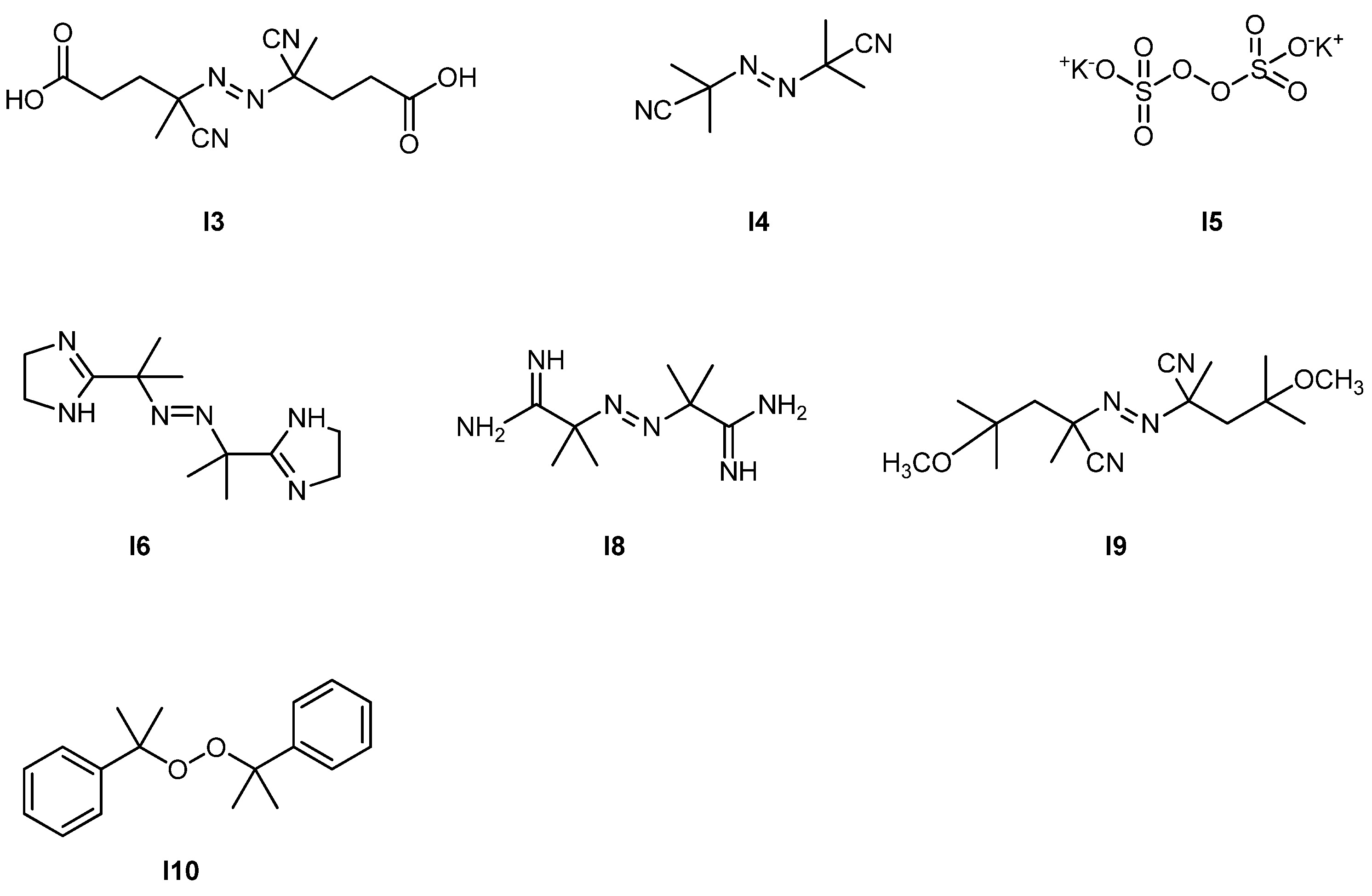
Synthesis of Glycopolymer Architectures by Reversible-Deactivation Radical Polymerization

Abstract
:1. Introduction
2. Glycopolymers and Reversible-Deactivation Radical Polymerization
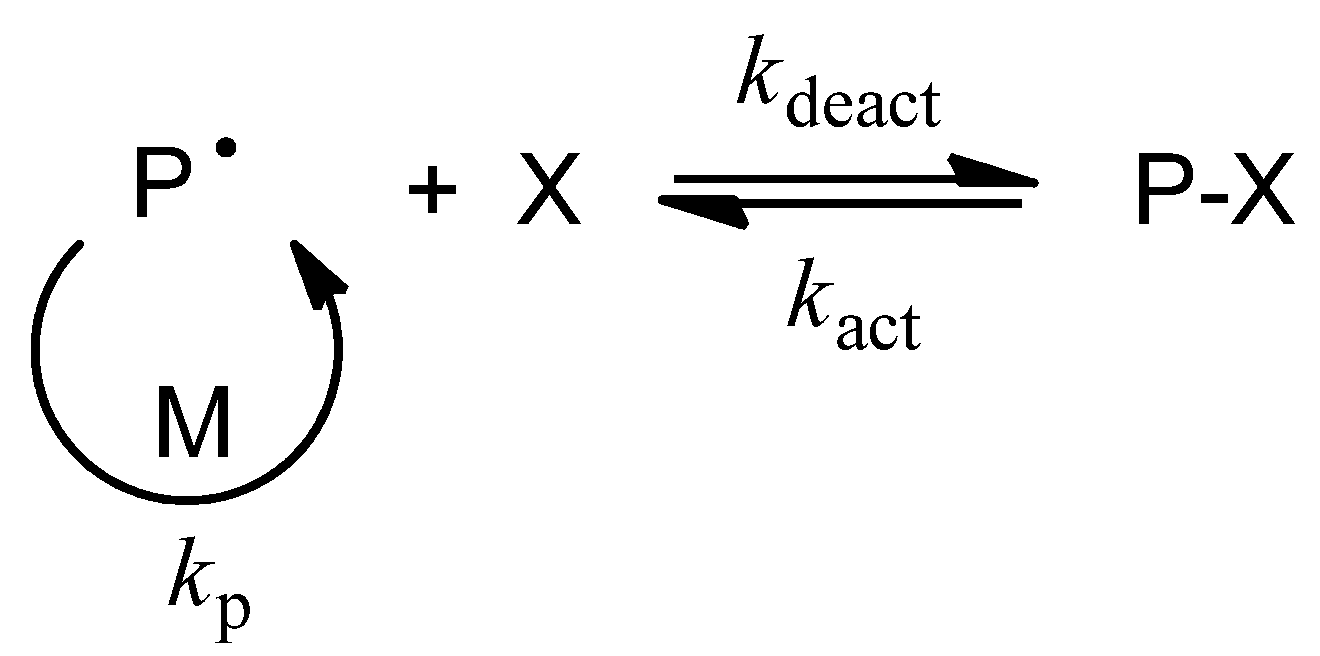
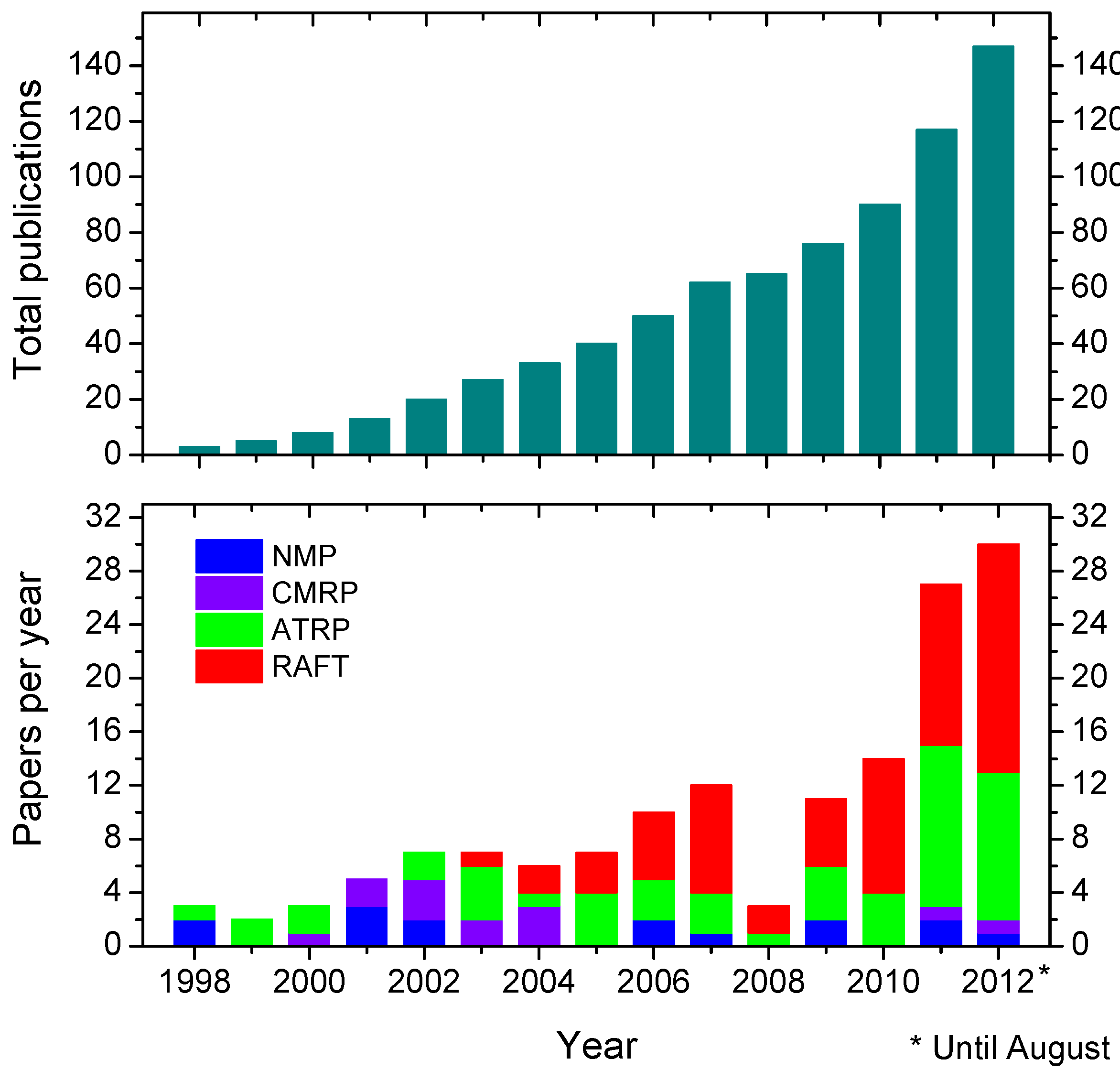
3. How to Consult the Review
4. Synthesis of Glycopolymers by Stable Free Radical Polymerization (SFRP)
4.1. SFRP Starting from Protected Glycomonomers/Control Agents
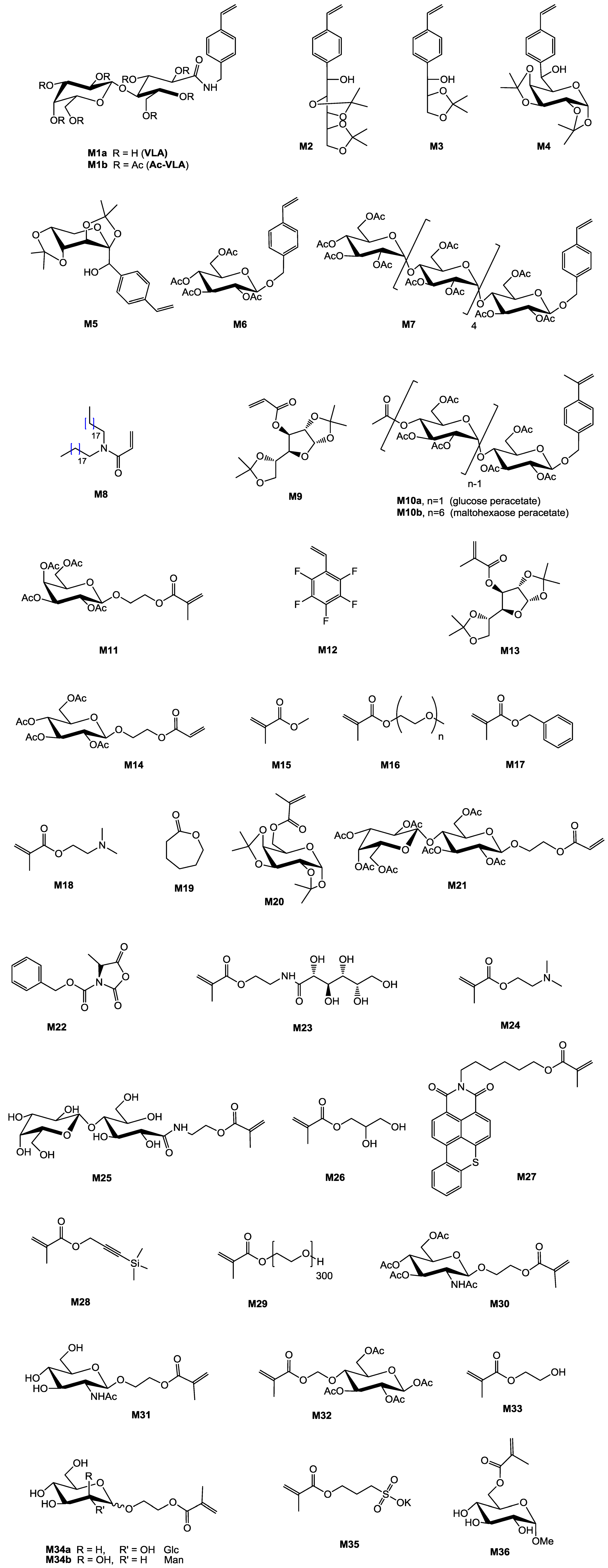
4.1.1. (Meth)acrylate Monomers
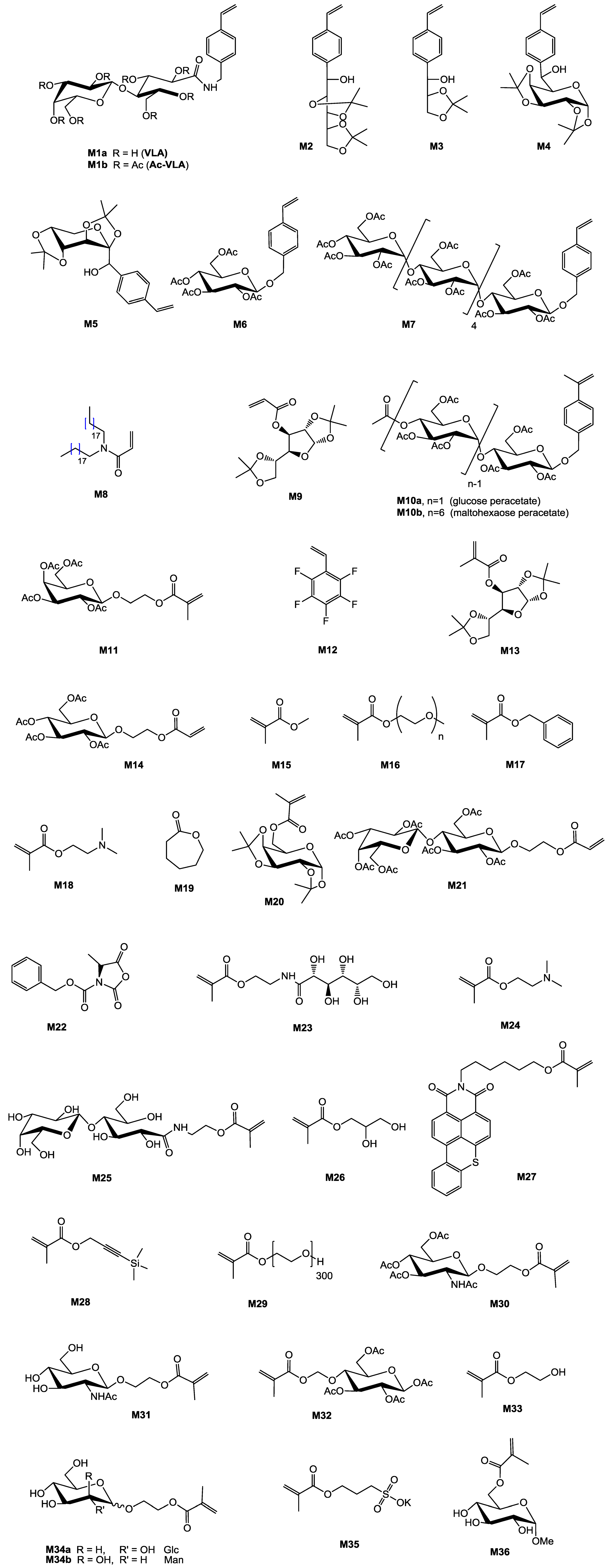
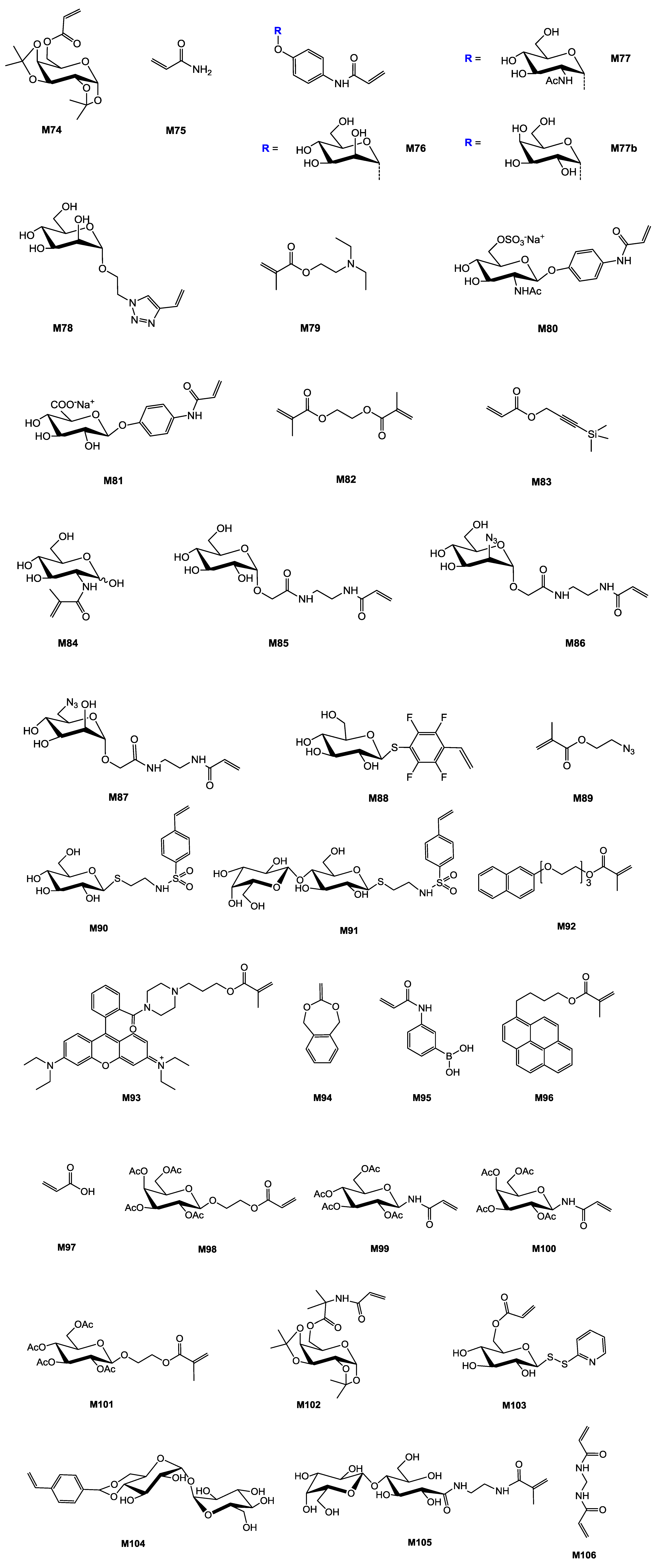

| Entry | Entry Carbohydrate | Entry Monomer(s) | Entry Initiator | Entry Additive | Entry Conv. a % | Entry Mn (×10−3) | Entry Mn/Mn,th b | Entry Đ c | Entry Structure | Entry Application sought/test | Entry Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Alkene manomers (unproctected) | |||||||||||
| 1 | lactose (α-O) | M119a/M75 | N12a | – | 15 | 28.8 | – | 1.31 | A-stat-B | promoter of binding of dFGF-2 to FGF receptor-1 | Baskaran et al. [76] |
| 2 | lactose (persulfated, α-O) | M119b/M75 | N12a | – | 30 | 38 | – | 1.5 | A-stat-B | promoter of binding of dFGF-2 to FGF receptor-1 | Baskaran et al. [76] |
| 3 | N-acetylglucosamine (α-O) | M117a/M75 | N12a | – | 30 | 43 | – | 1.47 | A-stat-B | – | Chaikof et al. [77,78] |
| 4 | N-acetylglucosamine (α-O) | M117c/M75 | N12a | – | 20 | 99.3 | – | 1.45 | A-stat-B | – | Chaikof et al. [77,78] |
| 5 | N-acetylglucosamine (persulfated, α-O) | M117b/M75 | N12a | – | 35 | 57.3 | – | 1.37 | A-stat-B | – | Chaikof et al. [77,78] |
| 6 | N-acetylglucosamine (persulfated, α-O) | M117d/M75 | N12a | – | 26 | 57.2 | – | 1.2 | A-stat-B | – | Chaikof et al. [77,78] |
| (Meth)acrylamide monomers (unprotected) | |||||||||||
| 7 | lactose (β-O) | M107a/M75 | N12a | – | 71 | 9 | – | 1.3 | A-stat-B | anticoagulant, antithrombin | Sun et al. [79] |
| 8 | lactose (persulfated, β-O) | M107b | N12a | – | 55 | 7.5 | – | 1.19 | homo | anticoagulant, antithrombin | Chaikof et al. [79,80] |
| 9 | lactose (persulfated, β-O) | M107b/M75 | N12a | – | 67 | 33.4 | – | 1.47 | homo | anticoagulant/antithrombin, promoter of binding of dFGF-2 to FGF receptor-1 | Chaikof et al. [79,80] |
| 10 | lactose (β-O) | M107a/M75 | N13a | – | 75 | 12 | – | 1.3 | A-stat-B | surface modification, lectin interaction | Chaikof et al. [81,82,83,84,85] |
| (Meth)acrylate monomers (protected) | |||||||||||
| 11 | galactose (β-O) | M11/St | N9 | – | 45 | 40.6 | – | 1.26 | A-stat-B | – | Ting et al. [86] |
| 12 | galactose (β-O) | M11/St | polySt·N9 | – | 48–79 | 21.7–79.9 | – | 1.34–1.50 | (A-stat-B)-block-C | micelles and structured films for lectin recognition | Ting et al. [86] |
| 13 | glucose (α/β, 3-O) | M9 | N7 | – | 58 | 9 | – | 1.17 | homo | film synthesis | Gotz et al. [87] |
| 14 | glucose (α/β, 3-O) | M9/M8 | N8 | – | 55 | 13.8 | – | 1.2 | A-stat-B | film synthesis | Gotz et al. [87] |
| (Meth)acrylate monomers (unprotected) | |||||||||||
| 15 | N-acetylglucosamine (α-O) | M118a | N12a | – | 25 | 15.4 | – | 1.26 | homo | – | Grande et al. [78] |
| 16 | N-acetylglucosamine (α-O) | M118a/M75 | N12a | – | 33 | 30.6 | – | 1.35 | A-stat-B | – | Grande et al. [78] |
| 17 | N-acetylglucosamine (persulfated, α-O) | M118b | N12a | – | 35 | 9.9 | – | 1.13 | homo | – | Grande et al. [78] |
| 18 | N-acetylglucosamine (persulfated, α-O) | M118b/M75 | N12a | – | – | 21.7 | – | 1.2 | A-stat-B | anticoagulant, antithrombin, promoter of binding of dFGF-2 to FGF receptor-1 | Chaikof et al. [79,80] |
| Styrenic monomers (protected) | |||||||||||
| 19 | fructose (pyranose, 1-C) | M5 | N4 | DCP | 79 | 16.7 | 0.58 | 2 | homo | – | Chen et al. [88] |
| 20 | galactose (α/β, 6-O) | M4 | N4 | DCP | 56 | 11 | 0.54 | 1.36 | homo | – | Chen et al. [88] |
| 21 | glucitol/mannitol | M2 | N4 | DCP | 82 | 16.8 | 0.61 | 1.37 | homo | – | Chen et al. [88] |
| 22 | glucitol/mannitol | M2 | polySt·N4 | – | – | 38 | – | 1.54 | block AB | film synthesis, surface modification | Chen et al. [89] |
| 23 | glucitol/mannitol | St | polyM2·N4 | – | – | 96.5 | – | 1.37 | block AB | film synthesis, surface modification | Chen et al. [89] |
| 24 | glucose (β-O) | M6 | N5 | – | ~50 | 12.7 | – | 1.13 | block AB | – | Narumi et al. [90] |
| 25 | glucose (β-O) | M6 | N6 | CSA | 21 | 4.2 | – | 1.09 | homo | – | Narumi et al. [91] |
| 26 | glucose (β-O) | St | polyM6·N6 | – | 10 | 12.5 | – | 1.14 | block ABA | – | Narumi et al. [91] |
| 27 | glucose (β-O) | St | polyM6·N6 | – | 18 | 17.9 | – | 1.12 | block ABA | – | Narumi et al. [91] |
| 28 | glucose (β-O) | St | polyM6·N6 | – | 17 | 29.4 | – | 1.17 | block ABA | – | Narumi et al. [91] |
| 29 | glucose (β-O) | M10a | N10a | DCP | 73 | 21 | – | 1.16 | block ABA | – | Narumi et al. [92] |
| 30 | glucose to maltohexaose (β-O) | St | N10a–f | – | ~40 | 5–25 | – | 1.07–1.14 | homo | – | Narumi et al. [33] |
| 31 | glyceraldehyde (1-C) | M3 | N4 | DCP | 88 | 13.1 | 0.63 | 1.26 | homo | – | Chen et al. [88] |
| 32 | lactobionic acid (amide) | M1a | N1 | DCP | 35 | 7.5 | – | 1.3 | homo | – | Ohno et al. [93] |
| 33 | lactobionic acid (amide) | M1b | N1 | DCP | 90 | 12.5 | – | 1.1 | homo | – | Ohno et al. [93] |
| 34 | lactobionic acid (amide) | M1b | N2 | DCP | 90 | 12 | – | ≤1.20 | homo | lectin recognition | Ohno et al. [94] |
| 35 | lactobionic acid (amide) | M1b | N3 | – | 36 | 17.5 | – | 1.36 | homo | lectin recognition | Miura et al. [95] |
| 36 | maltohexaose (β-O) | M7 | N5 | – | ≅50 | 16.2 | – | 1.21 | block AB | – | Narumi et al. [90] |
| 37 | maltohexaose (β-O) | M10b | N11 | DCP | 84 | 31.8 | – | 1.11 | block ABA | – | Narumi et al. [92] |
| Styrenic monomers (unprotected) | |||||||||||
| 38 | glucose (β-S) | M88 | N9 | – | 70 | 24 | – | 1.16 | homo | cytotoxicity | Babiuch et al. [96] |
| Glycopolymers from post-polymerization reactions | |||||||||||
| 39 | galactose (β-S) | M12 | N9 | – | 78 | 5.7 | 0.79 | 1.06 | homo | – | Babiuch et al. [97] |
| 40 | galactose (β-S) | M12 | polySt ·N9 | – | 52 | 14.3 | 1.15 | 1.16 | block BA | biocompatible films and nanoparticles | Babiuch et al. [97] |
| 41 | galactose (β-S) | M12 | N9 | – | – | 6.3 | – | 1.07 | homo | – | Wild et al. [98] |
| 42 | glucose (β-S) | M12 | N9 | – | 78 | 3.5 | 0.44 | 1.03 | homo | – | Becer et al. [99] |
| 43 | glucose (β-S) | St | polyM12·N9 | – | 66 | 17.8 | 1.02 | 1.21 | block AB | biocompatible films and nanoparticles | Becer et al. [99] |
| 44 | glucose (β-S) | M12 | polySt ·N9 | – | 76 | 7.1 | 0.56 | 1.16 | block AB | biocompatible films and nanoparticles | Becer et al. [99] |
| 45 | α2,3-sialyllactose (β-O) | M107a | N12a | – | 60 | 7 | – | – | A-stat-B | SPR, lectin binding | Narla et al. [100] |
| 46 | α2,6-sialyllactose (β-O) | M107a | N12a | – | 60 | 7 | – | – | A-stat-B | SPR, lectin binding | Narla et al. [100] |
4.1.2. Styrenic Monomers
4.2. SFRP Starting from Unprotected Glycomonomers/Control Agents
4.2.1. Alkene Monomers
4.2.2. (Meth)acrylamide Monomers
4.2.3. (Meth)acrylate Monomers
4.2.4. Styrenic Monomers
4.3. Glycopolymers from Post-Polymerization Reactions
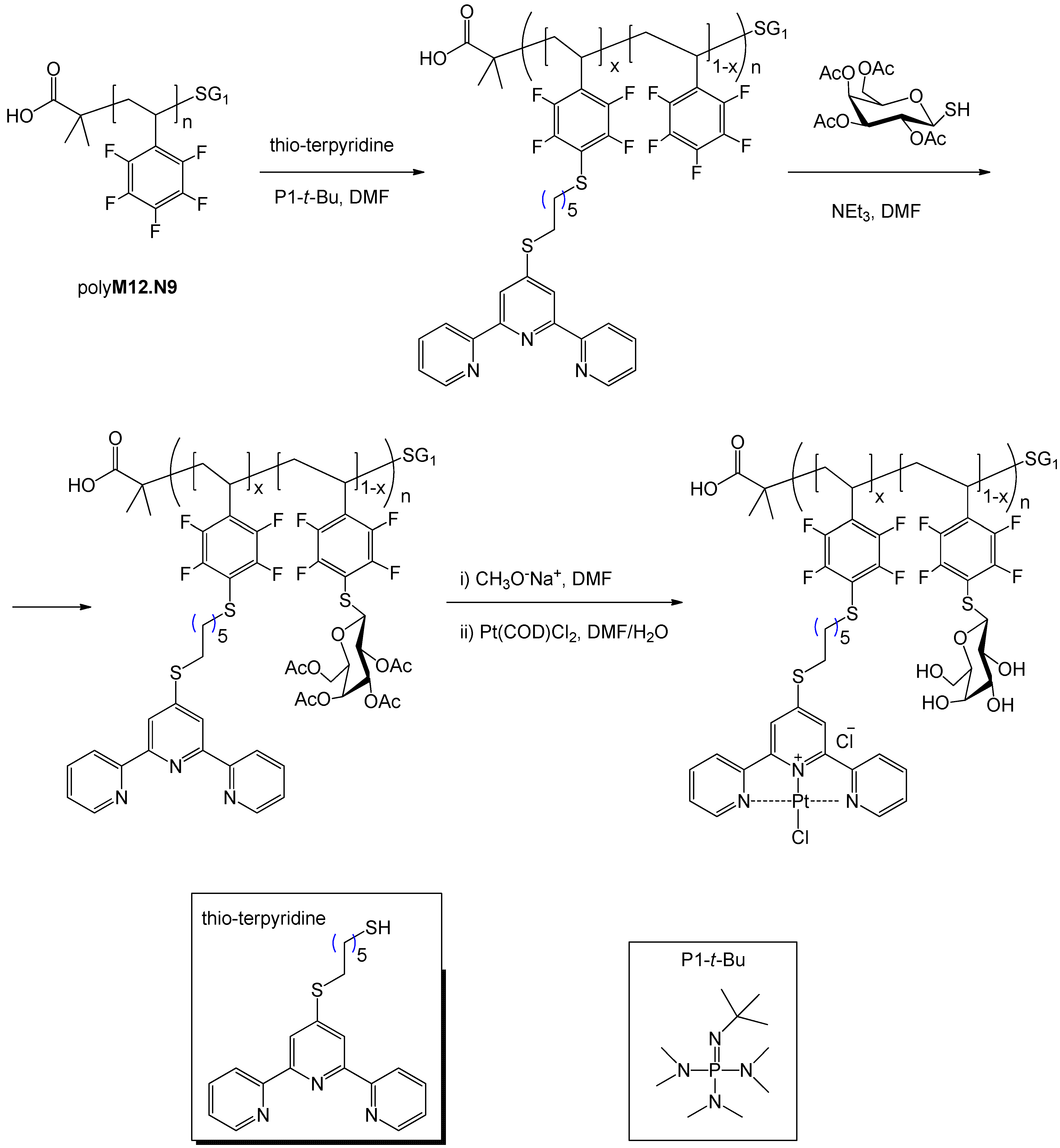
5. Synthesis of Glycopolymers by Atom Transfer Radical Polymerization (ATRP)

5.1. ATRP Starting from Protected Glycomonomers/Glycoinitiators
5.1.1. (Meth)acrylate Monomers
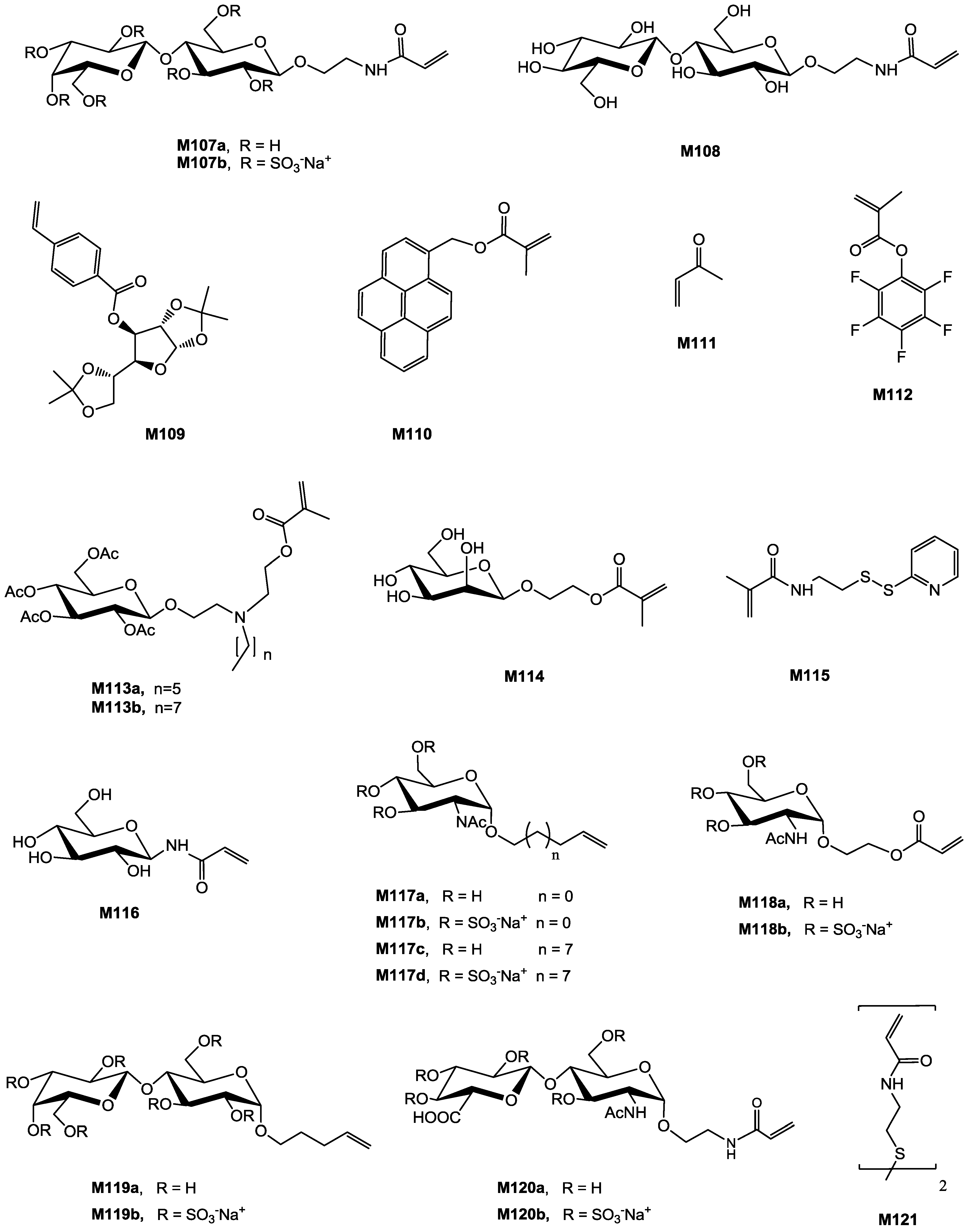
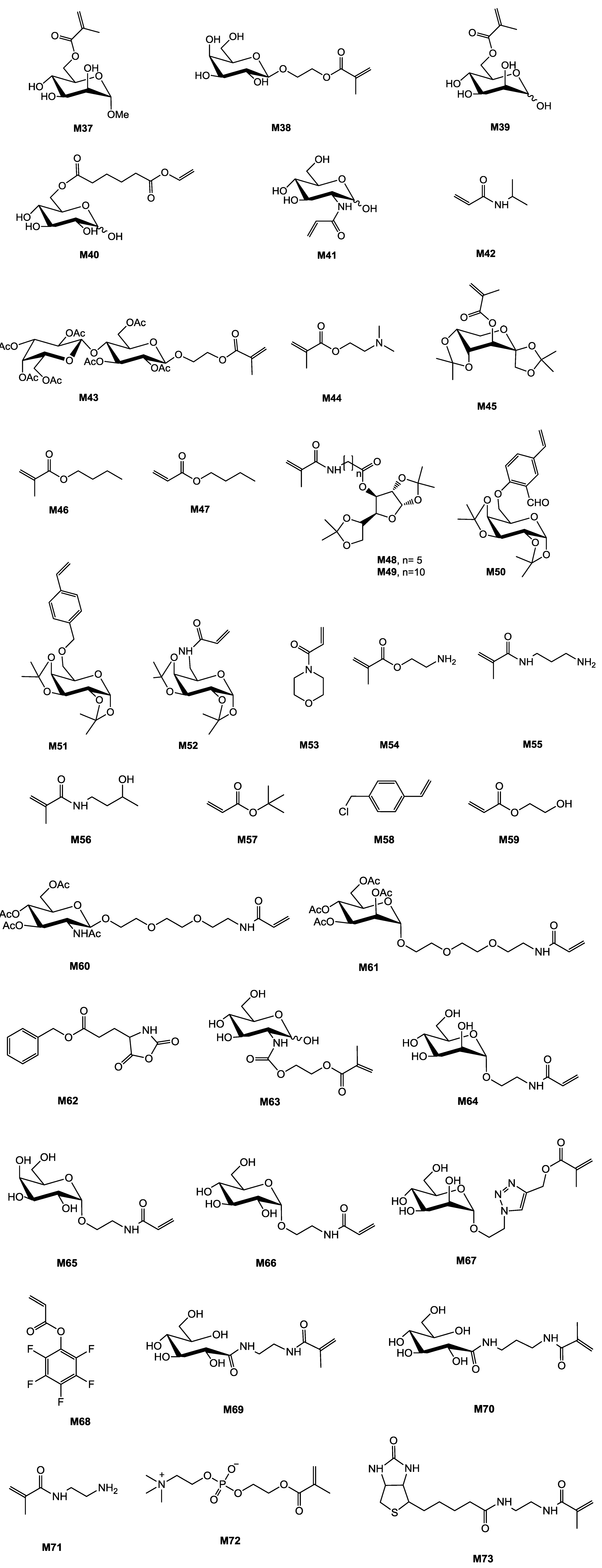
| Entry | Carbohydrate | Monomer(s) | Initiator | Additive | Conv. % | Mn (×10−3) | Mn/Mn,th | Đ b | Structure | Application sought/tested | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (Meth)acrylamide monomers (unprotected) | |||||||||||
| 47 | mannose (α-O) | M64 | A28/A20b | CuCl(L11), CuCl2 | – | 51 | – | 1.5 | brush | lectin recognition | Yu et al. [104] |
| (Meth)acrylate monomers (protected) | |||||||||||
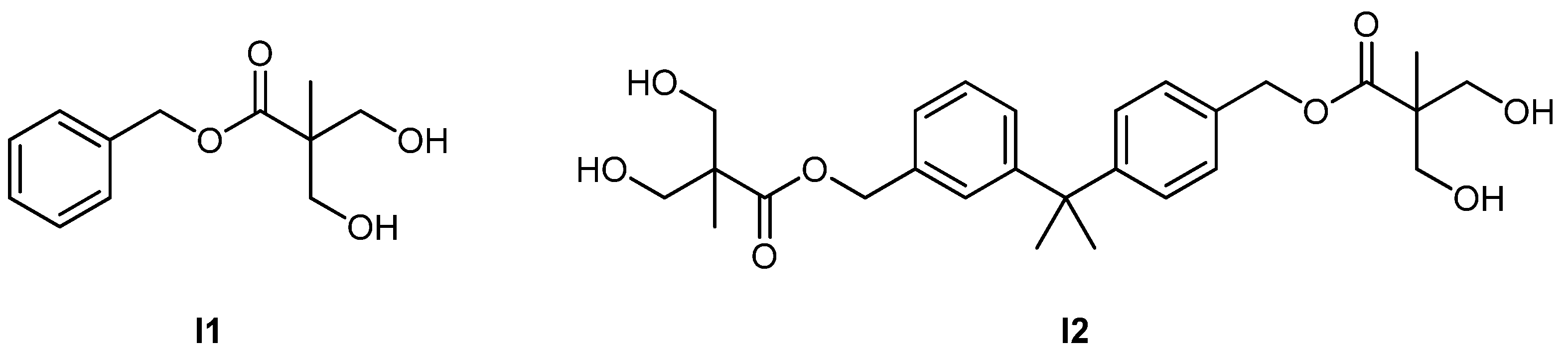
| 48 | – | M19 | I1 | Sn(Oct)2 | – | 6.6 | 1.12 | 1.14 | homo | – | Chen et al. [105] |
| 49 | – | M19 | I2 | Sn(Oct)2 | – | 13.7 | 1.15 | 1.12 | homo | – | Chen et al. [105] |
| 50 | – | M33 | A23 | CuBr(L3) | 45 | 20 | 0.99 | 1.14 | block AB | – | Ke et al. [106] |
| 51 | galactose (α/β, 6-O) | M20 | A12 | CuBr(L6) | 95 | 7.5 | – | 1.08 | homo | – | Ladmiral et al. [107] |
| 52 | galactose (α/β, 6-O) | M20 | A12 | CuBr(L6) | 99 | 13.4 | – | 1.1 | homo | – | Ladmiral et al. [107] |
| 53 | galactose (α/β, 6-O) | M20/M27 | A12 | CuBr(L6) | 87 | 6.1 | – | 1.08 | A-stat-B | – | Ladmiral et al. [107] |
| 54 | galactose (α/β, 6-O) | M20 | A72 | CuBr(L5) | 65 | 20.1 | – | 1.19 | block ABA | – | Chen et al. [105] |
| 55 | galactose (α/β, 6-O) | M20 | A74 | CuBr(L5) | 51 | 35 | – | 1.17 | star (4 arm) | – | Chen et al. [105] |
| 56 | galactose (β-O) | M98 | A8 | CuCl(L2) | 50 | 5.5 | 1.19 | 1.17 | homo | – | Wang et al. [11] |
| 57 | galactose (β-O) | M57 | A8·polyM98·Br | CuCl(L3) | 60 | 21.6 | 1.08 | 1.36 | block ABA | insulin release | Wang et al. [11] |
| 58 | galactose (α/β, 6-O) | M16, M17 | A5 | CuBr(L4) | – | 10.5 | – | 1.21 | block AB | – | Bes et al. [108] |
| 59 | glucose (α/β, 3-O) | M13 | A1 | CuBr(L1) | 83 | 75 | 0.45 | 1.82 | homo | – | Ohno et al. [109] |
| 60 | glucose (α/β, 3-O) | M13 | A1·polySt·Br | CuBr(L1) | – | 14.4 | – | 1.34 | block AB | nanostructured film | Ohno et al. [109] |
| 61 | glucose (α/β, 3-O) | M13 | A36 | CuBr(L1), A37 | – | – | – | – | brush (homo) | – | Ejaz et al. [110] |
| 62 | glucose (α/β, 3-O) | M13 | A12 | CuBr(L6) | 90 | 7.1 | – | 1.14 | homo | – | Ladmiral et al. [107] |
| 63 | glucose (α/β, 3-O) | M13 | A12 | CuBr(L6) | 90 | 14.7 | – | 1.31 | homo | – | Ladmiral et al. [107] |
| 64 | glucose (α/β, 3-O) | M13/M27 | A12 | CuBr(L6) | 93 | 6.1 | – | 1.18 | A-stat-B | – | Ladmiral et al. [107] |
| 65 | glucose (α/β, 3-O) | M13 | A14 | CuBr(L8) | 8 | 416 | – | 1.17 | star | – | Muthukrishnan et al. [111] |
| 66 | glucose (α/β, 3-O) | M13 | A14 | CuBr(L8) | 6 | 601 | – | 1.26 | star | – | Muthukrishnan et al. [111] |
| 67 | glucose (α/β, 3-O) | M9 | A1 | CuBr(L3) | 88 | 6.6 | 1.2 | 1.13 | homo | – | Muthukrishnan et al. [112] |
| 68 | glucose (α/β, 3-O) | M9 | A1 | CuBr(L3) | 93 | 18.5 | 1.3 | 1.25 | homo | – | Muthukrishnan et al. [112] |
| 69 | glucose (α/β, 3-O) | M9 | A1 | CuBr(L3) | 84 | 31 | – | 1.37 | homo | – | Muthukrishnan et al. [112] |
| 70 c | glucose (α/β, 3-O) | M9 | A15 | CuBr(L3) | 98 | 6.6 | – | 1.92 | hyper branched | – | Muthukrishnan et al. [112] |
| 71 d | glucose (α/β, 3-O) | M9 | A15 | CuBr(L3) | 96 | 13 | – | 1.95 | hyper branched | – | Muthukrishnan et al. [112] |
| 72 c | glucose (α/β, 3-O) | M13 | A16 | (PPh3)2NiBr2 | > 98 | 17.6 | – | 2.12 | hyper branched | – | Muthukrishnan et al. [113] |
| 73 d | glucose (α/β, 3-O) | M13 | A16 | (PPh3)2NiBr2 | > 98 | 23.3 | – | 1.57 | hyper branched | – | Muthukrishnan et al. [113] |
| 74 | glucose (α/β, 3-O) | M13 | polyA16 | CuBr(L8) | 10 | 58.6 | – | 1.07 | brush (cylindrical) | – | Muthukrishnan et al. [114] |
| 75 | glucose (α/β, 3-O) | M13 | A19 | CuBr(L8) | 85 | 37.4 | 1.16 | 1.45 | homo | bio-nanotechnology | Gao et al. [115] |
| 76 | glucose (α/β, 3-O) | M13 | A16/A19 | (PPh3)2NiBr2 | 90 | 4.37 | – | 1.81 | hyperbranched | bio-nanotechnology | Gao et al. [115] |
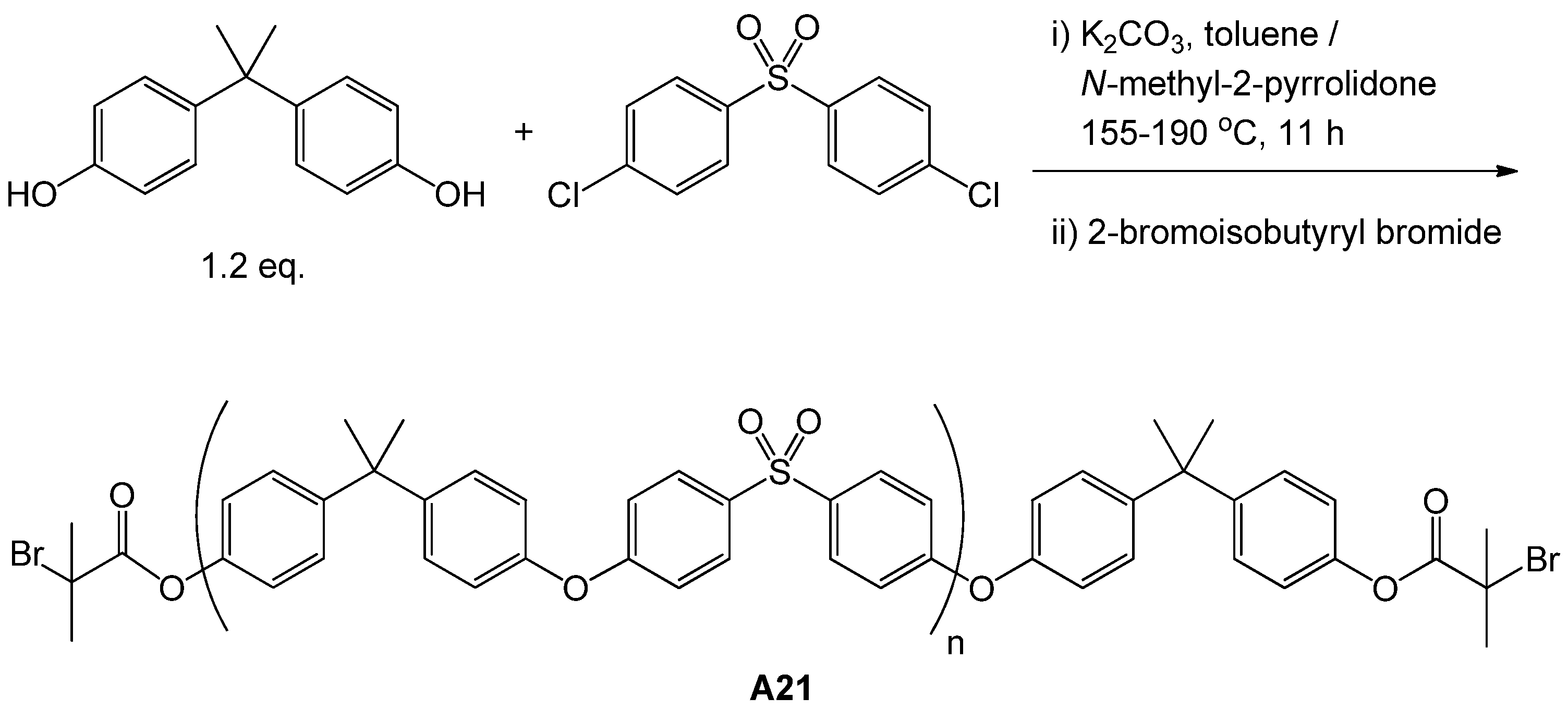
| 77 | glucose (α/β, 3-O) | M13 | A21 | CuCl(L10) | 51 | 12.5 | – | 1.18 | block ABA | biomedical | Wang et al. [116] |
| 78 | glucose (α/β, 4-O) | M32 | A23 | CuBr(L3) | 62 | 27.6 | 0.82 | 1.32 | homo | – | Ke et al. [106] |
| 79 | glucose (α/β, 4-O) | St/M32 | A22 | CuBr(L3) | 83 | 23.7 | 0.65 | 1.22 | A-stat-B | lectin recognition; film preparation | Ke et al. [106] |
| 80 | glucose (α/β, 4-O) | M32 | A24 | CuBr(L3) | 53 | 25.2 | 0.69 | 1.43 | graft AB | lectin recognition; film preparation | Ke et al. [106] |
| 81 | glucose (α/β, 3-O) | M16, M17 | A6 | CuBr(L4) | – | 11 | – | 1.18 | block AB | – | Bes et al. [108] |
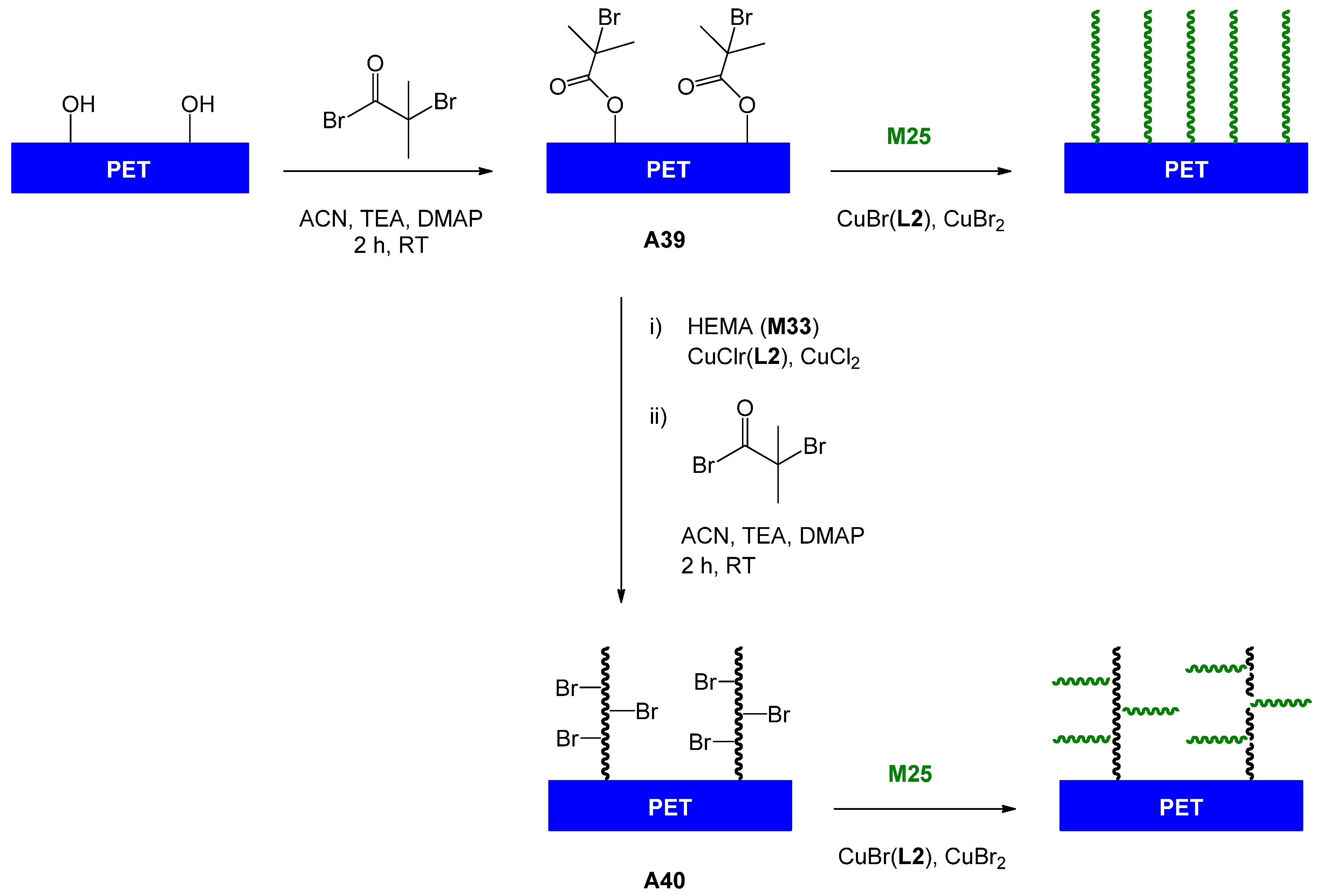
| 82 | glucose (β-O) | M14 | A2 | CuBr(L2) | 55 | 24.8 | 1 | 1.34 | homo | – | Liang et al. [117] |
| 83 | glucose (β-O) | M14 | A3 | CuBr(L3) | – | – | – | 1.12 | block AB | lectin interaction | You et al. [118] |
| 84 | lactose (β-O) | M21 | A8 | CuBr(L2) | 58 | 20.6 | 1.22 | 1.29 | homo | – | Dong et al. [119] |
| 85 | lactose (β-O) | M21 | A8 | CuBr(L2) | 96 | 9.3 | 1.26 | 1.24 | homo | – | Dong et al. [119] |
