Charge-Transfer Complexes Studied by Dynamic Force Spectroscopy

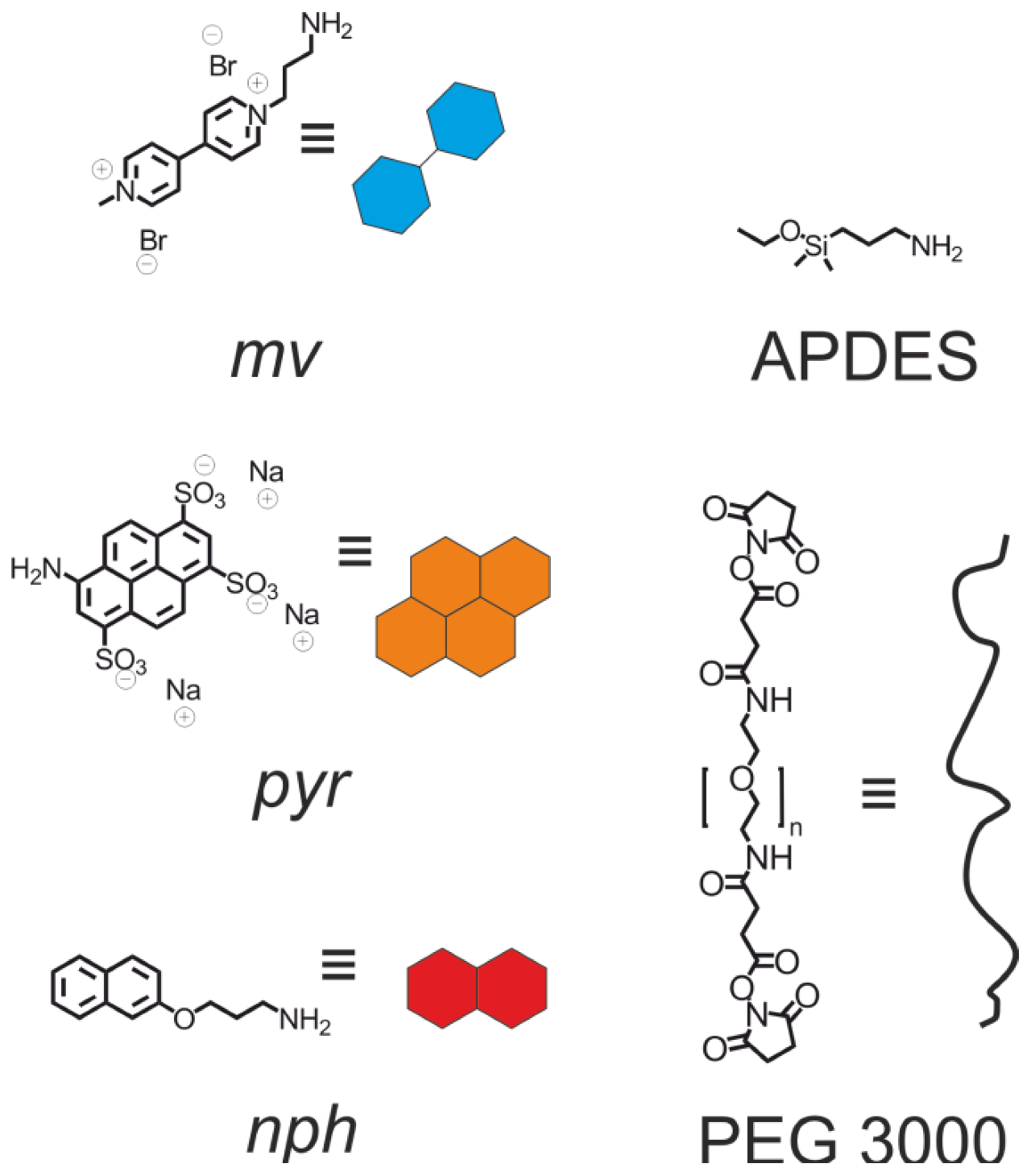{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
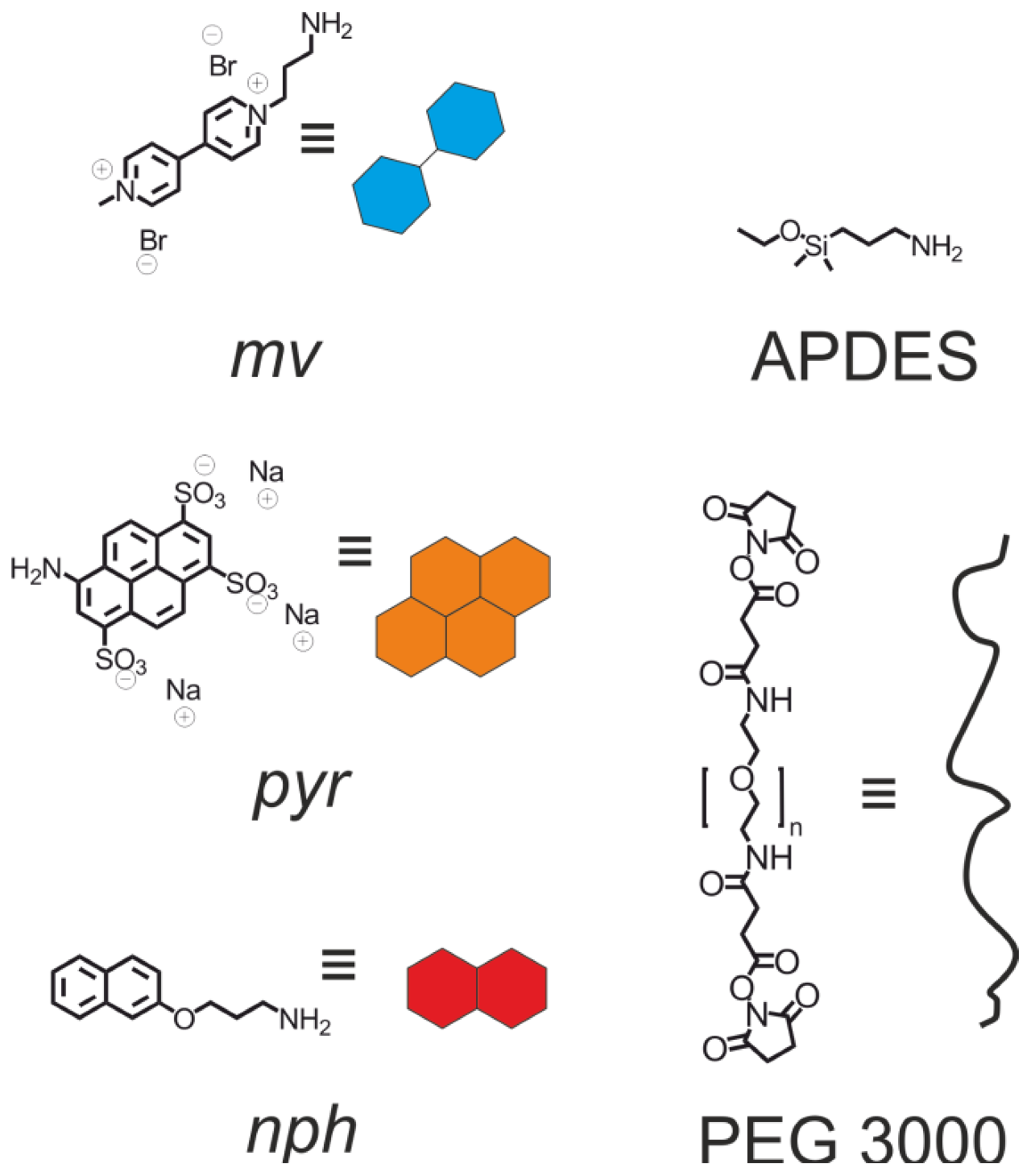
2. Results and Discussion
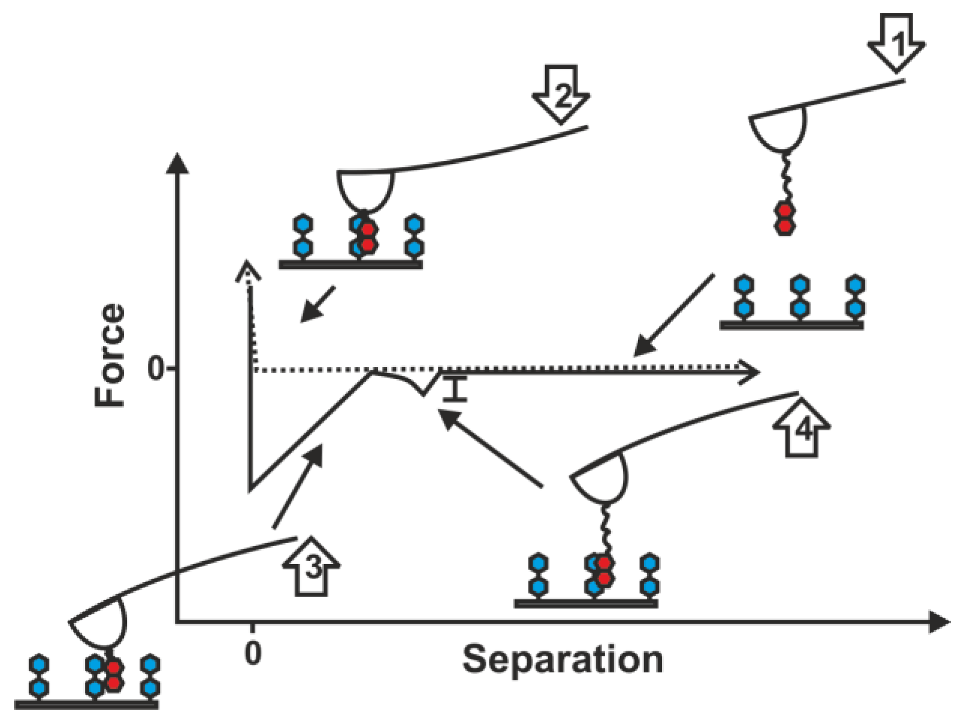
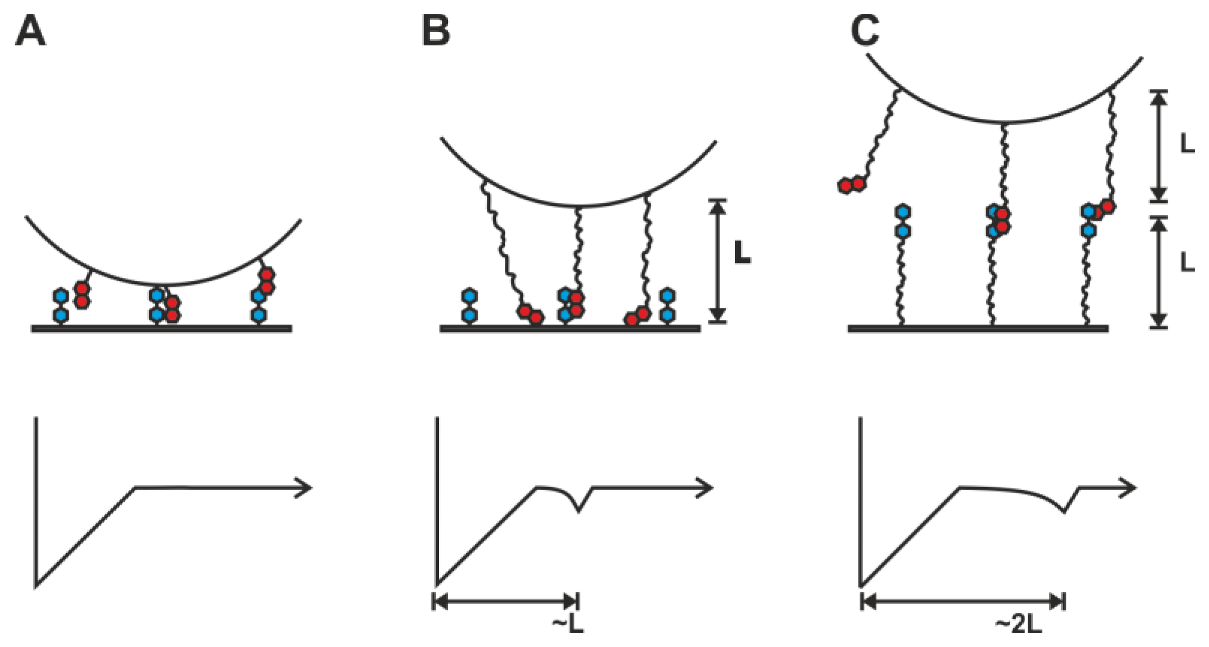
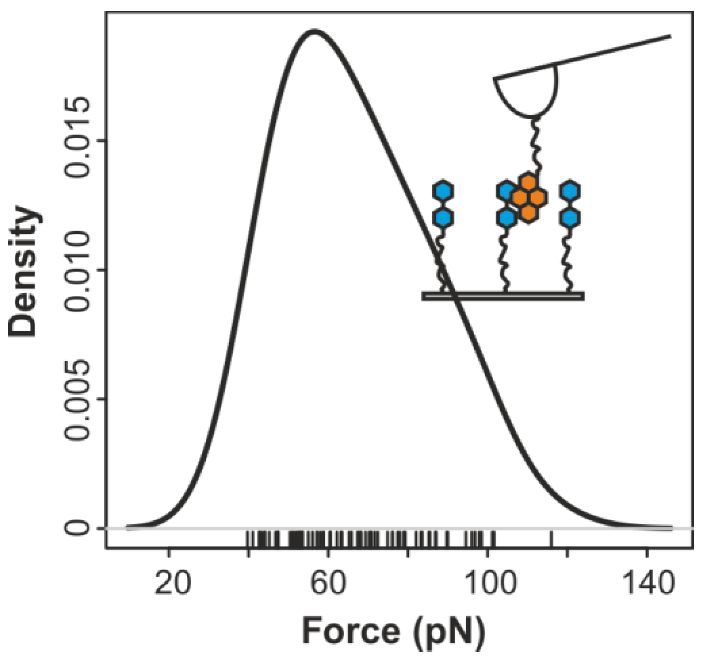
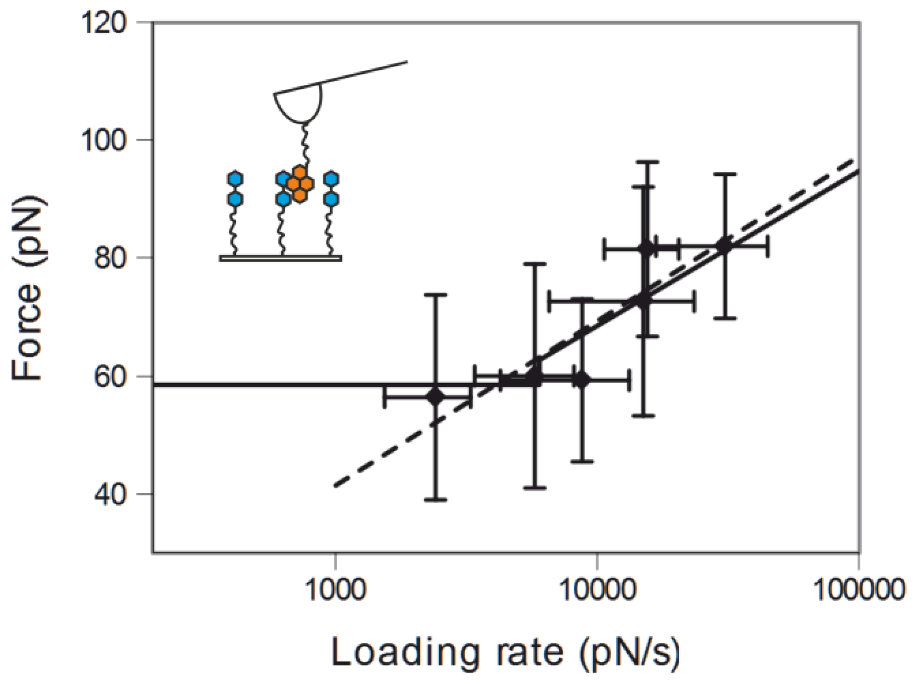
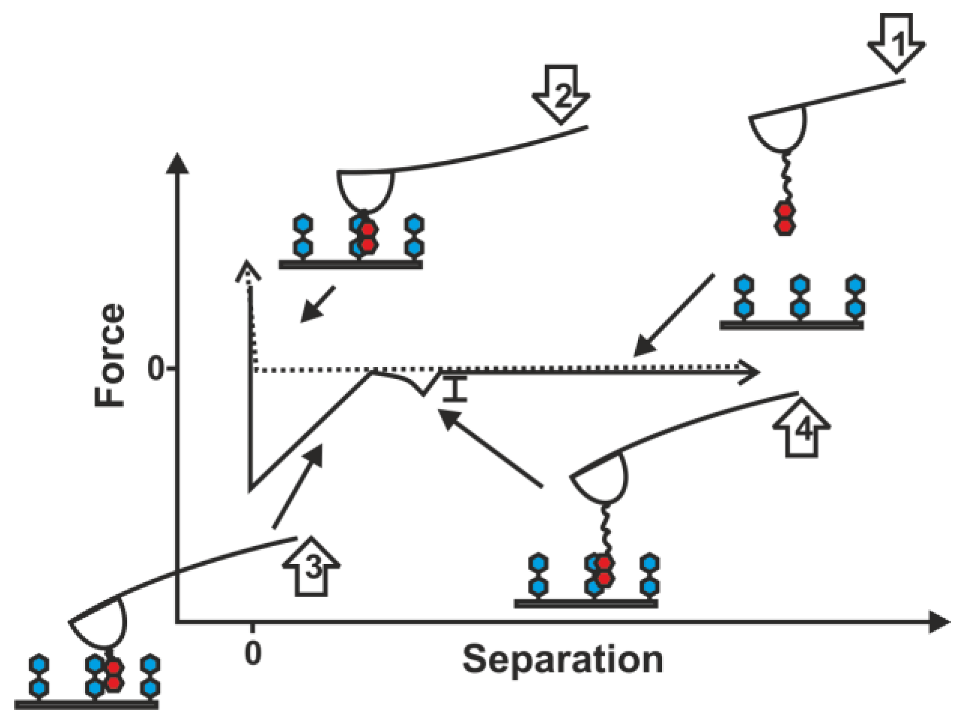
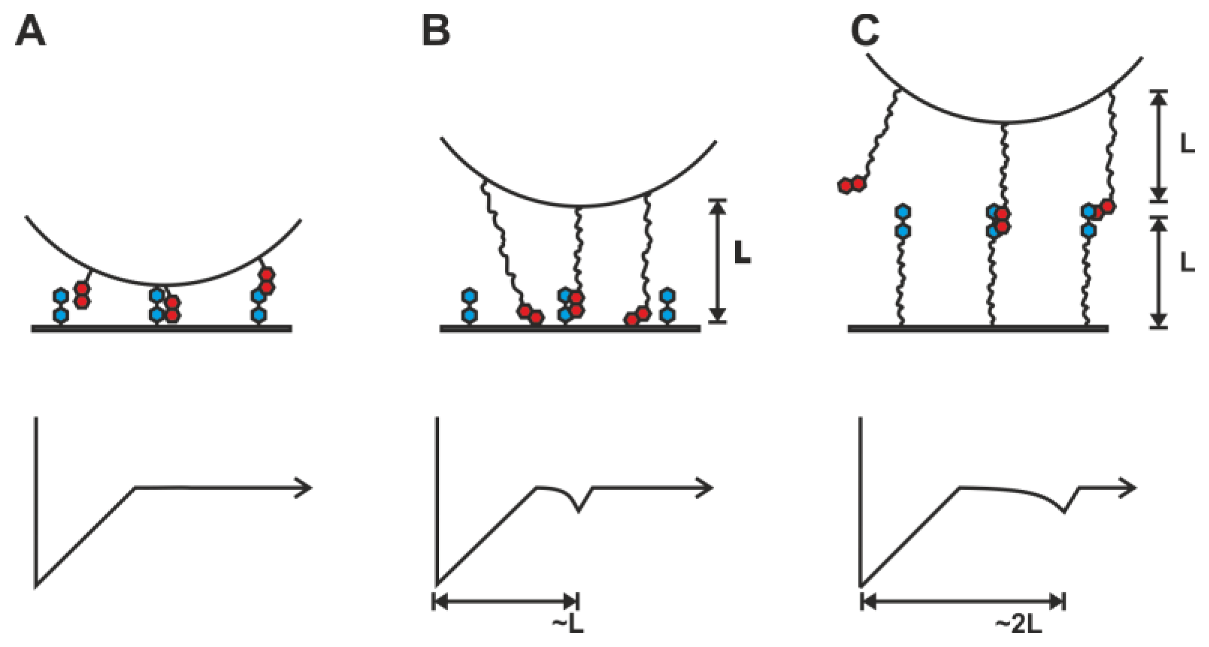
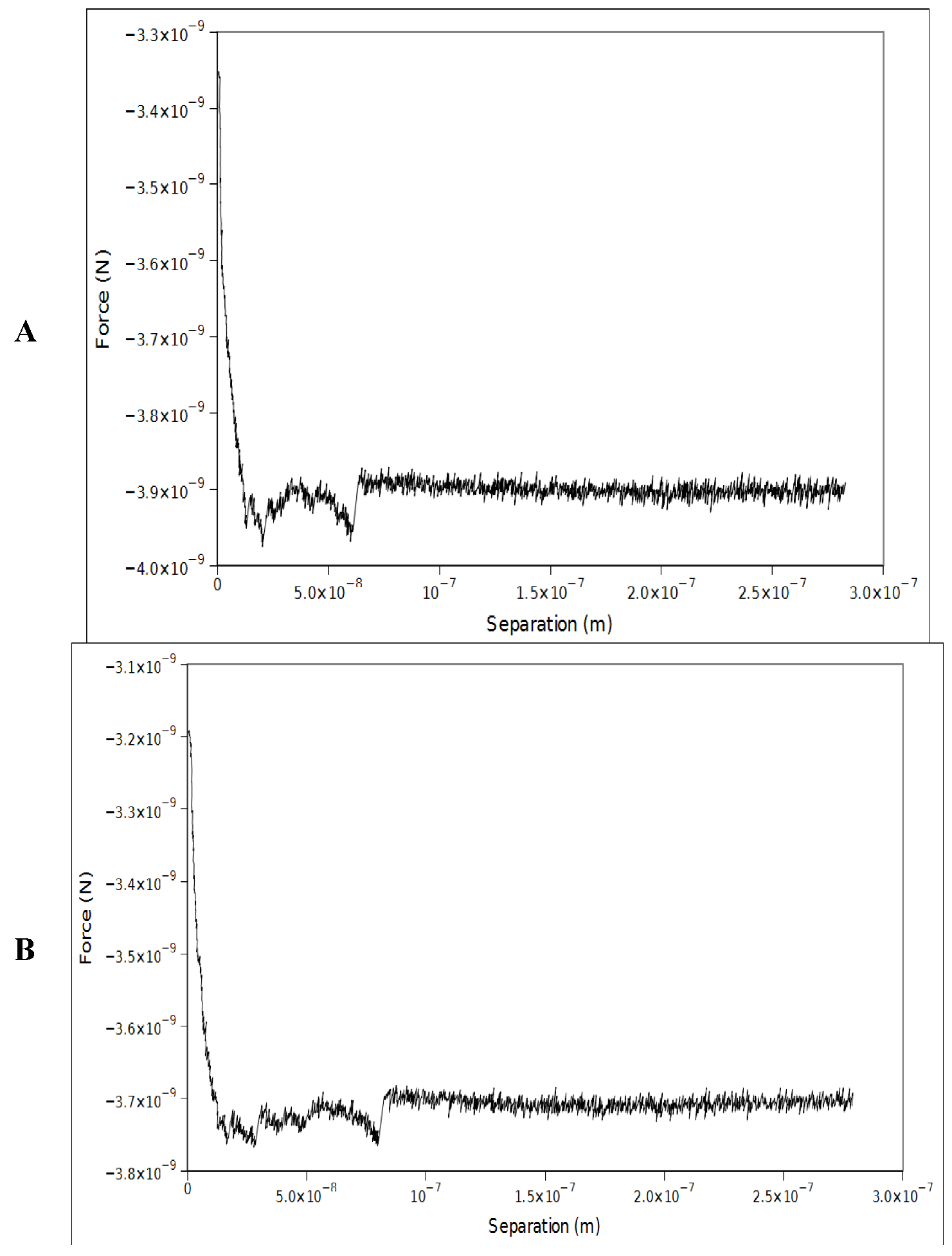
2.1. Double-Tethered DFS
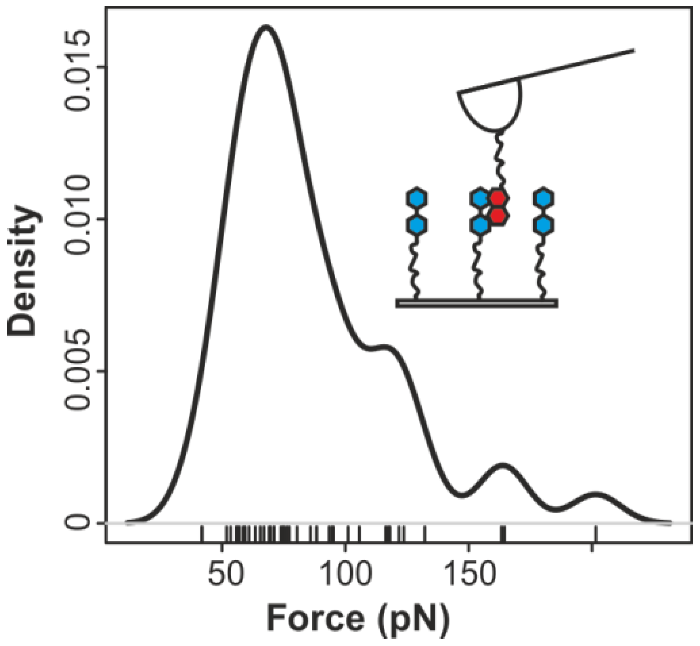
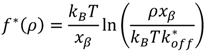
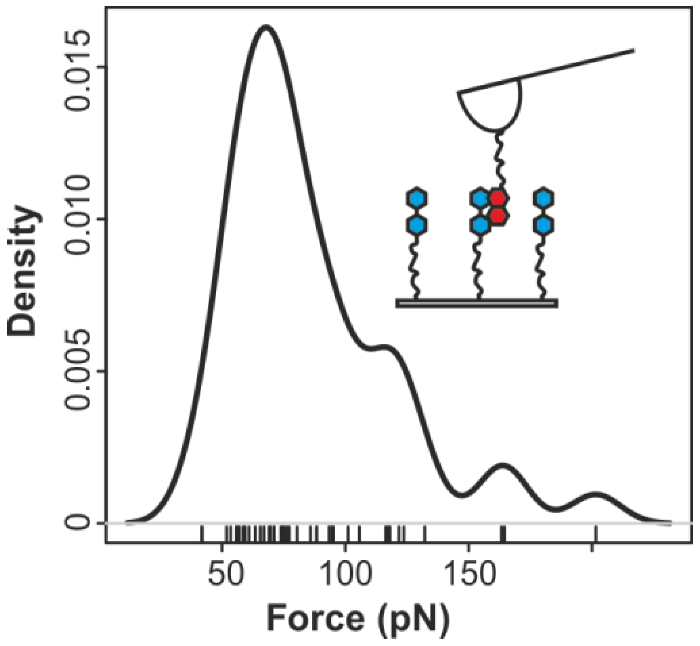
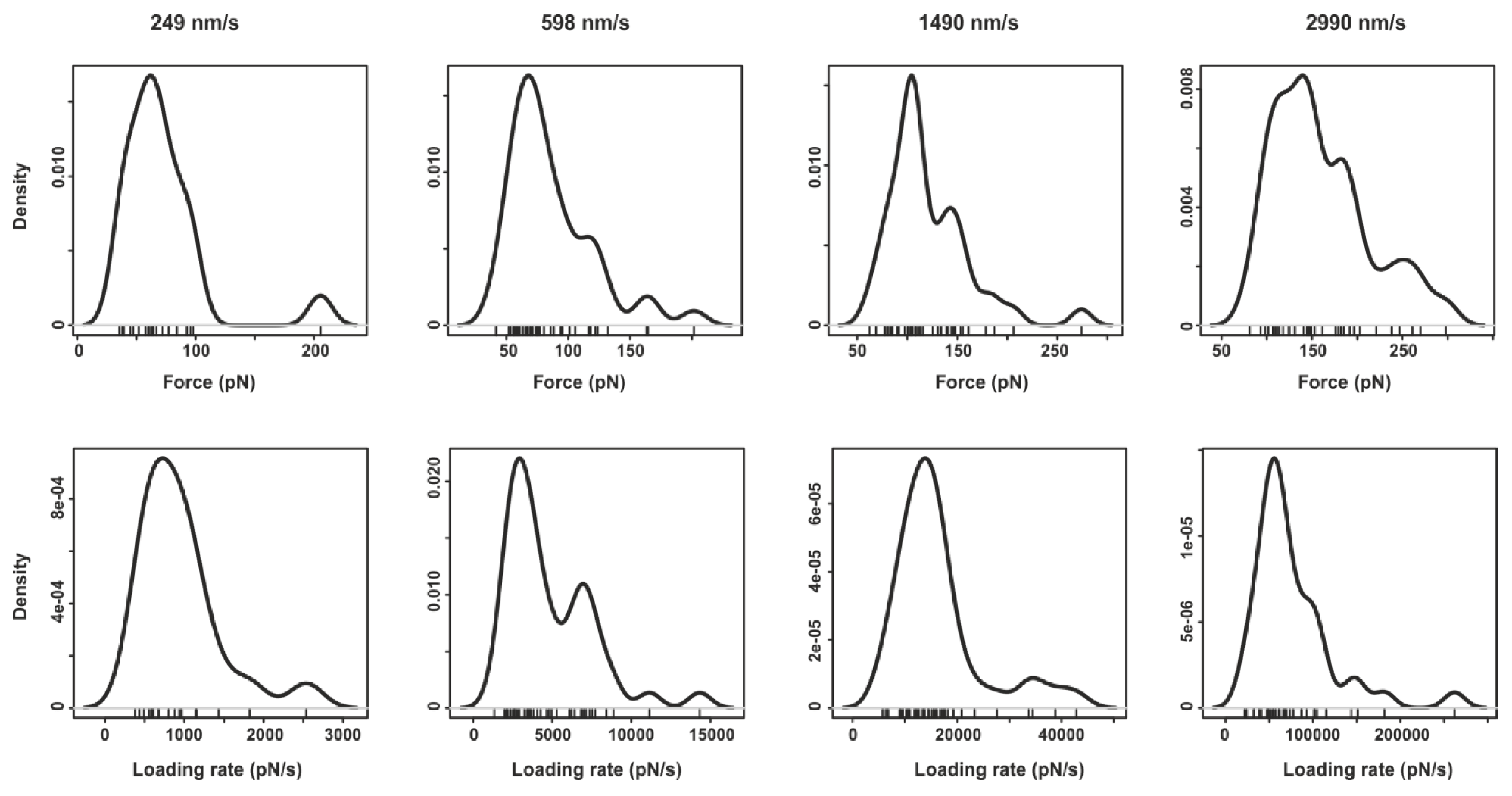
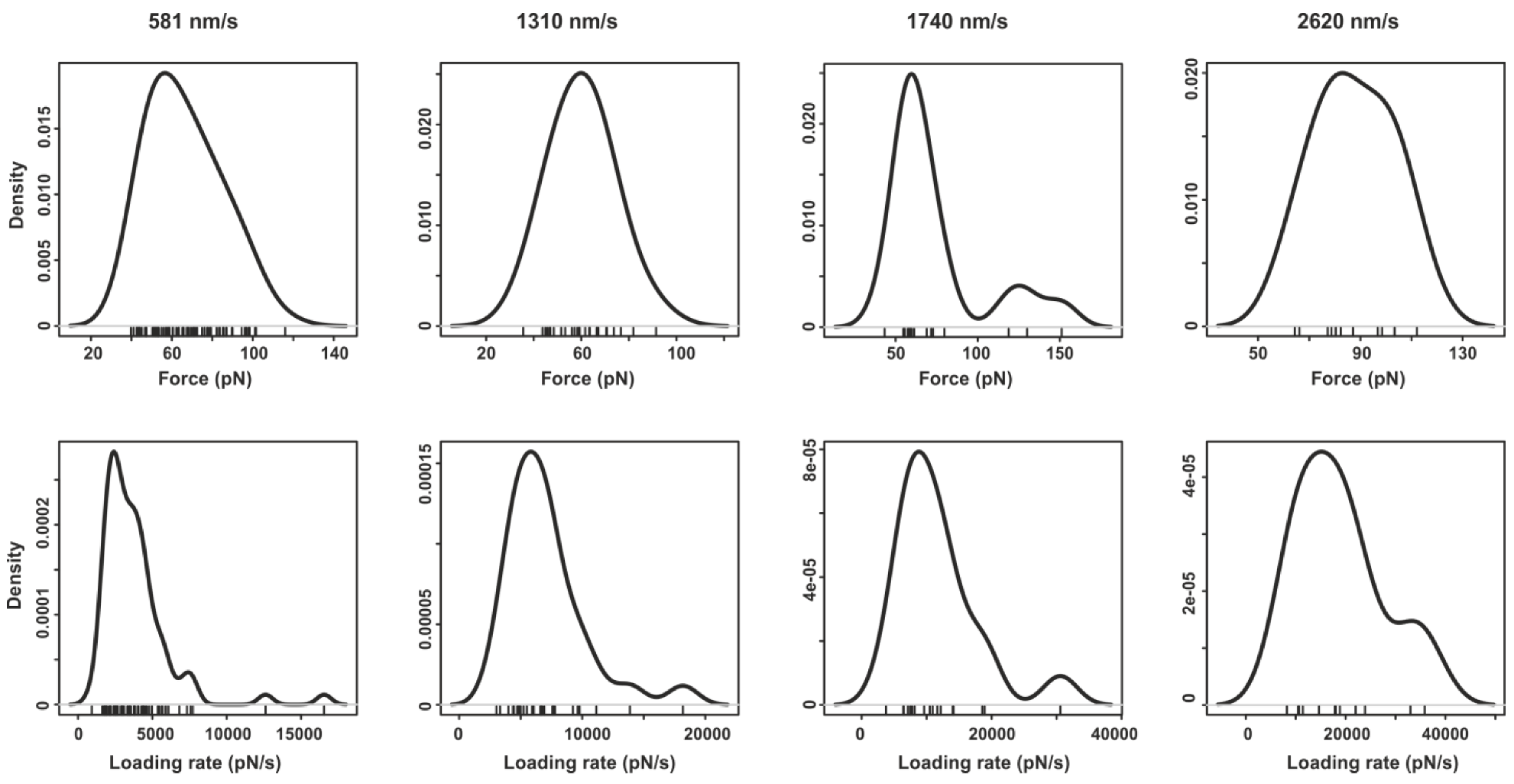
2.2. Pyrene-Methylviologen Complex
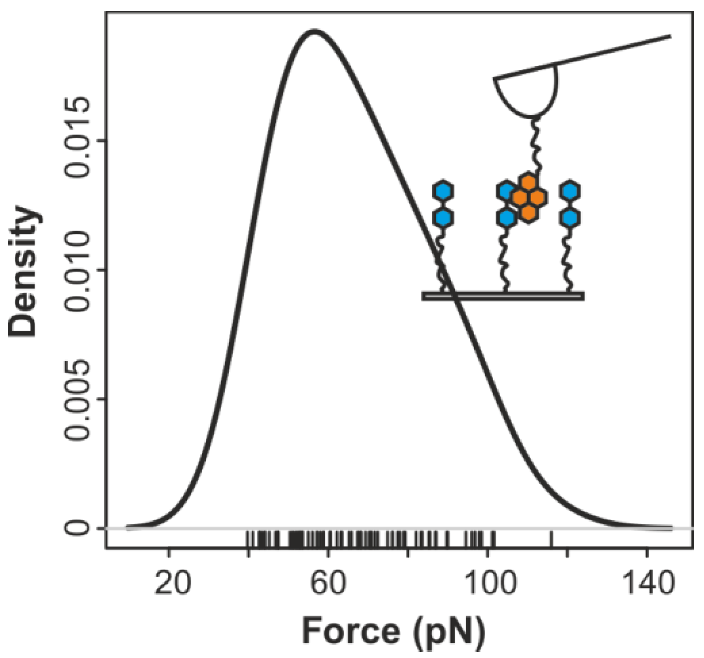
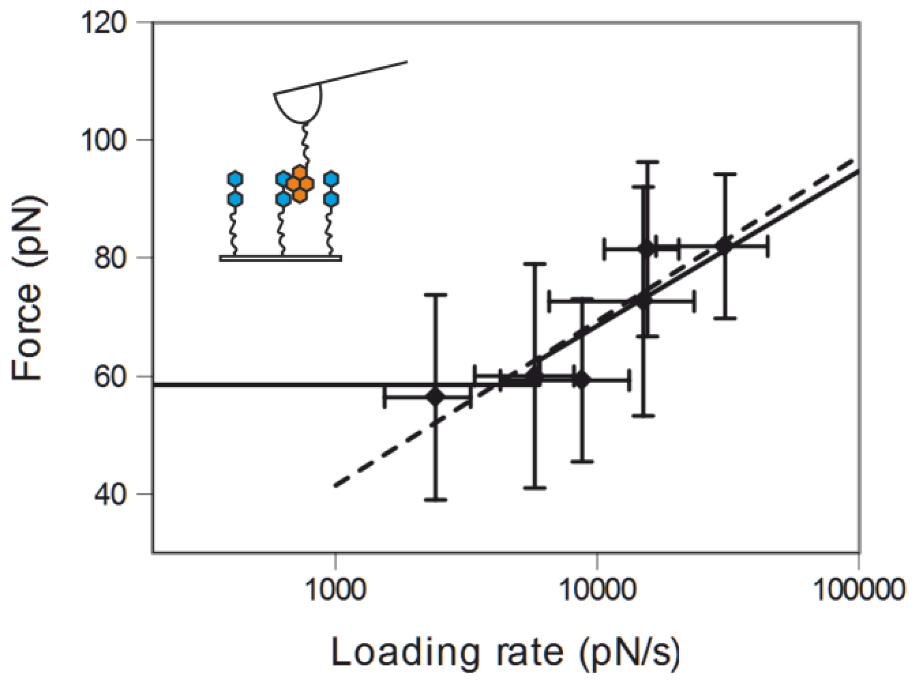
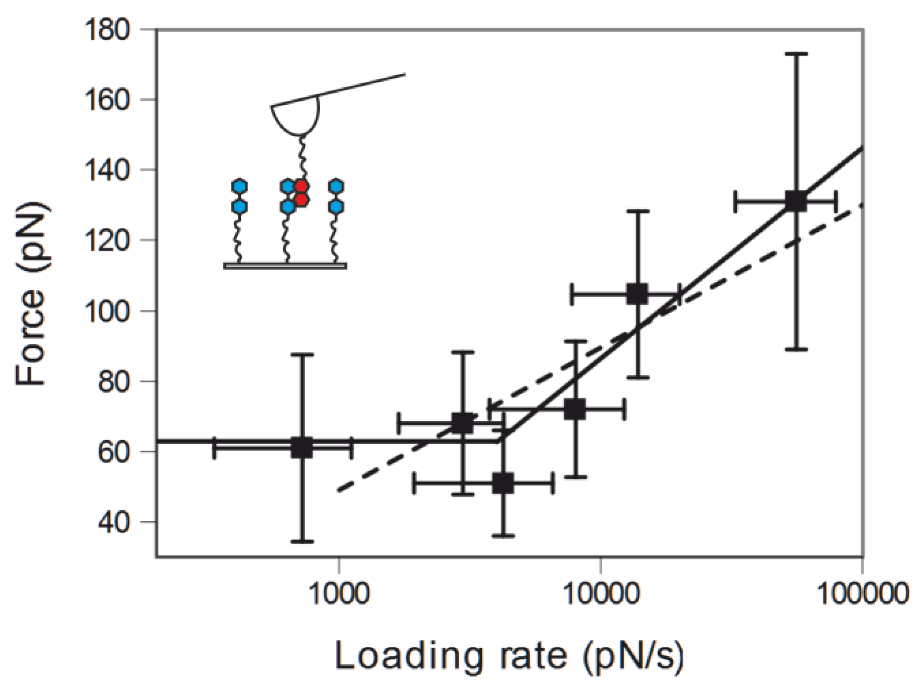
2.3. Comparing nph-mv and pyr-mv Complexes
3. Experimental Section
3.1. 3-(2-Naphthyl)-oxy-aminopropane (nph)
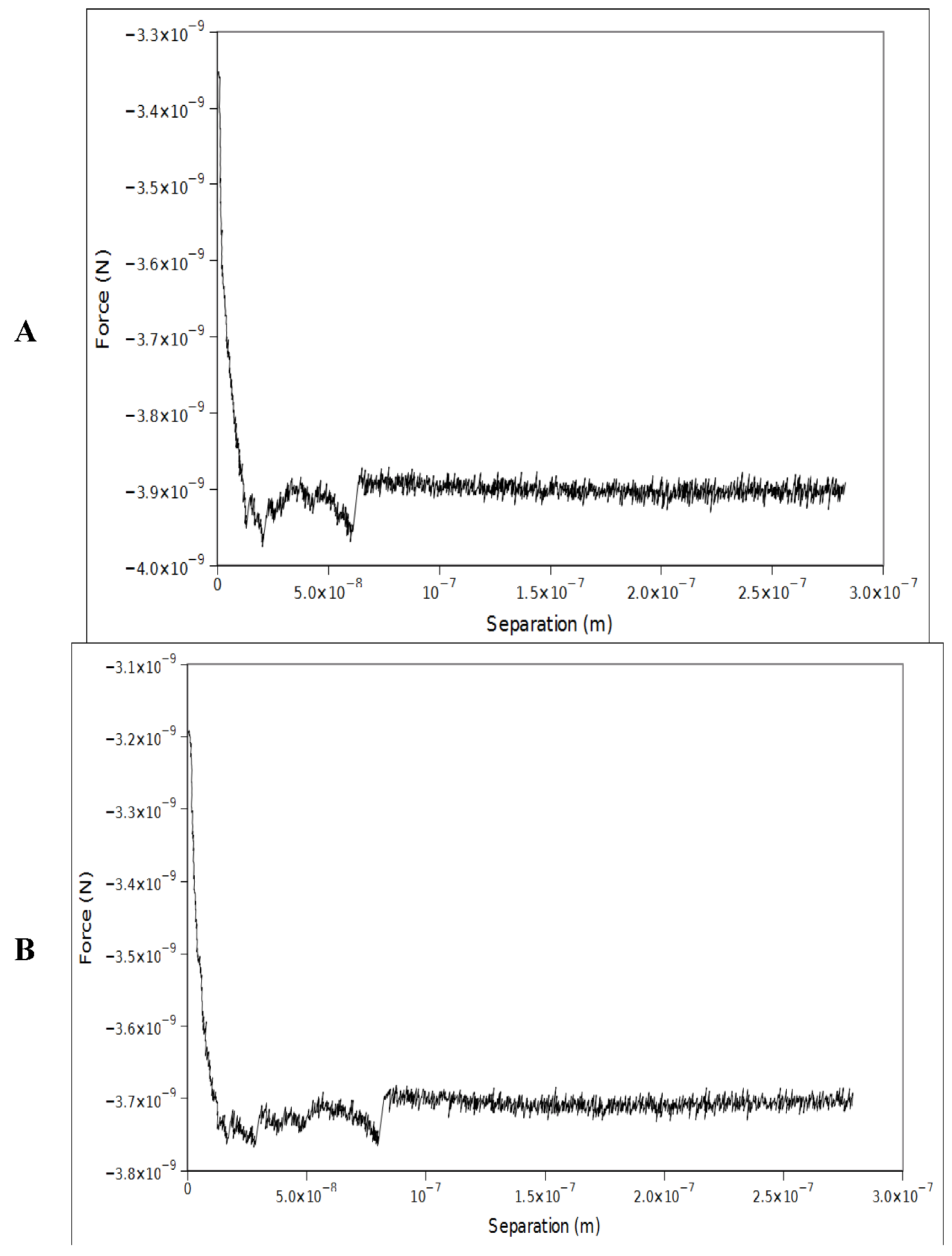
3.2. Substrate and AFM Cantilever Functionalization
3.3. AFM Force Spectroscopy Measurements
3.4. Data Analysis
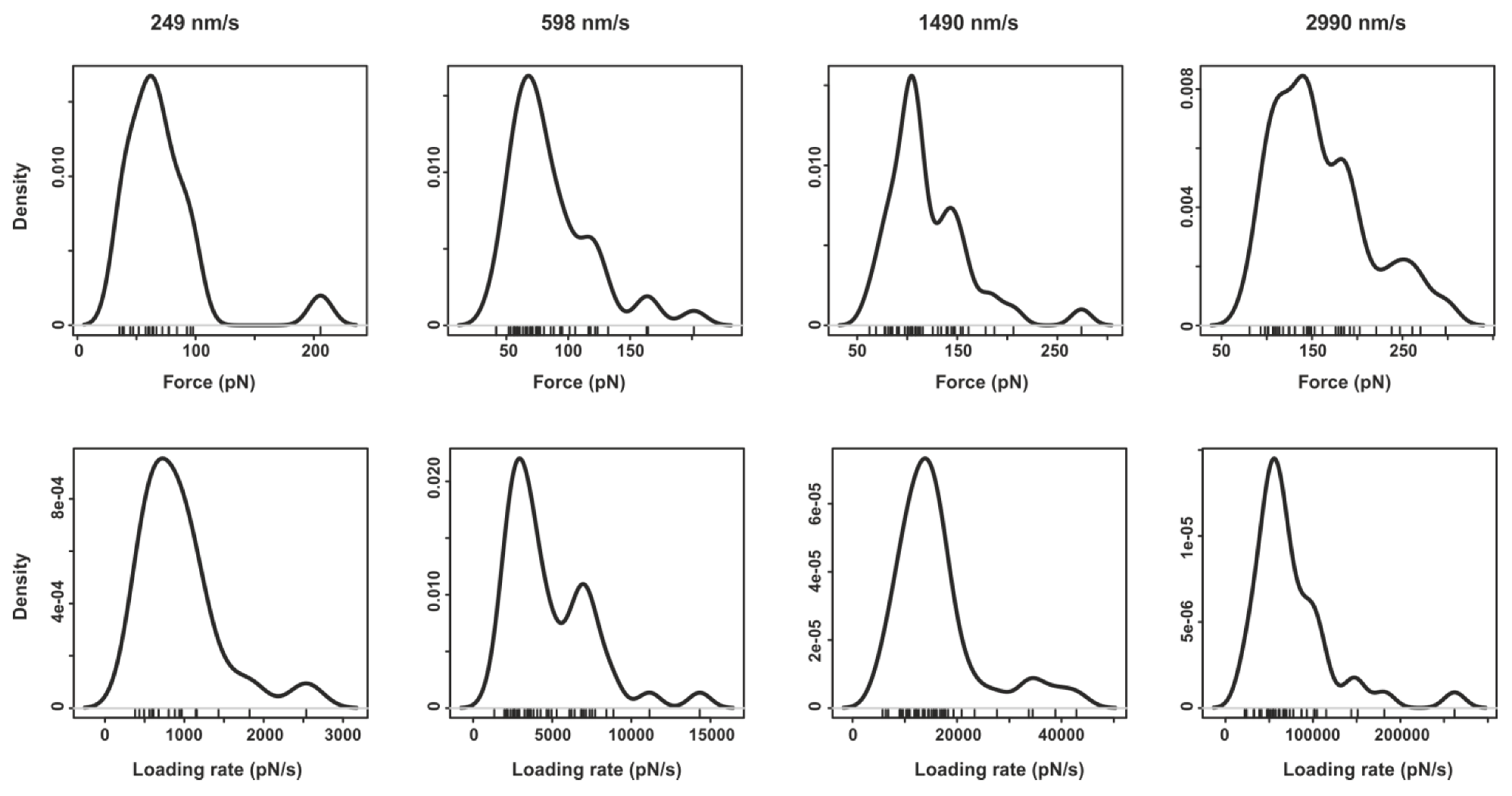
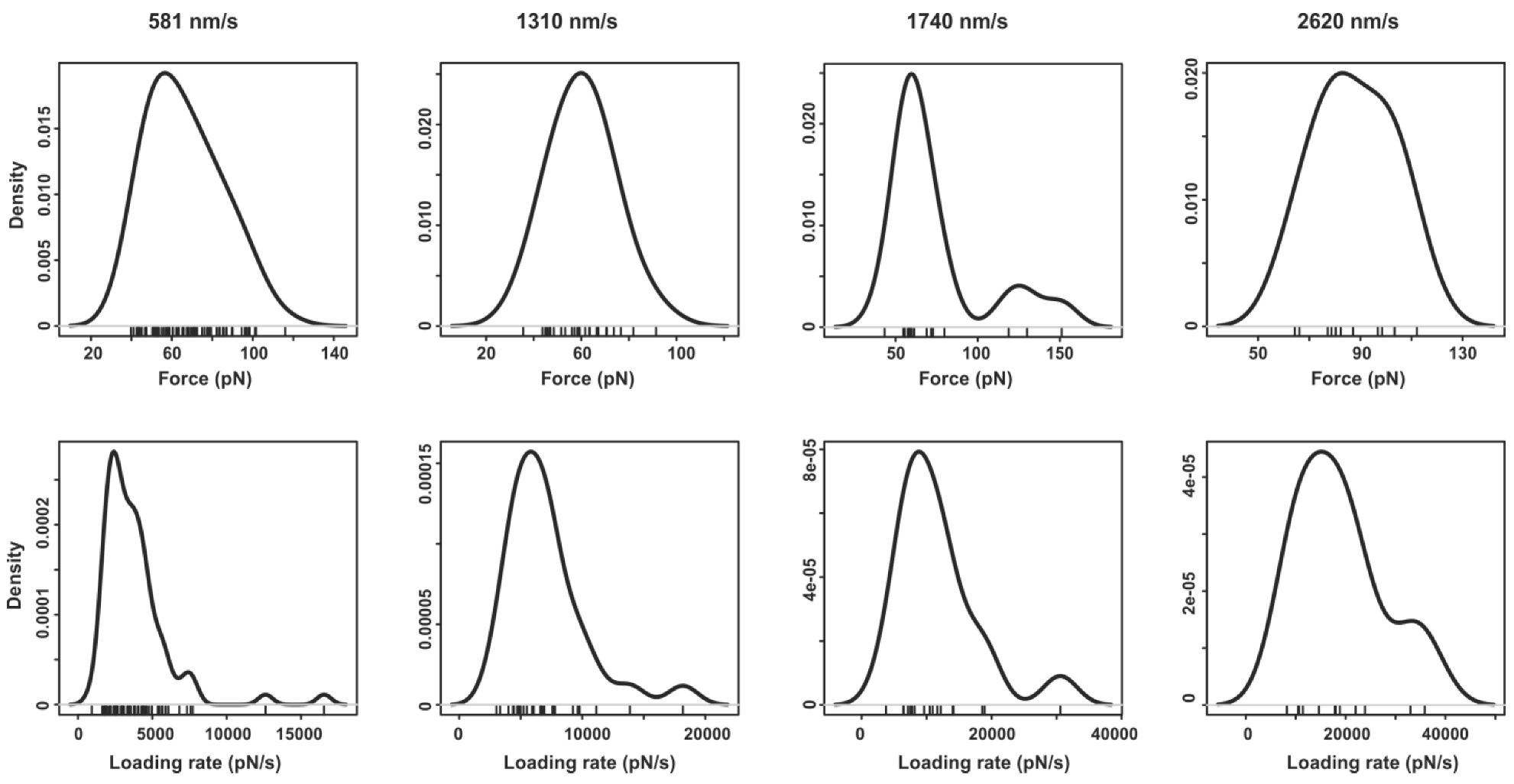
4. Conclusions
Acknowledgments
References
- Shifrin, S. Charge-transfer complexes in biological systems. Ann. N.Y. Acad. Sci. 1969, 158, 148–160. [Google Scholar] [CrossRef]
