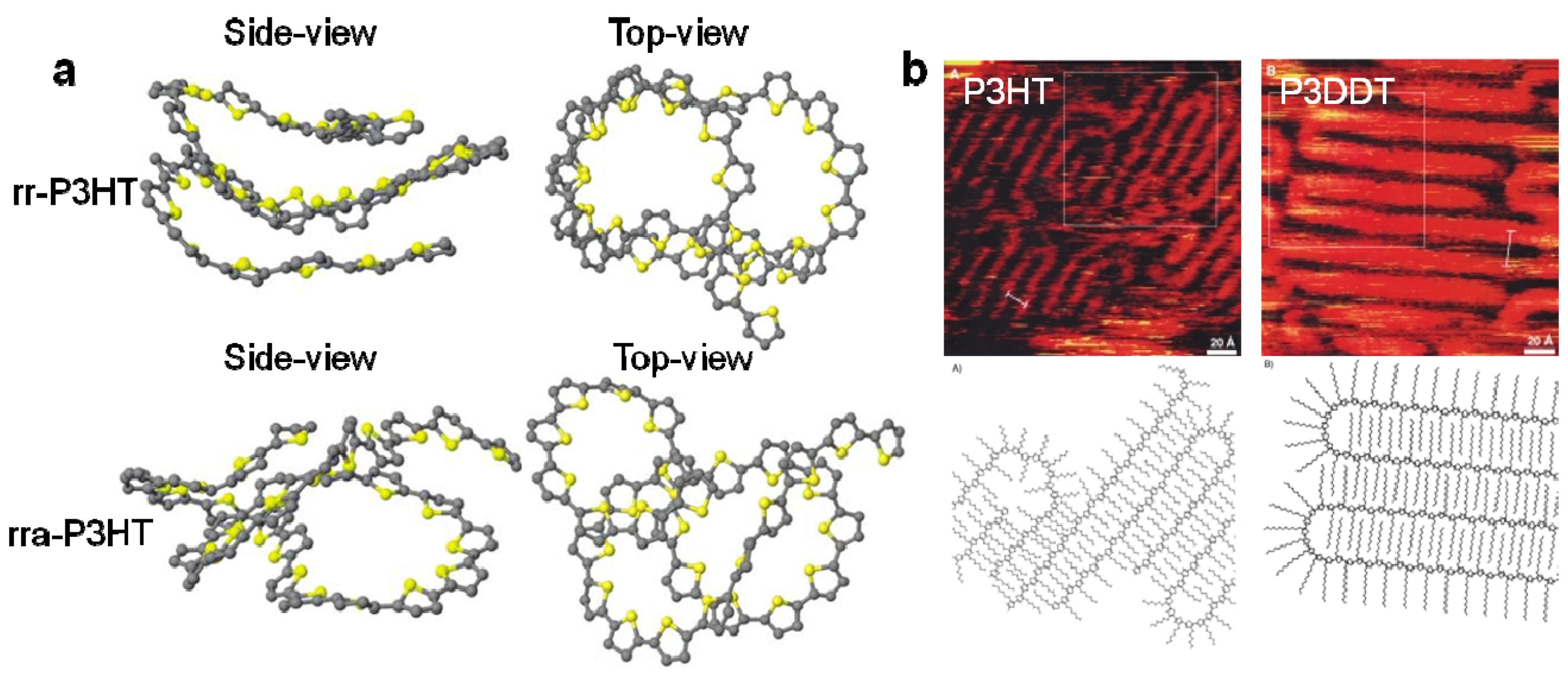
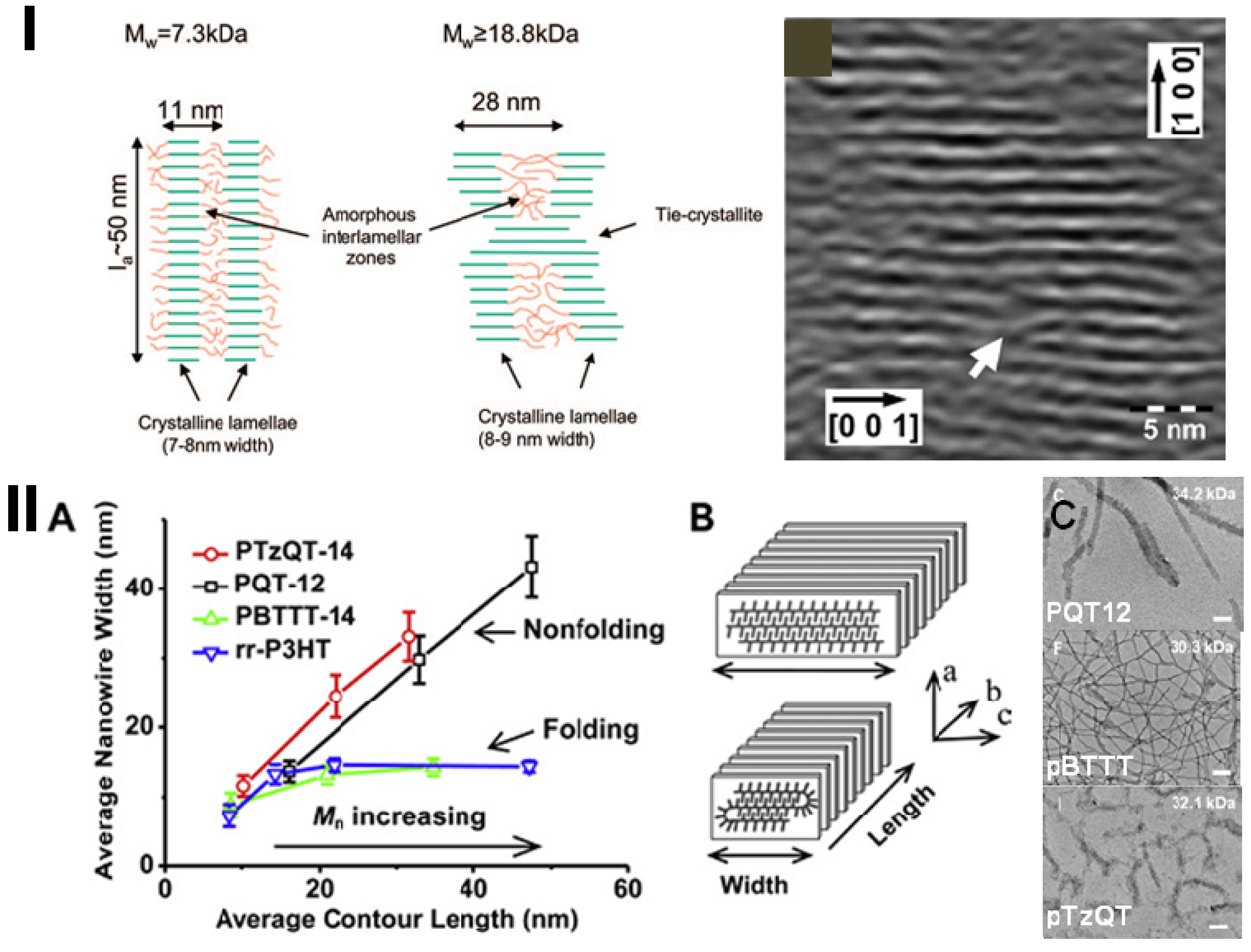
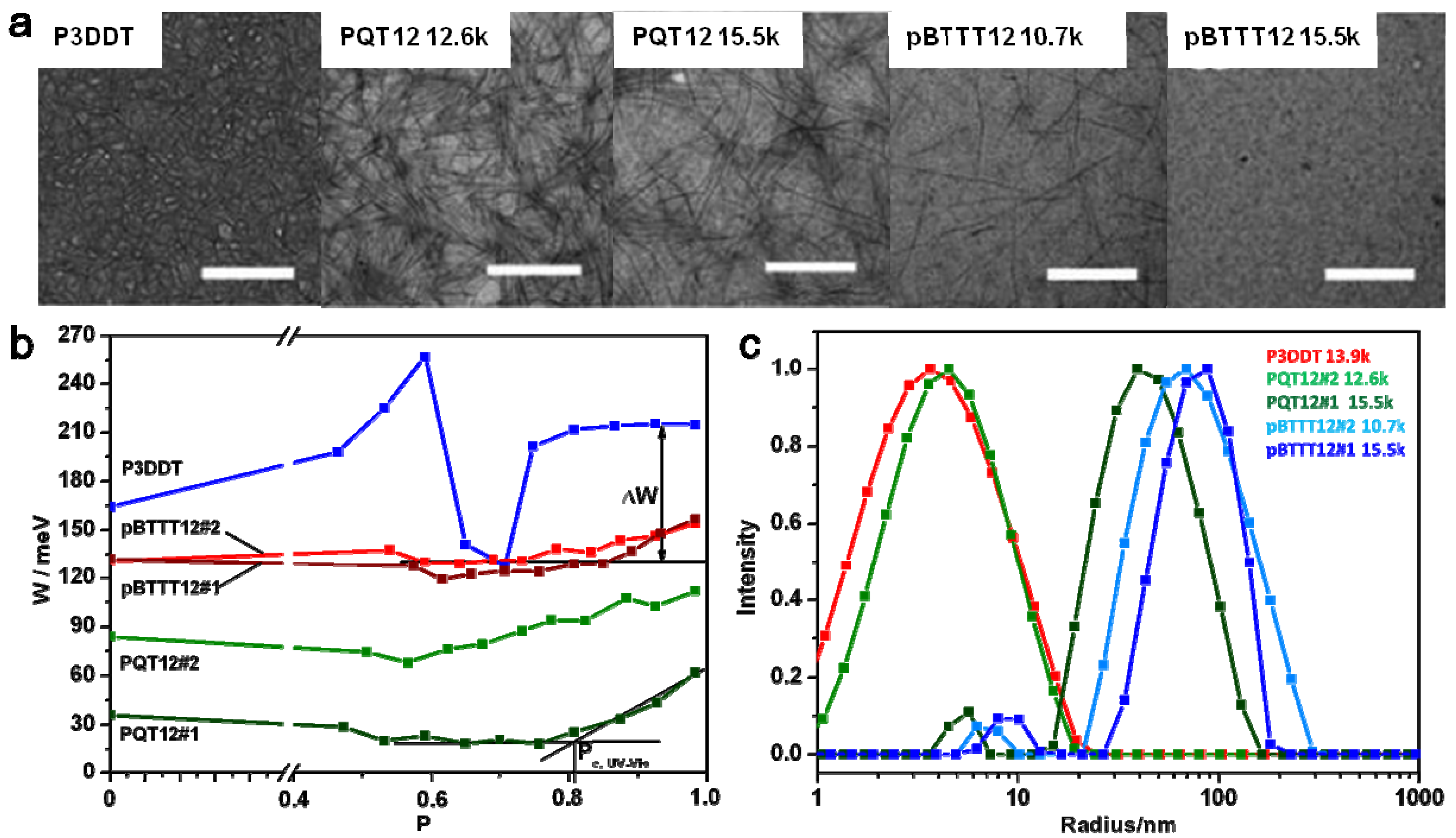
3.2. The Effect of Planarity and Rigidity on Structure, Morphology and Charge Transport

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Structures | Mw/Mn (kDa/kDa) | μmax (cm2∙V−1∙s−1) | Ref. |
|---|---|---|---|---|
| P1 |  a R = C6H13; b R = C12H25 | a 48/40 b 26.4/22 | a 0.13 b 0.097 | [108] |
| P2 |  a R1 = C6H13, R2 = C12H25; b R1 = R2 = C12H25 | a 25/14.7 b 53.3/24.9 | a 0.47 b 1.05 | [83] |
| P3 |  | 22.9/17.3 | 0.2 | [87] |
| P4 |  a R = C12H25; b R = C14H29 | a 54/29.4 b 59.6/33 | a 0.3 b 0.72 | [88] |
| P5 |  | 62.1/16.3 | 0.25 | [89] |
| P6 |  | 10.3/7.9 | 0.3 | [90] |
| P7 |  | 45.8/28.9 | 0.54 | [91] |
| P8 |  | 15.8/8.7 | 0.3 | [92] |
| P9 |  | 300.3/33 | 0.42 | [96] |
| P10 |  | 37.8/17.2 | 0.3 | [97] |
| P11 |  | 108/38 | 1.0 | [98] |
| P12 |  | 310/104 | 0.60 | [[37] |
| P13 |  | 197/59 | 1.04 | [104] |
| P14 |  | 194/50 | 1.36 for holes 1.56 for electrons | [105] |
| P15 |  a R1 = C8H17, R2 = C10H21; b R1 = C10H21, R2 = C12H25 | a 180/70 b 183/74 | a 4.7 b 8.2 | [106] |
| P16 |  | 58.0/35.8 | 12.04 | [17] |
| P17 |  | 89/38 | 1.1 for electrons | [107] |
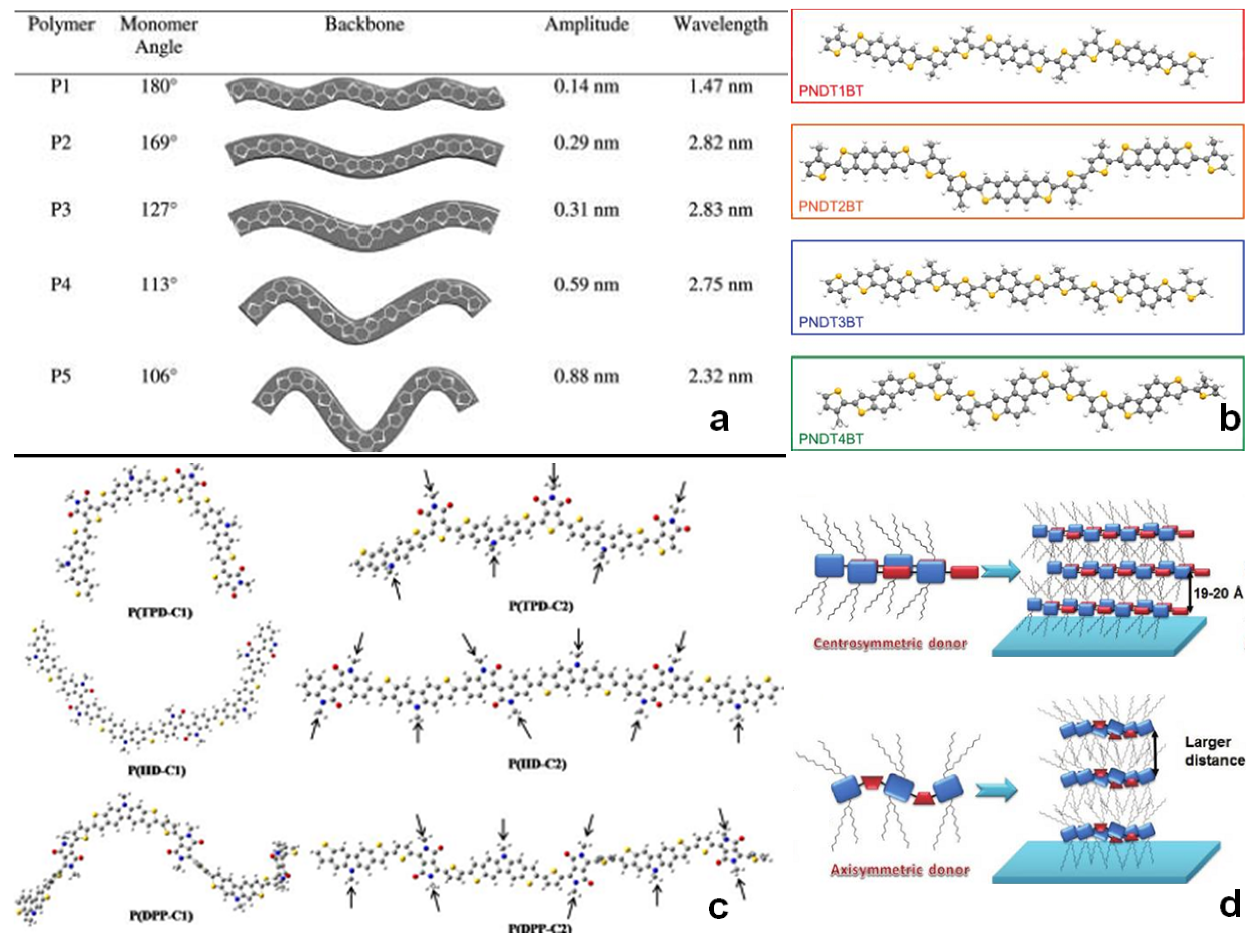
3.3. Backbone Shape
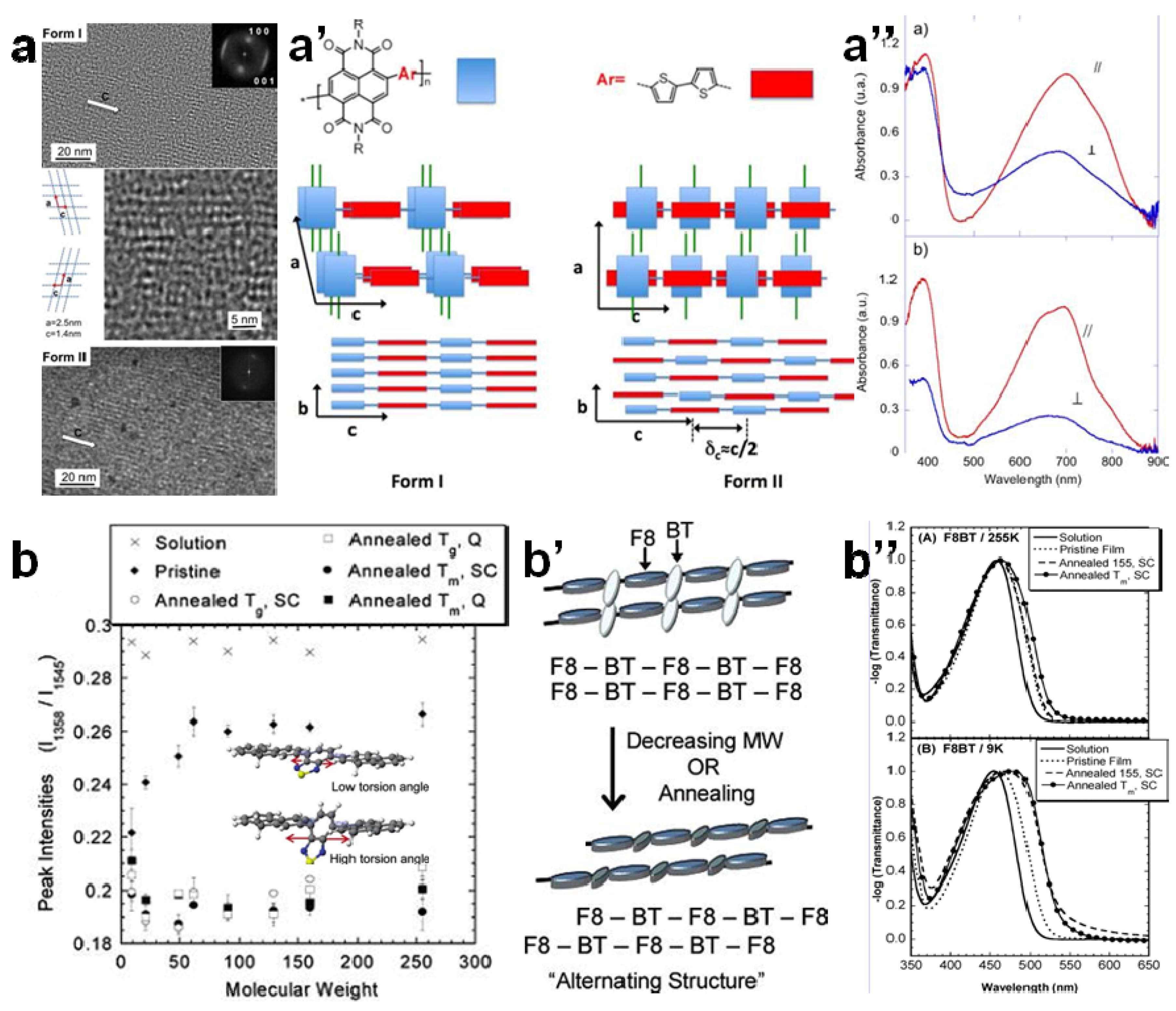


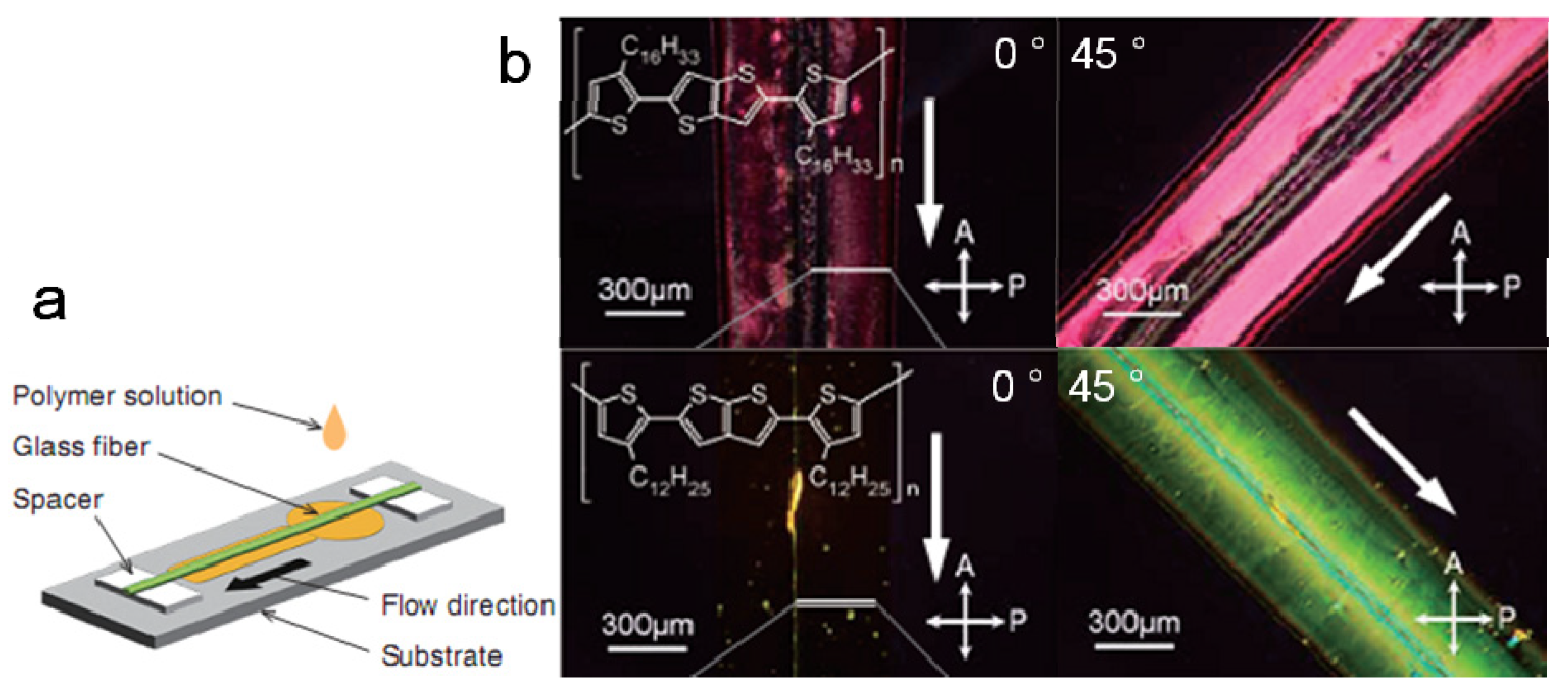
3.4. Side-Chains
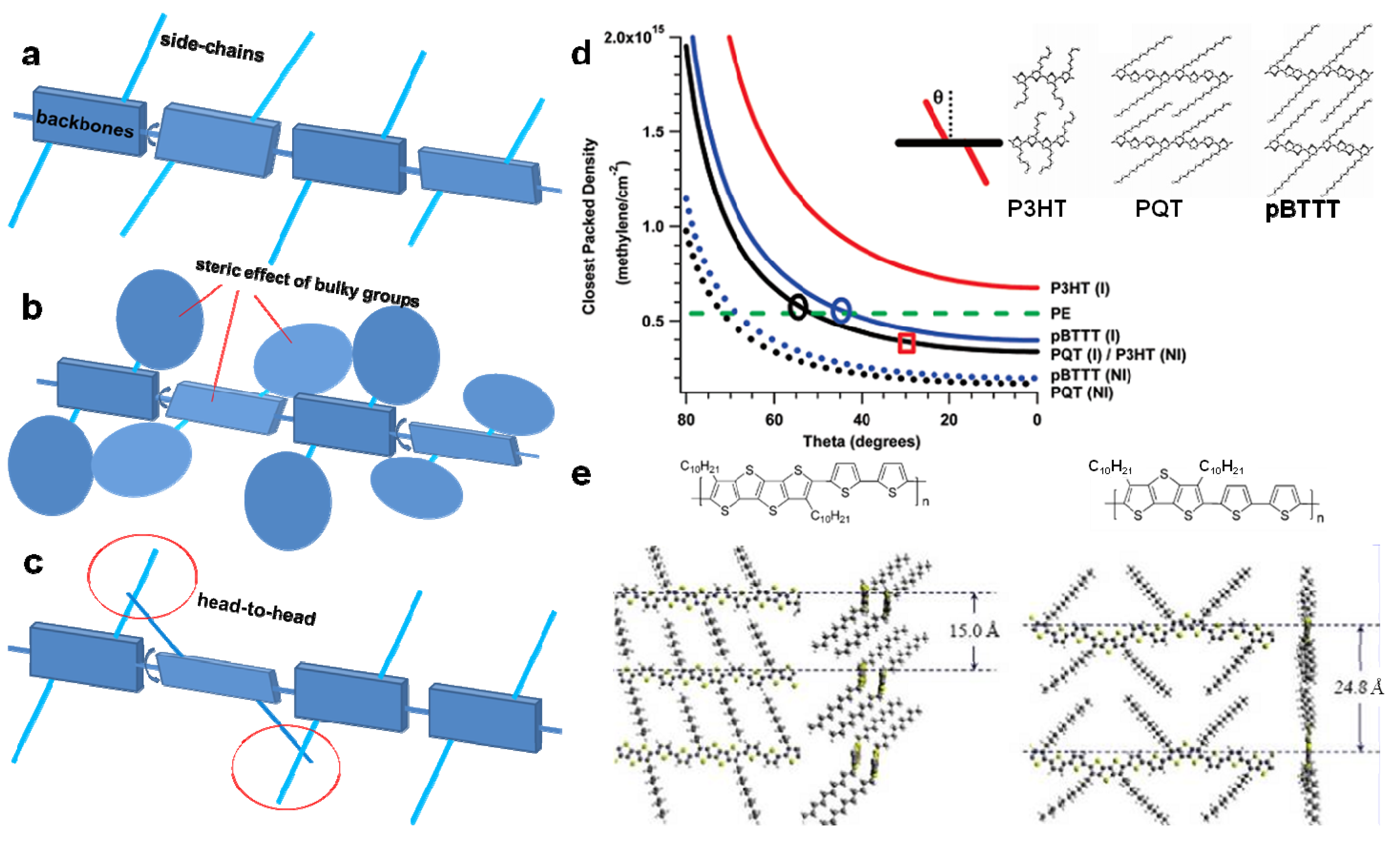
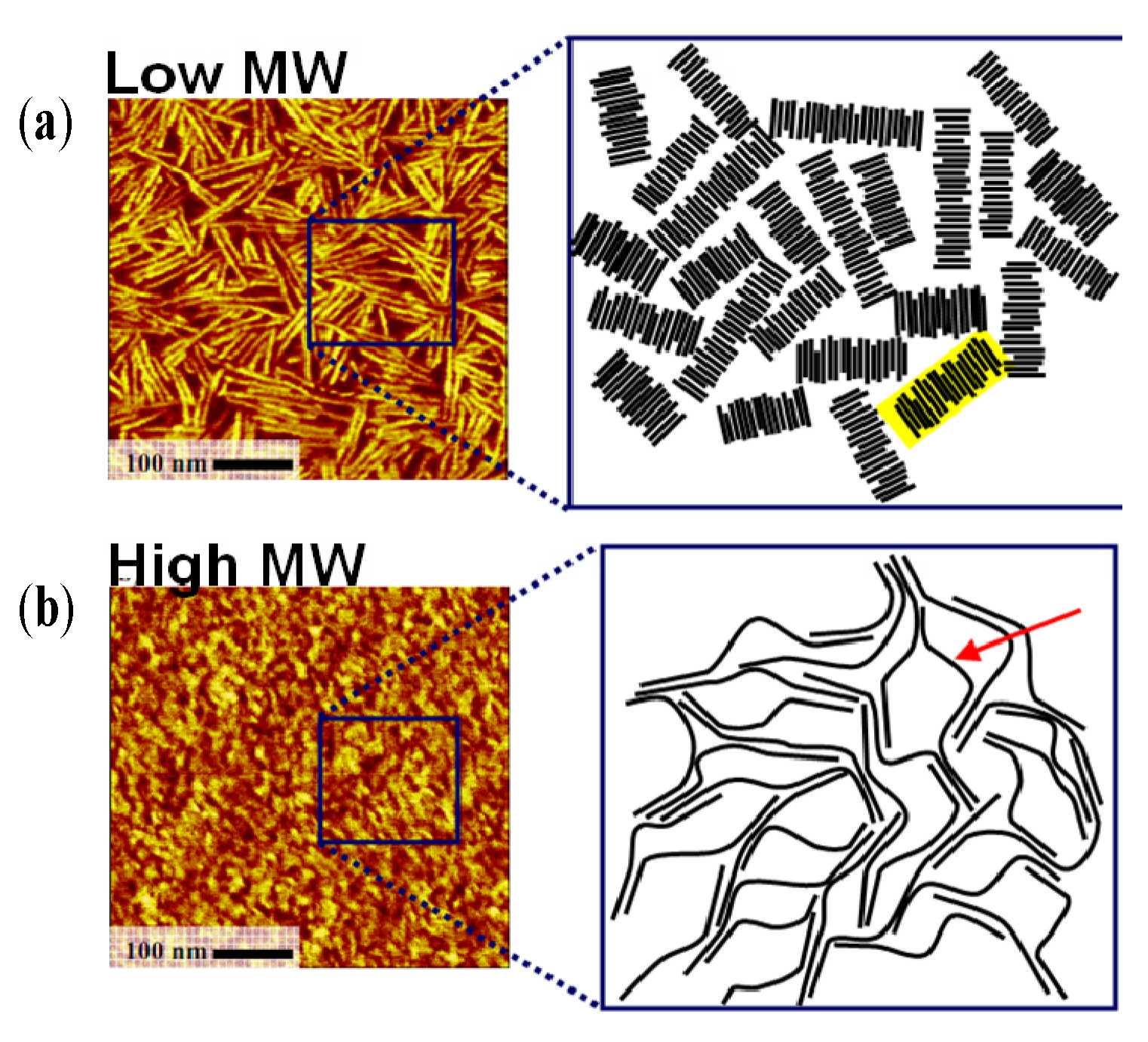
3.5. Molecular Weight

4. The Optimization of Solution State of Conjugated Polymers to Promote Order in Films
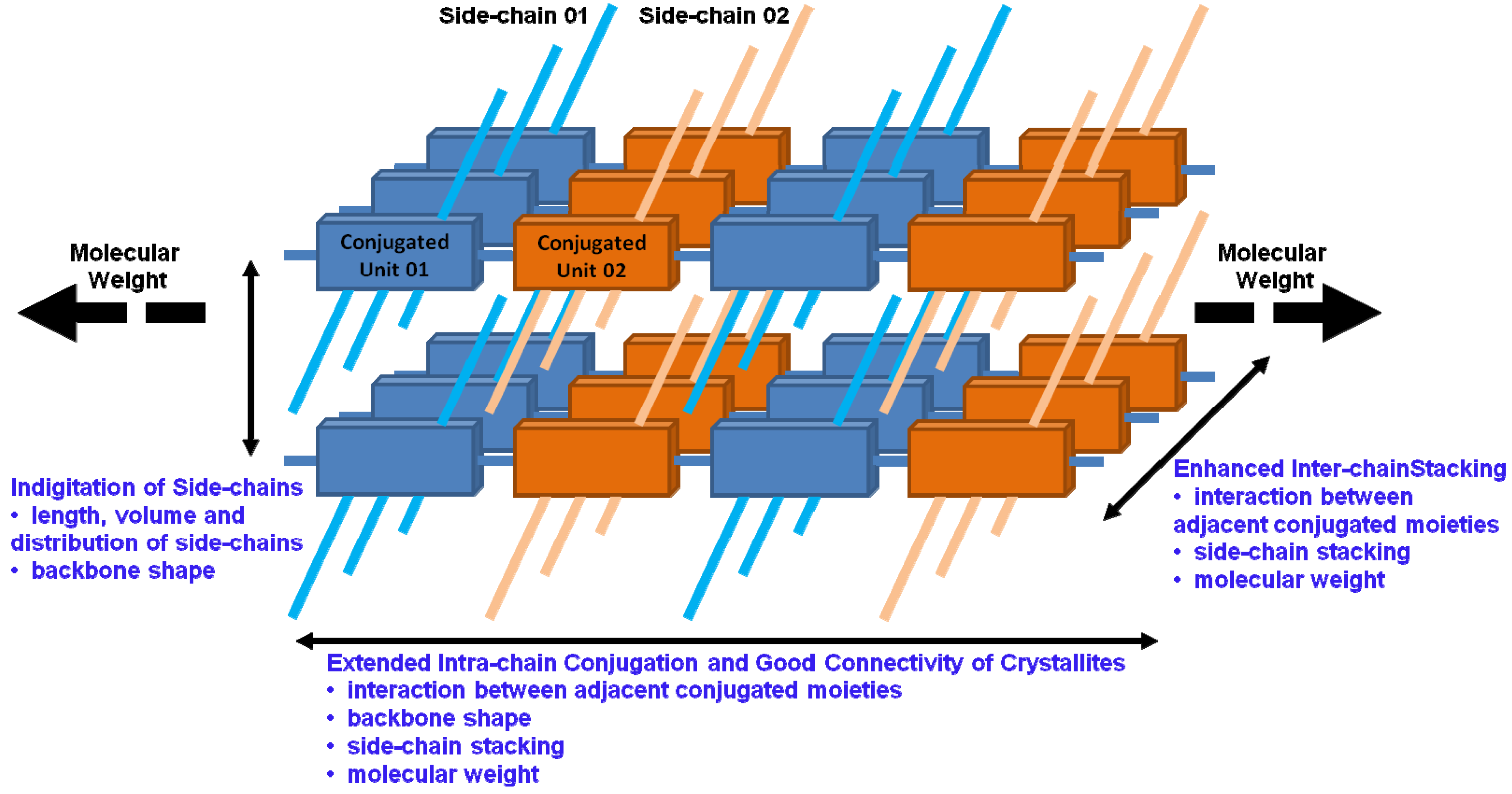
4.1. The Influences of Solution State on the Morphology and Performance of Conjugated Polymer Films
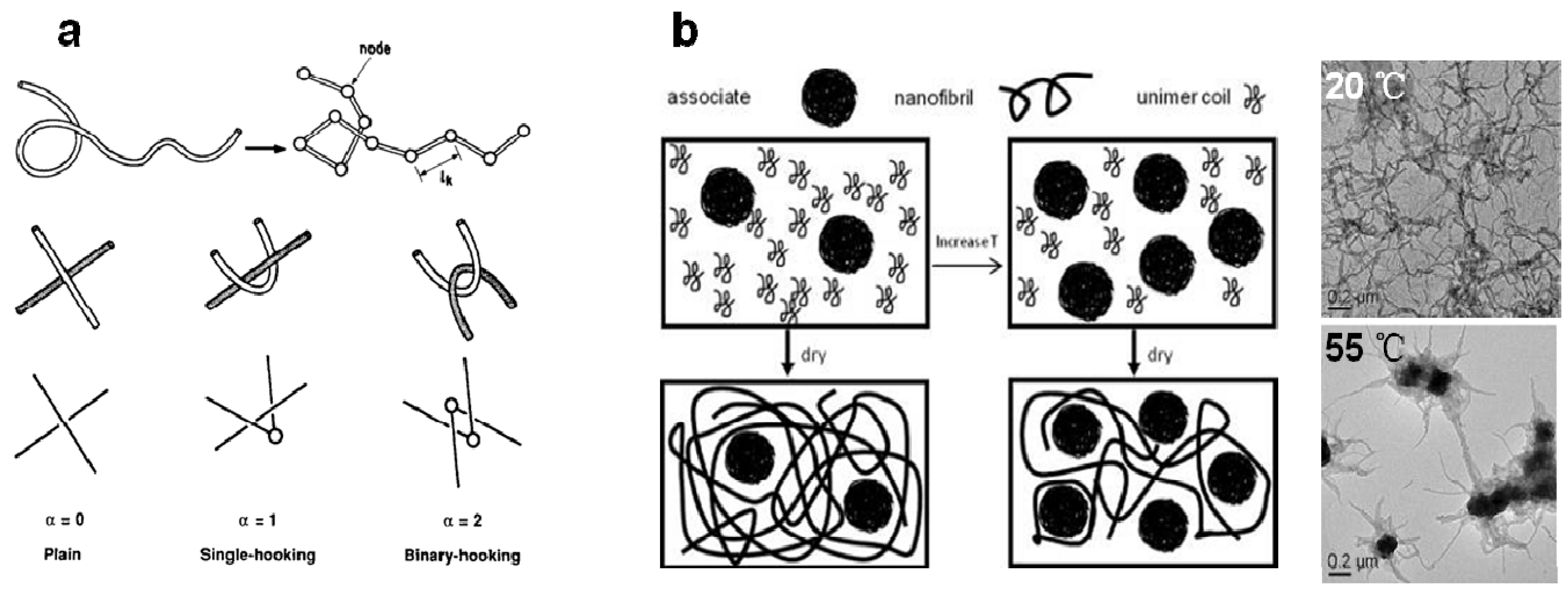
4.1.1. Equilibrium between Coil Unimers and Disordered Aggregates

4.1.2. The Conformation Transition and Ordered Aggregation in Solution
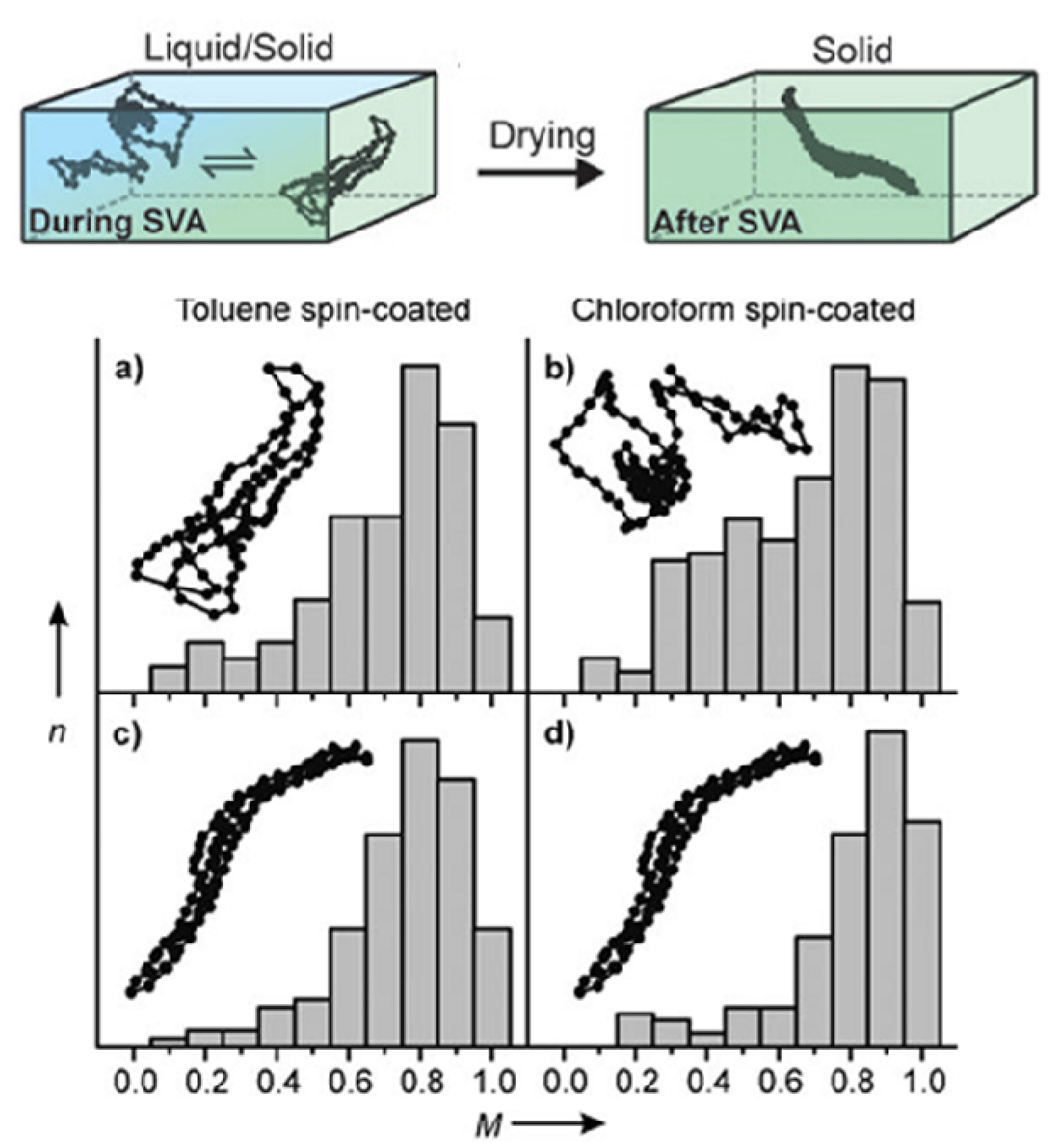
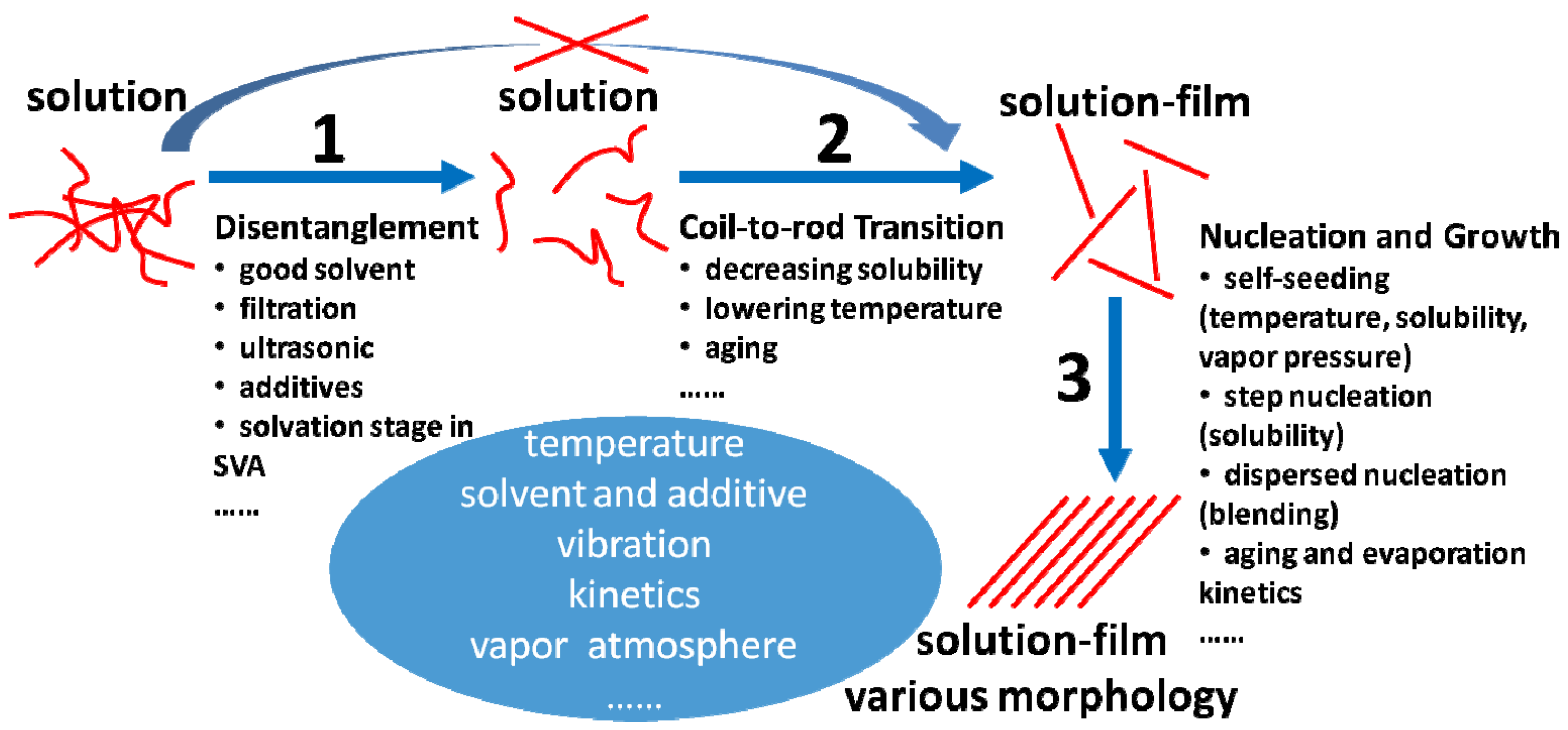
4.2. The Strategy to Optimize the Solution State of Conjugated Polymers
4.2.1. Disentanglement
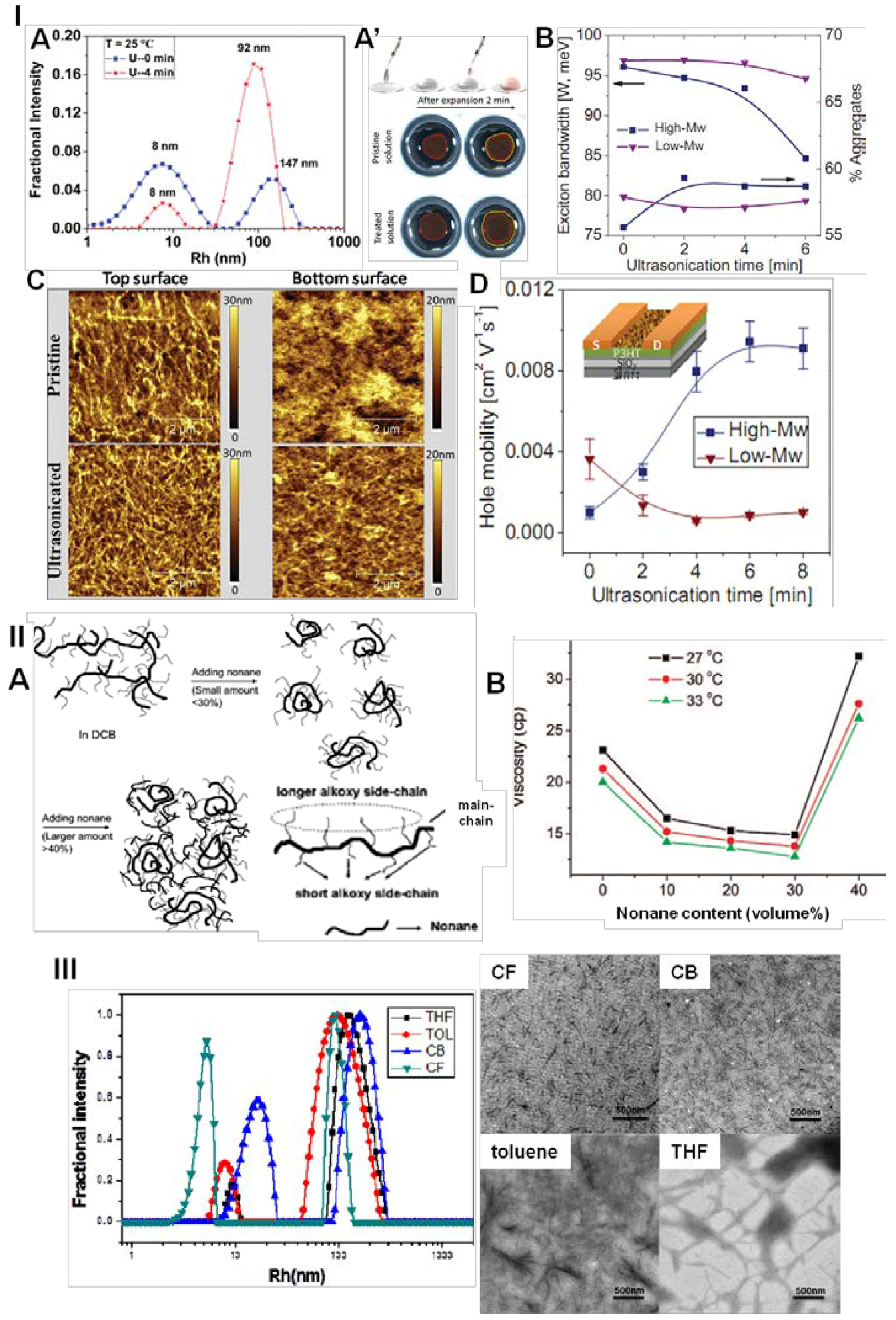
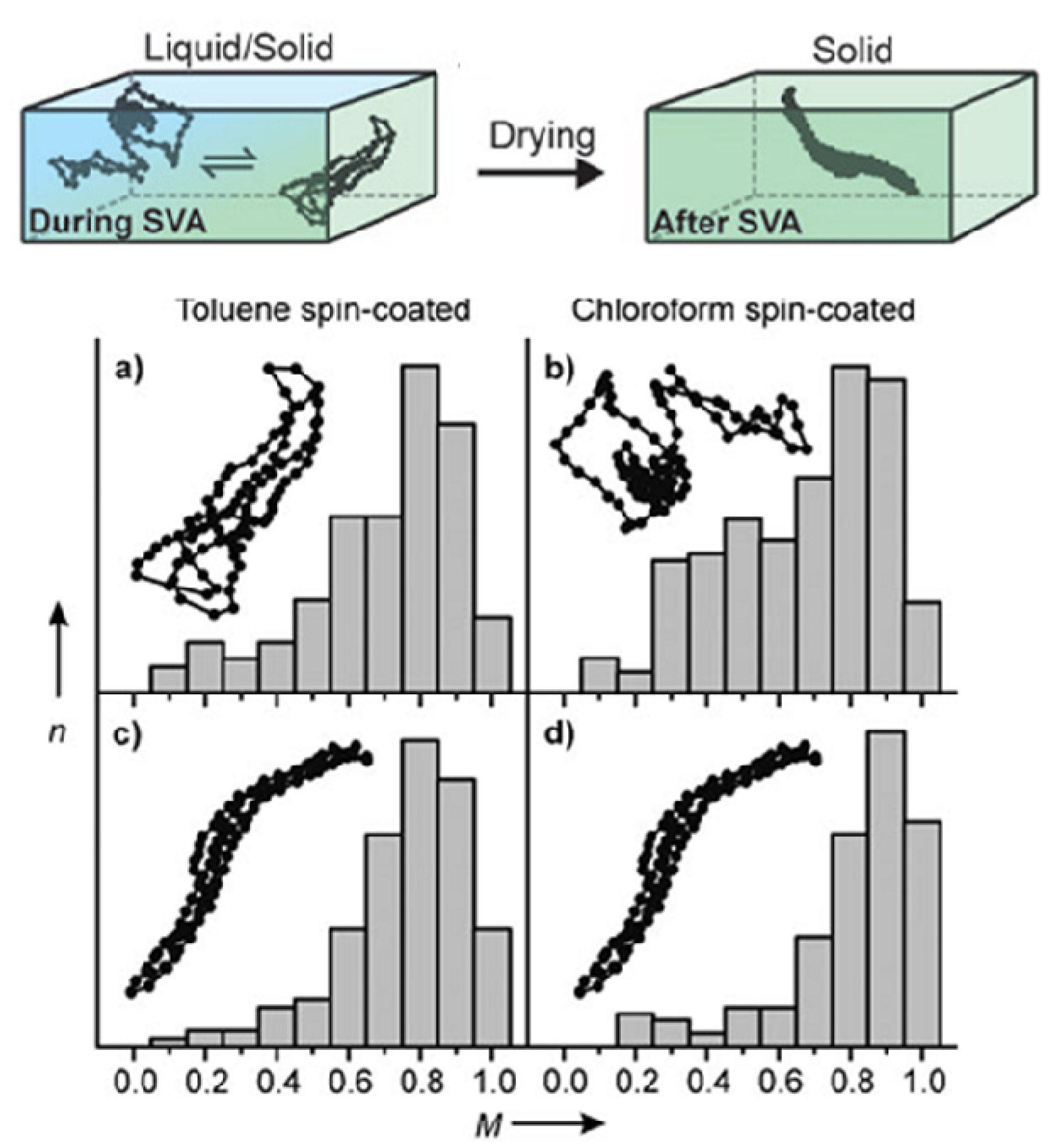
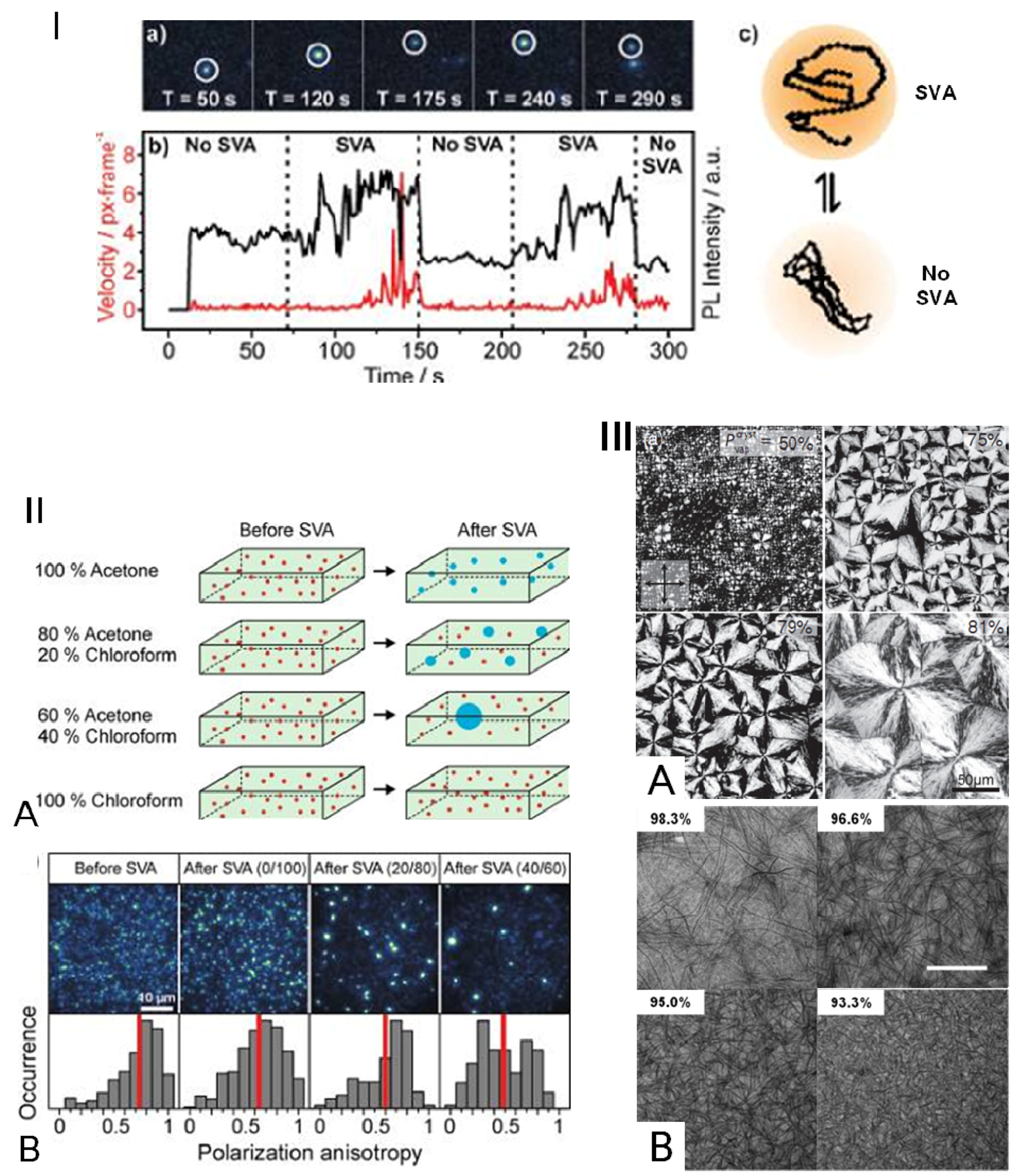
4.2.2. Promotion of Coil-to-Rod Transition and Ordered Rod-Rod Stacking


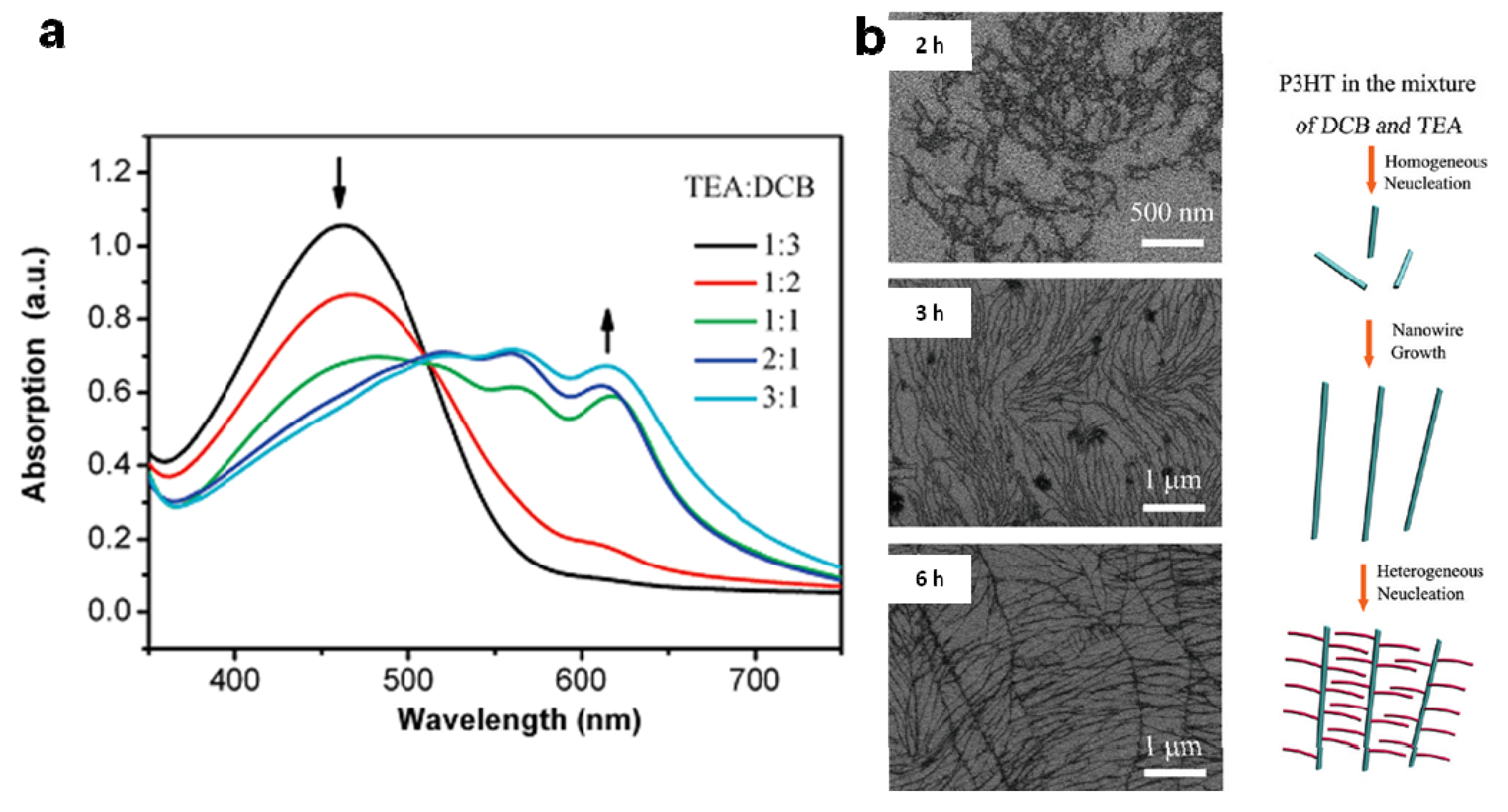


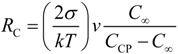

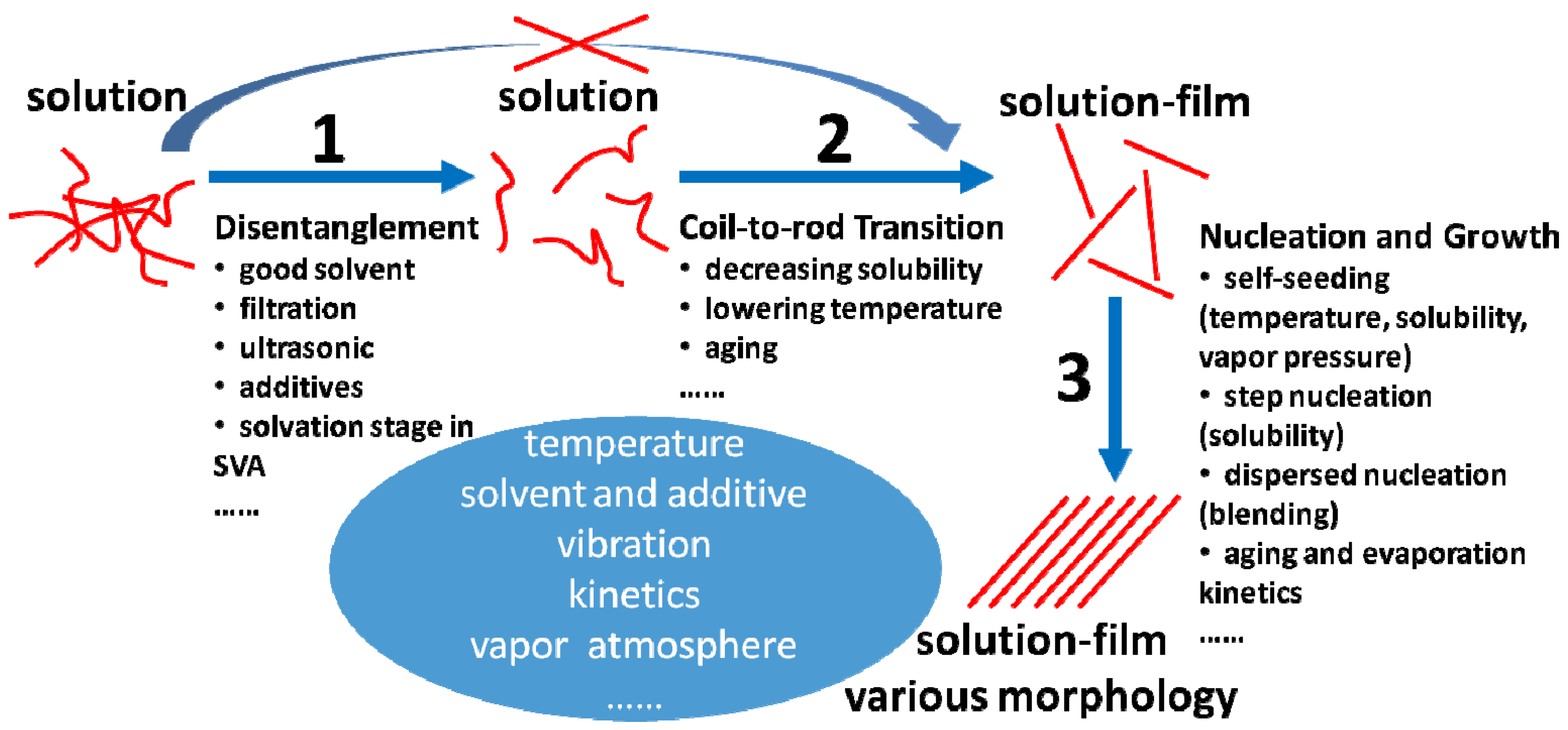
5. Concluding Remarks and Outlook
Acknowledgments
Conflicts of Interest
References
- Wang, C.L.; Dong, H.L. Semiconducting pi-conjugated systems in field-effect transistors: A material odyssey of organic electronics. Chem. Rev. 2012, 112, 2208–2267. [Google Scholar] [CrossRef]
- Kim, F.S.; Ren, G.Q. One-dimensional nanostructures of pi-conjugated molecular systems: Assembly, properties, and applications from photovoltaics, sensors, and nanophotonics to nanoelectronics. Chem. Mater. 2011, 23, 682–732. [Google Scholar] [CrossRef]
- Facchetti, A. Π-conjugated polymers for organic electronics and photovoltaic cell applications. Chem. Mater. 2011, 23, 733–758. [Google Scholar] [CrossRef]
- Mei, J.G.; Diao, Y. Integrated materials design of organic semiconductors for field-effect transistors. J. Am. Chem. Soc. 2013, 135, 6724–6746. [Google Scholar] [CrossRef]
- AlSalhi, M.S.; Alam, J. Recent advances in conjugated polymers for light emitting devices. Int. J. Mol. Sci. 2011, 12, 2036–2054. [Google Scholar] [CrossRef]
- Tang, C.; Liu, X.D. Recent progress in polymer white light-emitting materials and devices. Macromol. Chem. Phys. 2013, 214, 314–342. [Google Scholar] [CrossRef]
- Heeger, A.J. Semiconducting polymers: The third generation. Chem. Soc. Rev. 2010, 39, 2354–2371. [Google Scholar] [CrossRef]
- Krebs, F.C. Polymer solar cell modules prepared using roll-to-roll methods: Knife-over-edge coating, slot-die coating and screen printing. Solar Energy Mater. Solar Cells 2009, 93, 465–475. [Google Scholar] [CrossRef]
- Amb, C.M.; Craig, M.R. Aesthetically pleasing conjugated polymer: Fullerene blends for blue-green solar cells via roll-to-roll processing. ACS Appl. Mater. Interfaces 2012, 4, 1847–1853. [Google Scholar] [CrossRef]
- Yue, W.; Larsen-Olsen, T.T. Synthesis and photovoltaic properties from inverted geometry cells and roll-to-roll coated large area cells from dithienopyrrole-based donor-acceptor polymers. J. Mater. Chem. A 2013, 1, 1785–1793. [Google Scholar] [CrossRef]
- Nielsen, C.B.; Turbiez, M. Recent advances in the development of semiconducting DPP-containing polymers for transistor applications. Adv. Mater. 2013, 25, 1859–1880. [Google Scholar] [CrossRef]