| 86 | lactose (β-O) | M22 | H2N·polyM21·NH2 | – | 73 | 14.3 | 1.14 | 1.38 | block ABA | – | Dong et al. [119] |
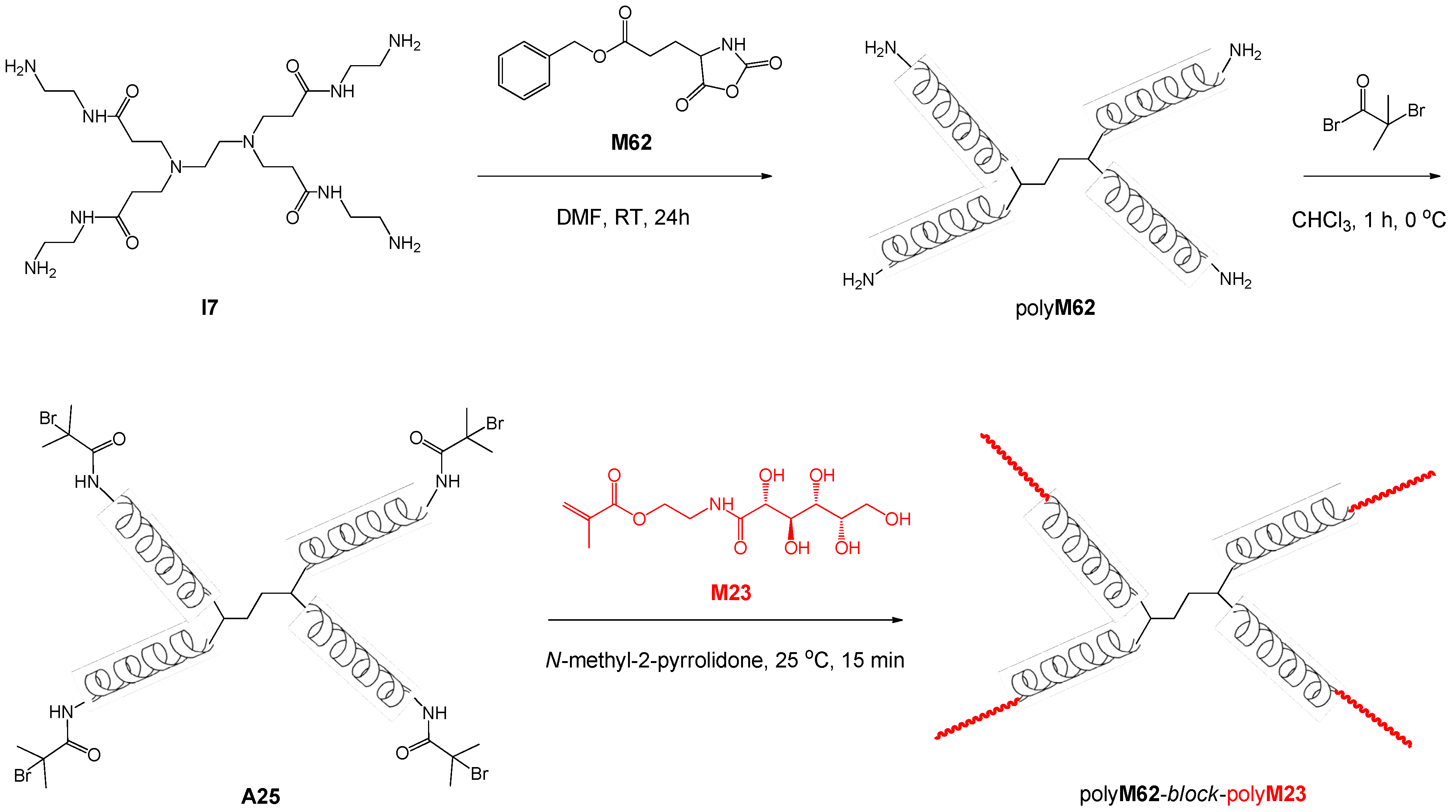
| 87 | lactose (β-O) | M62 | H2N·polyM21·NH2 | – | – | 15.9 | 1.03 | 1.33 | block ABA | – | Dong et al. [120] |
| 88 | maltoheptaose (α/β-O) | M16 | A4 | CuBr(L4) | 80 | 11.5 | 0.84 | 1.15 | block AB | – | Haddleton et al. [30] |
| 89 | maltoheptaose (α/β-O) | M18 | A4 | CuBr(L4) | 82 | – | – | – | block AB | – | Haddleton et al. [30] |
| 90 | maltoheptaose (α/β-O)/glucose (α/β, 3-O) | M13 | A4 | CuBr(L4) | 88 | 16.5 | 0.65 | 1.21 | block AB | – | Haddleton et al. [30] |
| 91 | maltoheptaose (α/β-O) | M15 | A4 | CuBr(L4) | 87 | 10.1 | 0.92 | 1.09 | block AB | – | Haddleton et al. [30] |
| 92 | N-acetylglucosamine (β-O) | M30 | A16 | CuCl(L8) | 95 | 11 | – | 1.29 | hyperbranched | lectin recognition | Pfaff et al. [121] |
| 93 | N-acetylglucosamine (β-O) | M30 | polySt·Br (latex)/A1 | CuCl(L8) | 95 | 96.7 | 0.45 | 1.12 | brush | – | Pfaff et al. [122] |
| (Meth)acrylate monomers (unprotected) | |||||||||||
| 94 e | gluconic acid (amide) | M23 | A923 | CuBr(L2) | > 97 | 11.4 | – | 1.23 | block AB | – | Narain et al. [123,124] |
| 95 f | gluconic acid (amide) | M23 | A923 | CuBr(L2) | > 97 | 12.6 | – | 1.48 | block AB | – | Narain et al. [123,124] |
| 96 g | gluconic acid (amide) | M23 | A923 | CuBr(L2) | > 97 | 13.4 | – | 1.82 | block AB | – | Narain et al. [123,124] |
| 97 | gluconic acid (amide) | M23 | A25 | CuBr(L2) | 64.5 i | 84.6 | – | 1.26 | star (4-arms) | lectin recognition and drug delivery | Qiu et al. [125] |
| 98 h | gluconic acid (amide)/ lactobionic acid (amide) | M23 | A10-polyM25·Br | CuBr(L2) | 68 i | 21.2 | – | 1.28 | block ABA | – | Narain et al. [126] |
| 99 | gluconic acid (amide) | M23 | Au-modified surface | CuBr(L2) | – | 19.7 | – | 1.6 | brush | lectin recognition, SPR | Mateescu et al. [127] |
| 100 | glucose (α/β-O) | M34a | A34 | CuBr(L2) | – | 8.6 | – | 1.44 | homo | amyloid β-peptide adsorption | Kitano et al. [128] |
| 101 | glucose (α-methyl, 6-O) | M36 | polyA16 | CuBr(L4) | 49 | 532 | – | 1.48 | brush (cylindrical) | – | Fleet et al. [129] |
| 102 | glucose (α-methyl, 6-O) | M36 | poly(A16-stat-M15) | CuBr(L4) | 30 | 196 | – | 1.49 | brush (cylindrical) | – | Fleet et al. [129] |
| 103 | glucose (α-methyl, 6-O) | M36 | poly(A16-block-M15) | CuBr(L4) | 41 | 320 | – | 1.52 | brush (cylindrical) | – | Fleet et al. [129] |
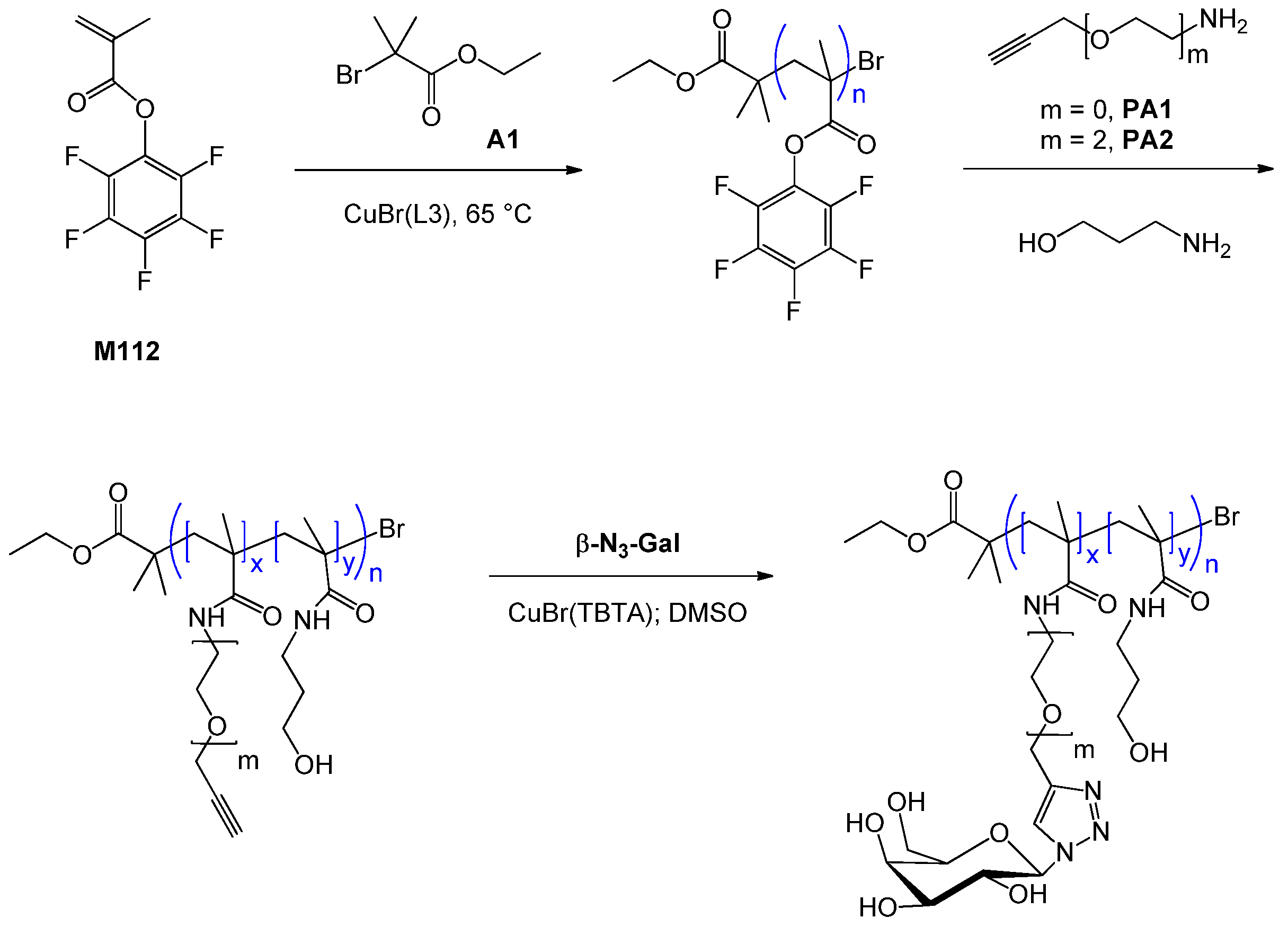
| 104 | glucose (α-methyl, 6-O) | M36 | poly(M58-alt-MAnh) | CuBr(L4) | 45 | 565 | – | 1.21 | brush (cylindrical) | – | Fleet et al. [129] |
| 105 h | lactobionic acid (amide) | M25 | A1023 | CuBr(L2) | - | 22.5 | – | 1.24 | block AB | – | Narain et al. [123] |
| 106 f | lactobionic acid (amide) | M25 | A1023 | CuBr(L2) | > 95 | 23.4 | – | 1.1 | block AB | – | Narain et al. [123] |
| 107 g | lactobionic acid (amide) | M25 | A1023 | CuBr(L2) | > 95 | 34.8 | – | 1.6 | block AB | – | Narain et al. [123] |
| 108 f | lactobionic acid (amide) | M24 | A10-polyM25·Br | CuBr(L2) | - | 17.9 | – | 1.34 | block ABC | – | Narain et al. [126] |
| 109 h | lactobionic acid (amide) | M26 | A10-polyM25·Br | CuBr(L2) | 72 i | 18.1 | – | 1.29 | block ABC | – | Narain et al. [126] |
| 110 | lactobionic acid (amide) | M25 | A17 | CuBr(L2) | 80 i | 24 | 1.02 | 1.32 | block AB | streptavidin binding | Narain et al. [130] |
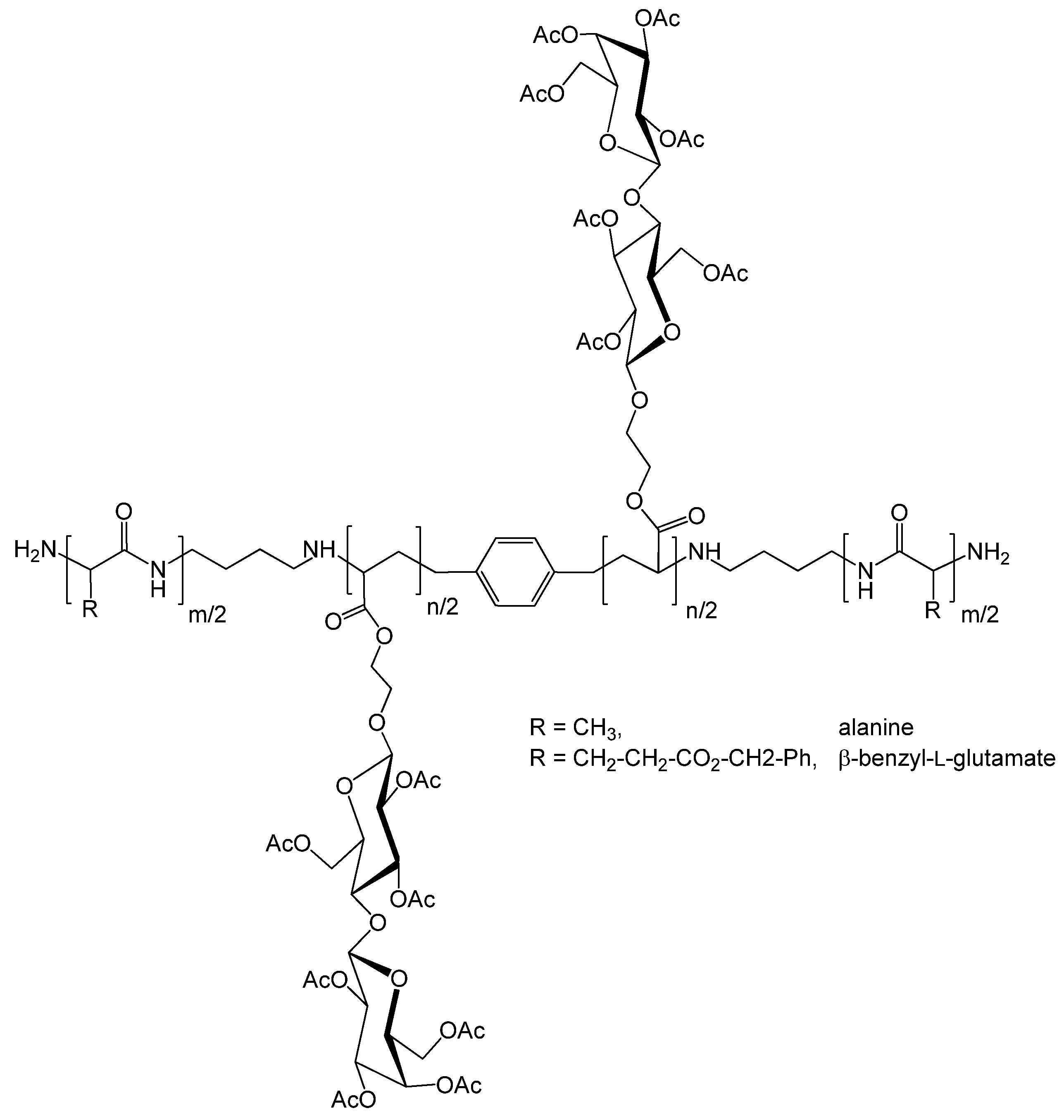
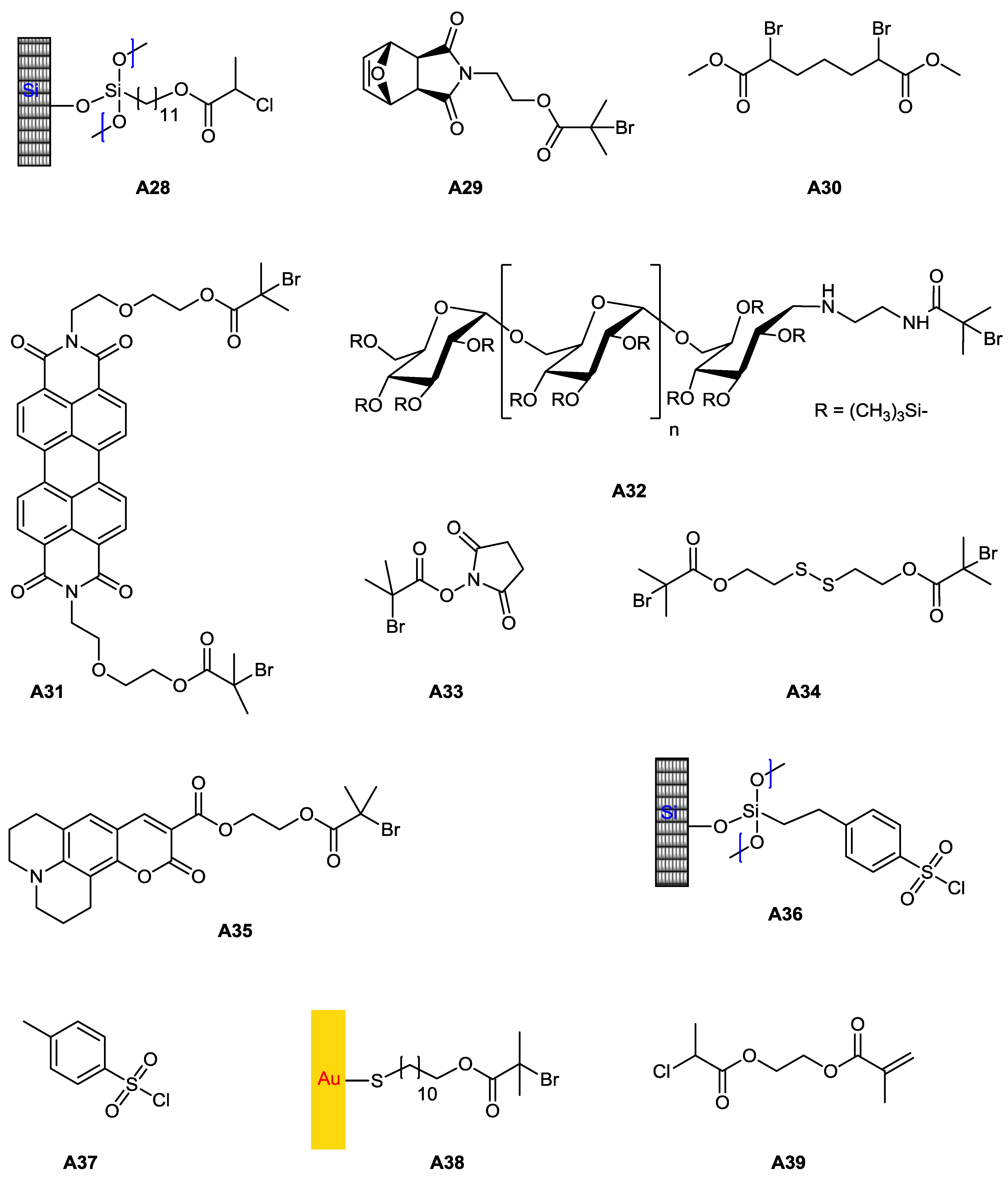
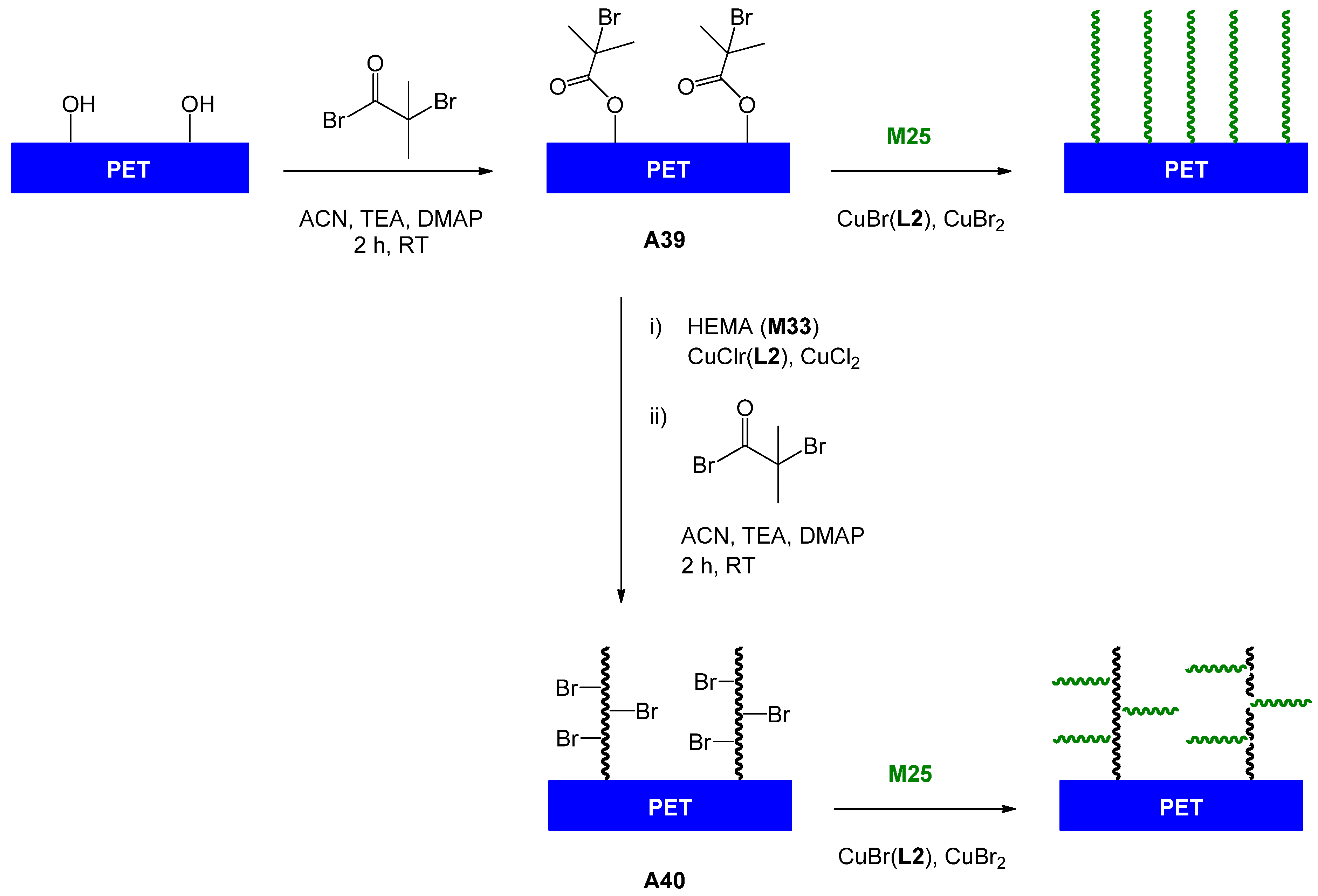
| 111 | lactobionic acid (amide) | M25 | A38, A1 or A38 | CuBr(L2), CuBr2 | – | 68 | 1.8 | – | brush | lectin recognition, SPR | Mateescu et al. [127] |
| 112 | lactobionic acid (amide) | M25 | A39 | CuBr(L2), CuBr2 | – | – | – | – | brush (linear) | lectin binding | Yang et al. [131] |
| 113 | lactobionic acid (amide) | M25 | A39 | CuBr(L2), CuBr2 | – | – | – | – | brush (comb) | lectin binding | Yang et al. [131] |
| 114 | mannose (α-O) | M67 | A35 | CuBr(L7) | 80 | 28.8 | – | 1.25 | homo | – | O’Connell et al. [132] |
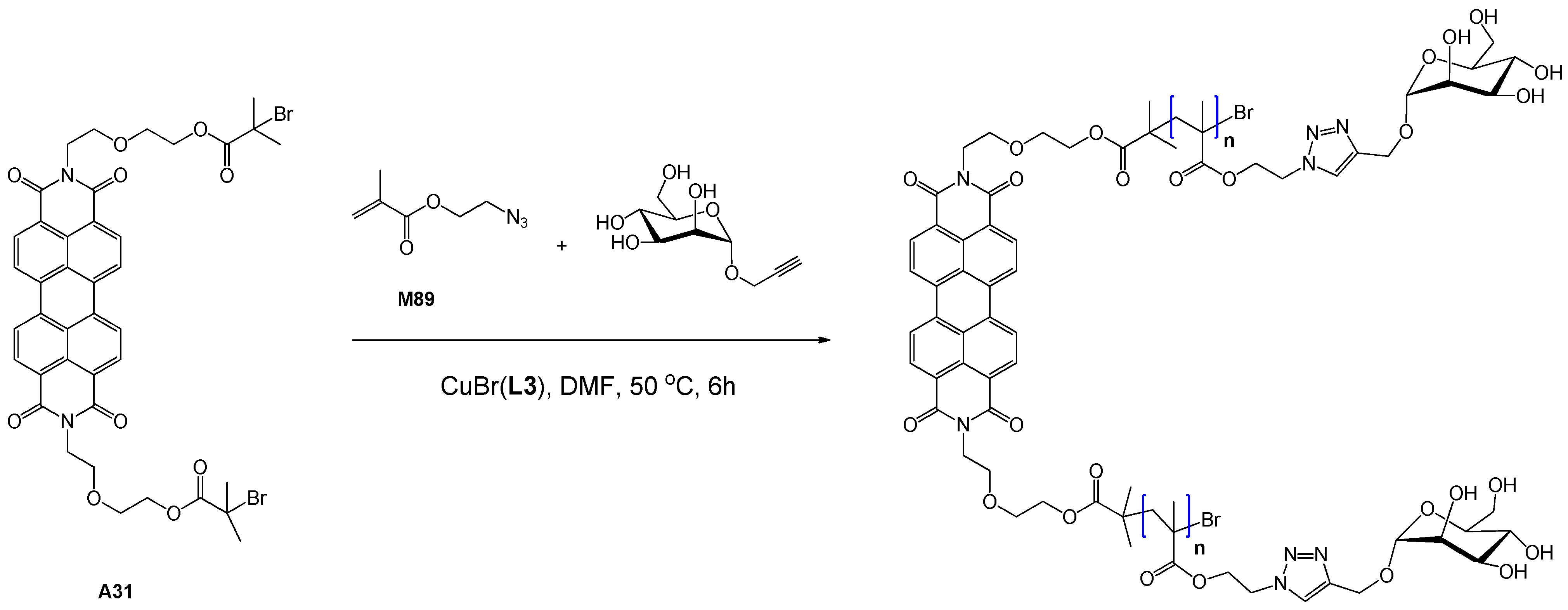
| 115 | mannose (α-O) | M89, 2-propynyl-α-Man | A31 | CuBr(L3) | – | 49.9 | – | 1.33 | homo | cell imaging | Xu et al. [133] |
| 116 | mannose (α/β-O) | M34b | A34 | CuBr(L2) | – | 7.8 | – | 1.2 | homo | lectin binding | Kitano et al. [134] |
| 117 | mannose (α-O) | M67 | A29 | CuBr(L7) | – | 26.1 | – | 1.2 | homo | lectin recognition | Geng et al. [135] |
| 118 | N-acetylglucosamine (β-O) | M30 | A18 | CuBr(L9) | 94 | 40.7 | 1.88 | 1.17 | homo | biotin-protein binding | Vazquez-Dorbatt et al. [136] |
| 119 | N-acetylglucosamine (β-O) | M31 | A18 | CuBr(L9) | 86 | 43.1 | 3.01 | 1.07 | homo | biotin-protein binding | Vazquez-Dorbatt et al. [136] |
| 120 | N-acetylglucosamine (β-O) | M31 | A26 | CuBr(L2), CuBr2 | 80 | 10.2 | – | 1.12 | homo | siRNA conjugation | Vazquez-Dorbatt et al. [137] |
| 121 | N-acetylglucosamine (α/β, N) | M63 | A1 | CuBr(L3) | 90 | 70 | – | 1.2 | homo | lectin recognition | Leon et al. [138] |
| 122 | N-acetylglucosamine (α/β, N) | M63 | A27 | CuBr(L3) | 75 | 27 | – | 1.15 | homo | lectin recognition | Leon et al. [138] |
| 123 | N-acetylglucosamine (α/β, N) | M47 | A1·polyM63·Br | CuCl(L3) | 90 | 15 | 0.87 | 1.31 | block AB | lectin recognition | Leon et al. [138] |
| 124 | N-acetylglucosamine (α/β, N) | M47 | A27·polyM63·Bt | CuCl(L3) | 93 | 17.6 | 0.98 | 1.38 | block ABA | lectin recognition | Leon et al. [138] |
| 125 | N-acetylglucosamine (α/β, N) | M63 | A20a·polyM47·Br | CuCl(L3) | 73 | 33.9 | 1.48 | 1.37 | block AB | lectin recognition | Leon et al. [139] |
| 126 | N-acetylglucosamine (α/β, N) | M63 | A30·polyM47·Br | CuCl(L3) | 93 | 38.5 | 1.23 | 1.32 | block ABA | lectin recognition | Leon et al. [139] |
| 127 | N-acetylglucosamine (α/β, N) | M46 | A1·polyM63·Br | CuCl(L3) | 45 | 32.7 | 1.2 | 1.3 | block AB | polymeric surfactant, lectin recognition | Munoz-Bonilla et al. [140] |
| 128 | N-acetylglucosamine (α/β, N) | M63 | A1·polyM15·Br | CuCl(L3) | 15 | 16.5 | 0.93 | 1.12 | block AB | lectin recognition; film preparation | de León et al. [141] |
| Styrenic monomers (protected) | |||||||||||
| 129 | dextran (1-deoxy-1-amide) | St | A32 | CuBr(L3) | – | 82.2 | – | 1.7 | block AB | carrier | Houga et al. [34] |
| 130 | glucose (α/β, 3-O) | M109 | A1 | CuCl(L3) | 68 i | 12.3 | – | 1.19 | homo | – | [142] |
| 131 | glucose (α/β, 3-O) | M110 | polyM109 | CuCl(L3) | 55 i | 21.2 | – | 1.46 | block AB | biomedical | Menon et al. [142] |
| 132 | maltoheptaose (α/β-O) | St | A4 | CuBr(L4) | 91 | 10.7 | 1.2 | 1.48 | block AB | – | Haddleton et al. [30] |
| Glycopolymers from post-polymerization reaction | |||||||||||
| 133 | – | M28/M15 | A13 | CuBr(L7) | >80 | 8.9 | 1.56 | 1.09 | A-stat-B | – | Ladmiral et al. [143] |
| 134 | – | M28/M29 | A13 | CuBr(L7) | >80 | 11.9 | 1.52 | 1.12 | A-stat-B | – | Ladmiral et al. [143] |
| 135 | galactose (β-N) | M112 | A1 | CuBr(L3) | 70 | 11.4 | 0.64 | 1.16 | homo, A-stat-B | lectin recognition j | Richards et al. [144] |
| 136 | galactose (α-O), mannose (α-O) | M28 | A13 | CuBr(L7) | >80 | 17.6 | 2.31 | 1.17 | homo | lectin recognition j | Ladmiral et al. [143] |
| 137 | mannose (α-O) | M28/M93 | A29 | CuBr(L7) | – | 16.4 | – | 1.28 | homo | lectin recognition j | Geng et al. [135] |
| 138 | mannose (α-O) | M28 | A34 | CuBr(L7) | – | 7.5 | – | 1.32 | homo | – | Gou et al. [145] |
5.1.2. Styrenic Monomers
5.2. ATRP Starting from Unprotected Glycomonomers/Glycoinitiators
5.2.1. (Meth)acrylamide Monomers
5.2.2. (Meth)acrylate Monomers
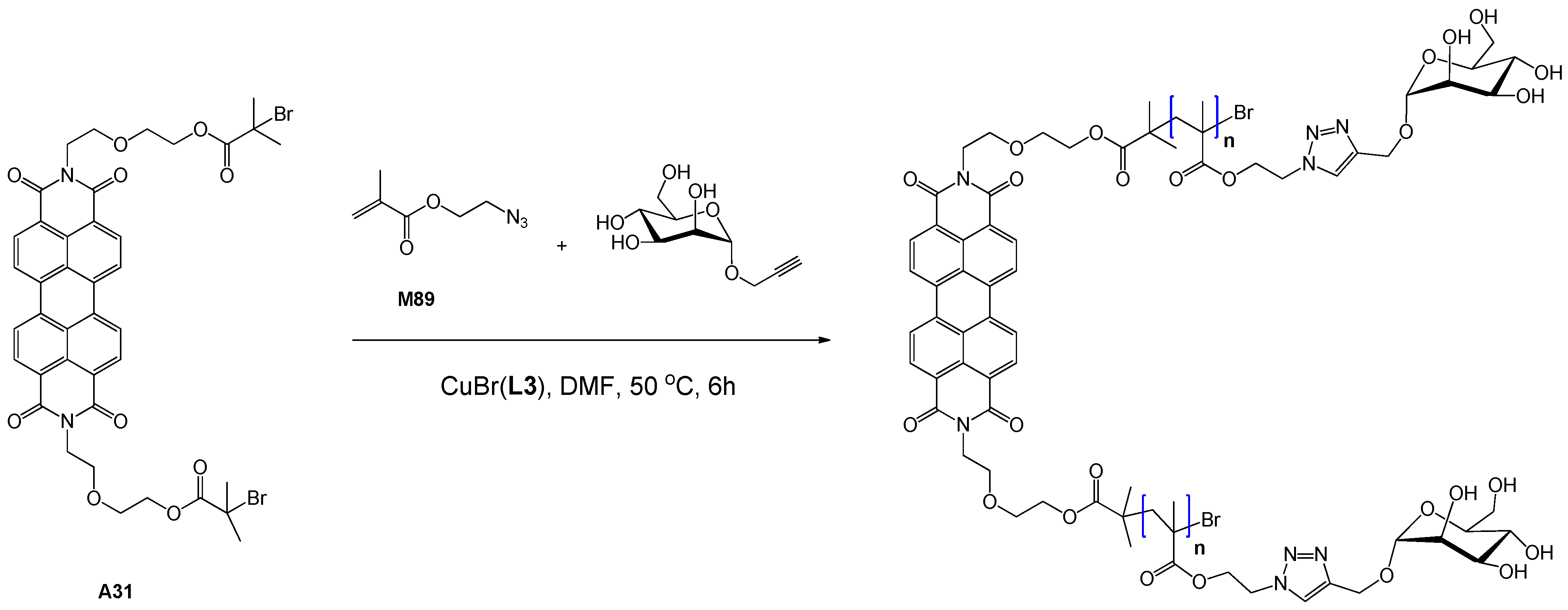
5.2.3. Styrenic Monomers
5.3. Glycopolymers from Post-Polymerization Reactions
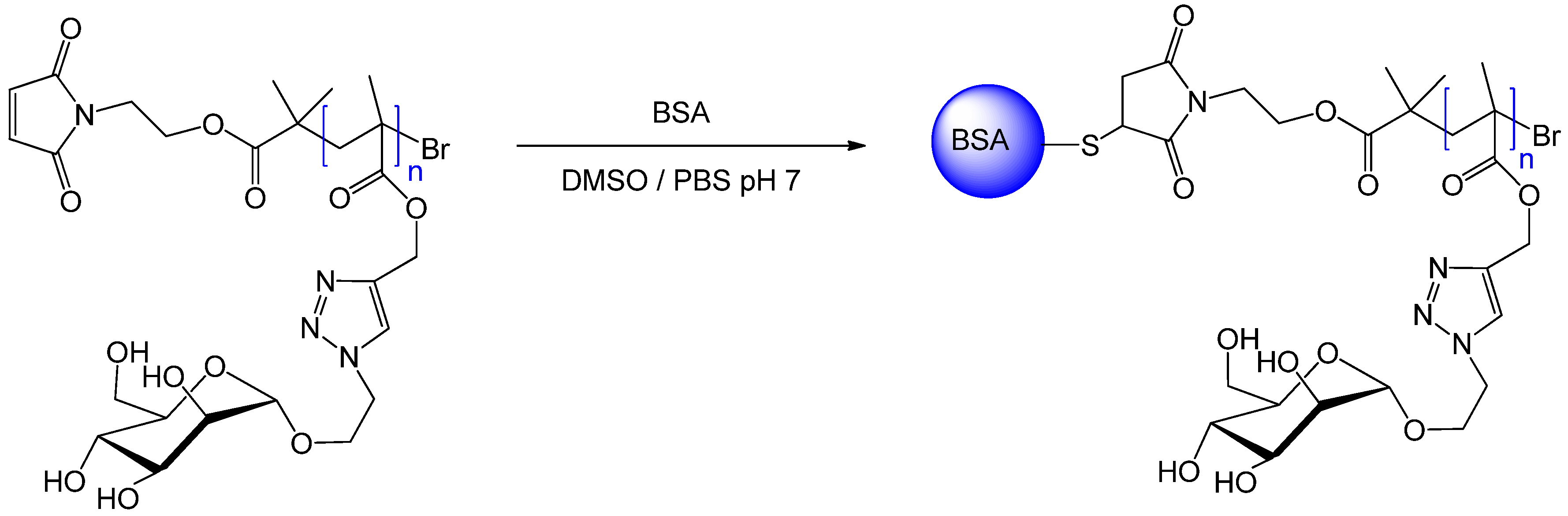
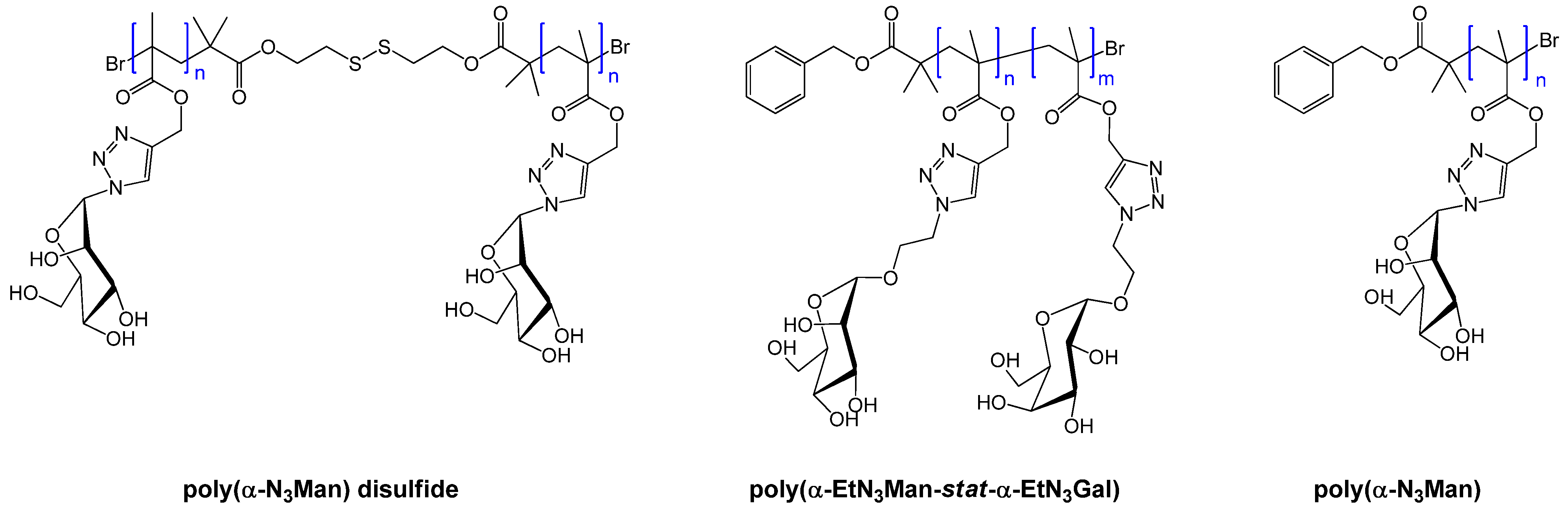
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
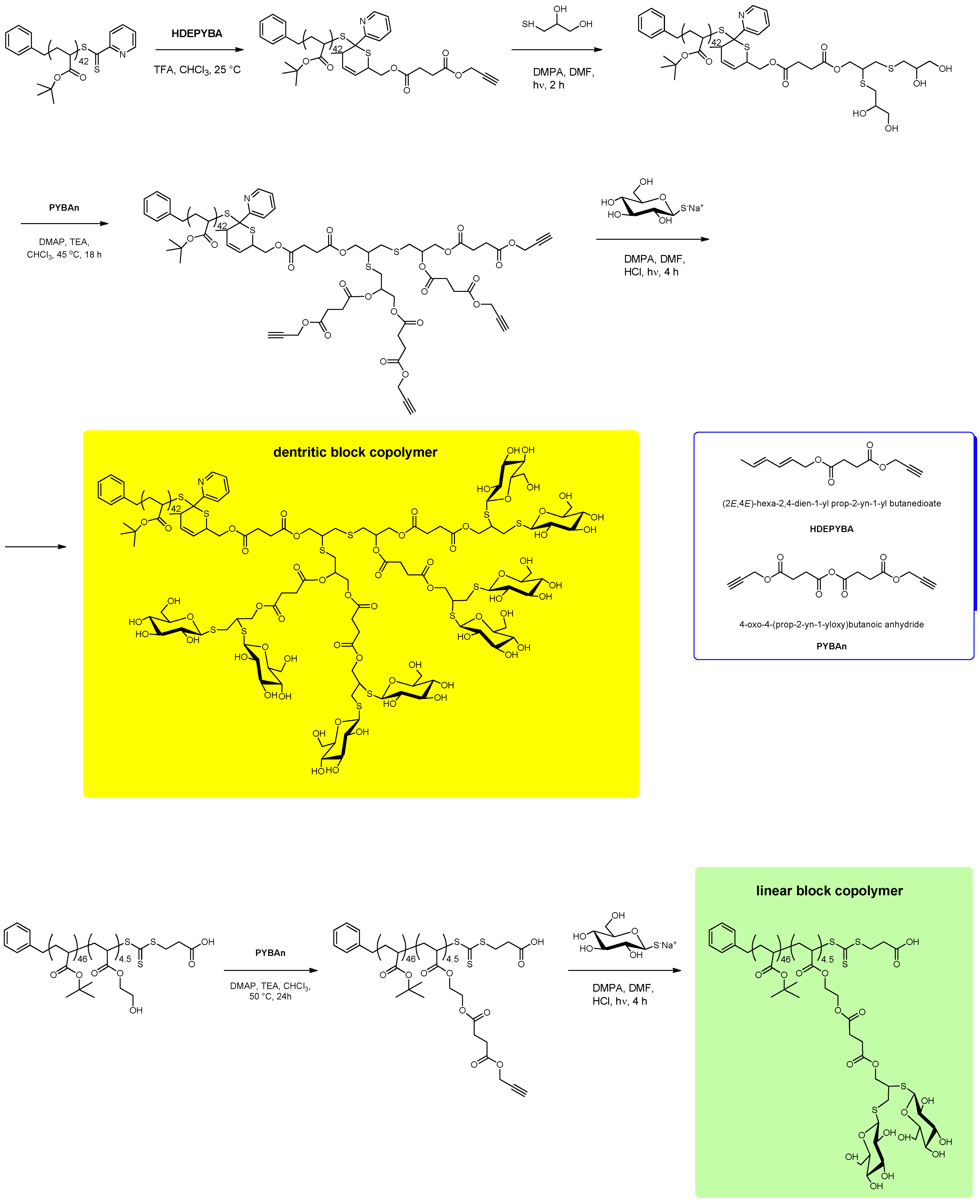{kind=link}
{kind=link}
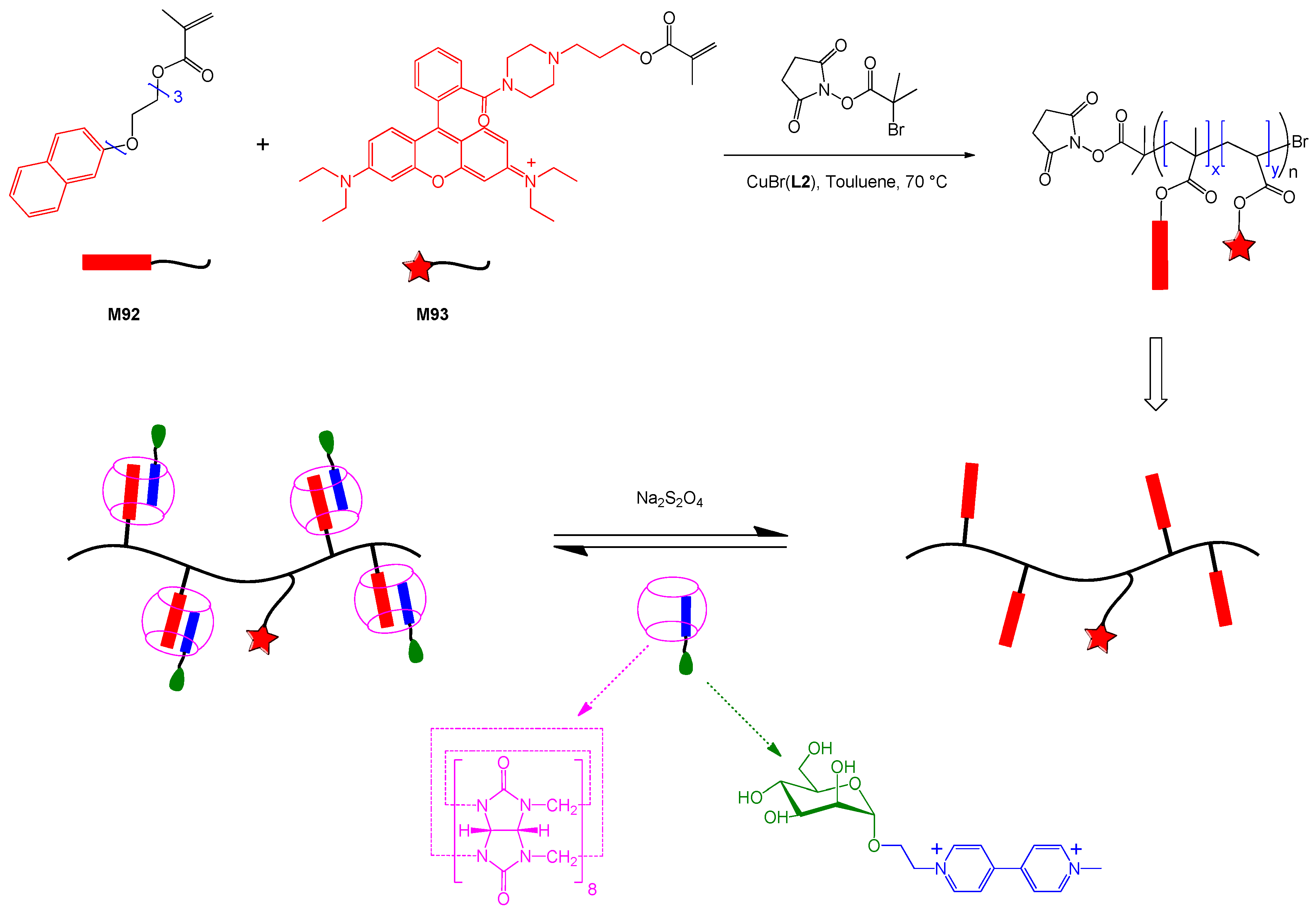
6. Reversible Addition-fragmentation Chain Transfer (RAFT) Polymerization
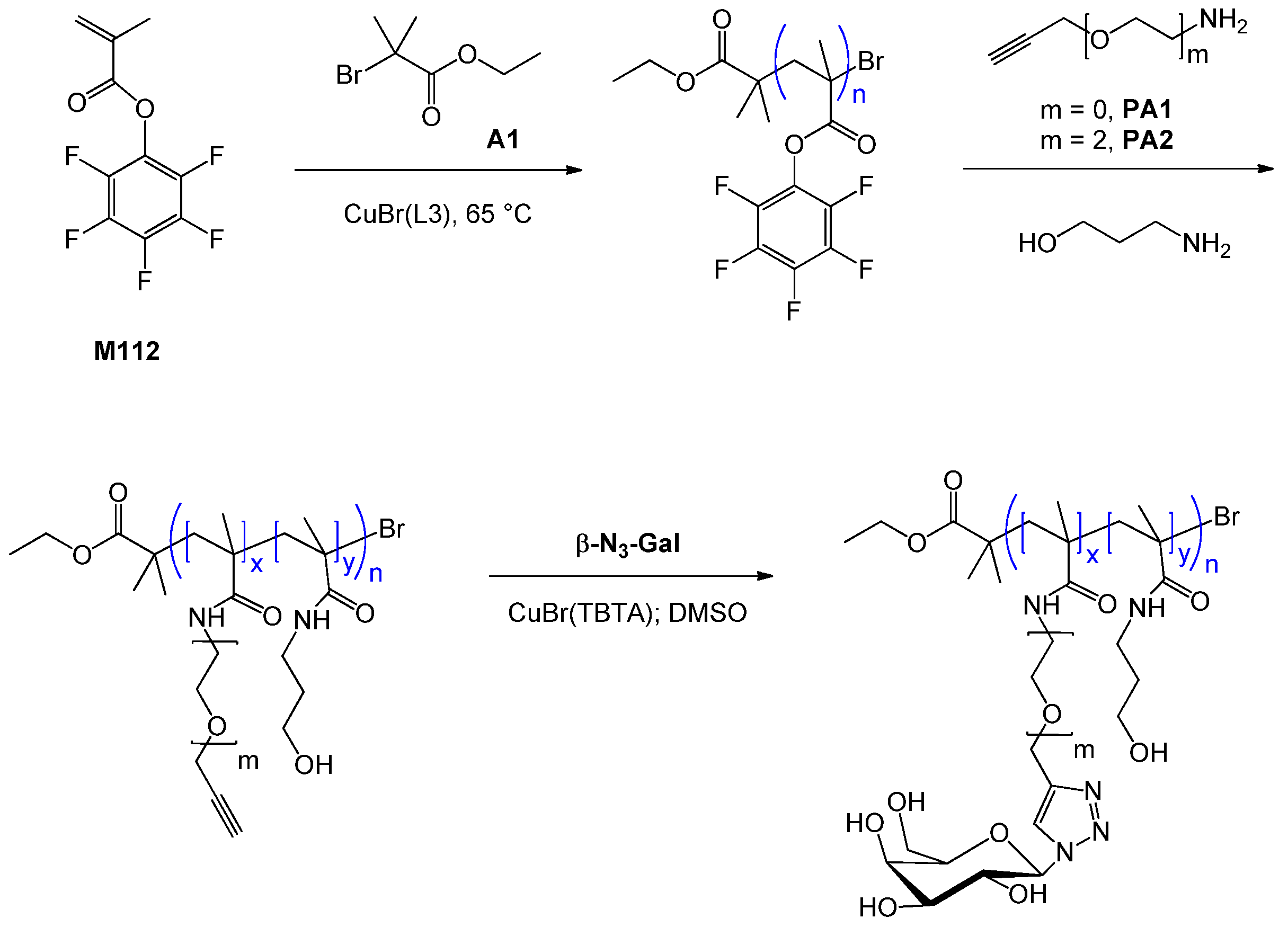
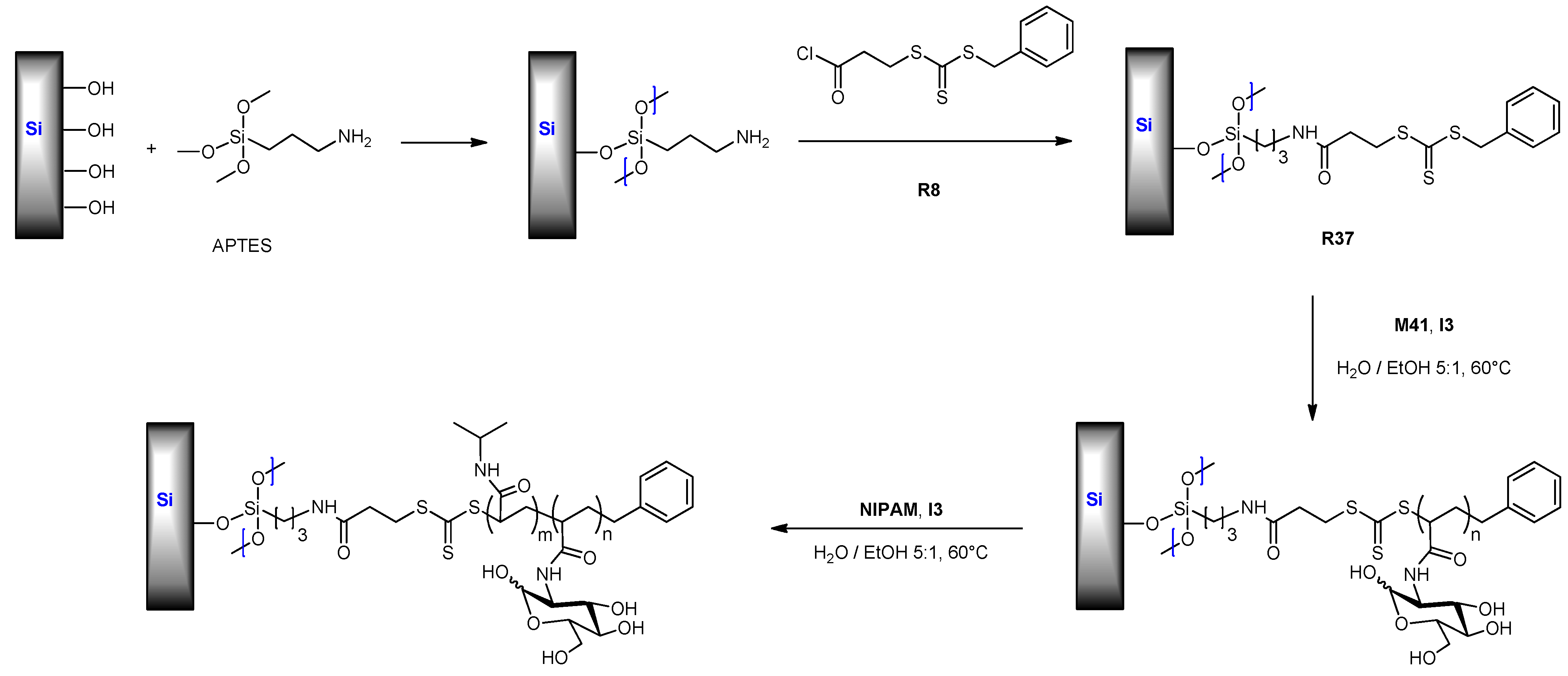
6.1. RAFT Starting from Protected Glycomonomers/GlycoRAFT Agents
6.1.1. (Meth)acrylamide Monomers
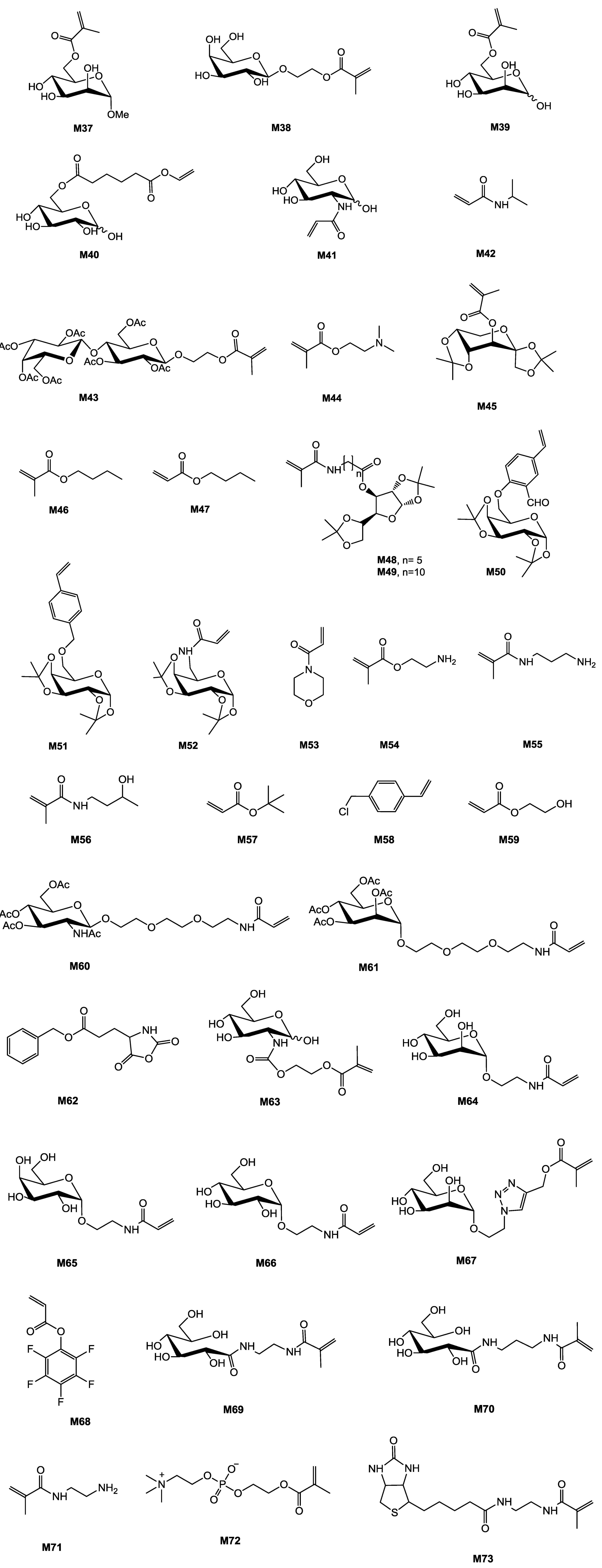
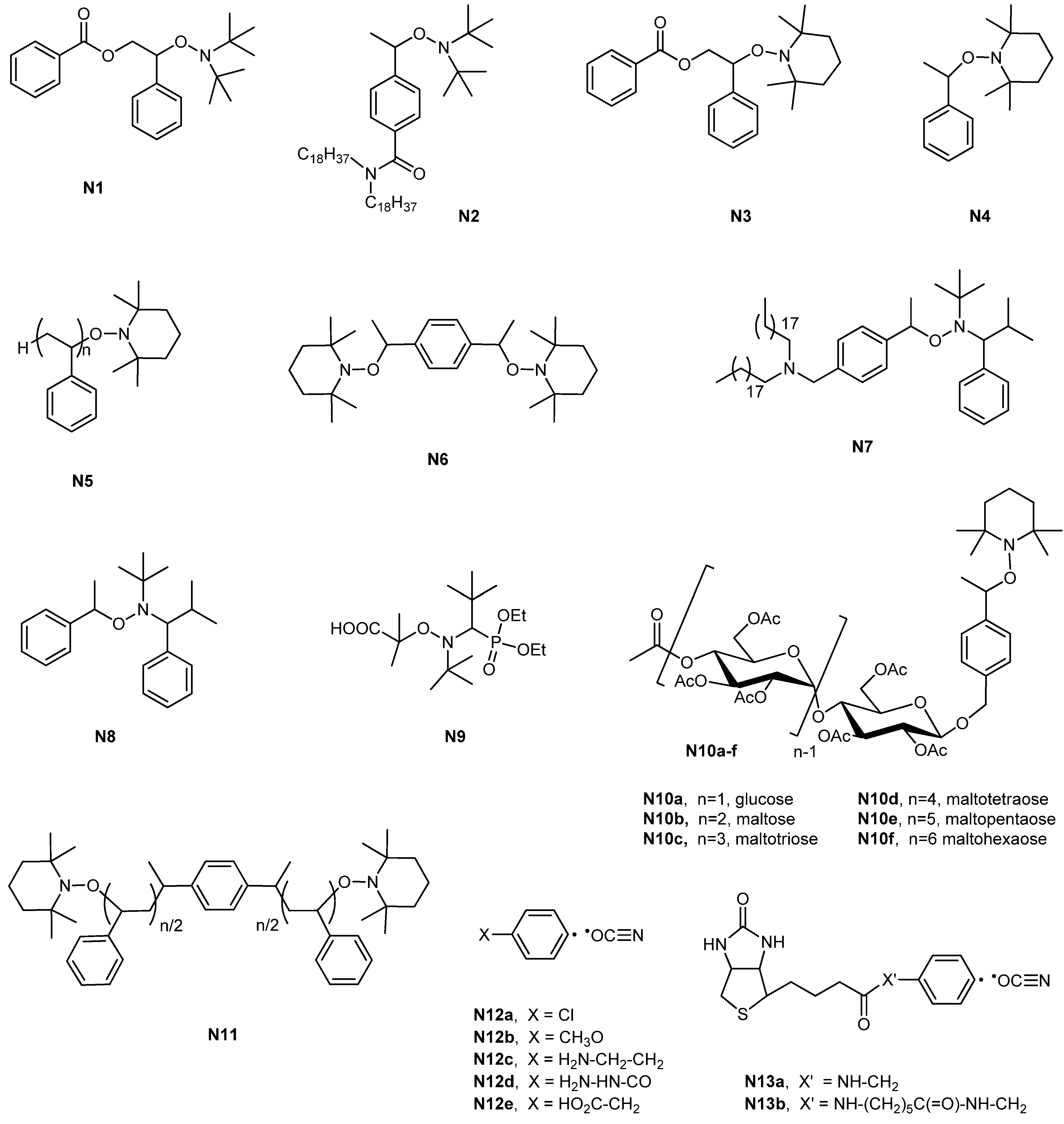
| Entry | Carbohydrate | Monomer(s) | RAFT agent | Initiator | Conv. % | Mn (×10−3) | Mn/Mn,th a | Đ | Structure | Application sought/tested | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (Meth)acrylamide monomers (protected) | |||||||||||
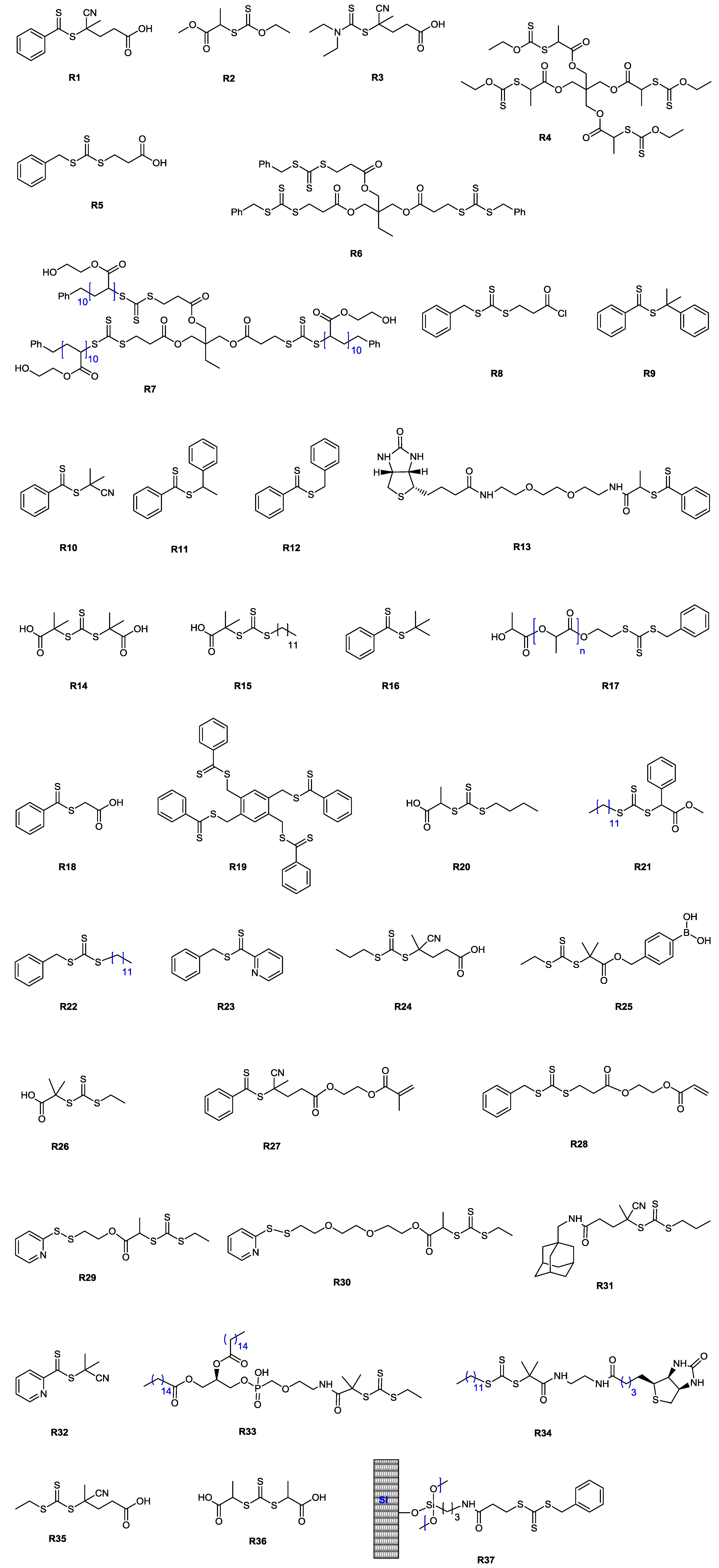
| 139 | galactose (α/β, 6-N) | M52/M53 | R13 | I4 | 80 | ~17 | – | ~1.5 | A-stat-B | – | Gody et al. [158] |
| 140 | galactose (β-N) | M100 | R26 | I4 | >70 | 11.9 | 1.18 | – | homo | lectin recognition | Su et al. [159] |
| 141 a | galactose (α/β, 6-O) | M102 | R5 | I4 | 93 | 5.2 | 0.7 | 1.14 | homo | – | Wei et al. [160] |
| 142 a | galactose (α/β, 6-O) | M102 | R28 | I4 | 100 | 28.6 | 1.43 | 1.49 | hyperbranched | – | Wei et al. [160] |
| 143 a | galactose (α/β, 6-O) | M20 | R27 | I4 | 91 | 9.7 | 1.61 | 1.48 | hyperbranched | – | Wei et al. [160] |
| 144 b | galactose (α/β, 6-O) | M20 | R27 | I4 | 82 | 23 | 1.6 | 1.44 | hyperbranched | – | Wei et al. [160] |
| 145 | glucose (α/β, 3-O) | M48 | R10 | I4 | 60 | 11.6 | 0.88 | 1.34 | homo | – | Ozyurek et al. [161] |
| 146 | glucose (α/β, 3-O) | M48/M42 | R10 | I4 | 80 | 20.5 c | – | 1.69 c | A-stat-B | – | Ozyurek et al. [161] |
| 147 | glucose (α/β, 3-O) | M49/M42 | R10 | I4 | 65 | 18.7 c | – | 1.29 c | A-stat-B | – | Ozyurek et al. [161] |
| 148 | glucose (α/β, 3-O) | M48 | polyM42·R10 | I4 | – | 15.2 | – | 1.57 | block AB | – | Ozyurek et al. [161] |
| 149 | glucose (α/β, 3-O) | M49 | polyM42·R10 | I4 | – | 27 | – | 1.69 | block AB | – | Ozyurek et al. [161] |
| 150 | glucose (β-N) | M99 | R26 | I4 | >70 | 11.9 | 1.2 | – | homo | lectin recognition | Su et al. [159] |
| 151 | mannose (α-O) | M53/M61 | R13 | I4 | 73 | 12.7 | 1.26 | 1.2 | A-stat-B | avidin and lectin recognition | Jiang et al. [162] |
| 152 | mannose (α-O) | M53/M61 | R16 | I4 | 83 | 9.7 | 1.21 | 1.14 | A-stat-B | lectin recognition | Jiang et al. [162] |
| 153 | N-acetylglucosamine (β-O) | M53/M60 | R13 | I4 | 85 | 53.7 | 0.91 | 1.6 | A-stat-B | avidin and lectin recognition | Jiang et al. [162] |
| 154 | N-acetylglucosamine (β-O) | M53/M60 | R16 | I4 | 93 | 62.3 | 1.02 | 1.46 | A-stat-B | lectin recognition | Jiang et al. [162] |
| (Meth)acrylamide monomers (unprotected) | |||||||||||
| 155 | cellobiose (β-O) | M108 | R33 | I3 | 98 | 22.5 | – | 1.22 | homo | – | Belardi et al. [163] |
| 156 | galactose (α-O) | M77b/M75 | R18 | I8 | 34 | 12 | – | 1.4 | A-stat-B | lectin recognition | Toyoshima et al. [164] |
| 157 | gluconic acid (amide) | M69 | R1 | I3 | 93 | 8.9 | 0.82 | 1.18 | homo | cytotoxicity tests | Deng et al. [165] |
| 158 | gluconic acid (amide) | M70 | R1 | I3 | 78 | 18.2 | 0.96 | 1.2 | homo | cytotoxicity tests | Deng et al. [165] |
| 159 | gluconic acid (amide) | M71 | polyM69·R1 | I3 | 70 | 15.3 | 0.83 | 1.44 | block AB | cytotoxicity tests | Deng et al. [165] |
| 160 | gluconic acid (amide) | M55 | polyM70·R1 | I3 | 70 | 18.4 | 1.47 | 1.4 | block AB | cytotoxicity tests | Deng et al. [165] |
| 161 | gluconic acid (amide) | M72 | polyM69·R1 | I3 | 70 | 25 | 1.05 | 1.39 | block AB | cytotoxicity tests | Deng et al. [165] |
| 162 | gluconic acid (amide) | M70/M55 | R1 | I3 | – | 29.5 | – | 1.29 | A-stat-B | gene delivery | Ahmed et al. [166] |
| 163 | gluconic acid (amide) | M70/M71 | R1 | I3 | – | 18.2 | – | 1.31 | A-stat-B | gene delivery | Ahmed et al. [166] |
| 164 | gluconic acid (amide) | M70/M106 | R1 | I3 | – | 38 | – | 2.5 | hyperbranched | biomedical | Ahmed et al. [167] |
| 165 | gluconic acid (amide) | M70/M71/ M106 | R1 | I3 | – | 27 | – | 2.5 | hyperbranched | gene delivery | Ahmed et al. [168] |
| 166 | glucosamine (α/β, 2-N) | M41 | R5 d | I3 | 89 | 100.8 | 1.2 | 1.26 | homo | – | Bernard et al. [169] |
| 167 | glucosamine (α/β, 2-N) | M41 | R5 e | I3 | 19 | 6.6 | 1.4 | 1.15 | homo | – | Bernard et al. [169] |
| 168 | glucosamine (α/β, 2-N) | M42 | polyM41·R5 | I3 | 88 | 88.4 | 1.07 | <1.25 | block AB | – | Bernard et al. [169] |
| 169 | glucosamine (α/β, 2-N) | M41 | R7 | I3 | 40 | 72.2 | 1.22 | 1.21 | star (3-arm) | – | Bernard et al. [169] |
| 170 | glucosamine (α/β, 2-N) | M41 | R21 | I4 | 79 | 13.5 | 2.14 | 1.3 | homo | – | Ting et al. [170] |