- Gutmann, F.; Johnson, C.; Keyzer, H.; Molnar, J. Charge Transfer Complexes in Biological Systems; CRC Press: New York, NY, USA, 1997. [Google Scholar]
- Liao, L.B.; Zhou, H.Y.; Xiao, X.M. Spectroscopic and viscosity study of doxorubicin interaction with DNA. J. Mol. Struct. 2005, 749, 108–113. [Google Scholar] [CrossRef]
- Shimazaki, Y.; Mitsuishi, M.; Ito, S.; Yamamoto, M. Preparation of the layer-by-layer deposited ultrathin film based on the charge-transfer interaction. Langmuir 1997, 13, 1385–1387. [Google Scholar] [CrossRef]
- Ju, Z.-F.; Yao, Q.-X.; Wu, W.; Zhang, J. Strong electron-accepting methylviologen dication confined in magnetic hosts: synthesis, structural characterization, charge-transfer and magnetic properties of {(MV)2[Ni(SCN)5]·Cl·2H2O}n and {(MV)[M(N3)2(SCN)2]}n (M = Mn, Co). Dalton Trans. 2008, 3, 355–362. [Google Scholar]
- Prasad, D.R.; Hoffman, M.Z. Charge-transfer complexation between methyl viologen and sacrificial electron donors EDTA, triethanolamine, and cysteine. J. Phys. Chem. 1984, 88, 5660–5665. [Google Scholar] [CrossRef]
- Nakahara, A.; Wang, J.H. Charge-transfer complexes of methylviologen. J. Phys. Chem. 1963, 67, 496–498. [Google Scholar] [CrossRef]
- Monk, P.M.S.; Hodgkinson, N.M. Charge-transfer complexes of the viologens: effects of complexation and the rate of electron transfer to methyl viologen. Electrochim. Acta 1998, 43, 245–55. [Google Scholar] [CrossRef]