- Usta, H.; Facchetti, A. N-channel semiconductor materials sesign for organic complementary circuits. Acc. Chem. Res. 2011, 44, 501–510. [Google Scholar] [CrossRef]
- Sista, P.; Biewer, M.C. Benzo[1,2-b:4,5-b′]dithiophene building block for the synthesis of semiconducting polymers. Macromol. Rapid Commun. 2012, 33, 9–20. [Google Scholar] [CrossRef]
- Huo, L.J.; Hou, J.H. Benzo[1,2-b:4,5-b′]dithiophene-based conjugated polymers: Band gap and energy level control and their application in polymer solar cells. Polym. Chem. 2011, 2, 2453–2461. [Google Scholar] [CrossRef]
- Balan, A.; Baran, D. Benzotriazole containing conjugated polymers for multipurpose organic electronic applications. Polym. Chem. 2011, 2, 1029–1043. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Y.Q. High-mobility conjugated polymers based on fused-thiophene building blocks. Macromol. Chem. Phys. 2011, 212, 428–443. [Google Scholar] [CrossRef]
- Kang, I.; Yun, H.J. Record high hole mobility in polymer semiconductors via side-chain engineering. J. Am. Chem. Soc. 2013, 135, 14896–14899. [Google Scholar] [CrossRef]
- Lu, L.Y.; Luo, Z.Q. Cooperative plasmonic effect of Ag and Au nanoparticles on enhancing performance of polymer solar cells. Nano Lett. 2013, 13, 59–64. [Google Scholar] [CrossRef]
- Salleo, A.; Kline, R.J. Microstructural characterization and charge transport in thin films of conjugated polymers. Adv. Mater. 2010, 22, 3812–3838. [Google Scholar] [CrossRef]
- Park, Y.W. Editorial for the conducting polymers for carbon electronics themed issue. Chem. Soc. Rev. 2010, 39, 2352–2353. [Google Scholar] [CrossRef]
- Giridharagopal, R.; Ginger, D.S. Characterizing morphology in bulk heterojunction organic photovoltaic systems. J. Phys. Chem. Lett. 2010, 1, 1160–1169. [Google Scholar] [CrossRef]
- Chen, W.; Nikiforov, M.P. Morphology characterization in organic and hybrid solar cells. Energy Environ. Sci. 2012, 5, 8045–8074. [Google Scholar] [CrossRef]
- Rivnay, J.; Mannsfeld, S.C.B. Quantitative determination of organic semiconductor microstructure from the molecular to device scale. Chem. Rev. 2012, 112, 5488–5519. [Google Scholar] [CrossRef]
- Brinkmann, M. Structure and morphology control in thin films of regioregular poly(3-hexylthiophene). J. Polym. Sci. B Polym. Phys. 2011, 49, 1218–1233. [Google Scholar] [CrossRef]
- Tsao, H.N.; Mullen, K. Improving polymer transistor performance via morphology control. Chem. Soc. Rev. 2010, 39, 2372–2386. [Google Scholar] [CrossRef]
- Virkar, A.A.; Mannsfeld, S. Organic semiconductor growth and morphology considerations for organic thin-film transistors. Adv. Mater. 2010, 22, 3857–3875. [Google Scholar] [CrossRef]
- Knaapila, M.; Monkman, A.P. Methods for controlling structure and photophysical properties in polyfluorene solutions and gels. Adv. Mater. 2013, 25, 1090–1108. [Google Scholar] [CrossRef]
- Lim, J.A.; Liu, F. Polymer semiconductor crystals. Mater. Today 2010, 13, 14–24. [Google Scholar]
- Vacha, M.; Habuchi, S. Conformation and physics of polymer chains: A single-molecule perspective. NPG Asia Mater. 2010, 2, 134–142. [Google Scholar] [CrossRef]
- Clark, J.; Silva, C. Role of intermolecular coupling in the photophysics of disordered organic semiconductors: Aggregate emission in regioregular polythiophene. Phys. Rev. Lett. 2007, 98, 206406:1–206406:4. [Google Scholar]
- Clark, J.; Chang, J.F. Determining exciton bandwidth and film microstructure in polythiophene films using linear absorption spectroscopy. Appl. Phys. Lett. 2009, 94, 163306:1–163306:4. [Google Scholar]
- Spano, F.C. The spectral signatures of frenkel polarons in H- and J-Aggregates. Acc. Chem. Res. 2010, 43, 429–439. [Google Scholar] [CrossRef]
- Oelkrug, D.; Egelhaaf, H.J. Electronic deactivation in single chains, nano-aggregates and ultrathin films of conjugated oligomers. Synth. Metals 1996, 76, 249–253. [Google Scholar] [CrossRef]
- Spano, F.C. Excitons in conjugated oligomer aggregates, films, and crystals. Annu. Rev. Phys. Chem. 2006, 57, 217–243. [Google Scholar] [CrossRef]
- Fidder, H.; Knoester, J. Superradiant emission and optical dephasing in J-Aggregates. Chem. Phys. Lett. 1990, 171, 529–536. [Google Scholar] [CrossRef]
- Niles, E.T.; Roehling, J.D. J-Aggregate behavior in poly-3-hexylthiophene nanofibers. J. Phys. Chem. Lett. 2012, 3, 259–263. [Google Scholar] [CrossRef]
- Zhang, X.R.; Richter, L.J. Molecular packing of high-mobility diketo pyrrolo-pyrrole polymer semiconductors with branched alkyl side chains. J. Am. Chem. Soc. 2011, 133, 15073–15084. [Google Scholar] [CrossRef]
- Lee, H.S.; Lee, J.S. Crystallinity-controlled naphthalene-alt-diketopyrrolopyrrole copolymers for high-performance ambipolar field effect transistors. J. Phys. Chem. C 2012, 116, 26204–26213. [Google Scholar] [CrossRef]
