Activity and Mechanism of Antimicrobial Peptide-Mimetic Amphiphilic Polymethacrylate Derivatives


 and
and
Abstract
: Cationic amphiphilic polymethacrylate derivatives (PMAs) have shown potential as a novel class of synthetic antimicrobials. A panel of PMAs with varied ratios of hydrophobic and cationic side chains were synthesized and tested for antimicrobial activity and mechanism of action. The PMAs are shown to be active against a panel of pathogenic bacteria, including a drug-resistant Staphylococcus aureus, compared to the natural antimicrobial peptide magainin which did not display any activity against the same strain. The selected PMAs with 47–63% of methyl groups in the side chains showed minimum inhibitory concentrations of ≤2–31 μg/mL, but cause only minimal harm to human red blood cells. The PMAs also exhibit rapid bactericidal kinetics. Culturing Escherichia coli in the presence of the PMAs did not exhibit any potential to develop resistance against the PMAs. The antibacterial activities of PMAs against E. coli and S. aureus were slightly reduced in the presence of physiological salts. The activity of PMAs showed bactericidal effects against E. coli and S. aureus in both exponential and stationary growth phases. These results demonstrate that PMAs are a new antimicrobial platform with no observed development of resistance in bacteria. In addition, the PMAs permeabilized the E. coli outer membrane at polymer concentrations lower than their MIC values, but they did not show any effect on the bacterial inner membrane. This indicates that mechanisms other than membrane permeabilization may be the primary factors determining their antimicrobial activity.1. Introduction
Host defense peptides have long attracted scientific and commercial interest because of their ability to kill bacteria without causing harm to human cells [1]. In general, these peptides display a broad spectrum of activity and rapid killing. Although the molecular details of their antimicrobial mechanism of action are a subject of ongoing debate, it is generally thought that these peptides bind to the bacterial surface and insert themselves into the hydrophobic region of the cell membrane. Subsequently, the bound/inserted peptides compromise membrane integrity, causing leakage of cytoplasmic contents and ultimately bacterial cell death. Alternatively, some peptides penetrate into the cytoplasm and interact with DNA, RNA, and/or cytoplasmic enzymes, resulting in the inhibition of macromolecule function [2]. These mechanisms of peptide antimicrobial activity seem to be especially advantageous because the development of resistance in bacteria is limited compared to conventional antibiotics [1]. Although there is significant diversity of antimicrobial peptides found in nature, the majority share common structural features, specifically in the formation of amphiphilic conformations. In addition, the mechanism does not rely on a specific receptor-ligand type binding, which is a likely contributing factor to the observed lack of resistance development. These properties support new peptidomimetic designs that incorporate the physiochemical parameters found in antimicrobial peptides such as their cationic charge, hydrophobicity, and amphiphilic structures which are key determinants in the mode of antimicrobial action [3-6]. This design approach has been successfully generalized to abiotic oligomers and macromolecules such as designed synthetic peptides [7-9] and peptidomimetics including β-peptides [10-12], arylamides [13], and peptoids [14,15]. These peptides and their mimics are a good basis for new antibiotics, however, issues associated with low stability in vivo, unknown systemic toxicity, and high manufacturing cost present challenges to their implementation as therapeutic agents or in biomedicine.
Synthetic polymers have been widely used as biocides in aqueous solutions [16-20] or tethered on surfaces [21-25]. These polymers have cationic, amphiphilic structures, which are designed to disrupt bacterial membranes with concomitant cell death [26]. However, these polymers often cause toxicity to human cells or their toxicity profiles are not fully characterized, which render these polymers suitable for disinfections or biocides, but not for therapeutic use. To address these issues, a newer approach has been to design synthetic polymers mimicking the functions and structural features of the less toxic host defense peptides. This approach was successfully applied to the development of new types of polymer antimicrobials over the last decade, including copolymers of β-lactams [27], polynorbornenes [28], polymethacrylates [29,30], and polystyrenes [31]. Further fine-tuning of the polymers' physiochemical properties by modifying the cationic functionality, amphiphilic balance, and polymer sizes has yielded favorable improvements in antimicrobial activity and selective toxicity to bacteria relative to human red blood cells [30,32-35]. In addition to these examples of peptidomimetic polymers, it has been recently reported that several other polymer scaffolds such as oligo(oxazoline)s [36], PEGylated poly(vinyl pyridine)s [37], and polyoxetanes with quaternary ammonium and PEG-like side chains [38] display antibacterial activity and selectivity. In contrast to the high cost associated with manufacturing peptides, the preparation of these polymers is inexpensive and facile, enabling production on the industrial scale. In addition, the versatility of polymer chemistry enables a wide exploration of the structural and chemical parameters which can be modified to optimize molecular properties for potent, selective activity.