- Atherton, S.J.; Hubig, S.M.; Callan, T.J.; Duncanson, J.A.; Snowden, P.T.; Rodgers, M.A.J. Photoinduced charge separation in a micelle-induced charge-transfer complex between methylviologen and ethidium ions: a picosecond absorption spectroscopy study. J. Phys. Chem. 1987, 91, 3137–3140. [Google Scholar] [CrossRef]
- Kinuta, T.; Kise, Y.; Kamon, K.; Tajima, N.; Sato, T.; Kuroda, R.; Matsubara, Y.; Imai, Y. Charge-transfer host complex with channel-like cavity using disubstituted-1,1′-bi-2-naphthol and benzylviologen. Tetrahedron Lett. 2009, 50, 5786–5789. [Google Scholar] [CrossRef]
- Wang, C.; Guo, Y.; Wang, Y.; Xu, H.; Wang, R.; Zhang, X. Supramolecular amphiphiles based on a water-soluble charge-transfer complex: fabrication of ultralong nanofibers with tunable straightness. Angew. Chem. Int. Ed. 2009, 48, 8962–8965. [Google Scholar]
- Hariharan, M.; Joseph, J.; Ramaiah, D. Novel bifunctional viologen-linked pyrene conjugates:Synthesis and study of their interactions with nucleosides and DNA. J. Phys. Chem. B 2006, 110, 24678–24686. [Google Scholar] [CrossRef]
- Kang, J.-K.; Hwang, I.; Ko, Y.H.; Jeon, W.S.; Kim, H.-J.; Kim, K. Electrochemically controllable reversible formation of cucurbit[8]uril-stabilized charge-transfer complex on surface. Supramol. Chem. 2008, 20, 149–155. [Google Scholar] [CrossRef]