- Kim, J.W.; Baeg, K.J. Optimal ambipolar charge transport of thienylenevinylene-based polymer semiconductors by changes in conformation for high-performance organic thin film transistors and inverters. Chem. Mater. 2013, 25, 1572–1583. [Google Scholar] [CrossRef]
- Gao, J.; Kamps, A. Encapsulation of poly(3-hexylthiophene) J-aggregate nanofibers with an amphiphilic block copolymer. Langmuir 2012, 28, 16401–16407. [Google Scholar] [CrossRef]
- Jimison, L.H.; Toney, M.F. Charge-transport anisotropy due to grain boundaries in directionally crystallized thin films of regioregular poly(3-hexylthiophene). Adv. Mater. 2009, 21, 1568–1572. [Google Scholar] [CrossRef]
- Lee, M.J.; Gupta, D. Anisotropy of charge transport in a uniaxially aligned and chain-extended, high-mobility, conjugated polymer semiconductor. Adv. Funct. Mater. 2011, 21, 932–940. [Google Scholar] [CrossRef]
- Lan, Y.K.; Huang, C.I. Charge mobility and transport behavior in the ordered and disordered states of the regioregular poly(3-hexylthiophene). J. Phys. Chem. B 2009, 113, 14555–14564. [Google Scholar] [CrossRef]
- Kline, R.J.; McGehee, M.D. Dependence of regioregular poly(3-hexylthiophene) film morphology and field-effect mobility on molecular weight. Macromolecules 2005, 38, 3312–3319. [Google Scholar] [CrossRef]
- Bolsee, J.C.; Oosterbaan, W.D. The importance of bridging points for charge transport in webs of conjugated polymer nanofibers. Adv. Funct. Mater. 2013, 23, 862–869. [Google Scholar] [CrossRef]
- Street, R.A.; Northrup, J.E. Transport in polycrystalline polymer thin-film transistors. Phys. Rev. B 2005, 71, 165202:1–165202:13. [Google Scholar]
- Crossland, E.J.W.; Tremel, K. Anisotropic charge transport in spherulitic poly(3-hexylthiophene) films. Adv. Mater. 2012, 24, 839–844. [Google Scholar] [CrossRef]
- Zhang, X.R.; Hudson, S.D. In-plane liquid crystalline texture of high-performance thienothiophene copolymer thin films. Adv. Funct. Mater. 2010, 20, 4098–4106. [Google Scholar] [CrossRef]
- Schuettfort, T.; Watts, B. Microstructure of polycrystalline PBTTT films: Domain mapping and structure formation. ACS Nano 2012, 6, 1849–1864. [Google Scholar] [CrossRef]
- Liu, J.Y.; Zhang, R. Highly disordered polymer field effect transistors: N-alkyl dithieno[3,2-b:2′,3′-d]pyrrole-based copolymers with surprisingly high charge carrier mobilities. J. Am. Chem. Soc. 2008, 130, 13167–13176. [Google Scholar] [CrossRef]
- Lee, J.B.; Kim, K.H. High-performance amorphous donor-acceptor conjugated polymers containing x-shaped anthracene-based monomer and 2,5-bis(2-octyldodecyl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione for organic thin-film transistors. J. Polym. Sci. A Polym. Chem. 2012, 50, 2809–2818. [Google Scholar] [CrossRef]
- Aiyar, A.R.; Hong, J.I. Tunable crystallinity in regioregular poly(3-hexylthiophene) thin films and its impact on field effect mobility. Adv. Funct. Mater. 2011, 21, 2652–2659. [Google Scholar] [CrossRef]
- Yu, Z.; Fang, J. Self-assembly of well-defined poly(3-hexylthiophene) nanostructures toward the structure-property relationship determination of polymer solar cells. J. Phys. Chem. C 2012, 116, 23858–23863. [Google Scholar] [CrossRef]
- Lee, J.S.; Son, S.K. Importance of solubilizing group and backbone planarity in low band gap polymers for high performance ambipolar field-effect transistors. Chem. Mater. 2012, 24, 1316–1323. [Google Scholar] [CrossRef]
- Kim, D.H.; Ayzner, A.L. Comparison of the photovoltaic characteristics and nanostructure of fullerenes blended with conjugated polymers with siloxane-terminated and branched aliphatic side chains. Chem. Mater. 2013, 25, 431–440. [Google Scholar] [CrossRef]
- Osaka, I.; Kakara, T. Naphthodithiophene-naphthobisthiadiazole copolymers for solar cells: Alkylation drives the polymer backbone flat and promotes efficiency. J. Am. Chem. Soc. 2013, 135, 8834–8837. [Google Scholar] [CrossRef]
- Liu, J.G.; Sun, Y. Oriented poly(3-hexylthiophene) nanofibril with the π–π stacking growth direction by solvent directional evaporation. Langmuir 2011, 27, 4212–4219. [Google Scholar] [CrossRef]
- Muller, C.; Aghamohammadi, M. One-step macroscopic alignment of conjugated polymer systems by epitaxial crystallization during spin-coating. Adv. Funct. Mater. 2013, 23, 2368–2377. [Google Scholar]
- Beniek, L.; Leclerc, N. Large scale alignment and charge transport anisotropy of pBTTT films oriented by high temperature rubbing. Macromolecules 2013, 46, 4014–4023. [Google Scholar] [CrossRef]
- Lu, G.H.; Li, L.G. Achieving perpendicular alignment of rigid polythiophene backbones to the substrate by using solvent-vapor treatment. Adv. Mater. 2007, 19, 3594–3598. [Google Scholar] [CrossRef]