| 171 | glucosamine (α/β, 2-N) | St | polyM41·R21 | I3 | 83 | 38.5 | 1.63 | 1.65 | lattex | lectin recognition | Ting et al. [170] |
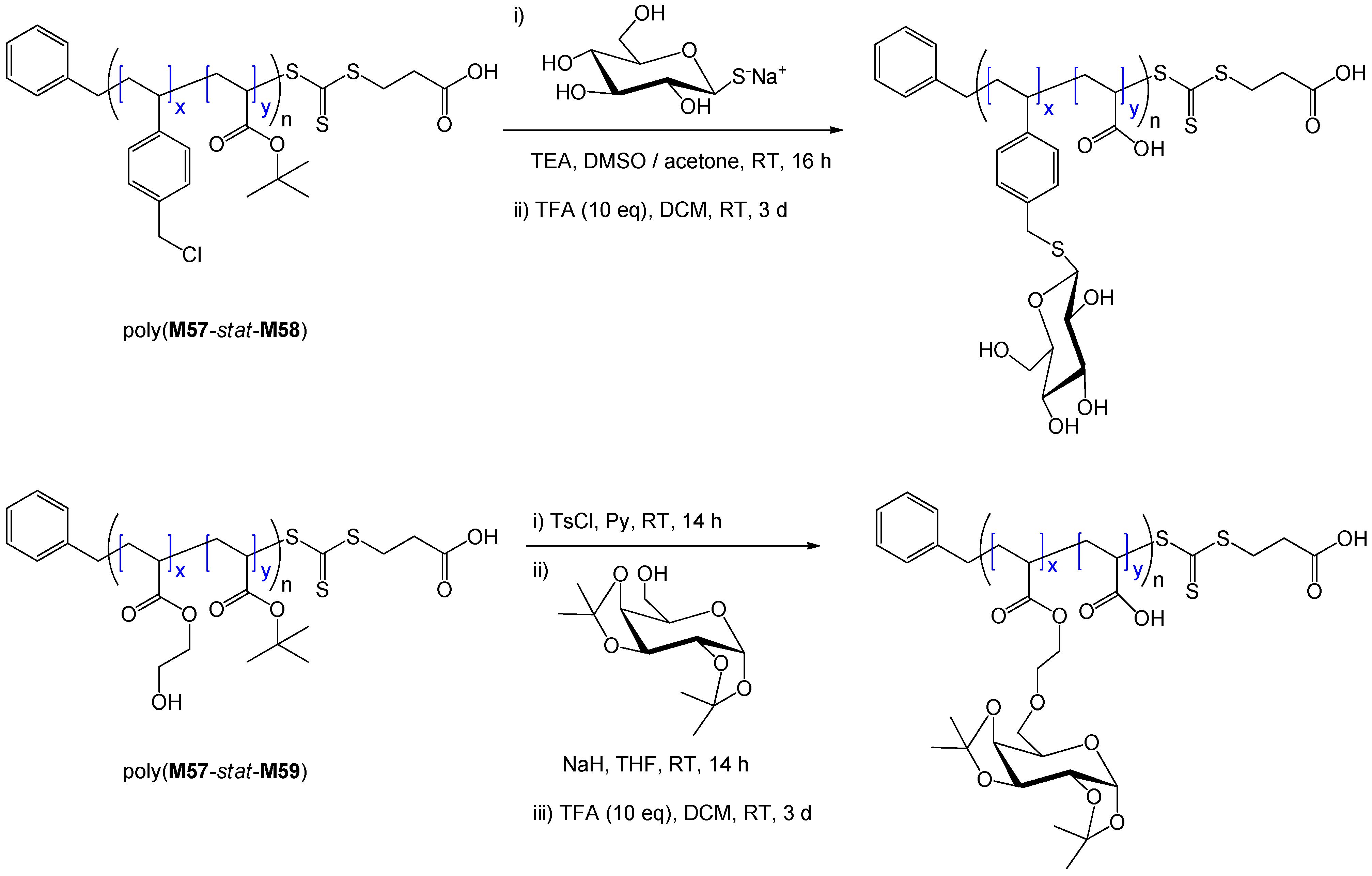
| 172 | glucosamine (α/β, 2-N) | St | polyM41·R21 | I3 | 81 | 660 | – | 1.33 | latex (cross-linked) | lectin recognition | Ting et al. [170] |
| 173 | glucosamine (α/β) | M84 | R24 | I4 | – | 11.7 | – | 1.24 | homo | – | Smith et al. [171] |
| 174 | glucosamine (α/β) | M71 | polyM84·R24 | I4 | – | 14.4 | – | 1.15 | block AB | gene delivery | Smith et al. [171] |
| 175 | glucosamine (α/β) | M71 | polyM84·R24 | I4 | – | 17.8 | – | 1.12 | block AB | gene delivery | Smith et al. [171] |
| 176 | glucose (α-O) | M85 | R5 | I3 | 85 | 113 | – | 1.08 | homo | – | Abdelkader et al. [172] |
| 177 | glucose (β-N) | M116 | polyM53 | I4 | – | 25.6 | 0.74 | 1.06 | block ABA | – | Albertin et al. [173] |
| 178 | glucuronic acid (β-O) | M81/M75 | R18 | I8 | 81 | 73 | – | 1.7 | A-stat-B | biomedical | Miura et al. [174] |
| 179 | lactobionic acid (amide) | M105/M106 | R1 | I3 | – | 36 | – | 2.2 | hyperbranched | biomedical | Ahmed et al. [167] |
| 180 | lactobionic acid (amide) | M105/M71/M106 | R1 | I3 | – | 31 | – | 2.1 | hyperbranched | gene delivery | Ahmed et al. [168] |
| 181 | lactose (β-O) | M107a | R33 | I3 | 95 | 20.7 | – | 1.21 | homo | – | Belardi et al. [163] |
| 182 | mannose (α-O) | M76 | R18 | I8 | 85 | 47.5 | – | 1.2 | homo | lectin recognition | Toyoshima et al. [175] |
| 183 | mannose (α-O) | M76/M75 | R18 | I8 | 82 | 9.3 | – | 1.5 | A-stat-B | lectin recognition | Toyoshima et al. [175] |
| 184 | mannose (α-O) | M76/M75 | R18 | I8 | – | 20 | – | 1.3 | A-stat-B | biosensor | Ishii et al. [176] |
| 185 | mannose (2-deoxy-2-azido, α-O) | M86 | R5 | I3 | 50 | 37 | – | 1.35 | homo | – | Abdelkader et al. [172] |
| 186 | mannose (2-deoxy-2-azido, α-O) | M86 | R5 | I9 | 75 | 56 | – | 1.15 | homo | – | Abdelkader et al. [172] |
| 187 | N-acetylglucosamine (α-O) | M77/M75 | R18 | I8 | 19 | 8.6 | – | 1.5 | A-stat-B | lectin recognition | Toyoshima et al. [175] |
| 188 | N-acetylglucosamine (6-sulfo, β-O) | M80/M75 | R18 | I8 | 67 | 210 | – | 1 | A-stat-B | biomedical | Miura et al. [174] |
| 189 | N-acetylglucosamine (6-sulfo, β-O) | M80/M81/M75 | R18 | I8 | 16 | 7.6 | – | 1.4 | A-stat-B-stat-C | biomedical | Miura et al. [174] |
| (Meth)acrylate monomers (protected) | |||||||||||
| 190 | fructose (α/β, 3-O) | M45 | R9 | I5 | 91 | 41.2 | 2.48 | 1.25 | homo | – | Al-Bagoury et al. [177] |
| 191 | galactose (α/β, 6-O) | M20 | R9 | I4 | 75 | 13.9 | – | 1.2 | homo | – | Lowe et al. [178] |
| 192 | galactose (α/β, 6-O) | M20 | R10 | I4 | – | 12.3 | – | 1.18 | homo | – | Lowe et al. [178] |
| 193 | galactose (α/β, 6-O) | M44 | polyM20 | I4 | – | 16.3 | – | 1.2 | block AB | – | Lowe et al. [178] |
| 194 | galactose (α/β, 6-O) | M74 | R22 | I4 | 86 | 51 | 1.11 | 1.17 | homo | – | Ting et al. [179] |
| 195 | galactose (α/β, 6-O) | M74 | R17 | I4 | 73 | 52 | 0.71 | 1.2 | block AB | biomedical | Ting et al. [179] |
| 196 | galactose (α/β, 3-O) | M13 | R1 | I3 | 40 | 4.1 | 0.94 | 1.12 | homo | – | Liu et al. [180] |
| 197 | galactose (α/β, 3-O) | M13 | polyM7918 | I3 | – | 7 | – | 1.19 | block AB | lectin recognition/ biomedical | Liu et al. [180] |
| 198 | galactose (α/β, 6-O) | M20 | R10 | I4 | 86 | 13.7 | – | 1.22 | homo | biosensor | Pfaff et al. [181] |
| 199 | galactose (α/β, 6-O) | M96/M20 | polyM20·R10 | I4 | – | 14.8 | – | 1.18 | A-block-(B-stat-C) | biosensor | Pfaff et al. [181] |
| 200 | galactose (β-O) | M101 | R1 | I3 | 50-60 | 6.3 | 0.96 | 1.1 | homo | lectin recognition, antioxidant | Shi et al. [182] |
| 201 | glucose (α/β, 3-O) | M13 | R11 | I5 | 90 | 213 | 17.8 | 1.9 | homo | – | Al-Bagoury et al. [177] |
| 202 | glucose (α/β, 3-O) | M13 | R11 | I4 | 60 | 313 | 64 | 1.58 | homo | – | Al-Bagoury et al. [177] |
| 203 | glucose (α/β, 3-O) | M13 | R10 | I5 | 30 | 20.9 | 4.01 | 1.32 | homo | – | Al-Bagoury et al. [177] |
| 204 | glucose (α/β, 3-O) | M13 | R9 | I5 | 99 | 27.7 | 2.64 | 1.1 | homo | – | Al-Bagoury et al. [177] |
| 205 | glucose (α/β, 3-O) | M13 | R1 | I3 | 43 | 6.4 | – | 1.21 | homo | lectin recognition | Luo et al. [183] |
| 206 | glucose (α/β, 3-O) | M33 | polyM13 | I3 | 45 | 7.5 | – | 1.21 | block AB | lectin recognition | Luo et al. [183] |
| 207 | glucose (α/β, 3-O) | M42 | poly(M13-block-M33) | I3 | 35 | 9.3 | – | 1.27 | block ABA | lectin recognition | Luo et al. [183] |
| 208 | glucose (α/β, 3-O) | M9 | R32 | I4 | – | 6.4 | – | 1.14 | homo | – | Glassner et al. [184] |
| 209 | glucose (α/β, 3-O) | M9 | R23 | I4 | 25 | 4.2 | – | 1.2 | homo | – | Kaupp et al. [185] |
| 210 | glucose (β-O) | M113a | R10 | I4 | 74 f | 21.9 | – | 1.09 | homo | lectin recognition | Dan et al. [186] |
| 211 | glucose (β-O) | M113b | R10 | I4 | 46 f | 13 | – | 1.12 | homo | lectin recognition | Dan et al. [186] |
| 212 | lactose (β-O) | M43 | R9 | I4 | 31 | 6.29 | 0.87 | 1.09 | homo | stationary phase | Guo et al. [187] |
| 213 | lactose (β-O) | M43 | R1 | I4 | >90 | 20 | – | 1.22 | homo | gene therapy | Zhou et al. [188] |
| (Meth)acrylate monomers (unprotected) | |||||||||||
| 214 | galactose (β-O) | M38 | R1 | I3 | 80 | 24 | 1.01 | 1.09 | homo | lectin recognition | Spain et al. [189] |
| 215 | galactose (β-O) | M38/M115 | R35 | I3 | > 99 | 12.2 | – | 1.2 | A-stat-B | biomedical | Song et al. [12] |
| 216 | gluconic acid (amide) | M23 | R14 | I3 | > 95 | 14.1 | – | 1.19 | homo | streptavidin binding | Housni et al. [190] |
| 217 | glucose (α/β-O) | M34a | R1 | I3 | 70 | 27.4 | 1.31 | 1.03 | homo | – | Lowe et al. [191] |
| 218 | glucose (α/β-O) | M34a | R1 | I3 | 40 | 14.2 | 1.18 | 1.07 | homo | – | Lowe et al. [191] |
| 219 | glucose (α/β-O) | M34a | polyM34a | I3 | – | 33.8 | 0.92 | 1.54 | block AB | – | Lowe et al. [191] |
| 220 | glucose (α/β-O) | M35 | polyM34a | I3 | – | 37.1 | – | 1.63 | block AB | – | Lowe et al. [191] |
| 221 | glucose (β-S, 6-O) | M103 | polyM59 | I4 | 53 | 15.2 | 0.94 | 1.45 | block AB | biomedical | Pearson et al. [192] |
| 222 | glucose (α/β-O) | M34a | polyM36·R1 | I3 | 71 | 52 | 0.82 | 1.2 | block AB | – | Albertin et al. [193] |
| 223 | glucose/mannose (α/β; or α-methyl, 6-O) | M37 | polyM34a·R1 | I3 | 52 | 61.3 | 0.75 | 1.16 | block AB | – | Albertin et al. [193] |
| 224 g | glucose (α-methyl, 6-O | M36 | R1 | I3 | 97 | 327 | 12.5 | 3.67 | homo | – | Albertin et al. [194] |
| 225 h | glucose (α-methyl, 6-O) | M36 | R1 | I3 | 99 | 174 | 6.6 | 1.75 | homo | – | Albertin et al. [194] |
| 226 i | glucose (α-methyl, 6-O) | M36 | R1 | I3 | 100 | 26.3 | 0.93 | 1.14 | homo | – | Albertin et al. [194] |
| 227 i | glucose (α-methyl, 6-O) | M33 | polyM36·R1 | I3 | – | 45 | – | 1.2 | block AB | – | Albertin et al. [195] |
| 228 | lactobionic acid (amide) | M25 | R15 | I3 | >95 | 24.7 | – | 1.22 | homo | streptavidin binding | Housni et al. [190] |
| 229 | mannose (α/β, 6-O) | M39 | R1 | I4 | 71 | 42.5 | – | 1.23 | homo | – | Pfaff et al. [196] |
| 230 | mannose (α/β, 6-O) | M39 | R1 | I3 | ≥95 | 85 | ≅1.13 | 1.06 | homo | lectin recognition | Granville et al. [197] |
| 231 | mannose (β-O) | M114/M115 | R35 | I3 | >99 | 11.4 | – | 1.2 | A-stat-B | biomedical | Song et al. [12] |
| 232 | N-acetylglucosamine (β-O) | M31/M115 | R35 | I3 | >99 | 13.1 | – | 1.2 | A-stat-B | biomedical | Song et al. [12] |
| Vinyl ester monomers (unprotected) | |||||||||||
| 233 | glucose (α/β, 6-O) | M40 | R2 | I3 | 14 | 17.1 | – | 1.1 | homo | – | Albertin et al. [198] |
| 234 | glucose (α/β, 6-O) | M40 | R3 | I3 | 27 | 19.6 | – | 1.19 | homo | – | Albertin et al. [198] |
| 235 | glucose (α/β, 6-O) | M40 | R4 | I3 | 50 | ~28 | – | 1.48 | star (4-arm) | – | Bernard et al. [199] |
| Diene-like monomers (unprotected) | |||||||||||
| 236 | mannose (α-O) | M78 | R5 | I3 | 48 | 51.5 | – | 1.16 | homo | lectin recognition | Hetzer et al. [200] |
| 237 | mannose (α-O) | M42 | polyM78 | I4 | – | 32.4 | – | 1.12 | block AB | lectin recognition | Hetzer et al. [200] |
| Styrenic monomers (protected) | |||||||||||
| 238 | galactose (α/β, 6-O) | M50 | R11 | I4 | 85 | 27 | ≅ 0.6 | 1.1 | homo | protein bioconjugation | Xiao et al. [201] |
| 239 | galactose (α/β, 6-O) | M50/M94 | R11 | I10 | 75 | 18 | – | 1.29 | A-stat-B | drug delivery | Xiao et al. [202] |
| 240 | galactose (α/β, 6-O) | M51 | R12 | I4 | 60 | 5.2 | – | 1.11 | homo | chiral recognition | Wang et al. [203] |
| 241 | galactose (α/β, 6-O) | M51 | polySty·R12 | I4 | – | 19.6 | – | 1.57 | block AB | – | Wang et al. [203] |
| 242 | galactose (α/β, 6-O) | M51 | polyMMA·R12 | I4 | – | 20.8 | – | 1.41 | block AB | – | Wang et al. [203] |
| 243 | galactose (α/β, 6-O) | M51 | polyMA·R12 | I4 | – | 24.6 | – | 1.35 | block AB | – | Wang et al. [203] |
| Styrenic monomers (unprotected) | |||||||||||
| 244 | trehalose | M104 | R29 | I4 | 77 | 9.4 | 0.98 | 1.07 | homo | protein formulation | Mancini et al. [204] |
| 245 | trehalose | M104 | R30 | I4 | 73 | 24.5 | 0.95 | 1.2 | homo | protein formulation | Mancini et al. [204] |
| 246 | trehalose | M104 | R30 | I4 | 81 | 49.5 | 1.1 | 1.47 | homo | protein formulation | Mancini et al. [204] |
| Glycopolymers from post-polymerization reaction | |||||||||||
| 247 | galactose (α/β, 6-O) | M57/M59 | R5 | I4 | – | 11 | 0.88 | 1.14 | A-stat-B | glycoGNP | Boyer et al. [205] |
| 248 | galactose (α-N), 2-deoxy-2-amino-d-glucose (N) | M68 | R5 | I4 | – | 2.8 | 0.84 | 1.2 | homo | lectin recognition | Boyer et al. [206] |
| 249 | galactose (α-N), 2-deoxy-2-amino-d-glucose (N) | M68, M121 | R5 | I4 | 70, 96 | 90 | 0.84 | 1.18 | star (16-arm) | – | Boyer et al. [207] |
| 250 | galactose (β-O), glucose (β-S) | M82/M83 | R20 | I4 | 93 | 29 | – | 1.9 | branched | – | Semsarilar et al. [208] |
| 251 | glucose (β-S) | M57/M58 | R5 | I4 | – | 10 | 0.91 | 1.12 | A-stat-B | glycoGNP | Boyer et al. [205] |
| 252 | glucose (β-S) | M58 | R19 | – | 85 | 51.4 | 0.49 | 1.9 | star (4-arm) | lectin recognition | Chen et al. [209] |
| 253 | glucose (β-S) | M82 | R10 | I4 | 25 | 15.4 | – | 11.8 | branched | - | Semsarilar et al. [208] |
| 254 | glucose (β-S) | M57 | R23 | I4 | – | 5.6 | – | 1.13 | homo | lectin recognition | Kumar et al. [210] |
| 255 | glucose (β-S) | M59 | polyM57·R5 | I4 | – | 6.1 | – | 1.15 | homo | lectin recognition | Kumar et al. [210] |
| 256 | glucuronic acid (1-amino-1-deoxy alditol) | M54 | R1 | I6 | 47 | 33 | 0.98 | 1.05 | homo | – | Alidedeoglu et al. [211] |
| 257 | glucuronic acid (1-amino-1-deoxy alditol) | M55 | R1 | I3 | 31 | 15 | 1.3 | 1.08 | homo | – | Alidedeoglu et al. [211] |
| 258 | glucuronic acid (1-amino-1-deoxy alditol) | M56 | polyM54 | I3 | 33 | 48.4 | 0.85 | 1.05 | block AB | – | Alidedeoglu et al. [211] |
| 259 | N-acetylglucosamine (α-O) | M111 | R34 | I3 | 82 | 15 | – | 1.12 | A-stat-B | biosensor | Godula et al. [212] |
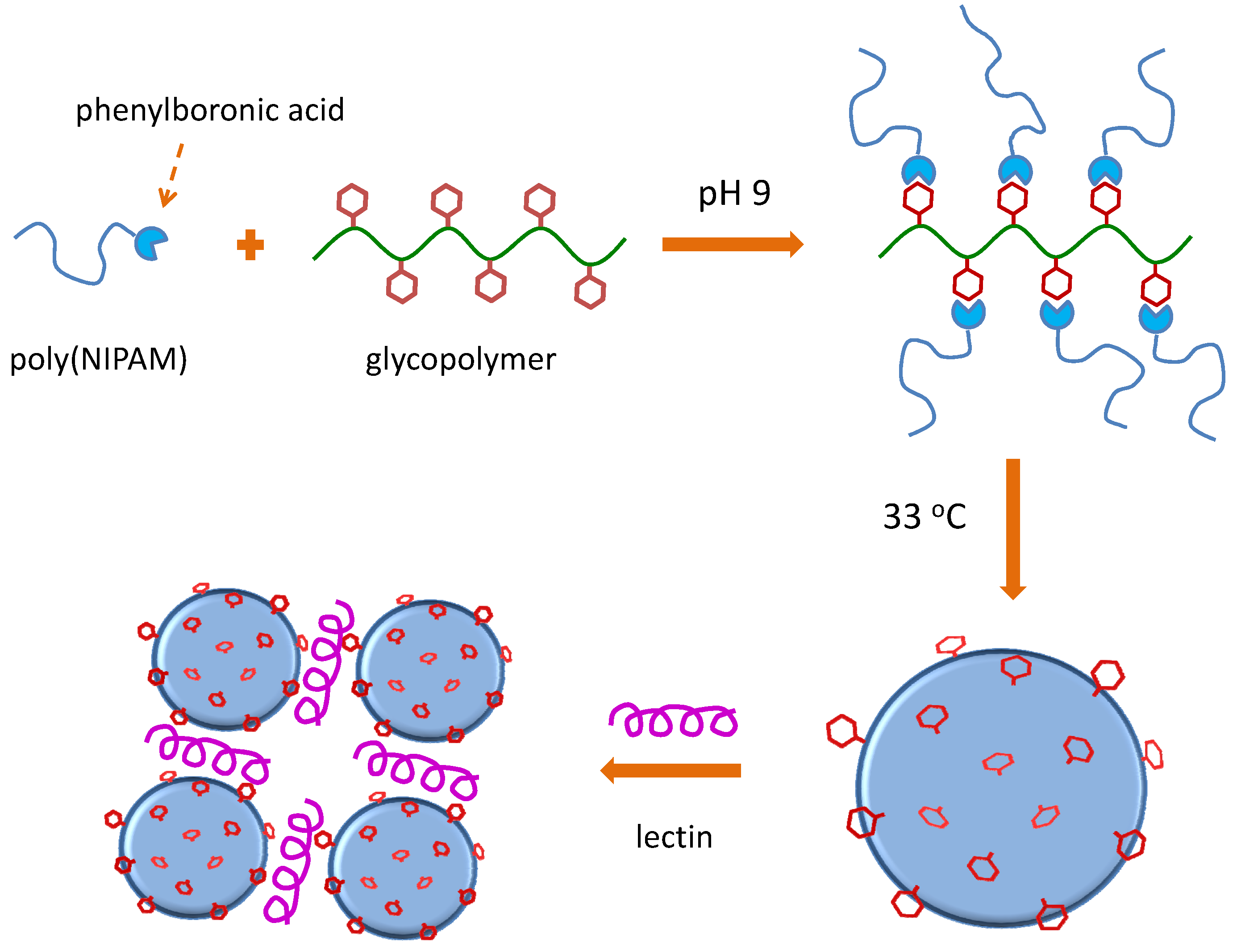
6.1.2. (Meth)acrylate Monomers
6.1.3. Styrenic Monomers
6.2. RAFT Starting from Unprotected Glycomonomers/GlycoRAFT Agents
6.2.1. Diene-Like Monomers
6.2.2. (Meth)acrylamide Monomers
6.2.3. (Meth)acrylate Monomers
6.2.4. Styrenic Monomers
6.2.5. Vinyl Ester Monomers
6.3. Glycopolymers from Post-Polymerization Reaction
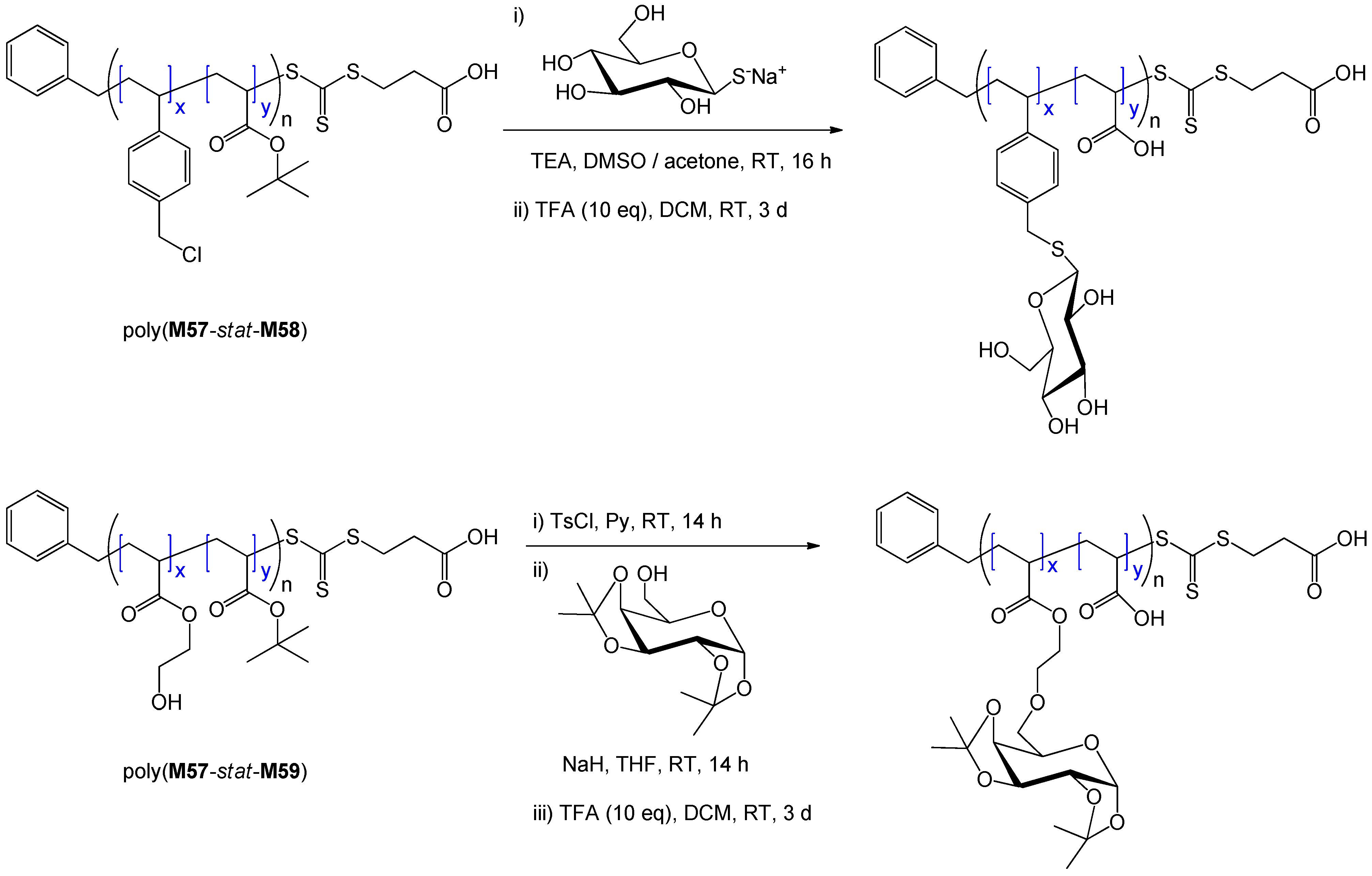
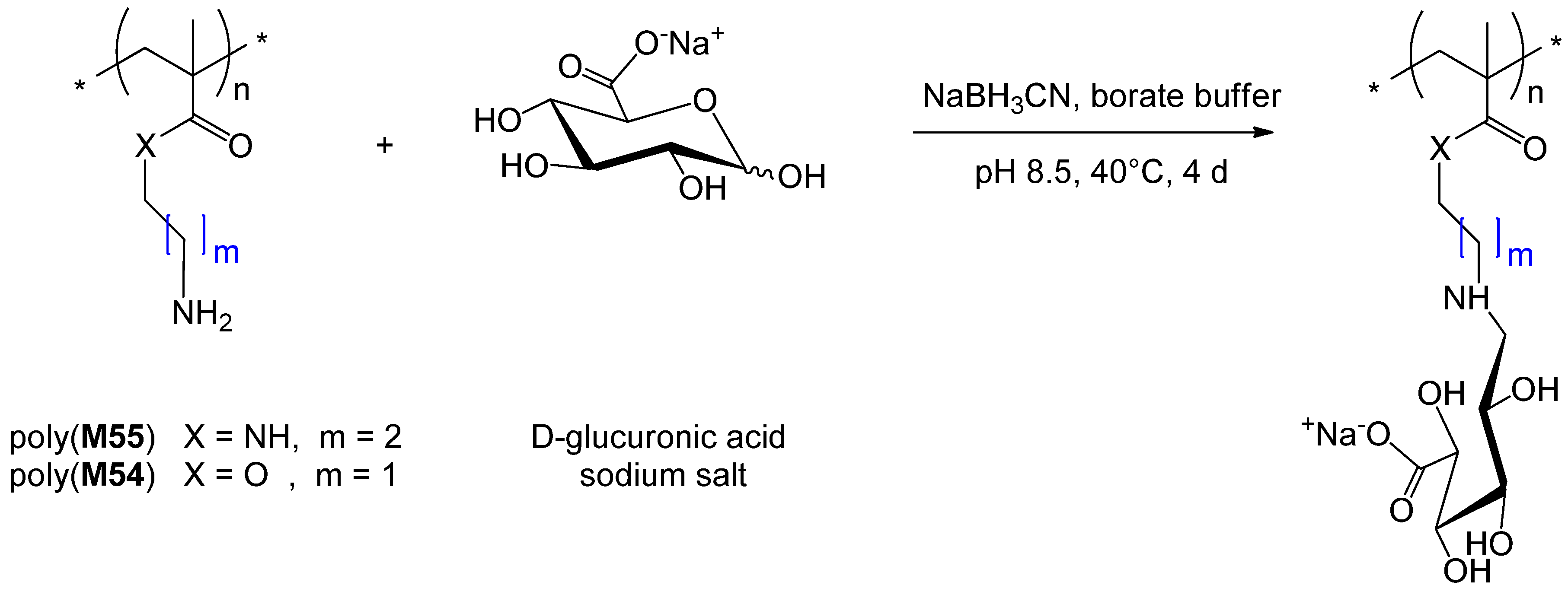
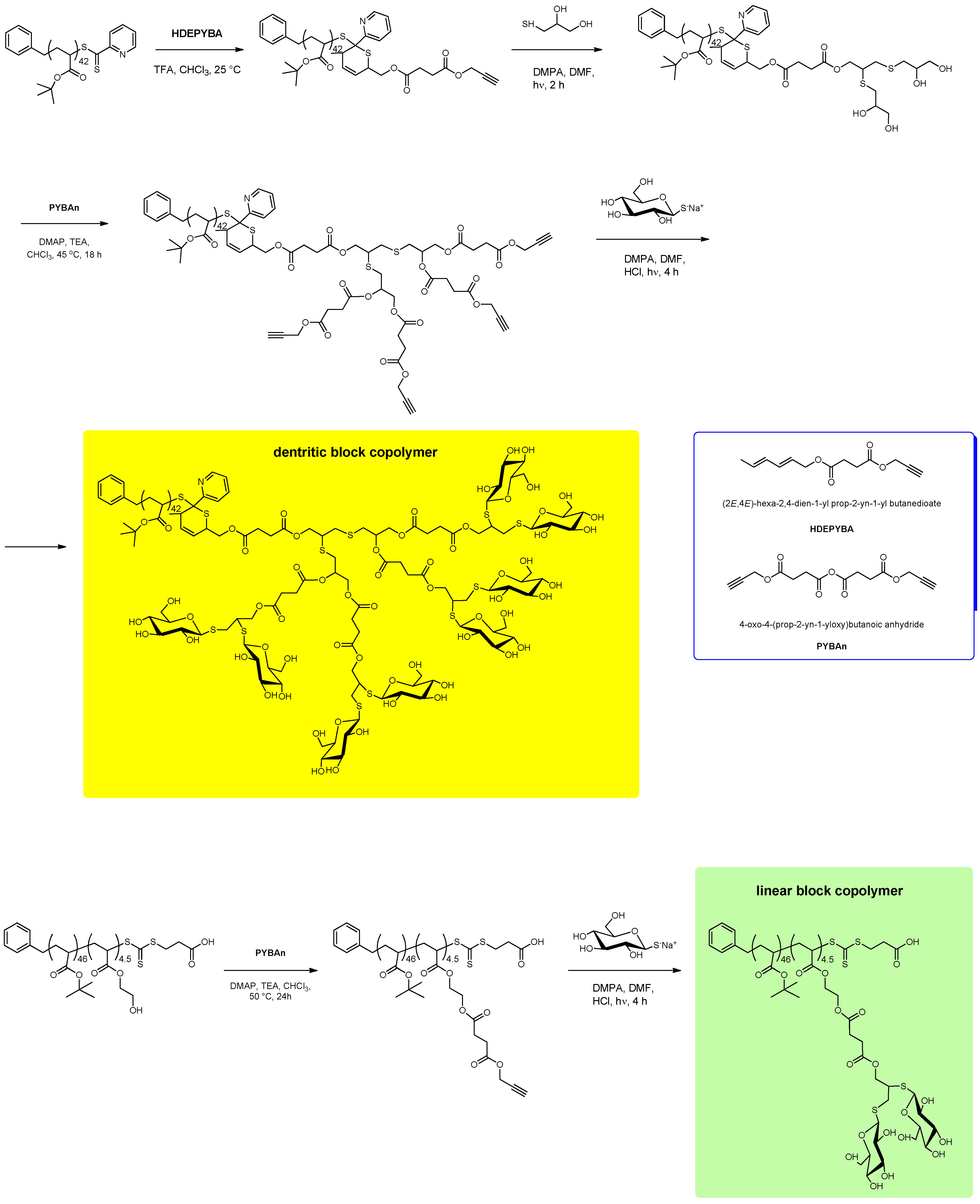

7. Conclusion and Perspectives
- functional groups likely to deactivate the catalyst (e.g., acid functions) need to be protected during the polymerization process [238];
- achieving a good degree of control in aqueous media is challenging due to the occurrence of several side reactions involving the catalytic system [239]. For instance, in water the CuI-based ATRP activator may disproportionate; the CuII-based deactivator is likely to lose its halide ligand; and the alkyl halide initiator may hydrolyze or react with the monomer if it contains basic or nucleophilic groups. In this case, better results are obtained by adding an organic co-solvent (e.g., methanol or DMF) and (or) a CuII halide complex to the catalyst;
- between 1000 ppm and 10,000 ppm of copper are present in a polymer prepared by classic ATRP and its removal adds to the complexity of the process.
| Symbols and Abbreviations | |
|---|---|
| Aβ | Aβ peptideamyloid β peptide |
| AFM | AFM atomic force microscopy |