- Rauwald, U.; Scherman, O.A. Supramolecular block copolymers with cucurbit[8]uril in water. Angew. Chem. Int. Ed. 2008, 47, 3950–3953. [Google Scholar] [CrossRef]
- Bizzarri, A.R.; Cannistraro, S. The application of atomic force spectroscopy to the study of biological complexes undergoing a biorecognition process. Chem. Soc. Rev. 2010, 39, 734–749. [Google Scholar] [CrossRef]
- Carvalho, F.A.; Connell, S.; Miltenberger-Miltenyi, G.; Pereira, S.n.V.; Tavares, A.; Ariens, R.A.S.; Santos, N.C. Atomic force microscopy-based molecular recognition of a fibrinogen receptor on human erythrocytes. ACS Nano 2010, 4, 4609–4620. [Google Scholar] [CrossRef]
- Embrechts, A.; Schönherr, H.; Vancso, G.J. Rupture force of single supramolecular bonds in associative polymers by AFM at fixed loading rates. J. Phys. Chem. B 2008, 112, 7359–7362. [Google Scholar] [CrossRef]
- Evans, E.; Ritchie, K. Dynamic strength of molecular adhesion bonds. Biophys. J. 1997, 72, 1541–1555. [Google Scholar] [CrossRef]
- Skulason, H.; Frisbie, C.D. Direct detection by atomic force microscopy of single bond forces associated with the rupture of discrete charge-transfer complexes. J. Am. Chem. Soc. 2002, 124, 15125–15133. [Google Scholar] [CrossRef]
- Gil, R.; Guillerez, M.G.; Poulin, J.C.; Schulz, E. Charge-transfer complex study by chemical force spectroscopy: a dynamic force spectroscopic approach. Langmuir 2007, 23, 542–548. [Google Scholar] [CrossRef]