- Lu, G.H.; Li, L.G. Morphology and crystalline transition of poly(3-butylthiophene) associated with its polymorphic modifications. Macromolecules 2008, 41, 2062–2070. [Google Scholar] [CrossRef]
- Aryal, M.; Trivedi, K. Nano-confinement induced chain alignment in ordered P3HT nanostructures defined by nanoimprint lithography. ACS Nano 2009, 3, 3085–3090. [Google Scholar] [CrossRef]
- Rivnay, J.; Toney, M.F. Unconventional face-on texture and exceptional in-plane order of a high mobility n-type polymer. Adv. Mater. 2010, 22, 4359–4363. [Google Scholar] [CrossRef]
- DeLongchamp, D.M.; Vogel, B.M. Variations in semiconducting polymer microstructure and hole mobility with spin-coating speed. Chem. Mater. 2005, 17, 5610–5612. [Google Scholar] [CrossRef]
- Yang, H.H.; LeFevre, S.W. Solubility-driven thin film structures of regioregular poly(3-hexyl thiophene) using volatile solvents. Appl. Phys. Lett. 2007. [Google Scholar] [CrossRef]
- Wang, S.; Kiersnowski, A. Microstructure evolution and device performance in solution-processed polymeric field-effect transistors: The key role of the first monolayer. J. Am. Chem. Soc. 2012, 134, 4015–4018. [Google Scholar] [CrossRef]
- Wang, S.H.; Pisula, W. Nanofiber growth and alignment in solution processed n-type naphthalene-diimide-based polymeric field-effect transistors. J. Mater. Chem. 2012, 22, 24827–24831. [Google Scholar] [CrossRef]
- Zhao, L.H.; Png, R.Q. Role of borderline solvents to induce pronounced extended-chain lamellar order in π-stackable polymers. Macromolecules 2011, 44, 9692–9702. [Google Scholar] [CrossRef]
- Adachi, T.; Brazard, J. Regioregularity and single polythiophene chain conformation. J. Phys. Chem. Lett. 2011, 2, 1400–1404. [Google Scholar] [CrossRef]
- Mena-Osteritz, E.; Meyer, A. Two-dimensional crystals of poly(3-alkylthiophene)s: Direct visualization of polymer folds in submolecular resolution. Angew. Chem. Int. Ed. 2000, 39, 2680–2684. [Google Scholar]
- Brinkmann, M.; Rannou, P. Molecular weight dependence of chain packing and semicrystalline structure in oriented films of regioregular poly(3-hexylthiophene) revealed by high-resolution transmission electron microscopy. Macromolecules 2009, 42, 1125–1130. [Google Scholar] [CrossRef]
- Liu, J.; Mikhaylov, I.A. Insight into how molecular structures of thiophene-based conjugated polymers affect crystallization behaviors. Polymer 2011, 52, 2302–2309. [Google Scholar] [CrossRef]
- Xiao, X.L.; Wang, Z.B. Single crystals of polythiophene with different molecular conformations obtained by tetrahydrofuran vapor annealing and controlling solvent evaporation. J. Phys. Chem. B 2010, 114, 7452–7460. [Google Scholar] [CrossRef]
- Liu, C.F.; Wang, Q.L. Extended-chain lamellar crystals of monodisperse polyfluorenes. Polymer 2013, 54, 2459–2465. [Google Scholar] [CrossRef]
- Kim, D.H.; Han, J.T. Single-crystal polythiophene microwires grown by self-assembly. Adv. Mater. 2006, 18, 719–723. [Google Scholar] [CrossRef]
- Liu, C.F.; Wang, Q.L. Insight into lamellar crystals of monodisperse polyfluorenes—Fractionated crystallization and the crystal’s stability. Polymer 2013, 54, 1251–1258. [Google Scholar] [CrossRef]
- Rahimi, K.; Botiz, I. Controllable processes for generating large single crystals of poly(3-hexylthiophene). Angew. Chem. Int. Ed. 2012, 51, 11131–11135. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, D.H. Novel polymer nanowire crystals of diketopyrrolopyrrole-based copolymer with excellent charge transport properties. Adv. Mater. 2013, 25, 4102–4106. [Google Scholar] [CrossRef]
- Wang, H.Y.; Liu, J.G. Fibrillar morphology of derivatives of poly(3-alkylthiophene)s by solvent vapor annealing: Effects of conformational transition and conjugate length. J. Phys. Chem. B 2013, 117, 5996–6006. [Google Scholar] [CrossRef]
- Hultell, M.; Stafstrom, S. Impact of ring torsion on the intrachain mobility in conjugated polymers. Phys. Rev. B 2007, 75, 104304:1–104304:7. [Google Scholar]
- Cho, H.H.; Kang, T.E. Effect of incorporated nitrogens on the planarity and photovoltaic performance of donor-acceptor copolymers. Macromolecules 2012, 45, 6415–6423. [Google Scholar] [CrossRef]
- Ong, B.S.; Wu, Y. Thiophene polymer semiconductors for organic thin-film transistors. Chem. Eur. J. 2008, 14, 4766–4778. [Google Scholar] [CrossRef]
- Kim, J.; Lim, B. Highly soluble poly(thienylenevinylene) derivatives with charge-carrier mobility exceeding 1 cm2∙V−1∙s−1. Chem. Mater. 2011, 23, 4663–4665. [Google Scholar] [CrossRef]
- Lee, W.H.; Kong, H. Field-effect transistors based on ppv derivatives as a semiconducting layer. J. Polym. Sci. A Polym. Chem. 2009, 47, 111–120. [Google Scholar] [CrossRef]