| AGET | AGET activator generated by electron transfer |
| Ai | Ai initiator “i” used in ATRP |
| AIBN | AIBN 2,2′-azobis-isobutyronitrile |
| ATRP | ATRP atom transfer radical polymerization |
| BIEM | BIEM 2-(2-bromoisobutyryloxy)ethyl methacrylate |
| BSA | BSA bovine serum albumin |
| cac | cac critical association concetration |
| CD | CD circular dichroism |
| CMC | CMC critical micelle concentration |
| CMPSF | CMPSF chloromethylated polysulfone |
| COD | COD 1,5-cyclooctadiene |
| ConA | ConA Concanavalin A |
| Conv | Conv conversion |
| Cp | Cp cyclopentadiene |
| CTA | CTA chain transfer agent |
| Ctx | Ctx cholera toxin |
| DCM | DCM dichloromethane |
| Đ | Đ molar mass dispersity index |
| Đdparticle | Đdparticle diameter dispersity index |
| DCP | DCP dicumyl peroxide |
| DLS | DLS dynamic light scattering |
| DMAc | DMAc dimethyl acetamide |
| DMF | DMF dimethylformamide |
| DMPA | DMPA 2,2-dimethoxy-2-phenylacetophenone |
| DMSO | DMSO dimethyl sulfoxide |
| DNA | DNA deoxyribonucleic acid |
| DP | DP degree of polymerization |
| DTT | DTT 1,4-dithiothreitol |
| ECA | ECA Erythrina cristagalli agglutinin |
| EDC | EDC 1-ethyl-3-(3-dimethylaminopropyl-carbodiimide) |
| EWCRDS | EWCRDS evanescent wave cavity ring-down spectroscopy |
| Fb | Fb Fibrinogen |
| FCS | FCS fluorescence correlation spectroscopy |
| FGF | FGF Fibroblast growth factor |
| FimH | FimH fimbrial lectin |
| FRET | FRET Förster resonance energy transfer |
| FTIR | FTIR Fourier transform infrared |
| Gal | Gal galactose |
| Glc | Glc glucose |
| GlcNAc | GlcNAc N-acetyl-d-glucosamine |
| GNP(s) | GNP(s) gold nanoparticle(s) |
| HDA | HDA hetero-Diels Alder |
| HEMA | HEMA 2-hydroxyethyl methacrylate |
| HIV | HIV human immunodeficiency virus |
| HOBT | HOBT 1-hydroxybenzotrizole |
| homo | homo homopolymer |
| HPA | HPA Helix pomatia agglutinin |
| IC50 | IC50 the half maximal inhibitory concentration, i.e., the concentration of a particular substance (inhibitor) needed to inhibit a given biological process by half |
| Lac | Lac lactose |
| LBL | LBL layer by layer |
| LCST | LCST lower critical solution temperature |
| Li | Li ligand “i” used in ATRP catalyst |
| MA | MA methyl acrylate |
| MALDI-ToF | MALDI-ToF matrix-assisted laser desorbtion ionization-time of flight |
| Man | Man mannose |
| MAnh | MAnh maleic anhydride |
| MHS | MHS Mark-Houwink-Sakurada |
| Mi | Mi monomer “i” |
| MMA | MMA methyl methacrylate |
| Mn | Mn number average molar mass |
| Mn,th | Mn,th theoretical number average molar mass |
| MS | MS mass spectroscopy |
| Mw | Mw weight average molar mass |
| MWNT | MWNT multiwalled carbon nanotube |
| NHS | NHS N-hydroxysuccinimide |
| Ni | Ni initiator/control agent “i” used in NMP |
| NIPAAm | NIPAAm N-isopropylacrylamide |
| NMP | NMP nitroxide mediated polymerization |
| NMR | NMR nuclear magnetic resonance |
| p | p monomer conversion |
| PDVB | PDVB poly(divinylbenzene) |
| PEG | PEG polyethylene glycol |
| PEO | PEO polyethylene oxide |
| PET | PET poly(ethyleneterephtalate) |
| PMMA | PMMA poly(methylmethacrylate) |
| polyMi | polyMi poly(monomer i) |
| polyMi·Ni(Ri) | polyMi·Ni(Ri) macro-initiator/macro-control agent poly(monomer i) obtained from the polymerization of monomer “i” with initiator Ni or RAFT agent Ri |
| PNA | PNA peanut agglutinin |
| PSF | PSF polysulfone |
| p-TsCl | p-TsCl p-toluenesulfonyl chloride (Tosyl chloride) |
| PVDF | PVDF poly(vinylidene difluoride) |
| QCM | QCM quartz crystal microbalance |
| QD | QD quatum dots |
| RAFT | RAFT reversible addition-fragmentation chain transfer |
| RAFTstab | RAFTstab reversible addition-fragmentation chain transfer colloidal stabilizer |
| RCA | RCA Ricinus communis agglutinin |
| RDRP | RDRP reversible deactivation radical polymerization |
| Ri | Ri chain transfer agent “i” used in RAFT polymerization |
| RNA | RNA ribonucleic acid |
| ROMP | ROMP ring Opening Metathesis Polymerization |
| ROP | ROP ring Opening Polymerization |
| RT | RT room temperature |
| SBA | SBA soybean agglutinin |
| SCVCP | SCVCP self-condensing vinyl copolymerization |
| SEC | SEC size exclusion chromatography |
| SEM | SEM scanning Electron Microscopy |
| SG1 | SG1 N-tert-butyl-N-(1-diethylphosphono-2,2-dimethylpropyl) |
| siRNA | siRNA small interfering RNA |
| SLS | SLS static light scattering |
| SPR | SPR surface plasmon resonance |
| Sty | Sty styrene |
| TBAF | TBAF tetra-n-butylammonium fluoride |
| TEM | TEM transmission electron microscopy |
| TEMPO | TEMPO 2,2,6,6-tetramethylpiperidine-1-oxyl |
| TFA | TFA trifluoroacetic acid |
| THF | THF tetrahydrofuran |
| ThT | ThT thioflavin T |
| TIPNO | TIPNO 2,2,5-trimethyl-4-phenyl-3-azahexane-3-oxyl |
| TsCl | TsCl p-toluenesulfonyl chloride |
| VVA | VVA Vicia villosa agglutinin |
| WFL | WFL Wisteria floribunda lectin |
| WGA | WGA wheat germ agglutinin |
References
- Horejsi, V.; Smolek, P.; Kocourek, J. Studies on lectins. XXXV. Water-soluble O-glycosyl polyacrylamide derivatives for specific precipitation of lectins. Biochim. Biophys. Acta Gen. Subj. 1978, 538, 293–298. [Google Scholar] [CrossRef]
- Choi, S.K.; Mammen, M.; Whitesides, G.M. Generation and in situ evaluation of libraries of poly(acrylic acid) presenting sialosides as side chains as polyvalent inhibitors of influenza-mediated hemagglutination. J. Am. Chem. Soc. 1997, 119, 4103–4111. [Google Scholar] [CrossRef]
- Gordon, E.J.; Strong, L.E.; Kiessling, L.L. Glycoprotein-inspired materials promote the proteolytic release of cell surface l-selectin. Bioorg. Med. Chem. 1998, 6, 1293–1299. [Google Scholar] [CrossRef]
- Kiessling, L.L.; Gestwicki, J.E.; Strong, L.E. Synthetic multivalent ligands in the exploration of cell-surface interactions. Curr. Opin. Chem. Biol. 2000, 4, 696–703. [Google Scholar] [CrossRef]
- David, A.; Kopeckova, P.; Kopecek, J.; Rubinstein, A. The role of galactose, lactose, and galactose valency in the biorecognition of N-(2-hydroxypropyl)methacrylamide copolymers by human colon adenocarcinoma cell. Pharm. Res. 2002, 19, 1114–1122. [Google Scholar] [CrossRef]
- Roy, R.; Baek, M.G. Glycodendrimers: Novel glycotope isosteres unmasking sugar coding. Case study with T-antigen markers from breast cancer MUC1 glycoprotein. Rev. Mol. Biotechnol. 2002, 90, 291–309. [Google Scholar] [CrossRef]
- Gambaryan, A.S.; Boravleva, E.Y.; Matrosovich, T.Y.; Matrosovich, M.N.; Klenk, H.D.; Moiseeva, E.V.; Tuzikov, A.B.; Chinarev, A.A.; Pazynina, G.V.; Bovin, N.V. Polymer-bound 6′ sialyl-N-acetyllactosamine protects mice infected by influenza virus. Antivir. Res. 2005, 68, 116–123. [Google Scholar] [CrossRef]
- Fleming, C.; Maldjian, A.; Da Costa, D.; Rullay, A.K.; Haddleton, D.M.; John, J.St.; Penny, P.; Noble, R.C.; Cameron, N.R.; Davis, B.G. A carbohydrate-antioxidant hybrid polymer reduces oxidative damage in spermatozoa and enhances fertility. Nat. Chem. Biol. 2005, 1, 270–274. [Google Scholar] [CrossRef]
- Palomino, E. Carbohydrate handles as natural resources in drug delivery. Adv. Drug Deliv. Rev. 1994, 13, 311–323. [Google Scholar] [CrossRef]
- Wadhwa, M.S.; Rice, K.G. Receptor mediated glycotargeting. J. Drug Target. 1995, 3, 111–127. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Han, Y.; Cheng, C.; Li, C. pH- and glucose-sensitive glycopolymer nanoparticles based on phenylboronic acid for triggered release of insulin. Carbohydr. Polym. 2012, 89, 124–131. [Google Scholar] [CrossRef]
- Song, E.-H.; Manganiello, M.J.; Chow, Y.-H.; Ghosn, B.; Convertine, A.J.; Stayton, P.S.; Schnapp, L.M.; Ratner, D.M. In vivo targeting of alveolar macrophages via RAFT-based glycopolymers. Biomaterials 2012, 33, 6889–6897. [Google Scholar] [CrossRef]
- Karamuk, E.; Mayer, J.; Wintermantel, E.; Akaike, T. Partially degradable film/fabric composites: Textile scaffolds for liver cell culture. Artif. Organs 1999, 23, 881–884. [Google Scholar] [CrossRef]
- Chaikof, E.L.; Grande, D.; Baskaran, S. Glycopolymers and Free Radical Polymerization Methods. U.S. Patent 7,244,830 B2, 17 July 2007. [Google Scholar]
- Liu, X.C.; Dordick, J.S. Sugar acrylate-based polymers as chiral molecularly imprintable hydrogels. J. Polym. Sci. A Polym. Chem. 1999, 37, 1665–1671. [Google Scholar]
- Gruber, H.; Knaus, S. Synthetic polymers based on carbohydrates: Preparation, properties and applications. Macromol. Symp. 2000, 152, 95–105. [Google Scholar] [CrossRef]
- Baek, M.G.; Roy, R. Design and synthesis of water-soluble glycopolymers bearing breast tumor marker and enhanced lipophilicity for solid-phase assays. Biomacromolecules 2000, 1, 768–770. [Google Scholar] [CrossRef]
- Miyata, T.; Uragami, T.; Nakamae, K. Biomolecule-sensitive hydrogels. Adv. Drug Deliv. Rev. 2002, 54, 79–98. [Google Scholar] [CrossRef]
- Novick, S.J.; Dordick, J.S. Preparation of active and stable biocatalytic hydrogels for use in selective transformations. Chem. Mater. 1998, 10, 955–958. [Google Scholar] [CrossRef]
- Wulff, G.; Schmid, J.; Venhoff, T.P. The preparation of new types of polymerizable vinyl sugars with C-C bonds between sugar and double bond. Macromol. Chem. Phys. 1996, 197, 1285–1299. [Google Scholar] [CrossRef]
- Wulff, G.; Zhu, L.; Schmidt, H. Investigations on surface-modified bulk polymers. 1. copolymers of styrene with a styrene moiety containing a sugar monomer. Macromolecules 1997, 30, 4533–4539. [Google Scholar] [CrossRef]
- Miyata, T.; Nakamae, K. Polymers with pendent saccharides: “glycopolymers”. Trends Polym. Sci. 1997, 5, 198–206. [Google Scholar]
- Wulff, G.; Schmidt, H.; Zhu, L.M. Generating hydrophilic surfaces on standard polymers after copolymerization with low amounts of protected vinyl sugars. Macromol. Chem. Phys. 1999, 200, 774–782. [Google Scholar] [CrossRef]
- Varki, A.; Cummings, R.; Esko, J.; Freeze, H.; Hart, G.; Marth, J. Essentials of Glycobiology, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Whittaker, G.R. Intracellular trafficking of influenza virus: Clinical implications for molecular medicine. Expert Rev. Mol. Med. 2001, 3, 1–13. [Google Scholar] [CrossRef]
- Weis, W.; Brown, J.H.; Cusack, S.; Paulson, J.C.; Skehel, J.J.; Wiley, D.C. Structure of the influenza-virus haemagglutinin complexed with its receptor, sialic-acid. Nature 1988, 333, 426–431. [Google Scholar] [CrossRef]
- Bertozzi, C.R.; Kiessling, L.L. Chemical glycobiology. Science 2001, 291, 2357–2364. [Google Scholar] [CrossRef]
- Vandamme, E.J.; de Baets, S.; Steinbüchel, A. Polysaccharides I: Polysaccharides from Prokaryotes; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Vandamme, E.J.; de Baets, S.; Steinbüchel, A. Polysaccharides II: Polysaccharides from Eukaryotes; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Haddleton, D.M.; Ohno, K. Well-Defined oligosaccharide-terminated polymers from living radical polymerization. Biomacromolecules 2000, 1, 152–156. [Google Scholar] [CrossRef]
- Halila, S.; Manguian, M.; Fort, S.; Cottaz, S.; Hamaide, T.; Fleury, E.; Driguez, H. Syntheses of well-defined glyco-polyorganosiloxanes by “click” chemistry and their surfactant properties. Macromol. Chem. Phys. 2008, 209, 1282–1290. [Google Scholar] [CrossRef]
- Chiron, S.; Labeau, M.-P.; Fleury, E.; Viet, D.; Cottaz, S.; Driguez, H.; Halila, S. Novel Glycopolymers, Uses Thereof, and Monomers Useful for Preparation Thereof. WO Patent 2006016063 A1, 13 November 2008. [Google Scholar]
- Narumi, A.; Miura, Y.; Otsuka, I.; Yamane, S.; Kitajyo, Y.; Satoh, T.; Hirao, A.; Kaneko, N.; Kaga, H.; Kakuchi, T. End-functionalization of polystyrene by malto-oligosaccharide generating aggregation-tunable polymeric reverse micelle. J. Polym. Sci. A Polym. Chem. 2006, 44, 4864–4879. [Google Scholar] [CrossRef]
- Houga, C.; Le Meins, J.-F.; Borsali, R.; Taton, D.; Gnanou, Y. Synthesis of ATRP-induced dextran-b-polystyrene diblock copolymers and preliminary investigation of their self-assembly in water. Chem. Commun. 2007, 3063–3065. [Google Scholar]
- Aissou, K.; Otsuka, I.; Rochas, C.; Fort, S.; Halila, S.; Borsali, R. Nano-Organization of Amylose-b-polystyrene block copolymer films doped with bipyridine. Langmuir 2011, 27, 4098–4103. [Google Scholar] [CrossRef]
- Bernard, J.; Save, M.; Arathoon, B.; Charleux, B. Preparation of a xanthate-terminated dextran by click chemistry: Application to the synthesis of polysaccharide-coated nanoparticles via surfactant-free ab initio emulsion polymerization of vinyl acetate. J. Polym. Sci. A Polym. Chem. 2008, 46, 2845–2857. [Google Scholar] [CrossRef]