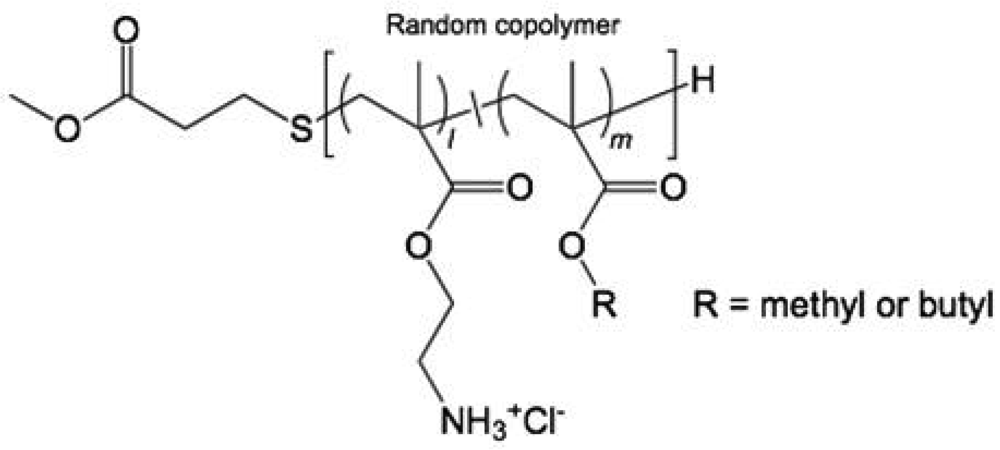
We have previously reported the antibacterial and hemolytic activities in a library of amphiphilic polymethacrylate derivatives (PMAs) with small molecular size (2–10 kDa), multiple cationic charges, and hydrophobic side chain groups (Figure 1) [30,39,40]. By tuning the composition of hydrophobic and cationic groups, the activity of PMAs was optimized for selective toxicity to bacteria over human cells: they displayed potent antibacterial activity against Escherichia coli in the low μM range and moderate to low hemolytic activity. Their activities are comparable to the natural antibacterial peptide magainin, suggesting that the PMAs have potential as new antibacterial platforms for further development. The previous structure-activity relationship studies, however, were limited to measurements of minimum inhibitory concentrations (MICs) against E. coli and hemolytic activity. More comprehensive studies of the PMAs biological activity are requisite for assessment of their potential usefulness as antimicrobials.
In this study, we investigated the bactericidal action of amphiphilic PMAs and assessed the potential usefulness of PMAs as a broad-spectrum antimicrobial through a more detailed characterization of the mechanism of action. Based on our previous studies with an extensive library of PMAs [30,39,40], we designed representative compounds with molecular size (2.2–2.8 kDa) similar to antimicrobial peptides which bear various ratios of primary amine groups and short alkyl groups in the side chains to investigate the optimal composition for potent activity. Previous work has shown that the ratio of hydrophobic to cationic groups is a critical factor in the balance between non-specific membrane disruption and selective antibacterial activity [39]. Herein, we report the in vitro activity of these PMAs against a panel of clinically relevant bacterial strains, their bactericidal kinetics, the propensity of bacteria to develop PMA-resistance, and their antibacterial activity in the presence of physiological salts. In addition, we also investigated the potential antimicrobial mechanism of these PMAs by examining the permeabilization of outer and inner membranes of E. coli as well as model lipid bilayers.
2. Experimental Section
2.1. Materials
2,2′-azobisisobutyronitrile (AIBN), Triton X-100 and the bee venom toxin melittin (purity > 85%) were purchased from Sigma-Aldrich and used without further purification. Methylmethacrylate (MMA), butylmethacrylate (BMA), methyl 3-mercaptopropionate (MMP), ethanolamine, di-tert-butyldicarbonate, methacryloyl chloride, and MgCl2 were purchased from Acros and used without further purification. Reagent grade solvents, NaCl, and CaCl2 were purchased from Fisher and used without further purification. The antimicrobial peptide magainin −2 (purity > 90%) was purchased from AnaSpec Inc. Antibiotics norfloxacin (NOR; purity ∼99.7%) and ciprofloxacin (CIP; purity ∼98%) were obtained from MP Biomedicals, and LKT Laboratories, respectively. Mueller-Hinton broth (MHB), brucella broth and agar were purchased from Difco Laboratories. Human red blood cells (Red Blood Cells Leukocytes Reduced Adenine Saline Added) were obtained from the American Red Cross Blood Services Southeastern Michigan Region.
2.2. Polymer Synthesis
Free radical polymerization of N-(tert-butoxycarbonyl)aminoethyl methacrylate (Boc-AEMA) with an alkyl methacrylate was carried out as previously described [40] with some modifications. Briefly, Boc-AEMA and alkyl methacrylates (various ratios, 0.5 mmol total), MMP (16.7 μL, 0.15 mmol) and AIBN (0.82 mg, 0.005 mmol) dissolved in acetonitrile (0.5 mL) in a sealed borosilicate glass test tube were deoxygenated with N2 bubbling for 2 min and then stirred at 60–70 °C in a mineral oil bath for 20 h. Solvent was evaporated and the crude polymer was purified by size exclusion chromatography (Sephadex LH-20 gel, methanol) monitored by thin layer chromatography (ethyl acetate:hexane 1:1). Fractions containing unreacted monomers and MMP were discarded. The remaining fractions were concentrated, dissolved in 1.25 M HCl in methanol (5–10 mL), and stirred at room temperature for 2 h to cleave the protecting groups. Excess acid was removed by N2 flushing and the polymers were twice precipitated from methanol into diethylether. The precipitates were collected by centrifugation and lyophilized to afford the random copolymers bearing primary amine groups in the form of ammonium chloride salts. The polymers were characterized by 1H NMR to determine the mole percentage of alkyl groups (MPalkyl) and degree of polymerization (DP) as previously described in detail [30,39,40].