- Stevens, D.M.; Qin, Y. Enhancement of the morphology and open circuit voltage in bilayer polymer/fullerene solar cells. J. Phys. Chem. C 2009, 113, 11408–11415. [Google Scholar] [CrossRef]
- Lei, T.; Cao, Y. Systematic investigation of isoindigo-based polymeric field-effect transistors: Design strategy and impact of polymer symmetry and backbone curvature. Chem. Mater. 2012, 24, 1762–1770. [Google Scholar] [CrossRef]
- Ong, B.S.; Wu, Y.L. Structurally ordered polythiophene nanoparticles for high-performance organic thin-film transistors. Adv. Mater. 2005, 17, 1141–1144. [Google Scholar] [CrossRef]
- McCulloch, I.; Heeney, M. Liquid-crystalline semiconducting polymers with high charge-carrier mobility. Nat. Mater. 2006, 5, 328–333. [Google Scholar] [CrossRef]
- Li, J.; Qin, F. High-performance thin-film transistors from solution-processed dithienothiophene polymer semiconductor nanoparticles. Chem. Mater. 2008, 20, 2057–2059. [Google Scholar] [CrossRef]
- Pan, H.L.; Li, Y.N. Low-temperature, solution-processed, high-mobility polymer semiconductors for thin-film transistors. J. Am. Chem. Soc. 2007, 129, 4112–4113. [Google Scholar] [CrossRef]
- Osaka, I.; Abe, T. High-mobility semiconducting naphthodithiophene copolymers. J. Am. Chem. Soc. 2010, 132, 5000–5001. [Google Scholar] [CrossRef]
- Osaka, I.; Zhang, R. High-lamellar ordering and amorphous-like π-network in short-chain thiazolothiazole-thiophene copolymers lead to high mobilities. J. Am. Chem. Soc. 2009, 131, 2521–2529. [Google Scholar] [CrossRef]
- Zhang, M.X.; Zhao, G.J. Heteroatomic effects on charge-transfer mobility of dianthra[2,3-b:2′,3′-f]thieno[3,2-b]thiophene (DATT) and its derivatives. J. Phys. Chem. C 2012, 116, 19197–19202. [Google Scholar] [CrossRef]
- Kang, I.; An, T.K. Effect of selenophene in a DPP copolymer incorporating a vinyl group for high-performance organic field-effect transistors. Adv. Mater. 2013, 25, 524–528. [Google Scholar] [CrossRef]
- Lin, Y.Z.; Fan, H.J. Thiazole-based organic semiconductors for organic electronics. Adv. Mater. 2012, 24, 3087–3106. [Google Scholar] [CrossRef]
- Osaka, I.; Saito, M. Drastic change of molecular orientation in a thiazolothiazole copolymer by molecular-weight control and blending with PC61BM leads to high efficiencies in solar cells. Adv. Mater. 2012, 24, 425–430. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Lemke, H. High mobility ambipolar charge transport in polyselenophene conjugated polymers. Adv. Mater. 2010, 22, 2371–2375. [Google Scholar] [CrossRef]
- Zhang, W.M.; Smith, J. Indacenodithiophene semiconducting polymers for high-performance, air-stable transistors. J. Am. Chem. Soc. 2010, 132, 11437–11439. [Google Scholar] [CrossRef]
- Guo, X.G.; Kim, F.S. Naphthalene diimide-based polymer semiconductors: Synthesis, structure-property correlations, and n-channel and ambipolar field-effect transistors. Chem. Mater. 2012, 24, 1434–1442. [Google Scholar] [CrossRef]
- Yan, H.; Chen, Z.H. A high-mobility electron-transporting polymer for printed transistors. Nature 2009, 457, 679–686. [Google Scholar] [CrossRef]
- Zhao, X.G.; Zhan, X.W. Electron transporting semiconducting polymers in organic electronics. Chem. Soc. Rev. 2011, 40, 3728–3743. [Google Scholar] [CrossRef]
- Li, Y.N.; Sonar, P. High mobility diketopyrrolopyrrole (DPP)-based organic semiconductor materials for organic thin film transistors and photovoltaics. Energy Environ. Sci. 2013, 6, 1684–1710. [Google Scholar] [CrossRef]
- Beaujuge, P.M.; Amb, C.M. Spectral engineering in π-conjugated polymers with intramolecular donor-acceptor interactions. Acc. Chem. Res. 2010, 43, 1396–1407. [Google Scholar] [CrossRef]
- Ha, J.S.; Kim, K.H. 2,5-Bis(2-octyldodecyl)pyrrolo[3,4-c]pyrrole-1,4-(2H,5H)-dione-based donor-acceptor alternating copolymer bearing 5,5′-di(thiophen-2-yl)-2,2′-biselenophene exhibiting 1.5 cm2∙V−1∙s−1 hole mobility in thin-film transistors. J. Am. Chem. Soc. 2011, 133, 10364–10367. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Lee, M.J. High-performance ambipolar diketopyrrolopyrrole-thieno[3,2-b]thiophene copolymer field-effect transistors with balanced hole and electron mobilities. Adv. Mater. 2012, 24, 647–652. [Google Scholar] [CrossRef]
- Chen, H.J.; Guo, Y.L. Highly π-extended copolymers with diketopyrrolopyrrole moieties for high-performance field-effect transistors. Adv. Mater. 2012, 24, 4618–4622. [Google Scholar] [CrossRef]
- Lei, T.; Dou, J.H. Electron-deficient poly(p-phenylene vinylene) provides electron mobility over 1 cm2∙V−1∙s−1 under ambient conditions. J. Am. Chem. Soc. 2013, 135, 12168–12171. [Google Scholar] [CrossRef]