- Ghadban, A.; Albertin, L.; Rinaudo, M.; Heyraud, A. Biohybrid glycopolymer capable of ionotropic gelation. Biomacromolecules 2012, 13, 3108–3119. [Google Scholar] [CrossRef]
- Sigal, G.B.; Mammen, M.; Dahmann, G.; Whitesides, G.M. Polyacrylamides bearing pendant alpha—sialoside groups strongly inhibit agglutination of erythrocytes by influenza virus-The strong inhibition reflects enhanced binding through cooperative polyvalent interactions. J. Am. Chem. Soc. 1996, 118, 3789–3800. [Google Scholar] [CrossRef]
- Kanai, M.; Mortell, K.H.; Kiessling, L.L. Varying the size of multivalent ligands-The dependence of concanavalin A binding on neoglycopolymer length. J. Am. Chem. Soc. 1997, 119, 9931–9932. [Google Scholar] [CrossRef]
- Georges, M.K.; Veregin, R.P.N.; Kazmaier, P.M.; Hamer, G.K. Narrow molecular weight resins by a free-radical polymerization process. Macromolecules 1993, 26, 2987–2988. [Google Scholar] [CrossRef]
- Kato, M.; Kamigaito, M.; Sawamoto, M.; Higashimura, T. Polymerization of methyl methacrylate with the carbon tetrachloride/dichlorotris- (triphenylphosphine)ruthenium(II)/Methylaluminum Bis(2,6-di-tert-butylphenoxide) initiating System: Possibility of living radical polymerization. Macromolecules 1995, 28, 1721–1723. [Google Scholar] [CrossRef]
- Chiefari, J.; Chong, Y.K.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T.P.T.; Mayadunne, R.T.A.; Meijs, G.F.; Moad, C.L.; Moad, G.; et al. Living free-radical polymerization by reversible addition-fragmentation chain transfer—The RAFT process. Macromolecules 1998, 31, 5559–5562. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Davis, T.P. Handbook of radical polymerization; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2002. [Google Scholar]
- Lowe, A.B.; McCormick, C.L. Homogeneous controlled free radical polymerization in aqueous media. Aust. J. Chem. 2002, 55, 367–379. [Google Scholar] [CrossRef]
- Xia, J.H.; Gaynor, S.G.; Matyjaszewski, K. Controlled/“living” radical polymerization. Atom transfer radical polymerization of acrylates at ambient temperature. Macromolecules 1998, 31, 5958–5959. [Google Scholar] [CrossRef]
- Robinson, K.L.; Khan, M.A.; Banez, M.V.D.; Wang, X.S.; Armes, S.P. Controlled polymerization of 2-hydroxyethyl methacrylate by ATRP at ambient temperature. Macromolecules 2001, 34, 3155–3158. [Google Scholar] [CrossRef]
- Quinn, J.F.; Rizzardo, E.; Davis, T.P. Ambient temperature reversible addition-fragmentation chain transfer polymerisation. Chem. Commun. 2001, 1044–1045. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living radical polymerization by the RAFT process. Aust. J. Chem. 2005, 58, 379–410. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living radical polymerization by the RAFT process—A first update. Aust. J. Chem. 2006, 59, 669–692. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living radical polymerization by the RAFT process—A second update. Aust. J. Chem. 2009, 62, 1402–1472. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Atom Transfer Radical Polymerization (ATRP): Current status and future perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar] [CrossRef]
- Grubbs, R.B. Nitroxide-Mediated radical polymerization: Limitations and versatility. Polym. Rev. 2011, 51, 104–137. [Google Scholar] [CrossRef]
- Nicolas, J.; Guillaneuf, Y.; Lefay, C.; Bertin, D.; Gigmes, D.; Charleux, B. Nitroxide-mediated polymerization. Prog. Polym. Sci. 2013, 63–235. [Google Scholar]
- Jenkins, A.D.; Jones, R.G.; Moad, G. Terminology for reversible-deactivation radical polymerization previously called “controlled” radical or “living” radical polymerization (IUPAC Recommendations 2010). Pure Appl. Chem. 2010, 82, 483–491. [Google Scholar] [CrossRef]
- Fukuda, T.; Goto, A.; Tsujii, Y. Kinetics of Living Radical Polymerisation. In Handbook of Radical Polymerization; Matyjaszewski, K., Davis, T.P., Eds.; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2002; pp. 407–462. [Google Scholar]
- Matyjaszewski, K. General Concepts and History of Living Radical Polymerisation. In Handbook of Radical Polymerization; Matyjaszewski, K., Davis, T.P., Eds.; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2002; pp. 361–406. [Google Scholar]
- Miura, Y. Design and synthesis of well-defined glycopolymers for the control of biological functionalities. Polym. J. 2012, 44, 679–689. [Google Scholar] [CrossRef]
- Becer, C.R. The glycopolymer code: Synthesis of glycopolymers and multivalent carbohydrate-lectin interactions. Macromol. Rapid Commun. 2012, 33, 742–752. [Google Scholar] [CrossRef]
- Spain, S.G.; Cameron, N.R. A spoonful of sugar: The application of glycopolymers in therapeutics. Polym. Chem. 2011, 2, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Pearson, S.; Chen, G.; Stenzel, M.H. Synthesis of Glycopolymers. In Engineered Carbohydrate-Based Materials for Biomedical Applications; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 1–118. [Google Scholar]
- Voit, B.; Appelhans, D. Glycopolymers of various architectures-more than mimicking nature. Macromol. Chem. Phys. 2010, 211, 727–735. [Google Scholar] [CrossRef]
- Ting, S.R.S.; Chen, G.; Stenzel, M.H. Synthesis of glycopolymers and their multivalent recognitions with lectins. Polym. Chem. 2010, 1, 1392–1412. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Xia, C.; Wang, P.G. Glycopolymers: The Future Antiadhesion Drugs. In Polymer Biocatalysis and Biomaterials II; American Chemical Society: Washington, DC, USA, 2008; Volume 999, pp. 342–361. [Google Scholar]
- Spain, S.G.; Gibson, M.I.; Cameron, N.R. Recent advances in the synthesis of well-defined glycopolymers. J. Polym. Sci. A Polym. Chem. 2007, 45, 2059–2072. [Google Scholar] [CrossRef]
- Rice, K.G.; Kim, J.-S.; Liu, D. Glycopolymer Tools for Studying Targeted Nonviral Gene Delivery. In Polymeric Gene Delivery Principles and Applications; Amiji, M.M., Ed.; CRC Press: Punta Gorda, FL, USA, 2005; pp. 509–521. [Google Scholar]
- Ladmiral, V.; Melia, E.; Haddleton, D.M. Synthetic glycopolymers: An overview. Eur. Polym. J. 2004, 40, 431–449. [Google Scholar] [CrossRef]
- Okada, M. Molecular design and syntheses of glycopolymers. Prog. Polym. Sci. 2001, 26, 67–104. [Google Scholar]
- Roy, R. Blue-prints, synthesis and applications of glycopolymers. Trends Glycosci. Glycotechnol. 1996, 8, 79–99. [Google Scholar] [CrossRef]
- Druliner, J.D. Living radical polymerization involving oxygen-centered species attached to propagating chain ends. Macromolecules 1991, 24, 6079–6082. [Google Scholar] [CrossRef]
- Gnanou, Y.; Grande, D.; Guerrero, R. Meth)Acrylate-based graft copolymers via cyanoxyl-mediated free-radical polymerization. Polym. Prepr. 1999, 40, 99–100. [Google Scholar]
- Grande, D.; Guerrero, R.; Gnanou, Y. Cyanoxyl-mediated free-radical polymerization of acrylic acid: Its scope and limitations. J. Polym. Sci. A Polym. Chem. 2005, 43, 519–533. [Google Scholar] [CrossRef]
- David, G.; Boyer, C.; Tonnar, J.; Ameduri, B.; Lacroix-Desmazes, P.; Boutevin, B. Use of iodocompounds in radical polymerization. Chem. Rev. 2006, 106, 3936–3962. [Google Scholar] [CrossRef]
- Yamago, S. Development of organotellurium-mediated and organostibine-mediated living radical polymerization reactions. J. Polym. Sci. A Polym. Chem. 2006, 44, 1–12. [Google Scholar] [CrossRef]
- Goto, A.; Hirai, N.; Nagasawa, K.; Tsujii, Y.; Fukuda, T.; Kaji, H. Phenols and carbon compounds as efficient organic catalysts for reversible chain transfer catalyzed living radical polymerization (RTCP). Macromolecules 2010, 43, 7971–7978. [Google Scholar] [CrossRef]
- Kickelbick, G.; Pintauer, T.; Matyjaszewski, K. Structural comparison of Cu-II complexes in atom transfer radical polymerization. New J. Chem. 2002, 26, 462–468. [Google Scholar] [CrossRef]
- Baskaran, S.; Grande, D.; Sun, X.L.; Yayon, A.; Chaikof, E.L. Glycosaminoglycan-Mimetic biomaterials. 3. Glycopolymers prepared from alkene-derivatized mono- and disaccharide-based glycomonomers. Bioconjug. Chem. 2002, 13, 1309–1313. [Google Scholar] [CrossRef]
- Grande, D.; Baskaran, S.; Baskaran, C.; Gnanou, Y.; Chaikof, E.L. Glycosaminoglycan-mimetic biomaterials. 1. Nonsulfated and sulfated glycopolymers by cyanoxyl-mediated free-radical polymerization. Macromolecules 2000, 33, 1123–1125. [Google Scholar] [CrossRef]
- Grande, D.; Baskaran, S.; Chaikof, E.L. Glycosaminoglycan mimetic biomaterials. 2. Alkene- and acrylate-derivatized glycopolymers via cyanoxyl-mediated free-radical polymerization. Macromolecules 2001, 34, 1640–1646. [Google Scholar] [CrossRef]
- Sun, X.L.; Grande, D.; Baskaran, S.; Hanson, S.R.; Chaikof, E.L. Glycosaminoglycan mimetic biomaterials. 4. Synthesis of sulfated lactose-based glycopolymers that exhibit anticoagulant activity. Biomacromolecules 2002, 3, 1065–1070. [Google Scholar] [CrossRef]
- Guan, R.; Sun, X.L.; Hou, S.; Wu, P.; Chaikof, E.L. A glycopolymer chaperone for fibroblast growth factor-2. Bioconjug. Chem. 2004, 15, 145–151. [Google Scholar]
- Narla, S.N.; Sun, X.-L. Orientated glyco-macroligand formation based on site-specific immobilization of O-cyanate chain-end functionalized glycopolymer. Org. Biomol. Chem. 2011, 9, 845–850. [Google Scholar] [CrossRef]
- Sun, X.-L.; Cui, W.; Haller, C.; Chaikof, E.L. Site-Specific multivalent carbohydrate labeling of quantum dots and magnetic beads. ChemBioChem 2004, 5, 1593–1596. [Google Scholar] [CrossRef]
- Hou, S.; Sun, X.-L.; Dong, C.-M.; Chaikof, E.L. Facile synthesis of chain-end functionalized glycopolymers for site-specific bioconjugation. Bioconjug. Chem. 2004, 15, 954–959. [Google Scholar] [CrossRef]
- Faucher, K.M.; Sun, X.L.; Chaikof, E.L. Fabrication and characterization of glycocalyx-mimetic surfaces. Langmuir 2003, 19, 1664–1670. [Google Scholar] [CrossRef]
- Sun, X.L.; Faucher, K.M.; Houston, M.; Grande, D.; Chaikof, E.L. Design and synthesis of biotin chain-terminated glycopolymers for surface glycoengineering. J. Am. Chem. Soc. 2002, 124, 7258–7259. [Google Scholar] [CrossRef]
- Ting, S.R.S.; Min, E.-H.; Escale, P.; Save, M.; Billon, L.; Stenzel, M.H. Lectin recognizable biomaterials synthesized via nitroxide-mediated polymerization of a methacryloyl galactose monomer. Macromolecules 2009, 42, 9422–9434. [Google Scholar] [CrossRef]
- Gotz, H.; Harth, E.; Schiller, S.M.; Frank, C.W.; Knoll, W.; Hawker, C.J. Synthesis of lipo-glycopolymer amphiphiles by nitroxide-mediated living free-radical polymerization. J. Polym. Sci. A Polym. Chem. 2002, 40, 3379–3391. [Google Scholar] [CrossRef]
- Chen, Y.M.; Wulff, G. Synthesis of poly(styryl sugar)s by TEMPO mediated free radical polymerization. Macromol. Chem. Phys. 2001, 202, 3426–3431. [Google Scholar] [CrossRef]
- Chen, Y.M.; Wulff, G. Amphiphilic block copolymers with pendent sugar as hydrophilic segments and their surface properties. Macromol. Chem. Phys. 2001, 202, 3273–3278. [Google Scholar] [CrossRef]
- Narumi, A.; Matsuda, T.; Kaga, H.; Satoh, T.; Kakuchi, T. Glycoconjugated polymer II. Synthesis of polystyrene-block-poly(4-vinylbenzyl glucoside) and polystyrene-block-poly (4-vinylbenzyl maltohexaoside) via 2,2,6,6-tetramethylpiperidine-1-oxyl-mediated living radical polymerization. Polym. J. 2001, 33, 939–945. [Google Scholar] [CrossRef]
- Narumi, A.; Matsuda, T.; Kaga, H.; Satoh, T.; Kakuchi, T. Synthesis of amphiphilic triblock copolymer of polystyrene and poly(4-vinylbenzyl glucoside) via TEMPO-mediated living radical polymerization. Polymer 2002, 43, 4835–4840. [Google Scholar] [CrossRef]
- Narumi, A.; Otsuka, I.; Matsuda, T.; Miura, Y.; Satoh, T.; Kaneko, N.; Kaga, H.; Kakuchi, T. Glycoconjugated polymer: synthesis and characterization of poly(vinyl saccharide)-block-polystyrene-block-poly(vinyl saccharide) as an amphiphilic ABA triblock copolymer. J. Polym. Sci. A Polym. Chem. 2006, 44, 3978–3985. [Google Scholar] [CrossRef]
- Ohno, K.; Tsujii, Y.; Miyamoto, T.; Fukuda, T.; Goto, M.; Kobayashi, K.; Akaike, T. Synthesis of a well-defined glycopolymer by nitroxide-controlled free radical polymerization. Macromolecules 1998, 31, 1064–1069. [Google Scholar] [CrossRef]
- Ohno, K.; Fukuda, T.; Kitano, H. Free radical polymerization of a sugar residue-carrying styryl monomer with a lipophilic alkoxyamine initiator: Synthesis of a well-defined novel glycolipid. Macromol. Chem. Phys. 1998, 199, 2193–2197. [Google Scholar] [CrossRef]