2.3. Antibacterial Testing
Antibacterial activity of polymers was determined in a standard microbroth dilution assay according to the Clinical and Laboratory Standards Institute guidelines (CLSI [41]) with suggested modifications by R.E.W. Hancock Laboratory (University of British Columbia, Vancouver, British Columbia, Canada [42]) and Giacometti et al. [43] for testing cationic agents. Each polymer was dissolved in 0.01% acetic acid and acetic acid was used as a solvent control. The bacterial strains Escherichia coli ATCC® 25922™, Staphylococcus aureus ATCC® 25923™, Pseudomonas aeruginosa ATCC® 27853™, Salmonella enterica subsp. enterica serovar Typhimurium ATCC® 14028™, and Bacillus subtilis ATCC® 6633™ were aerobically cultured in MHB. MHB was prepared according to manufacturer's instructions and contained approximately 130 mM Na+, 110 mM Cl−, 0.4 mM Ca2+ and 0.15 mM Mg2+ (pH 7.3 [44]). Because of slow growth of Enterococcus faecalis ATCC® 29212™ in MHB, tryptic soy broth was used for cultivating of this strain. An overnight culture of bacterial strains was regrown to exponential phase (OD600 of 0.5–0.6) and diluted to give the final concentration of bacteria on the microplate approximately 5 × 105 CFU/mL. After addition of the test compounds at a 1/10 volume into a 96-well sterile assay plate (Corning #3359), the assay plate was incubated at 37 °C for 18 h. Bacterial growth was detected at OD600 using Varioskan Flash microplate reader (Thermo Fisher). Where indicated, fixed concentrations of NaCl, MgCl2, and CaCl2 were added into MHB. Heat-inactivated serum was used to examine the effect of serum on antibacterial activity of polymers in the absence potential problems with complement-mediated cell lysis. Anaerobic strain Propionibacterium acnes ATCC® 6919™ was grown in brucella broth supplemented with hemin (5 μg/mL) and vitamin K1 (10 μg/mL) in the anaerobic chamber with an atmosphere of 10% CO2, 10% H2, and 80% N2. Activity of polymers against P. acnes was determined by the broth dilution method according to CLSI M11-A6 guidelines [45]. The inoculum was 106 CFU/mL. The inoculated microplates (Falcon #3077) were incubated for 72 h at 37 °C in the anaerobic chamber. Each MIC experiment was independently repeated at least three times in triplicate on different days. The minimum inhibitory concentration (MIC) was defined as the lowest polymer concentration to completely inhibit bacterial growth. Because biological activity of compounds often depends on the assay conditions and the selected model organisms, it would be impractical to compare biological activity for different compounds reported in the literature. Therefore, we selected the natural host-defense peptide magainin-2 and the bee venom toxin melittin as reference standards in all biological assays.
2.4. Bactericidal Kinetics
Time-kill studies were performed for the Gram-negative E. coli ATCC 25922 and the Gram-positive S. aureus ATCC 25923 to measure the time dependence of bactericidal activity by PMAs. Cells in exponential or stationary phase were diluted in MHB to give the final concentration of bacteria approximately 5 × 105 CFU/mL and treated with 2 × MIC of PMAs. Aliquots from the assay were removed at certain time intervals and immediately diluted with 0.9% saline to remove the effects of the polymer. Viability was enumerated by serial dilution plating. After 24-h incubation, no regrowth of E. coli or S. aureus cultures containing PMAs was observed. Experiments were carried out two times and produced similar results.
2.5. Resistance Study
The first MIC determination of polymers and two antibiotics CIP and NOR against E. coli ATCC 25922 was performed as described above. Bacteria samples from duplicate wells at the concentration of one-half MIC were removed and used to prepare the bacterial inoculation (5 × 105 CFU/mL) for the next experiment. This bacteria solution was then replaced on new 96-well plates with fresh dilutions of compounds. After incubation for 18 h at 37 °C, the change of MIC values was determined. This experiment was repeated each day for 21 successive passages.
2.6. Hemolysis
Human red blood cells (RBCs; 1 mL) were suspended in 9 mL of TBS buffer (10 mM Tris buffer, 150 mM NaCl, pH 7.3) and rinsed three times. The cell suspension (1 mL) was diluted by 29 mL of TBS to give 3.3% RBC (v/v). Cells were counted using a hematocytometer and ∼2 × 107 cells were seeded per well. Polymer solutions (10 μL) and the RBCs (90 μL) were mixed on a 96-well assay plate (Corning #3359), and incubated with orbital shaking at 37 °C. After 1 h, the plate was centrifuged at 780 × g for 5 min, and an aliquot of supernatant (8 μL) from each well was diluted within TBS buffer (92 μL), and the absorbance of the released hemoglobin at 415 nm was measured. Hemolysis was determined relative to the positive lysis control TRITON-X (0.1 % v/v in water) and negative control buffer. HC50 was defined as the polymer concentration causing 50% hemolysis, which was estimated with 95% confidence intervals by a curve fitting with the following equation: H = 100/(1 + (HC50/[polymer])n) where H is the percentage of hemolysis measured, and [polymer] is the total concentration of polymer. The fitting parameters were n and HC50. Final HC50 is reported as the average value from at least three experiments independently performed in triplicate on different days.