- Sauvé, G.; Javier, A.E. Well-defined, high molecular weight poly(3-alkylthiophene)s in thin-film transistors: side chain invariance in field-effect mobility. J. Mater. Chem. 2010, 20, 3195–3201. [Google Scholar] [CrossRef]
- Hu, Z.J.; Liu, J.H. Influence of backbone rigidness on single chain conformation of thiophene-based conjugated polymers. J. Phys. Chem. B 2013, 117, 4461–4467. [Google Scholar] [CrossRef]
- Beaujuge, P.M.; Pisula, W. Tailoring structure-property relationships in dithienosilole-benzothiadiazole donor-acceptor copolymers. J. Am. Chem. Soc. 2009, 131, 7514–7515. [Google Scholar] [CrossRef]
- Deng, Y.F.; Chen, Y.G. Donor-acceptor conjugated polymers with dithienocarbazoles as donor units: Effect of structure on semiconducting properties. Macromolecules 2012, 45, 8621–8627. [Google Scholar] [CrossRef]
- Osaka, I.; Abe, T. Impact of isomeric structures on transistor performances in naphthodithiophene semiconducting polymers. J. Am. Chem. Soc. 2011, 133, 6852–6860. [Google Scholar] [CrossRef]
- Brinkmann, M.; Gonthier, E. Segregated vs. mixed interchain stacking in highly oriented films of naphthalene diimide bithiophene copolymers. ACS Nano 2012, 6, 10319–10326. [Google Scholar] [CrossRef]
- Donley, C.L.; Zaumseil, J. Effects of packing structure on the optoelectronic and charge transport properties in poly(9,9-di-n-octylfluorene-alt-benzothiadiazole). J. Am. Chem. Soc. 2005, 127, 12890–12899. [Google Scholar] [CrossRef]
- Park, J.W.; Lee, D.H. Conformationally twisted semiconducting polythiophene derivatives with alkylthiophene side chain: High solubility and air stability. Macromolecules 2010, 43, 2118–2123. [Google Scholar] [CrossRef]
- Schroeder, B.C.; Nielsen, C.B. Benzotrithiophene co-polymers with high charge carrier mobilities in field-effect transistors. Chem. Mater. 2011, 23, 4025–4031. [Google Scholar] [CrossRef]
- Ren, X.K.; Wu, Y.C. Crystal structure and molecular packing behavior of poly(2,3-diphenyl-1,4-phenylenevinylene) derivatives containing alkyl side-chains. Macromolecules 2013, 46, 155–163. [Google Scholar] [CrossRef]
- Bolognesi, A.; Botta, C. Oriented thin films from soluble polythiophenes. Polym. Adv. Technol. 2003, 14, 537–543. [Google Scholar] [CrossRef]
- Nagamatsu, S.; Misaki, M. Side-chain effects on friction-transferred polymer orientation. Polym. J. 2007, 39, 1300–1305. [Google Scholar] [CrossRef]
- Higashi, T.; Yamasaki, N. Anisotropic properties of aligned π-conjugated polymer films fabricated by capillary action and their post-annealing effects. Appl. Phys. Express 2011. [Google Scholar] [CrossRef]
- He, M.Q.; Li, J.F. Alkylsubstituted thienothiophene semiconducting materials: Structure-property relationships. J. Am. Chem. Soc. 2009, 131, 11930–11938. [Google Scholar] [CrossRef]
- Rieger, R.; Beckmann, D. Backbone curvature in polythiophenes. Chem. Mater. 2010, 22, 5314–5318. [Google Scholar] [CrossRef]
- Yazawa, K.; Inoue, Y. Molecular dynamics of regioregular poly(3-hexylthiophene) investigated by nmr relaxation and an interpretation of temperature dependent optical absorption. J. Phys. Chem. B 2010, 114, 1241–1248. [Google Scholar] [CrossRef]
- Oosterbaan, W.D.; Bolsee, J.C. Alkyl-chain-length-independent hole mobility via morphological control with poly(3-alkylthiophene) nanofibers. Adv. Funct. Mater. 2010, 20, 792–802. [Google Scholar] [CrossRef]
- Pankaj, S.; Beiner, M. Confined dynamics and crystallization in self-assembled alkyl nanodomains. J. Phys. Chem. B 2010, 114, 15459–15465. [Google Scholar] [CrossRef]
- Malik, S.; Nandi, A.K. Crystallization mechanism of regioregular poly(3-alkyl thiophene)s. J. Polym. Sci. B Polym. Phys. 2002, 40, 2073–2085. [Google Scholar] [CrossRef]
- Li, Z.; Tsang, S.W. Alternating copolymers of cyclopenta[2,1-b;3,4-b′] dithiophene and thieno[3,4-c]pyrrole-4,6-dione for high-performance polymer solar cells. Adv. Funct. Mater. 2011, 21, 3331–3336. [Google Scholar] [CrossRef]
- Yamamoto, T.; Ikai, T. Synthesis and characterization of thieno[3,4-b]thiophene-based copolymers bearing 4-substituted phenyl ester pendants: Facile fine-tuning of HOMO energy levels. Macromolecules 2011, 44, 6659–6662. [Google Scholar] [CrossRef]
- Kline, R.J.; DeLongchamp, D.M. Critical role of side-chain attachment density on the order and device performance of polythiophenes. Macromolecules 2007, 40, 7960–7965. [Google Scholar] [CrossRef]
- Wang, C.C.; Jimison, L.H. Microstructural origin of high mobility in high-performance poly(thieno-thiophene) thin-film transistors. Adv. Mater. 2010, 22, 697–701. [Google Scholar] [CrossRef]