- Miura, Y.; Koketsu, D.; Kobayashi, K. Synthesis and properties of a well-defined glycopolymer via living radical polymerization. Polym. Adv. Technol. 2007, 18, 647–651. [Google Scholar] [CrossRef]
- Babiuch, K.; Wyrwa, R.; Wagner, K.; Seemann, T.; Hoeppener, S.; Becer, C.R.; Linke, R.; Gottschaldt, M.; Weisser, J.; Schnabelrauch, M.; et al. Functionalized, biocompatible coating for superparamagnetic nanoparticles by controlled polymerization of a thioglycosidic monomer. Biomacromolecules 2011, 12, 681–691. [Google Scholar] [CrossRef]
- Babiuch, K.; Becer, C.R.; Gottschaldt, M.; Delaney, J.T.; Weisser, J.; Beer, B.; Wyrwa, R.; Schnabelrauch, M.; Schubert, U.S. Adhesion of Preosteoblasts and fibroblasts onto Poly(pentafluorostyrene)-based glycopolymeric films and their biocompatibility. Macromol. Biosci. 2011, 11, 535–548. [Google Scholar] [CrossRef]
- Wild, A.; Babiuch, K.; Konig, M.; Winter, A.; Hager, M.D.; Gottschaldt, M.; Prokop, A.; Schubert, U.S. Synthesis of a glycopolymeric PtII carrier and its induction of apoptosis in resistant cancer cells. Chem. Commun. 2012, 48, 6357–6359. [Google Scholar] [CrossRef]
- Becer, C.R.; Babiuch, K.; Pilz, D.; Hornig, S.; Heinze, T.; Gottschaldt, M.; Schubert, U.S. Clicking pentafluorostyrene copolymers: Synthesis, nanoprecipitation, and glycosylation. Macromolecules 2009, 42, 2387–2394. [Google Scholar] [CrossRef]
- Narla, S.N.; Sun, X.-L. Immobilized sialyloligo-macroligand and its protein binding specificity. Biomacromolecules 2012, 13, 1675–1682. [Google Scholar] [CrossRef]
- Mammen, M.; Choi, S.K.; Whitesides, G.M. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. 1998, 37, 2755–2794. [Google Scholar]
- Chaikof, E.L.; Sun, X.L. Multivalent Polymers with Chain-Terminating Binding Groups. WO Patent 2003099835 A1, 2003. [Google Scholar]
- Grande, D.; Baskaran, S.; Chaikof, E.L. Mono- and disaccharide-based glycopolymers by free-radical polymerization processes. Polym. Mater. Sci. Eng. 2001, 84, 141–142. [Google Scholar]
- Yu, K.; Kizhakkedathu, J.N. Synthesis of functional polymer brushes containing carbohydrate residues in the pyranose form and their specific and nonspecific interactions with proteins. Biomacromolecules 2010, 11, 3073–3085. [Google Scholar] [CrossRef]
- Chen, Y.M.; Wulff, G. ABA and star amphiphilic block copolymers composed of polymethacrylate bearing a galactose fragment and poly(ε-caprolactone). Macromol. Rapid Commun. 2002, 23, 59–63. [Google Scholar] [CrossRef]
- Ke, B.-B.; Wan, L.-S.; Zhang, W.-X.; Xu, Z.-K. Controlled synthesis of linear and comb-like glycopolymers for preparation of honeycomb-patterned films. Polymer 2010, 51, 2168–2176. [Google Scholar] [CrossRef]
- Ladmiral, V.; Monaghan, L.; Mantovani, G.; Haddleton, D.M. Functional glycopolymers: New materials for (poly)peptide conjugation. Polymer 2005, 46, 8536–8545. [Google Scholar] [CrossRef]
- Bes, L.; Angot, S.; Limer, A.; Haddleton, D.M. Sugar-coated amphiphilic block copolymer micelles from living radical polymerization: Recognition by immobilized lectins. Macromolecules 2003, 36, 2493–2499. [Google Scholar] [CrossRef]
- Ohno, K.; Tsujii, Y.; Fukuda, T. Synthesis of a well-defined glycopolymer by atom transfer radical polymerization. J. Polym. Sci. A Polym. Chem. 1998, 36, 2473–2481. [Google Scholar] [CrossRef]
- Ejaz, M.; Ohno, K.; Tsujii, Y.; Fukuda, T. Controlled grafting of a well-defined glycopolymer on a solid surface by surface-initiated atom transfer radical polymerization. Macromolecules 2000, 33, 2870–2874. [Google Scholar] [CrossRef]
- Muthukrishnan, S.; Plamper, F.; Mori, H.; Muller, A.H.E. Synthesis and characterization of glycomethacrylate hybrid stars from silsesquioxane nanoparticles. Macromolecules 2005, 38, 10631–10642. [Google Scholar] [CrossRef]
- Muthukrishnan, S.; Jutz, G.; Andre, X.; Mori, H.; Muller, A.H.E. Synthesis of hyperbranched glycopolymers via self-condensing atom transfer radical copolymerization of a sugar-carrying acrylate. Macromolecules 2005, 38, 9–18. [Google Scholar] [CrossRef]
- Muthukrishnan, S.; Mori, H.; Muller, A.H.E. Synthesis and characterization of methacrylate-type hyperbranched glycopolymers via self-condensing atom transfer radical copolymerization. Macromolecules 2005, 38, 3108–3119. [Google Scholar] [CrossRef]
- Muthukrishnan, S.; Zhang, M.; Burkhardt, M.; Drechsler, M.; Mori, H.; Muller, A.H.E. Molecular sugar sticks: Cylindrical glycopolymer brushes. Macromolecules 2005, 38, 7926–7934. [Google Scholar] [CrossRef]
- Gao, C.; Muthukrishnan, S.; Li, W.W.; Yuan, J.Y.; Xu, Y.Y.; Muller, A.H.E. Linear and hyperbranched glycopolymer-functionalized carbon nanotubes: Synthesis, kinetics, and characterization. Macromolecules 2007, 40, 1803–1815. [Google Scholar] [CrossRef]
- Wang, J.; Qian, Y.; Zhang, F.; Zhu, B.; Xu, Y. Synthesis and self-assembly of amphiphilic ABA type triblock copolymer with well-defined glycopolymer segments. Chin. Sci. Bull. 2008, 53, 1343–1351. [Google Scholar] [CrossRef]
- Liang, Y.-Z.; Li, Z.-C.; Chen, G.-Q.; Li, F.-M. Synthesis of well-defined poly[(2-β-d-glucopyranosyloxy)ethyl acrylate] by atom transfer radical polymerization. Polym. Int. 1999, 48, 739–742. [Google Scholar] [CrossRef]
- You, L.-C.; Lu, F.-Z.; Li, Z.-C.; Zhang, W.; Li, F.-M. Glucose-Sensitive aggregates formed by Poly(ethylene oxide)-block-poly(2-glucosyloxyethyl acrylate) with concanavalin A in dilute aqueous medium. Macromolecules 2003, 36, 1–4. [Google Scholar] [CrossRef]
- Dong, C.-M.; Sun, X.-L.; Faucher, K.M.; Apkarian, R.P.; Chaikof, E.L. Synthesis and characterization of glycopolymer-polypeptide triblock copolymers. Biomacromolecules 2004, 5, 224–231. [Google Scholar] [CrossRef]
- Dong, C.-M.; Faucher, K.M.; Chaikof, E.L. Synthesis and properties of biomimetic poIy(L-glutamate)-b-poly(2-acryloyloxyethyllactoside)-b-poly(L-glutamate) triblock copolymers. J. Polym. Sci. A Polym. Chem. 2004, 42, 5754–5765. [Google Scholar] [CrossRef]
- Pfaff, A.; Muller, A.H.E. Hyperbranched glycopolymer grafted microspheres. Macromolecules 2011, 44, 1266–1272. [Google Scholar] [CrossRef]
- Pfaff, A.; Shinde, V.S.; Lu, Y.; Wittemann, A.; Ballauff, M.; Muller, A.H.E. Glycopolymer-grafted polystyrene nanospheres. Macromol. Biosci. 2011, 11, 199–210. [Google Scholar] [CrossRef]
- Narain, R.; Armes, S.P. Direct synthesis and aqueous solution properties of well-defined cyclic sugar methacrylate polymers. Macromolecules 2003, 36, 4675–4678. [Google Scholar] [CrossRef]
- Narain, R.; Armes, S.P. Synthesis of low polydispersity, controlled-structure sugar methacrylate polymers under mild conditions without protecting group chemistry. Chem. Commun. 2002, 2776–2777. [Google Scholar]
- Qiu, S.; Huang, H.; Dai, X.-H.; Zhou, W.; Dong, C.-M. Star-shaped polypeptide/glycopolymer biohybrids: Synthesis, self-assembly, biomolecular recognition, and controlled drug release behavior. J. Polym. Sci. A Polym. Chem. 2009, 47, 2009–2023. [Google Scholar] [CrossRef]
- Narain, R.; Armes, S.P. Synthesis and aqueous solution properties of novel sugar methacrylate-based homopolymers and block copolymers. Biomacromolecules 2003, 4, 1746–1758. [Google Scholar] [CrossRef]
- Mateescu, A.; Ye, J.; Narain, R.; Vamvakaki, M. Synthesis and characterization of novel glycosurfaces by ATRP. Soft Matter 2009, 5, 1621–1629. [Google Scholar] [CrossRef]
- Kitano, H.; Saito, D.; Kamada, T.; Gemmei-Ide, M. Binding of β-amyloid to sulfated sugar residues in a polymer brush. Colloids Surf. B 2012, 93, 219–225. [Google Scholar]
- Fleet, R.; van den Dungen, E.T.A.; Klumperman, B. Novel glycopolymer brushes via ATRP: 1. synthesis and characterization. Macromol. Chem. Phys. 2011, 212, 2191–2208. [Google Scholar] [CrossRef]
- Narain, R. Tailor-made protein-glycopolymer bioconjugates. React. Funct. Polym. 2006, 66, 1589–1595. [Google Scholar] [CrossRef]
- Yang, Q.; Ulbricht, M. Cylindrical membrane pores with well-defined grafted linear and comblike glycopolymer layers for lectin binding. Macromolecules 2011, 44, 1303–1310. [Google Scholar] [CrossRef]
- O’Connell, M.A.; de Cuendias, A.; Gayet, F.; Shirley, I.M.; Mackenzie, S.R.; Haddleton, D.M.; Unwin, P.R. Evanescent Wave Cavity Ring-Down Spectroscopy (EW-CRDS) as a probe of macromolecule adsorption kinetics at functionalized interfaces. Langmuir 2012, 28, 6902–6910. [Google Scholar] [CrossRef]
- Xu, L.Q.; Huang, C.; Wang, R.; Neoh, K.-G.; Kang, E.-T.; Fu, G.D. Synthesis and characterization of fluorescent perylene bisimide-containing glycopolymers for Escherichia coli conjugation and cell imaging. Polymer 2011, 52, 5764–5771. [Google Scholar] [CrossRef]
- Kitano, H.; Takahashi, Y.; Mizukami, K.; Matsuura, K. Kinetic study on the binding of lectin to mannose residues in a polymer brush. Colloids Surf. B 2009, 70, 91–97. [Google Scholar]
- Geng, J.; Mantovani, G.; Tao, L.; Nicolas, J.; Chen, G.; Wallis, R.; Mitchell, D.A.; Johnson, B.R.G.; Evans, S.D.; Haddleton, D.M. Site-Directed conjugation of “clicked” glycopolymers to form glycoprotein mimics: Binding to mammalian lectin and induction of immunological function. J. Am. Chem. Soc. 2007, 129, 15156–15163. [Google Scholar] [CrossRef]
- Vazquez-Dorbatt, V.; Maynard, H.D. Biotinylated glycopolymers synthesized by atom transfer radical polymerization. Biomacromolecules 2006, 7, 2297–2302. [Google Scholar] [CrossRef]
- Vazquez-Dorbatt, V.; Tolstyka, Z.P.; Chang, C.-W.; Maynard, H.D. Synthesis of a pyridyl disulfide end-functionalized glycopolymer for conjugation to biomolecules and patterning on gold surfaces. Biomacromolecules 2009, 10, 2207–2212. [Google Scholar] [CrossRef]
- Leon, O.; Bordege, V.; Munoz-Bonilla, A.; Sanchez-Chaves, M.; Fernandez-Garcia, M. Well-controlled amphiphilic block glycopolymers and their molecular recognition with lectins. J. Polym. Sci. A Polym. Chem. 2010, 48, 3623–3631. [Google Scholar] [CrossRef]
- Leon, O.; Munoz-Bonilla, A.; Bordege, V.; Sanchez-Chaves, M.; Fernandez-Garcia, M. Amphiphilic block glycopolymers via atom transfer radical polymerization: Synthesis, self-assembly and biomolecular recognition. J. Polym. Sci. A Polym. Chem. 2011, 49, 2627–2635. [Google Scholar] [CrossRef]
- Munoz-Bonilla, A.; Heuts, J.P.A.; Fernandez-Garcia, M. Glycoparticles and bioactive films prepared by emulsion polymerization using a well-defined block glycopolymer stabilizer. Soft Matter 2011, 7, 2493–2499. [Google Scholar] [CrossRef]
- De, L.A.S.; Munoz-Bonilla, A.; Fernandez-Garcia, M.; Rodriguez-Hernandez, J. Breath figures method to control the topography and the functionality of polymeric surfaces in porous films and microspheres. J. Polym. Sci. A Polym. Chem. 2012, 50, 851–859. [Google Scholar] [CrossRef]
- Menon, S.; Das, S. A photoresponsive fluorescent glycopolymer. Polym. Chem. 2012, 3, 2619–2624. [Google Scholar] [CrossRef]
- Ladmiral, V.; Mantovani, G.; Clarkson, G.J.; Cauet, S.; Irwin, J.L.; Haddleton, D.M. Synthesis of neoglycopolymers by a combination of “click chemistry” and living radical polymerization. J. Am. Chem. Soc. 2006, 128, 4823–4830. [Google Scholar]
- Richards, S.-J.; Jones, M.W.; Hunaban, M.; Haddleton, D.M.; Gibson, M.I. Probing bacterial-toxin inhibition with synthetic glycopolymers prepared by tandem post-polymerization modification: Role of linker length and carbohydrate density. Angew. Chem. Int. Ed. 2012, 51, 7812–7816. [Google Scholar]
- Gou, Y.; Slavin, S.; Geng, J.; Voorhaar, L.; Haddleton, D.M.; Becer, C.R. Controlled alternate layer-by-layer assembly of lectins and glycopolymers using QCM-D. ACS Macro Lett. 2011, 1, 180–183. [Google Scholar]
- Houga, C.; Giermanska, J.; Lecommandoux, S.; Borsali, R.; Taton, D.; Gnanou, Y.; Le, M.J.-F. Micelles and polymersomes obtained by self-assembly of dextran and polystyrene based block copolymers. Biomacromolecules 2009, 10, 32–40. [Google Scholar] [CrossRef]
- Yu, K.; Lai, B.F.L.; Kizhakkedathu, J.N. Carbohydrate structure dependent hemocompatibility of biomimetic functional polymer brushes on surfaces. Adv. Healthc. Mater. 2012, 1, 199–213. [Google Scholar] [CrossRef]
- Fleet, R.; van den Dungen, E.T.A.; Klumperman, B. Novel glycopolymer brushes via ATRP: 2. thermal and mechanical properties. Macromol. Chem. Phys. 2011, 212, 2209–2216. [Google Scholar] [CrossRef]