- Yu, Y.; Yao, Y.; Wang, L.; Li, Z. Charge-transfer interaction between poly(9-vinylcarbazole) and 3,5-dinitrobenzamido group or 3-nitrobenzamido group. Langmuir 2010, 26, 3275–3279. [Google Scholar] [CrossRef]
- Hutter, J.L.; Bechhoefer, J. Calibration of atomic-force microscope tips. Rev. Sci. Instrum. 1993, 64, 1868–1873. [Google Scholar]
- Sader, J.E.; Chon, J.W.M.; Mulvaney, P. Calibration of rectangular atomic force microscope cantilevers. Rev. Sci. Instrum. 1999, 70, 3967–3969. [Google Scholar] [CrossRef]
- Hinterdorfer, P.; Kienberger, F.; Raab, A.; Gruber, H.J.; Baumgartner, W.; Kada, G.; Riener, C.; Wielert-Badt, S.; Borken, C.; Schindler, H. Poly(ethylene glycol): an ideal spacer for molecular recognition force microscopy/spectroscopy. Single Mol. 2000, 1, 99–103. [Google Scholar] [CrossRef]
- Christian, K.R.; Borken, C.; Kienberger, K.; Schindler, H.; Hinterdorfer, P.; Gruber, H.J. Collection of long, flexible PEG-crosslinkers for molecular recognition force microscopy on single molecules. Single Mol. 2001, 2, 127–128. [Google Scholar] [CrossRef]
- Evans, E. Probing the relation between force—lifetime—and chemistry in single molecular bonds. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 105–128. [Google Scholar] [CrossRef]
- Ratto, T.V.; Langry, K.C.; Rudd, R.E.; Balhorn, R.L.; Allen, M.J.; McElfresh, M.W. Force spectroscopy of the double-tethered concanavalin-A mannose bond. Biophys. J. 2004, 86, 2430–2437. [Google Scholar] [CrossRef]