- Keg, P.; Lohani, A. Direct observation of alkyl chain interdigitation in conjugated polyquarterthiophene self-organized on graphite surfaces. Macromole. Rapid Commun. 2008, 29, 1197–1202. [Google Scholar] [CrossRef]
- Meager, L.; Ashraf, R.S. Photocurrent Enhancement from diketopyrrolopyrrole polymer solar cells through alkyl-chain branching point manipulation. J. Am. Chem. Soc. 2013, 135, 11537–11540. [Google Scholar] [CrossRef]
- Kim, D.H.; Lee, J. Molecular weight-induced structural transition of liquid-crystalline polymer semiconductor for high-stability organic transistor. Adv. Funct. Mater. 2011, 21, 4442–4447. [Google Scholar] [CrossRef]
- Wu, S. Chain structure and entanglement. J. Polym. Sci. B Polym. Phys. 1989, 27, 723–741. [Google Scholar] [CrossRef]
- Wen, Y.H.; Lin, P.C. Dynamic structure factor for large aggregate clusters with internal motions: A self-consistent light-scattering study on conjugated polymer solutions. J. Phys. Chem. B 2011, 115, 14369–14380. [Google Scholar] [CrossRef]
- Huang, Y.; Cheng, H. Temperature induced structure evolution of regioregular poly(3-hexylthiophene) in dilute solution and its influence on thin film morphology. Macromolecules 2010, 43, 10031–10037. [Google Scholar] [CrossRef]
- Huang, Y.; Cheng, H. Unimer–aggregate equilibrium to large scale association of regioregular poly(3-hexylthiophene) in THF solution. Macromolecules 2011, 44, 5020–5026. [Google Scholar] [CrossRef]
- Rughooputh, S.D.D.V.; Hotta, S. Chromism of soluble polythienylenes. J. Polym. Sci. B Polym. Phys. 1987, 25, 1071–1078. [Google Scholar] [CrossRef]
- Vogelsang, J.; Brazard, J. Watching the annealing process one polymer chain at a time. Angew. Chem. Int. Ed. 2011, 50, 2257–2261. [Google Scholar] [CrossRef]
- Steyrleuthner, R.; Schubert, M. Aggregation in a high-mobility n-type low-bandgap copolymer with implications on semicrystalline morphology. J. Am. Chem. Soc. 2012, 134, 18303–18317. [Google Scholar] [CrossRef]
- Kohler, A.; Hoffmann, S.T. An order-disorder transition in the conjugated polymer MEH-PPV. J. Am. Chem. Soc. 2012, 134, 11594–11601. [Google Scholar] [CrossRef]
- Malik, S.; Nandi, A.K. Influence of alkyl chain length on the gelation mechanism of thermoreversible gels of regioregular poly(3-alkyl thiophenes) in xylene. J. Appl. Polym. Sci. 2007, 103, 2528–2537. [Google Scholar] [CrossRef]
- Xu, Y.Z.; Liu, J.G. Formation of parallel aligned nano-fibrils of poly(3,3′′′-didodecylquaterthiophene) induced by the unimer coils in solution. RSC Adv. 2013, 3, 12069–12074. [Google Scholar] [CrossRef]
- Samitsu, S.; Shimomura, T. Effective production of poly(3-alkylthiophene) nanofibers by means of whisker method using anisole solvent: structural, optical, and electrical properties. Macromolecules 2008, 41, 8000–8010. [Google Scholar] [CrossRef]
- Xue, L.J.; Gao, X. The formation of different structures of poly(3-hexylthiophene) film on a patterned substrate by dip-coating from aged solution. Nanotechnology 2010. [Google Scholar] [CrossRef]
- Zhao, K.; Xue, L. A new method to improve poly(3-hexyl thiophene) (P3HT) crystalline behavior: Decreasing chains entanglement to promote order−disorder transformation in solution. Langmuir 2010, 26, 471–477. [Google Scholar] [CrossRef]
- Zhao, K.; Khan, H.U. Entanglement of conjugated polymer chains influences molecular self-assembly and carrier transport. Adv. Funct. Mater. 2013. [Google Scholar] [CrossRef]
- Wang, P.S.; Lu, H.H. Gel formation via physical cross-linking in the soluble conjugated polymer, poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene], in solution by addition of alkalies. Macromolecules 2008, 41, 6500–6504. [Google Scholar] [CrossRef]
- Liu, J.; Shao, S. The mechanisms for introduction of n-dodecylthiol to modify the P3HT/PCBM morphology. Org. Electron. 2010, 11, 775–783. [Google Scholar] [CrossRef]
- Wang, H.Y.; Liu, J.G. Nano-fibrils formation of pBTTT via adding alkylthiol into solutions: Control of morphology and crystalline structure. Polymer 2013, 54, 948–957. [Google Scholar] [CrossRef]
- Traiphol, R.; Charoenthai, N. Chain organization and photophysics of conjugated polymer in poor solvents: Aggregates, agglomerates and collapsed coils. Polymer 2007, 48, 813–826. [Google Scholar] [CrossRef]
- Oh, J.Y.; Shin, M. Self-seeded growth of poly(3-hexylthiophene) (P3HT) nanofibrils by a cycle of cooling and heating in solutions. Macromolecules 2012, 45, 7504–7513. [Google Scholar] [CrossRef]
- Li, L.G.; Lu, G.H. Improving performance of polymer photovoltaic devices using an annealing-free approach via construction of ordered aggregates in solution. J. Mater. Chem. 2008, 18, 1984–1990. [Google Scholar] [CrossRef]