- Fleet, R.; van den Dungen, E.T.A.; Klumperman, B. Synthesis of novel glycopolymer brushes via a combination of RAFT-mediated polymerization and ATRP. S. Afr. J. Sci. 2011, 107, 101–111. [Google Scholar]
- Yuan, J.; Meng, J.-Q.; Kang, Y.-L.; Du, Q.-Y.; Zhang, Y.-F. Facile surface glycosylation of PVDF microporous membrane via direct surface-initiated AGET ATRP and improvement of antifouling property and biocompatibility. Appl. Surf. Sci. 2012, 258, 2856–2863. [Google Scholar] [CrossRef]
- Meng, J.; Yuan, J.; Kang, Y.; Zhang, Y.; Du, Q. Surface glycosylation of polysulfone membrane towards a novel complexing membrane for boron removal. J. Colloid Interface Sci. 2012, 368, 197–207. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the Road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Kohri, M.; Sato, M.; Abo, F.; Inada, T.; Kasuya, M.; Taniguchi, T.; Nakahira, T. Preparation and lectin binding specificity of polystyrene particles grafted with glycopolymers bearing S-linked carbohydrates. Eur. Polym. J. 2011, 47, 2351–2360. [Google Scholar] [CrossRef]
- Becer, C.R.; Gibson, M.I.; Geng, J.; Ilyas, R.; Wallis, R.; Mitchell, D.A.; Haddleton, D.M. High-Affinity glycopolymer binding to human DC-SIGN and disruption of DC-SIGN interactions with HIV envelope glycoprotein. J. Am. Chem. Soc. 2010, 132, 15130–15132. [Google Scholar]
- Gou, Y.; Richards, S.-J.; Haddleton, D.M.; Gibson, M.I. Investigation of glycopolymer-lectin interactions using QCM-d: Comparison of surface binding with inhibitory activity. Polym. Chem. 2012, 3, 1634–1640. [Google Scholar] [CrossRef]
- Geng, J.; Biedermann, F.; Zayed, J.M.; Tian, F.; Scherman, O.A. Supramolecular glycopolymers in water: A reversible route toward multivalent carbohydrate-lectin conjugates using Cucurbit[8]uril. Macromolecules 2011, 44, 4276–4281. [Google Scholar] [CrossRef]
- Muñoz-Bonilla, A.; León, O.; Cerrada, M.L.; Rodríguez-Hernández, J.; Sánchez-Chaves, M.; Fernández-García, M. Glycopolymers obtained by chemical modification of well-defined block copolymers. J. Polym. Sci. A Polym. Chem. 2012, 50, 2565–2577. [Google Scholar] [CrossRef]
- Gody, G.; Boullanger, P.; Ladaviere, C.; Charreyre, M.T.; Delair, T. Biotin alpha-end-functionalized gradient glycopolymers synthesized by RAFT copolymerization. Macromol. Rapid Commun. 2008, 29, 511–519. [Google Scholar] [CrossRef]
- Su, L.; Zhao, Y.; Chen, G.; Jiang, M. Polymeric vesicles mimicking glycocalyx (PV-Gx) for studying carbohydrate-protein interactions in solution. Polym. Chem. 2012, 3, 1560–1566. [Google Scholar] [CrossRef]
- Wei, Z.; Hao, X.; Gan, Z.; Hughes, T.C. One-pot synthesis of hyperbranched glycopolymers by RAFT polymerization. J. Polym. Sci. A Polym. Chem. 2012, 50, 2378–2388. [Google Scholar] [CrossRef]
- Ozyurek, Z.; Komber, H.; Gramm, S.; Schmaljohann, D.; Muller, A.H.E.; Voit, B. Thermoresponsive glycopolymers via controlled radical polymerization. Macromol. Chem. Phys. 2007, 208, 1035–1049. [Google Scholar] [CrossRef]
- Jiang, X.; Housni, A.; Gody, G.; Boullanger, P.; Charreyre, M.-T.; Delair, T.; Narain, R. Synthesis of Biotinylated α-d-Mannoside or N-Acetyl β-d-Glucosaminoside decorated gold nanoparticles: Study of their biomolecular recognition with ConA and WGA lectins. Bioconjug. Chem. 2010, 21, 521–530. [Google Scholar]
- Belardi, B.; O’Donoghue Geoff, P.; Smith Adam, W.; Groves Jay, T.; Bertozzi Carolyn, R. Investigating cell surface galectin-mediated cross-linking on glycoengineered cells. J. Am. Chem. Soc. 2012, 134, 9594–9552. [Google Scholar]
- Toyoshima, M.; Oura, T.; Fukuda, T.; Matsumoto, E.; Miura, Y. Biological specific recognition of glycopolymer- modified interfaces by RAFT living radical polymerization. Polym. J. 2009, 42, 172–178. [Google Scholar]
- Deng, Z.; Ahmed, M.; Narain, R. Novel well-defined glycopolymers synthesized via the reversible addition fragmentation chain transfer process in aqueous media. J. Polym. Sci. A Polym. Chem. 2009, 47, 614–627. [Google Scholar] [CrossRef]
- Ahmed, M.; Narain, R. The effect of polymer architecture, composition, and molecular weight on the properties of glycopolymer-based non-viral gene delivery system. Biomaterials 2011, 32, 5279–5290. [Google Scholar] [CrossRef]
- Ahmed, M.; Lai, B.F.L.; Kizhakkedathu, J.N.; Narain, R. Hyperbranched glycopolymers for blood biocompatibility. Bioconjug. Chem. 2012, 23, 1050–1058. [Google Scholar] [CrossRef]
- Ahmed, M.; Narain, R. The effect of molecular weight, compositions and lectin type on the properties of hyperbranched glycopolymers as non-viral gene delivery systems. Biomaterials 2012, 33, 3990–4001. [Google Scholar] [CrossRef]
- Bernard, J.; Hao, X.J.; Davis, T.P.; Barner-Kowollik, C.; Stenzel, M.H. Synthesis of various glycopolymer architectures via RAFT polymerization: From block copolymers to stars. Biomacromolecules 2006, 7, 232–238. [Google Scholar] [CrossRef]
- Ting, S.R.S.; Min, E.H.; Zetterlund, P.B.; Stenzel, M.H. Controlled/Living ab initio emulsion polymerization via a glucose RAFTstab: degradable cross-linked glyco-particles for concanavalin A/FimH conjugations to cluster E. coli bacteria. Macromolecules 2010, 43, 5211–5221. [Google Scholar] [CrossRef]
- Smith, A.E.; Sizovs, A.; Grandinetti, G.; Xue, L.; Reineke, T.M. Diblock glycopolymers promote colloidal stability of polyplexes and effective pDNA and siRNA delivery under physiological salt and serum conditions. Biomacromolecules 2011, 12, 3015–3022. [Google Scholar] [CrossRef]
- Abdelkader, O.; Moebs-Sanchez, S.; Queneau, Y.; Bernard, J.; Fleury, E. Generation of well-defined clickable glycopolymers from aqueous RAFT polymerization of isomaltulose-derived acrylamides. J. Polym. Sci. A Polym. Chem. 2011, 49, 1309–1318. [Google Scholar] [CrossRef]
- Albertin, L.; Wolnik, A.; Ghadban, A.; Dubreuil, F. Aqueous RAFT Polymerization of N-Acryloylmorpholine, synthesis of an ABA triblock glycopolymer and study of its self-association behavior. Macromol. Chem. Phys. 2012, 213, 1768–1782. [Google Scholar] [CrossRef]
- Miura, Y.; Mizuno, H. Interaction analyses of amyloid β peptide (1-40) with glycosaminoglycan model polymers. Bull. Chem. Soc. Jpn. 2010, 83, 1004–1009. [Google Scholar] [CrossRef]
- Toyoshima, M.; Miura, Y. Preparation of glycopolymer-substituted gold nanoparticles and their molecular recognition. J. Polym. Sci. A Polym. Chem. 2009, 47, 1412–1421. [Google Scholar] [CrossRef]
- Ishii, J.; Toyoshima, M.; Chikae, M.; Takamura, Y.; Miura, Y. Preparation of glycopolymer-modified gold nanoparticles and a new approach for a lateral flow assay. Bull. Chem. Soc. Jpn. 2011, 84, 466–470. [Google Scholar] [CrossRef]
- Al-Bagoury, M.; Buchholz, K.; Yaacoub, E.J. Synthesis of well-designed polymers carrying saccharide moieties via RAFT miniemulsion polymerization. Polym. Adv. Technol. 2007, 18, 313–322. [Google Scholar] [CrossRef]
- Lowe, A.B.; Wang, R. Synthesis of controlled-structure AB diblock copolymers of 3-O-methacryloyl-1,2:3,4-di-O-isopropylidene-d-galactopyranose and 2-(dimethylamino)ethyl methacrylate. Polymer 2007, 48, 2221–2230. [Google Scholar] [CrossRef]
- Ting, S.R.S.; Gregory, A.M.; Stenzel, M.H. Polygalactose containing nanocages: The RAFT process for the synthesis of hollow sugar balls. Biomacromolecules 2009, 10, 342–352. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, J.; Lv, W.; Luo, Y.; Wang, X. Well-defined pH-sensitive block glycopolymers via reversible addition-fragmentation chain transfer radical polymerization: Synthesis, characterization, and recognition with lectin. J. Polym. Sci. A Polym. Chem. 2010, 48, 3350–3361. [Google Scholar] [CrossRef]
- Pfaff, A.; Schallon, A.; Ruhland, T.M.; Majewski, A.P.; Schmalz, H.; Freitag, R.; Muller, A.H.E. Magnetic and fluorescent glycopolymer hybrid nanoparticles for intranuclear optical imaging. Biomacromolecules 2011, 12, 3805–3811. [Google Scholar] [CrossRef]
- Shi, H.; Liu, L.; Wang, X.; Li, J. Glycopolymer-peptide bioconjugates with antioxidant activity via RAFT polymerization. Polym. Chem. 2012, 3, 1182–1188. [Google Scholar] [CrossRef]
- Luo, Y.; Liu, L.; Wang, X.; Shi, H.; Lv, W.; Li, J. Sugar-installed thermoresponsive micellar aggregates self-assembled from “coil-comb-coil” triblock glycopolymers: Preparation and recognition with Concanavalin A. Soft Matter 2012, 8, 1634–1642. [Google Scholar] [CrossRef]
- Glassner, M.; Delaittre, G.; Kaupp, M.; Blinco, J.P.; Barner-Kowollik, C. Ultra)Fast catalyst-free macromolecular conjugation in aqueous environment at ambient temperature. J. Am. Chem. Soc. 2012, 134, 7274–7277. [Google Scholar]
- Kaupp, M.; Vogt, A.P.; Natterodt, J.C.; Trouillet, V.; Gruendling, T.; Hofe, T.; Barner, L.; Barner-Kowollik, C. Modular design of glyco-microspheres via mild pericyclic reactions and their quantitative analysis. Polym. Chem. 2012, 3, 2605–2614. [Google Scholar] [CrossRef]
- Dan, K.; Ghosh, S. pH-Responsive Aggregation of amphiphilic glyco-homopolymer. Macromol. Rapid Commun. 2012, 33, 127–132. [Google Scholar] [CrossRef]
- Guo, T.Y.; Liu, P.; Zhu, J.W.; Song, M.D.; Zhang, B.H. Well-Defined lactose-containing polymer grafted onto silica particles. Biomacromolecules 2006, 7, 1196–1202. [Google Scholar] [CrossRef]
- Zhou, D.; Li, C.; Hu, Y.; Zhou, H.; Chen, J.; Zhang, Z.; Guo, T. Glycopolymer modification on physicochemical and biological properties of poly(l-lysine) for gene delivery. Int. J. Biol. Macromol. 2012, 50, 965–973. [Google Scholar] [CrossRef]
- Spain, S.G.; Albertin, L.; Cameron, N.R. Facile in situ preparation of biologically active multivalent glyconanoparticles. Chem. Commun. 2006, 4198–4200. [Google Scholar]
- Housni, A.; Cai, H.J.; Liu, S.Y.; Pun, S.H.; Narain, R. Facile preparation of glyconanoparticles and their bioconjugation to streptavidin. Langmuir 2007, 23, 5056–5061. [Google Scholar] [CrossRef]
- Lowe, A.B.; Sumerlin, B.S.; McCormick, C.L. The direct polymerization of 2-methacryloxyethyl glucoside via aqueous reversible addition-fragmentation chain transfer (RAFT) polymerization. Polymer 2003, 44, 6761–6765. [Google Scholar] [CrossRef]
- Pearson, S.; Scarano, W.; Stenzel, M.H. Micelles based on gold-glycopolymer complexes as new chemotherapy drug delivery agents. Chem. Commun. 2012, 48, 4695–4697. [Google Scholar] [CrossRef]
- Albertin, L.; Stenzel, M.H.; Barner-Kowollik, C.; Foster, L.J.R.; Davis, T.P. Well-defined diblock glycopolymers from RAFT polymerization in homogeneous aqueous medium. Macromolecules 2005, 38, 9075–9084. [Google Scholar] [CrossRef]
- Albertin, L.; Stenzel, M.H.; Barner-Kowollik, C.; Davis, T.P. Effect of an added base on (4-cyanopentanoic acid)-4-dithiobenzoate mediated RAFT polymerization in water. Polymer 2006, 47, 1011–1019. [Google Scholar] [CrossRef]
- Albertin, L.; Stenzel, M.; Barner-Kowollik, C.; Foster, L.J.R.; Davis, T.P. Well-defined glycopolymers from RAFT polymerization: Poly(methyl 6-O-methacryloyl-a-d-glucoside) and its block copolymer with 2-hydroxyethyl methacrylate. Macromolecules 2004, 37, 7530–7537. [Google Scholar] [CrossRef]
- Pfaff, A.; Barner, L.; Muller, A.H.E.; Granville, A.M. Surface modification of polymeric microspheres using glycopolymers for biorecognition. Eur. Polym. J. 2011, 47, 805–815. [Google Scholar] [CrossRef]
- Granville, A.M.; Quemener, D.; Davis, T.P.; Barner-Kowollik, C.; Stenzel, M.H. Chemo-enzymatic synthesis and RAFT polymerization of 6-O-methacryloyl mannose: a suitable glycopolymer for binding to the tetrameric lectin concanavalin A? Macromol. Symp. 2007, 255, 81–89. [Google Scholar] [CrossRef]
- Albertin, L.; Kohlert, C.; Stenzel, M.; Foster, L.J.R.; Davis, T.P. Chemoenzymatic synthesis of narrow-polydispersity glycopolymers: Poly(6-O-vinyladipoyl-d-glucopyranose). Biomacromolecules 2004, 5, 255–260. [Google Scholar] [CrossRef]
- Bernard, J.; Favier, A.; Zhang, L.; Nilasaroya, A.; Davis, T.P.; Barner-Kowollik, C.; Stenzel, M.H. Poly(vinyl ester) star polymers via xanthate-mediated living radical polymerization: From poly(vinyl alcohol) to glycopolymer stars. Macromolecules 2005, 38, 5475–5484. [Google Scholar] [CrossRef]
- Hetzer, M.; Chen, G.; Barner-Kowollik, C.; Stenzel, M.H. Neoglycopolymers based on 4-vinyl-1,2,3-triazole monomers prepared by click chemistry. Macromol. Biosci. 2010, 10, 119–126. [Google Scholar] [CrossRef]
- Xiao, N.-Y.; Li, A.-L.; Liang, H.; Lu, J. A Well-defined novel aldehyde-functionalized glycopolymer: synthesis, micelle formation, and its protein immobilization. Macromolecules 2008, 41, 2374–2380. [Google Scholar] [CrossRef]