- Sandal, M.; Benedetti, F.; Brucale, M.; Gomez-Casado, A.; Samori, B. Hooke: an open software platform for force spectroscopy. Bioinformatics 2009, 25, 1428–1430. [Google Scholar] [CrossRef]
- Gomez-Casado, A.; Dam, H.H.; Yilmaz, M.D.; Florea, D.; Jonkheijm, P.; Huskens, J. Probing multivalent interactions in a synthetic host–guest complex by dynamic force spectroscopy. J. Am. Chem. Soc. 2011, 133, 10849–10857. [Google Scholar]
- Raible, M.; Evstigneev, M.; Bartels, F.W.; Eckel, R.; Nguyen-Duong, M.; Merkel, R.; Ros, R.; Anselmetti, D.; Reimann, P. Theoretical analysis of single-molecule force spectroscopy experiments: heterogeneity of chemical bonds. Biophys. J. 2006, 90, 3851–3864. [Google Scholar] [CrossRef]
- Gu, C.; Kirkpatrick, A.; Ray, C.; Guo, S.; Akhremitchev, B.B. Effects of multiple-bond ruptures in force spectroscopy measurements of interactions between fullerene C60-molecules in water. J. Phys. Chem. C 2008, 112, 5085–5092. [Google Scholar]
- Guo, S.; Ray, C.; Kirkpatrick, A.; Lad, N.; Akhremitchev, B.B. Effects of multiple-bond ruptures on kinetic parameters extracted from force spectroscopy measurements: Revisiting biotin-streptavidin interactions. Biophys. J. 2008, 95, 3964–3976. [Google Scholar] [CrossRef]
- Hsiao, J.-S.; Eckert, A.R.; Webber, S.E. Excited-state electron transfer from anthracene and pyrene covalently end-tagged onto poly(ethylene oxide). J. Phys. Chem. 1994, 98, 12032–12039. [Google Scholar]