- Park, Y.D.; Lee, H.S. Solubility-induced ordered polythiophene precursors for high-performance organic thin-film transistors. Adv. Funct. Mater. 2009, 19, 1200–1206. [Google Scholar] [CrossRef]
- Yan, H.; Yan, Y. Self-assembling branched and hyperbranched nanostructures of poly(3-hexylthiophene) by a solution process. J. Phys. Chem. C 2011, 115, 3257–3262. [Google Scholar] [CrossRef]
- Xu, W.; Li, L. Solvent-induced crystallization of poly(3-dodecylthiophene): Morphology and kinetics. J. Phys. Chem. B 2011, 115, 6412–6420. [Google Scholar] [CrossRef]
- Berson, S.; de Bettignies, R. Poly(3-hexylthiophene) fibers for photovoltaic applications. Adv. Funct. Mater. 2007, 17, 1377–1384. [Google Scholar] [CrossRef]
- He, M.; Ge, J. Fabricating polythiophene into highly aligned microwire film by fast evaporation of its whisker solution. Polymer 2010, 51, 2236–2243. [Google Scholar] [CrossRef]
- Crossland, E.J.W.; Rahimi, K. Systematic control of nucleation density in poly(3-hexylthiophene) thin films. Adv. Funct. Mater. 2011, 21, 518–524. [Google Scholar] [CrossRef]
- Lu, G.H.; Tang, H.W. Enhanced charge transportation in semiconducting polymer/insulating polymer composites: The role of an interpenetrating bulk interface. Adv. Funct. Mater. 2010, 20, 1714–1720. [Google Scholar] [CrossRef]
- Benetti, E.M.; Causin, V. Conjugated polymers in cages: Templating poly(3-hexylthiophene) nanocrystals by inert gel matrices. Adv. Mater. 2012, 24, 5636–5641. [Google Scholar] [CrossRef]
- Kim, F.S.; Jenekhe, S.A. Charge transport in poly(3-butylthiophene) nanowires and their nanocomposites with an insulating polymer. Macromolecules 2012, 45, 7514–7519. [Google Scholar] [CrossRef]
- Qiu, L.Z.; Lee, W.H. Organic thin-film transistors based on polythiophene nanowires embedded in insulating polymer. Adv. Mater. 2009, 21, 1349–1353. [Google Scholar] [CrossRef]
- Qiu, L.Z.; Wang, X. Organic thin-film transistors based on blends of poly(3-hexylthiophene) and polystyrene with a solubility-induced low percolation threshold. Chem. Mater. 2009, 21, 4380–4386. [Google Scholar] [CrossRef]
- Kim, D.H.; Park, Y.D. Solvent vapor-induced nanowire formation in poly(3-hexylthiophene) thin films. Macromol. Rapid Commun. 2005, 26, 834–839. [Google Scholar] [CrossRef]
- Chang, J.F.; Sun, B.Q. Enhanced mobility of poly(3-hexylthiophene) transistors by spin-coating from high-boiling-point solvents. Chem. Mater. 2004, 16, 4772–4776. [Google Scholar] [CrossRef]
- Wang, S.H.; Kappl, M. Organic field-effect transistors based on highly ordered single polymer fibers. Adv. Mater. 2012, 24, 417–420. [Google Scholar] [CrossRef]
- Dong, H.L.; Jiang, S.D. Nanowire crystals of a rigid rod conjugated polymer. J. Am. Chem. Soc. 2009, 131, 17315–17320. [Google Scholar] [CrossRef]
- Liu, C.F.; Wang, Q.L. Morphology and structure of the β phase crystals of monodisperse polyfluorenes. Macromolecules 2013, 46, 3025–3030. [Google Scholar] [CrossRef]
- Liu, Y.; Dong, H.L. High performance nanocrystals of a donor-acceptor conjugated polymer. Chem. Mater. 2013, 25, 2649–2655. [Google Scholar] [CrossRef]
- Jo, J.; Kim, S.S. Time-dependent morphology evolution by annealing processes on polymer: Fullerene blend solar cells. Adv. Funct. Mater. 2009, 19, 866–874. [Google Scholar] [CrossRef]
- Vogelsang, J.; Lupton, J.M. Solvent vapor annealing of single conjugated polymer chains: Building organic optoelectronic materials from the bottom up. J. Phys. Chem. Lett. 2012, 3, 1503–1513. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, Q.Q. Solvent-vapor induced self-assembly of a conjugated polymer: A correlation between solvent nature and transistor performance. Org. Electron. 2012, 13, 2372–2378. [Google Scholar] [CrossRef]
- Liu, J.G.; Chen, L. Constructing the nanointerpenetrating structure of PCDTBT:PC70BM bulk heterojunction solar cells induced by aggregation of PC70BM via mixed-solvent vapor annealing. J. Mater. Chem. A 2013, 1, 6216–6225. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, H.; Xu, Y.; Yu, X.; Xing, R.; Liu, J.; Han, Y. Structure and Morphology Control in Thin Films of Conjugated Polymers for an Improved Charge Transport. Polymers 2013, 5, 1272-1324. https://doi.org/10.3390/polym5041272
Wang H, Xu Y, Yu X, Xing R, Liu J, Han Y. Structure and Morphology Control in Thin Films of Conjugated Polymers for an Improved Charge Transport. Polymers. 2013; 5(4):1272-1324. https://doi.org/10.3390/polym5041272
Chicago/Turabian StyleWang, Haiyang, Yaozhuo Xu, Xinhong Yu, Rubo Xing, Jiangang Liu, and Yanchun Han. 2013. "Structure and Morphology Control in Thin Films of Conjugated Polymers for an Improved Charge Transport" Polymers 5, no. 4: 1272-1324. https://doi.org/10.3390/polym5041272