- Xiao, N.; Liang, H.; Lu, J. Degradable and biocompatible aldehyde-functionalized glycopolymer conjugated with doxorubicin via acid-labile Schiff base linkage for pH-triggered drug release. Soft Matter 2011, 7, 10834–10840. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, X.; Cheng, Z.; Zhang, Z.; Zhu, J. Preparation, characterization, and chiral recognition of optically active polymers containing pendent chiral units via reversible addition-fragmentation chain transfer polymerization. J. Polym. Sci. A Polym. Chem. 2007, 45, 3788–3797. [Google Scholar] [CrossRef]
- Mancini, R.J.; Lee, J.; Maynard, H.D. Trehalose glycopolymers for stabilization of protein conjugates to environmental stressors. J. Am. Chem. Soc. 2012, 134, 8474–8479. [Google Scholar] [CrossRef]
- Boyer, C.; Bousquet, A.; Rondolo, J.; Whittaker, M.R.; Stenzel, M.H.; Davis, T.P. Glycopolymer decoration of gold nanoparticles using a lbl approach. Macromolecules 2010, 43, 3775–3784. [Google Scholar] [CrossRef]
- Boyer, C.; Davis, T.P. One-pot synthesis and biofunctionalization of glycopolymers via RAFT polymerization and thiol-ene reactions. Chem. Commun. 2009, 6029–6031. [Google Scholar] [CrossRef]
- Boyer, C.; Whittaker, M.; Davis, T.P. Synthesis and postfunctionalization of well-defined star polymers via “double” click chemistry. J. Polym. Sci. A Polym. Chem. 2011, 49, 5245–5256. [Google Scholar] [CrossRef]
- Semsarilar, M.; Ladmiral, V.; Perrier, S. Highly branched and hyperbranched glycopolymers via reversible addition-fragmentation chain transfer polymerization and click chemistry. Macromolecules 2010, 43, 1438–1443. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, G.; Stenzel, M.H. Synthesis and lectin recognition of glyco star polymers prepared by “Clicking” thiocarbohydrates onto a reactive scaffold. Macromolecules 2010, 43, 8109–8114. [Google Scholar] [CrossRef]
- Kumar, J.; Bousquet, A.; Stenzel, M.H. Thiol-alkyne chemistry for the preparation of micelles with glycopolymer corona: Dendritic surfaces versus linear glycopolymer in their ability to bind to lectins. Macromol. Rapid Commun. 2011, 32, 1620–1626. [Google Scholar] [CrossRef]
- Alidedeoglu, A.H.; York, A.W.; Rosado, D.A.; McCormick, C.L.; Morgan, S.E. Bioconjugation of -glucuronic acid sodium salt to well-defined primary amine-containing homopolymers and block copolymers. J. Polym. Sci. A Polym. Chem. 2010, 48, 3052–3061. [Google Scholar] [CrossRef]
- Godula, K.; Bertozzi, C.R. Density variant glycan microarray for evaluating cross-linking of mucin-like glycoconjugates by lectins. J. Am. Chem. Soc. 2012, 134, 15732–15742. [Google Scholar] [CrossRef]
- Narain, R.; Housni, A.; Gody, G.; Boullanger, P.; Charreyre, M.-T.; Delair, T. Preparation of biotinylated glyconanoparticles via a photochemical process and study of their bioconjugation to streptavidin. Langmuir 2007, 23, 12835–12841. [Google Scholar] [CrossRef]
- Saricilar, S.; Knott, R.; Barner-Kowollik, C.; Davis, T.P.; Heuts, J.P.A. Reversible addition fragmentation chain transfer polymerization of 3-[tris(trimethylsilyloxy) silyl] propyl methacrylate. Polymer 2003, 44, 5169–5176. [Google Scholar] [CrossRef]
- Barner-Kowollik, C.; Buback, M.; Charleux, B.; Coote, M.L.; Drache, M.; Fukuda, T.; Goto, A.; Klumperman, B.; Lowe, A.B.; McLeary, J.B.; et al. Mechanism and kinetics of dithiobenzoate-mediated RAFT polymerization. I. The current situation. J. Polym. Sci. A Polym. Chem. 2006, 44, 5809–5831. [Google Scholar] [CrossRef]
- Konkolewicz, D.; Hawkett, B.S.; Gray-Weale, A.; Perrier, S. RAFT polymerization kinetics: Combination of Apparently conflicting models. Macromolecules 2008, 41, 6400–6412. [Google Scholar] [CrossRef]
- Konkolewicz, D.; Hawkett, B.S.; Gray-Weale, A.; Perrier, S. RAFT polymerization kinetics: How long are the cross-terminating oligomers? J. Polym. Sci. A Polym. Chem. 2009, 47, 3455–3466. [Google Scholar] [CrossRef]
- Ting, S.R.S.; Davis, T.P.; Zetterlund, P.B. Retardation in RAFT polymerization: Does cross-termination occur with short radicals only? Macromolecules 2011, 44, 4187–4193. [Google Scholar] [CrossRef]
- Yang, H.; Guo, T.-Y.; Zhou, D. Surface hydrophilic modification with well-defined glycopolymer for protein imprinting matrix. Int. J. Biol. Macromol. 2011, 48, 432–438. [Google Scholar] [CrossRef]
- Stenzel, M.H.; Zhang, L.; Huck, W.T.S. Temperature-responsive glycopolymer brushes synthesized via RAFT polymerization using the Z-group approach. Macromol. Rapid Commun. 2006, 27, 1121–1126. [Google Scholar] [CrossRef]
- Jiang, X.; Ahmed, M.; Deng, Z.; Narain, R. Biotinylated glyco-functionalized quantum dots: Synthesis, characterization, and cytotoxicity studies. Bioconjug. Chem. 2009, 20, 994–1001. [Google Scholar]
- Ishii, J.; Chikae, M.; Toyoshima, M.; Ukita, Y.; Miura, Y.; Takamura, Y. Electrochemical assay for saccharide-protein interactions using glycopolymer-modified gold nanoparticles. Electrochem. Commun. 2011, 13, 830–833. [Google Scholar]
- Miura, Y.; Yasuda, K.; Yamamoto, K.; Koike, M.; Nishida, Y.; Kobayashi, K. Inhibition of alzheimer amyloid aggregation with sulfated glycopolymers. Biomacromolecules 2007, 8, 2129–2134. [Google Scholar] [CrossRef]
- Min, E.H.; Ting, S.R.S.; Billon, L.; Stenzel, M.H. Thermo-responsive glycopolymer chains grafted onto honeycomb structured porous films via RAFT polymerization as a thermo-dependentswitcher for lectin Concanavalin a conjugation. J. Polym. Sci. A Polym. Chem. 2010, 48, 3440–3455. [Google Scholar] [CrossRef]
- Buckwalter, D.J.; Sizovs, A.; Ingle, N.P.; Reineke, T.M. MAG versus PEG: Incorporating a Poly(MAG) layer to promote colloidal stability of nucleic acid/“click cluster” complexes. ACS Macro Lett. 2012, 1, 609–613. [Google Scholar] [CrossRef]
- Albertin, L.; Cameron, N.R. RAFT Polymerization of Methyl 6-O-Methacryloyl-a-d-glucoside in homogeneous aqueous medium. A detailed kinetic study at the low molecular weight limit of the process. Macromolecules 2007, 40, 6082–6093. [Google Scholar] [CrossRef]
- Cheng, C.; Zhang, X.; Wang, Y.; Li, C. Synthesis of glucose-sensitive block glycopolymers based on phenylboronic acid via raft polymerization. J. Control. Release 2011, 152, e267–e269. [Google Scholar] [CrossRef]
- Chiellini, E.; Corti, A.; D’Antone, S.; Solaro, R. Biodegradation of poly (vinyl alcohol) based materials. Prog. Polym. Sci. 2003, 28, 963–1014. [Google Scholar] [CrossRef]
- DeMerlis, C.C.; Schoneker, D.R. Review of the oral toxicity of polyvinyl alcohol (PVA). Food Chem. Toxicol. 2003, 41, 319–326. [Google Scholar] [CrossRef]
- Nikolaev, A.F.; Mosyagina, L.P. Poly(vinyl alcohol) and vinyl alcohol copolymers in medicine. Int. Polym. Sci. Technol. 2000, 27, 47–58. [Google Scholar]
- Yamaoka, T.; Tabata, Y.; Ikada, Y. Comparison of body distribution of poly(vinyl alcohol) with other water-soluble polymers after intravenous administration. J. Pharm. Pharmacol. 1995, 47, 479–486. [Google Scholar]
- Frush, H.L.; Isbell, H.S. Mutorotation, hydrolysis, and structure of d-galactosamin. J. Res. Natl. Bur. Stand. 1951, 47, 239–247. [Google Scholar] [CrossRef]
- Brusseau, S.; D’Agosto, F.; Magnet, S.; Couvreur, L.; Chamignon, C.; Charleux, B. Nitroxide-Mediated copolymerization of methacrylic acid and sodium 4-styrenesulfonate in water solution and one-pot synthesis of amphiphilic block copolymer nanoparticles. Macromolecules 2011, 44, 5590–5598. [Google Scholar] [CrossRef]
- Phan, T.N.T.; Bertin, D. Synthesis of water-soluble homopolymers and block copolymers in homogeneous aqueous solution via nitroxide-mediated polymerization. Macromolecules 2008, 41, 1886–1895. [Google Scholar] [CrossRef]
- Rigolini, J.; Grassl, B.; Billon, L.; Reynaud, S.; Donard, O.F.X. Microwave-assisted nitroxide-mediated radical polymerization of acrylamide in aqueous solution. J. Polym. Sci. A Polym. Chem. 2009, 47, 6919–6931. [Google Scholar] [CrossRef]
- Rigolini, J.; Grassl, B.; Reynaud, S.; Billon, L. Microwave-assisted nitroxide-mediated polymerization for water-soluble homopolymers and block copolymers synthesis in homogeneous aqueous solution. J. Polym. Sci. A Polym. Chem. 2010, 48, 5775–5782. [Google Scholar] [CrossRef]
- Chenal, M.; Mura, S.; Marchal, C.; Gigmes, D.; Charleux, B.; Fattal, E.; Couvreur, P.; Nicolas, J. Facile synthesis of innocuous comb-shaped polymethacrylates with PEG side chains by nitroxide-mediated radical polymerization in hydroalcoholic solutions. Macromolecules 2010, 43, 9291–9303. [Google Scholar] [CrossRef]
- Ashford, E.J.; Naldi, V.; O’Dell, R.; Billingham, N.C.; Armes, S.P. First example of the atom transfer radical polymerisation of an acidic monomer: Direct synthesis of methacrylic acid copolymers in aqueous media. Chem. Commun. 1999, 14, 1285–1286. [Google Scholar]
- Tsarevsky, N.V.; Matyjaszewski, K. “Green” atom transfer radical polymerization: From process design to preparation of well-defined environmentally friendly polymeric materials. Chem. Rev. 2007, 107, 2270–2299. [Google Scholar] [CrossRef]
- Jakubowski, W.; Matyjaszewski, K. Activators regenerated by electron transfer for atom-transfer radical polymerization of (meth)acrylates and related block copolymers. Angew. Chem. Int. Ed. 2006, 45, 4482–4486. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Jakubowski, W.; Min, K.; Tang, W.; Huang, J.; Braunecker, W.A.; Tsarevsky, N.V. Diminishing catalyst concentration in atom transfer radical polymerization with reducing agents. Proc. Natl. Acad. Sci. USA 2006, 103, 15309–15314. [Google Scholar]
- Rosen, B.M.; Percec, V. Single-Electron transfer and single-electron transfer degenerative chain transfer living radical polymerization. Chem. Rev. 2009, 109, 5069–5119. [Google Scholar] [CrossRef]
- Percec, V.; Guliashvili, T.; Ladislaw, J.S.; Wistrand, A.; Stjerndahl, A.; Sienkowska, M.J.; Monteiro, M.J.; Sahoo, S. Ultrafast synthesis of ultrahigh molar mass polymers by metal-catalyzed living radical polymerization of acrylates, methacrylates, and vinyl chloride mediated by SET at 25 °C. J. Am. Chem. Soc. 2006, 128, 14156–14165. [Google Scholar] [CrossRef]
- Nicol, E.; Derouineau, T.; Puaud, F.; Zaitsev, A. Synthesis of double hydrophilic poly(ethylene oxide)-b-poly(2-hydroxyethyl acrylate) by single-electron transfer-living radical polymerization. J. Polym. Sci. A Polym. Chem. 2012, 50, 3885–3894. [Google Scholar] [CrossRef]
- Voepel, J.; Edlund, U.; Albertsson, A.C. A versatile single-electron-transfer mediated living radical polymerization route to galactoglucomannan graft-copolymers with tunable hydrophilicity. J. Polym. Sci. A Polym. Chem. 2011, 49, 2366–2372. [Google Scholar] [CrossRef]
- Zetterlund, P.B.; Kagawa, Y.; Okubo, M. Controlled/Living radical polymerization in dispersed systems. Chem. Rev. 2008, 108, 3747–3794. [Google Scholar] [CrossRef]
- Vyas, S.P.; Singh, A.; Sihorkar, V. Ligand-receptor-mediated drug delivery: An emerging paradigm in cellular drug targeting. Crit. Rev. Ther. Drug Carrier Syst. 2001, 18, 1–76. [Google Scholar] [CrossRef]
- Jones, M.N. Carbohydrate-mediated liposomal targeting and drug delivery. Adv. Drug Deliv. Rev. 1994, 13, 215–249. [Google Scholar] [CrossRef]
- Muñoz-Bonilla, A.; Marcelo, G.; Casado, C.; Teran, F.J.; Fernández-García, M. Preparation of glycopolymer-coated magnetite nanoparticles for hyperthermia treatment. J. Polym. Sci. A Polym. Chem. 2012, 50, 5087–5096. [Google Scholar] [CrossRef]
- Duncan, R. Development of HPMA copolymer-anticancer conjugates: Clinical experience and lessons learnt. Adv. Drug Deliv. Rev. 2009, 61, 1131–1148. [Google Scholar] [CrossRef]
- Duncan, R. The dawning era of polymer therapeutics. Nat. Rev. Drug Discov. 2003, 2, 347–360. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ghadban, A.; Albertin, L. Synthesis of Glycopolymer Architectures by Reversible-Deactivation Radical Polymerization. Polymers 2013, 5, 431-526. https://doi.org/10.3390/polym5020431
Ghadban A, Albertin L. Synthesis of Glycopolymer Architectures by Reversible-Deactivation Radical Polymerization. Polymers. 2013; 5(2):431-526. https://doi.org/10.3390/polym5020431
Chicago/Turabian StyleGhadban, Ali, and Luca Albertin. 2013. "Synthesis of Glycopolymer Architectures by Reversible-Deactivation Radical Polymerization" Polymers 5, no. 2: 431-526. https://doi.org/10.3390/polym5020431