- Hwang, J.H.; Lee, K.S.; Lee, S.; Park, W.J. An NMR study on the conformation of naphthalene–viologen linked compounds: effect of flexible spacer length. J. Chem. Soc. Perkin Trans. 1999, 2, 1081–1086. [Google Scholar]
- Odorico, M.; Teulon, J.M.; Bessou, T.; Vidaud, C.; Bellanger, L.; Chen, S.W.; Quemeneur, E.; Parot, P.; Pellequer, J.L. Energy landscape of chelated uranyl: antibody interactions by dynamic force spectroscopy. Biophys. J. 2007, 93, 645–654. [Google Scholar] [CrossRef]
- Evans, E. Looking inside molecular bonds at biological interfaces with dynamic force spectroscopy. Biophys. Chem. 1999, 82, 83–97. [Google Scholar] [CrossRef]
- Merkel, R.; Nassoy, P.; Leung, A.; Ritchie, K.; Evans, E. Energy landscapes of receptor-ligand bonds explored with dynamic force spectroscopy. Nature 1999, 397, 50–53. [Google Scholar]
- Zhang, Y.; Liu, C.; Shi, W.; Wang, Z.; Dai, L.; Zhang, X. Direct measurements of the interaction between pyrene and graphite in aqueous media by single molecule force spectroscopy: Understanding the π−π interactions. Langmuir 2007, 23, 7911–7915. [Google Scholar] [CrossRef]
- Zou, S.; Schönherr, H.; Vancso, G.J. Force spectroscopy of quadruple H-bonded dimers by AFM: Dynamic bond rupture and molecular time-temperature superposition. J. Am. Chem. Soc. 2005, 127, 11230–11231. [Google Scholar]