2.7. Inner Membrane Permeability Assay
A colony of Escherichia coli D31 was inoculated in a solution of Mueller-Hinton Broth (Difco) and Isopropyl β-D-1-thiogalactopyranoside (IPTG) (2 mM). The culture was incubated at 37° for 18 h. The overnight culture was then diluted 1:100 in fresh media and incubated at 37° until the OD600 was 0.200–0.500. Using a 96-well polystyrene plate, PMAs and CTAB (cetyl trimethyl ammonium bromide) were serially diluted from stocks. Next Z-buffer (56.25 μL) was added to each well followed the E. coli culture (18.75 μL). Just prior to reading, ortho-Nitrophenyl-β-galactoside (ONPG) dissolved in Z-buffer (15 μL, 4 mg/mL) was added to the wells. The absorbance at 420 nm was then monitored using a ThermoSkan plate reader for 90 min with shaking between individual readings to prevent cell settling. All assays were performed at least in triplicate.
2.8. Outer Membrane Permeability Assay
A colony of Escherichia coli D31 was inoculated in a solution of Mueller-Hinton Broth (Difco) and ampicillin (100 μg/mL). The culture was incubated at 37° for 18 h and then diluted 1:100 in fresh media and incubated at 37° until the OD600 was ≈0.200. The culture was then centrifuged at 2,500 rpm for 15 min, and the pellet resuspended in an equal volume of PBS buffer (10 mM phosphate, 200 mM NaCl, pH 7.0). The resuspended bacterial cells were added to the wells of a 96-well plate containing serially diluted PMAs or polymyxin B as a control. Nitrocefin was added to the solution to a concentration of 50 μg/mL. The plate was then monitored on a plate reader in the same format as the IM permeability procedure, except the sample absorbance was measured at 486 nm. All assays were performed at least in triplicate.
3. Results and Discussion
3.1. Activity Spectrum
Amphiphilic polymethacrylate derivatives (PMAs) containing cationic and hydrophobic alkyl side chains (methyl or butyl; Figure 1) in random sequence (Table 1) were prepared by free radical polymerization in the presence of a chain transfer agent, according to our previous report [40]. The polymers have low molecular weights of 2.2–2.8 kDa, which are comparable to those of the natural peptides magainin-2 and melittin. The hydrophobicity of polymers was varied by altering mole percentage of alkyl groups relative to the total number of monomeric units (MPalkyl) and by the length of the alkyl chains (methyl or butyl groups). The polydispersities of these polymers were unable to be determined due to the low solubility to a standard GPC solvent (THF). In our previous study, the boc-protected precursors of selected polymers prepared by the same polymerization method displayed a polydispersity index (PDI) of <1.5. We speculate that the polymers studied in this report also have a similar PDI. The PMA polymers are denoted as PMx or PBx (PM: copolymers with methyl groups, PB: copolymer with butyl groups, subscript x: mole percentage of alkyl side chains relative to the total number of monomeric units in a polymer chain, MPalkyl); for example, the PMA containing 47% methyl groups is referred to as PM47.
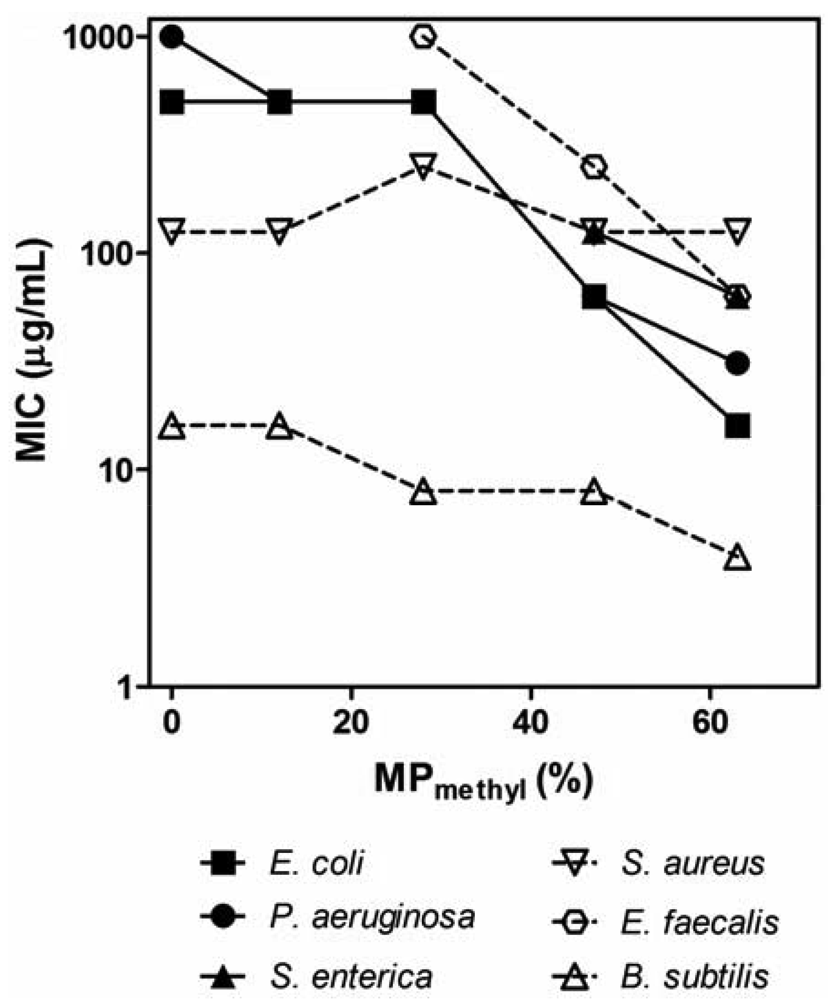
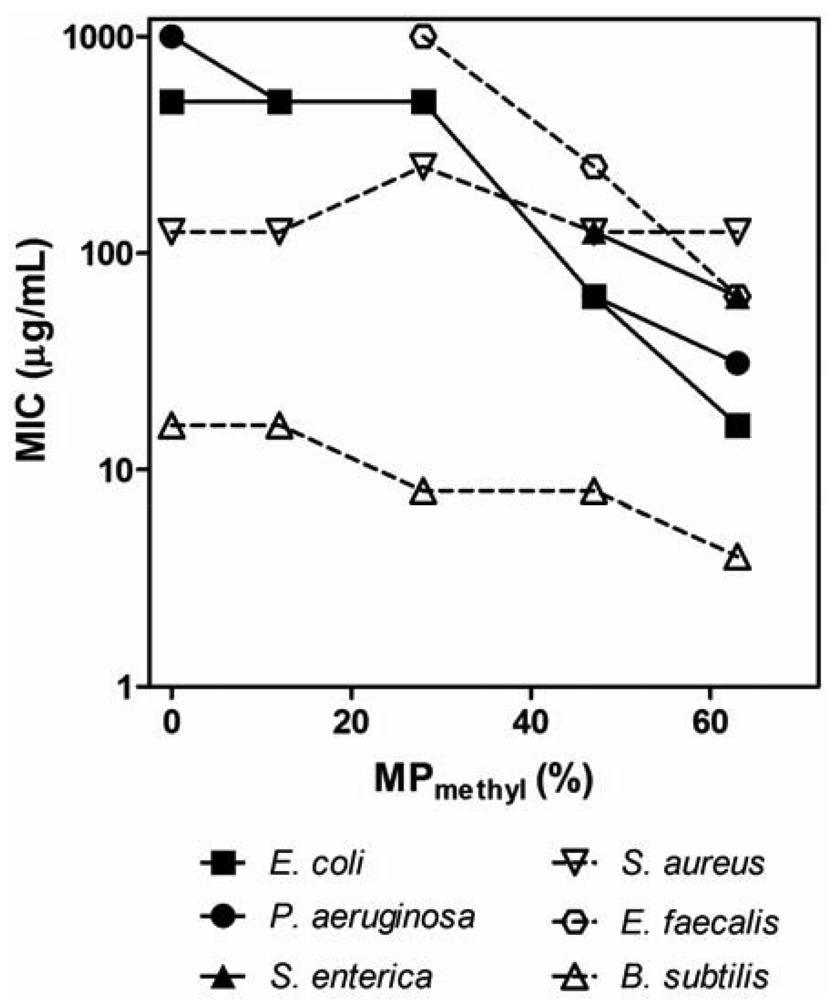
The PMAs were tested against a panel of clinically relevant bacterial pathogens (Table 2). We determined the minimum inhibitory concentration (MIC) by a turbidity-based broth microdilution assay as a first assessment of antibacterial activity of polymers. In general, the activity of PMAs critically depends on the hydrophobicity of the PMAs and the species of bacteria. In general, the MIC values of PM polymers (copolymers with methyl groups) for Gram-negative bacteria tested here decreased as the MPalkyl was increased (Figure 2). The homopolymer P0 (no hydrophobic groups) displayed a high MIC of ≥500 μg/mL, and the MIC of PM copolymers started to decrease by an order of magnitude around MP = 30% and reached low MIC (16–31 μg/mL) at the higher MPalkyl contents. This indicates that the polymer hydrophobicity enhances the antimicrobial activity, which is in agreement with the previous reports on PMAs [39,40]. As the MPalkyl is increased, the number of cationic amine groups decrease, which should reduce the electrostatic binding of polymers to negatively charged bacterial surfaces, potentially resulting in lower activity. However, the activity was enhanced with increasing MPalkyl, which indicates that the effect of hydrophobicity appears to be more influential factor in the polymers' antimicrobial activity. Although PMAs with MPalkyl higher than 63% were not examined, the activity could potentially be increased; however, the PMAs with high MPalkyl often precipitate in MH broth due to non-specific binding to the components in the broth [30]. The PMAs also likely form solution aggregates due to association of alkyl groups [39] which may result in no activity enhancement or even a decrease in activity. The MIC values at high MPs are lower or compatible to those of magainin-2 and melittin, indicating that the antibacterial activities of PMAs are comparable to or even higher than those of the natural peptides. The MIC for Gram-positive Enterococcus faecalis and Bacillus subtilis also decreased with increasing the MP in parallel with the results for Gram-negative bacteria. However, increasing MPmethyl from 10% to 63% had no or little effect on the MIC against S. aureus, suggesting that the activity of PMAs against S. aureus is not strongly sensitive to changes in polymer hydrophobicity or the number of cationic groups in contrast to other bacteria. Notably, all polymers tested were active against the methicillin-resistant S. aureus (MRSA) with the same MICs as against the susceptible strain, although the control peptide magainin-2 was not active against MRSA (MIC > 500 μg/mL). The P0 and other PMAs with low MPmethyl tend to be more selective to S. aureus and B. subtilis over other bacteria tested. Lienkamp et al. previously reported that cationic polynorbornenes showed significantly higher activity against S. aureus compared to E. coli [47,48]. The authors demonstrated that the double membrane structure of E. coil cell wall likely limits the access of the polymers to the cytoplasmic membrane [48]. Epand et al. reported that an acyl-Lys oligomer is bacteriostatic, and it does not compromise the cell membrane of S. aureus [49]. The authors speculate that the cell wall binding of acyl-Lys oligomer limits the diffusion of nutrients from extracellular environment and causes starvation. The same molecular mechanisms may explain the activity of PMAs in part; however, the basis of this effect by PMAs remains unclear. In addition, the selected PMAs were also highly active (MIC ≤ 2 μg/mL) against anaerobic bacteria P. acnes which is a major etiological agent of acne vulgaris; increasing resistance of this bacteria to standard antibiotic therapies reduces the efficacy of therapeutic agents and often leads to failure of dermatological treatment [50,51].