- Oesterhelt, F.; Rief, M.; Gaub, H.E. Single molecule force spectroscopy by AFM indicates helical structure of poly(ethylene-glycol) in water. New J. Phys. 1999, 1, 1–11. [Google Scholar] [CrossRef]
- RDC-Team. Available online: http://www.R-project.org (accessed on 1 April 2012).
- Sheather, S.J.; Jones, M.C. A reliable data-based bandwidth selection method for Kernel density estimation. J. R. Statist. Soc. B 1991, 53, 683–690. [Google Scholar]
- Lin, C.; Chinnappan, R.; Acharya, K.; Pellequer, J.L.; Jankowiak, R. On stabilization of a neutral aromatic ligand by π-cation interactions in monoclonal antibodies. Biophys. Chem. 2011, 154, 35–40. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gomez-Casado, A.; Gonzalez-Campo, A.; Zhang, Y.; Zhang, X.; Jonkheijm, P.; Huskens, J. Charge-Transfer Complexes Studied by Dynamic Force Spectroscopy. Polymers 2013, 5, 269-283. https://doi.org/10.3390/polym5010269
Gomez-Casado A, Gonzalez-Campo A, Zhang Y, Zhang X, Jonkheijm P, Huskens J. Charge-Transfer Complexes Studied by Dynamic Force Spectroscopy. Polymers. 2013; 5(1):269-283. https://doi.org/10.3390/polym5010269
Chicago/Turabian StyleGomez-Casado, Alberto, Arántzazu Gonzalez-Campo, Yiheng Zhang, Xi Zhang, Pascal Jonkheijm, and Jurriaan Huskens. 2013. "Charge-Transfer Complexes Studied by Dynamic Force Spectroscopy" Polymers 5, no. 1: 269-283. https://doi.org/10.3390/polym5010269