As a first assessment of polymer toxicity, we evaluated the hemolytic activity of PMAs against human red blood cells (RBCs). The hemolytic activity was determined by monitoring release of hemoglobin into solution from RBCs, which reflects membrane permeabilization and cell lysis induced by the polymers [46]. The homopolymer and PM polymers did not exhibit adverse hemolytic activities (HC50 > 2,000 μg/mL), except for PM63 which showed an HC50 value of 114 μg/mL (Table 2) [46]. To quantify the selectivity of polymers against bacteria over RBCs, selectivity index, defined as HC50/MIC, for Gram-negative E. coli and Gram-positive S. aureus as representatives, was summarized in Table 3. Examining the selectivity indexes, the polymers PM47 and PM63 are relatively selective against bacteria over RBCs (Table 3).
The PMA with longer alkyl chains (butyl), PB27, exhibited MIC at low μg/mL against all tested bacteria. PB27 displayed an order of magnitude lower MIC value (MIC = 16 μg/mL against E. coli) than PM28 (MIC = 500 μg/mL) while they have the similar mole percentages of hydrophobic groups (27–28 mol %) in a polymer chain and polymer length (DP = 15–16). This result indicates that polymers with more hydrophobic side chains are more active. Considering the activity enhancement with increasing MPalkyl for the PM polymers, these results support the notion that the overall hydrophobicity of PMAs increases the activity. PB27 also caused significant hemolysis with an HC50 value of 13 μg/mL. The hemolytic activity of PB27 likely reflects the hydrophobicity of butyl side chains; increasing the hydrophobicity beyond a threshold results in the loss of selectivity because the hydrophobic nature of polymers causes non-specific binding of polymers to RBCs and disruption of the cell membrane. This result is in agreement with our previous studies [39,40].
It has been previously reported that synthetic cationic polymers including random nylon copolymers [27,52], cationic polynorbones [53], poly(2-(dimethylaminoethyl)methacrylate derivatives [54], and oligo(oxazoline)s [36] are broad spectrum antimicrobials. In addition, a recent report from Wynne and coworkers indicated that random copolyoxetanes with PEG-like groups and C12-modified quaternary ammonium groups in the side chains showed MICs of low μg/mL against selected Gram-positive and negative bacteria [38]. These studies also support the notion that the polymer design using amphiphilic polycationic structures would have potential for new development of antimicrobials. It should be noted that the MIC and HC50 values strongly depend on the assay conditions including bacterial strains, broth, assay plates, incubation time, etc. [55]. Therefore, the quantitative comparison of MIC and HC50 values may be ambiguous. Accordingly, we limited the discussion in the qualitative comparison of activity profiles between other reports and the results in this study.
3.2. Bactericidal Kinetics
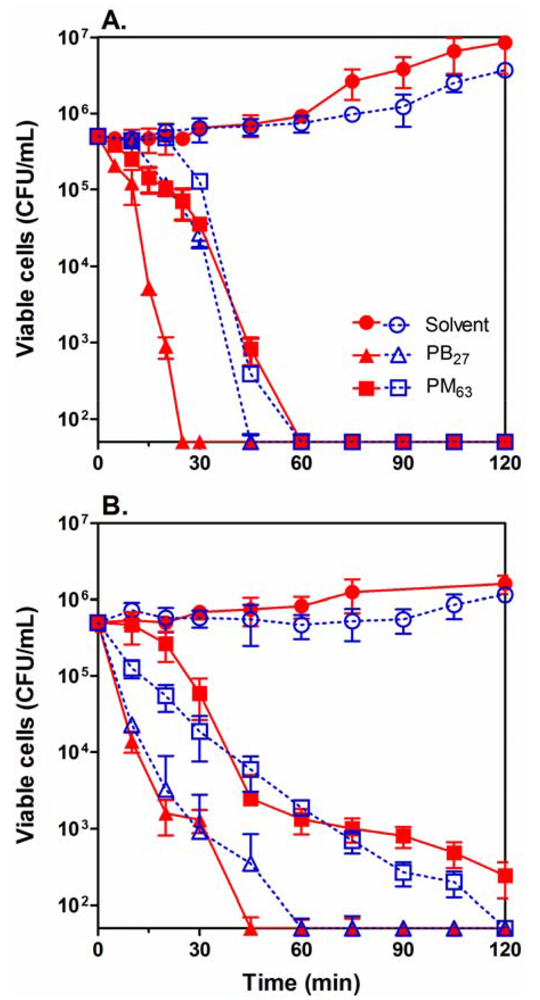
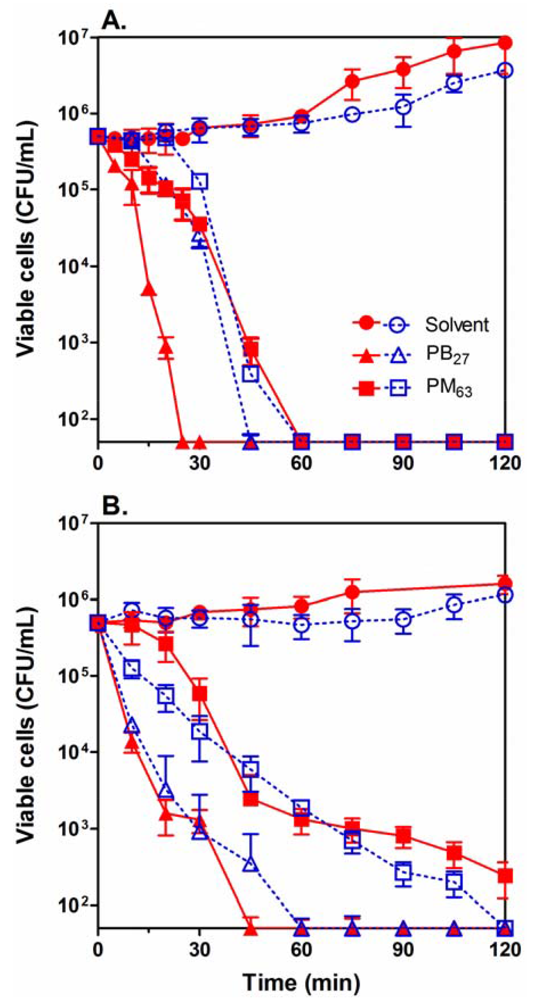
We next determined bactericidal kinetics for selected PMAs, PM63 and PB27, against Gram-negative E. coli and Gram-positive S. aureus (Figure 3). As shown in Table 3, PM63 is a relatively selective antimicrobial agent against E. coli over RBCs, and PB27 exhibits non-selective biocidal characteristics which make these two polymers interesting models to examine with respect to their mechanism and characteristics. In order to examine the effect of bacterial growth phase on the activity of the polymers, the bacteria were subcultured from either the exponential or stationary growth phases. At a concentration of two times MIC, the polymers reduced the number of viable cells by 4-log or 99.99% within 60 min for E. coli and 60–120 min for S. aureus in both cultures. This implies that the killing kinetics of the PMAs is independent of the growth phase of the bacteria (Figure 3). It should be noted that PB27 showed somewhat faster killing of E. coli in the exponential phase relative to the stationary phase. It is, however, not clear at this point why PB27 displayed the different behavior. These results indicate that the PMAs are bactericidal against bacteria in both exponential and stationary phases, which suggests that the mechanism may not rely on any growth phase-specific metabolic activity or cellular physiology of the bacterial cells.
3.3. PMA Resistance
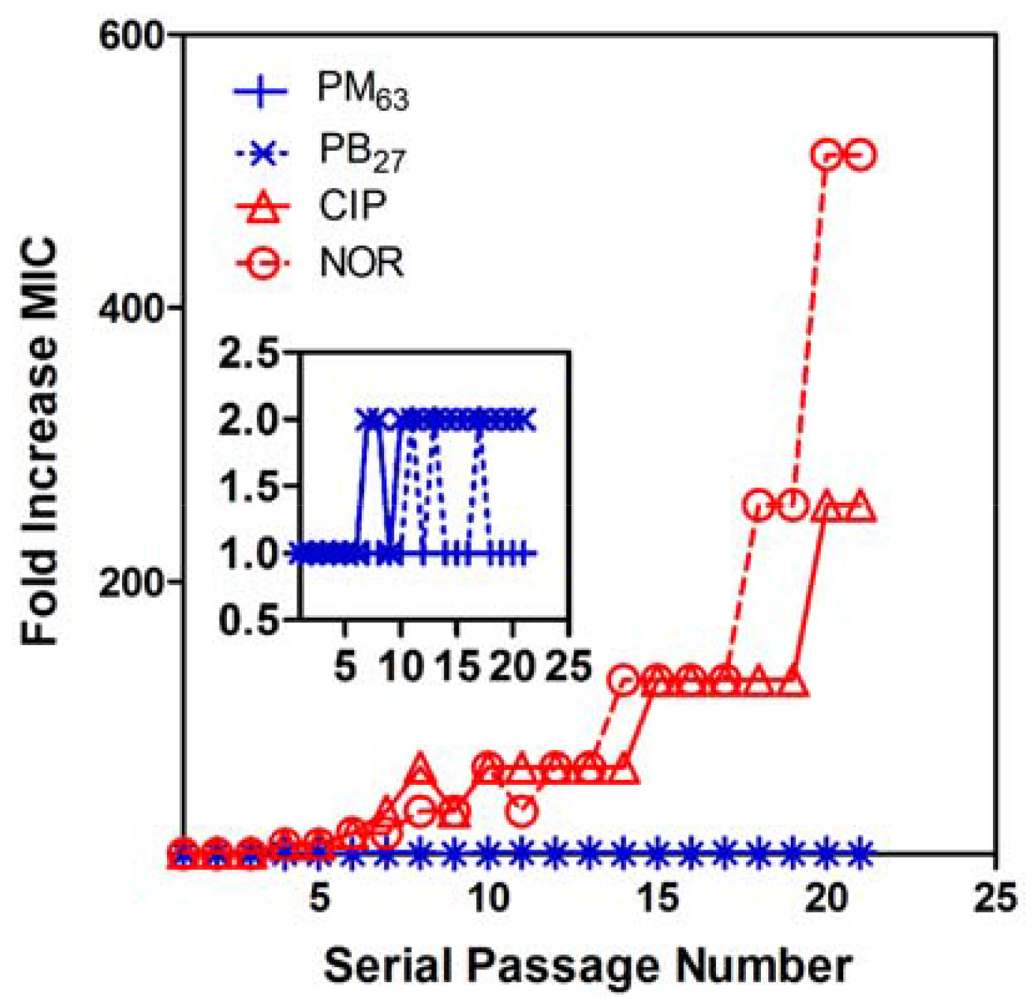
One of the hallmarks of natural antimicrobial peptides is low level of resistance development in bacteria. In order to examine the likelihood of development of PMA-resistant bacteria, E. coli were serially subcultured in MHB containing sub-lethal concentrations (one-half MIC) of selected polymers of the selective PM63 and biocidal PB27 for up to 21 passages. The MIC values of PM63 and PB27 did not increase by more than a single two-fold dilution throughout the experiment (Figure 4). In contrast, the MIC values of the antibiotic fluoroquinolones norfloxacin (NOR) and ciprofloxacin (CIP), which inhibit DNA synthesis [56,57], started to increase after as few as four passages and finally increased 512-fold and 256-fold, respectively. After the 21st passage, the obtained NOR- and CIP-resistant strains of E. coli were subcultured in antibiotic-free medium. After 15 antibiotic-free passages, the acquired resistance was persistent, indicating that the antibiotic resistance was not simply a physiological adaption. These CIP- and NOR-resistant strains were both still fully susceptible to PMAs, with the same MIC values as the CIP- and NOR-susceptible strains. Also, the E. coli incubated with PM63 and PB27 were still susceptible to CIP and NOR, with no increase in their MIC values after 21 passages. These results indicate that no cross-resistance developed between these fluoroquinolone antibiotics and the selected antibacterial PMAs tested here. It has been also reported that membrane-active cationic oligomers displayed no development of resistance in S. aureus [58,59]. These results highlight the potential of amphiphilic PMAs as an attractive class of new antimicrobials.
3.4. Salt Effects
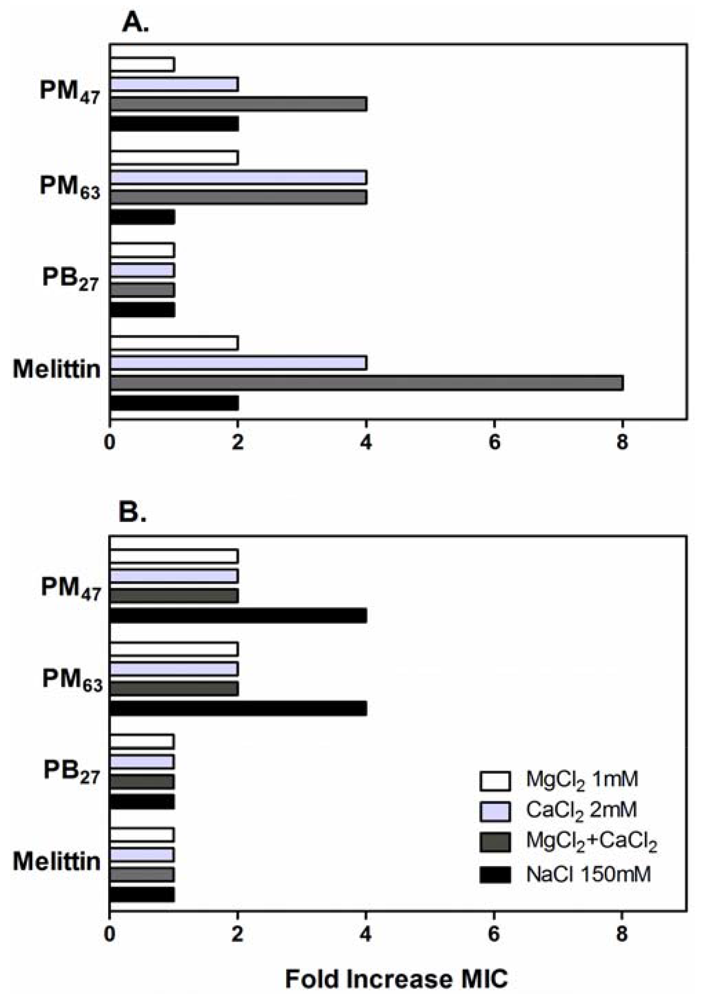
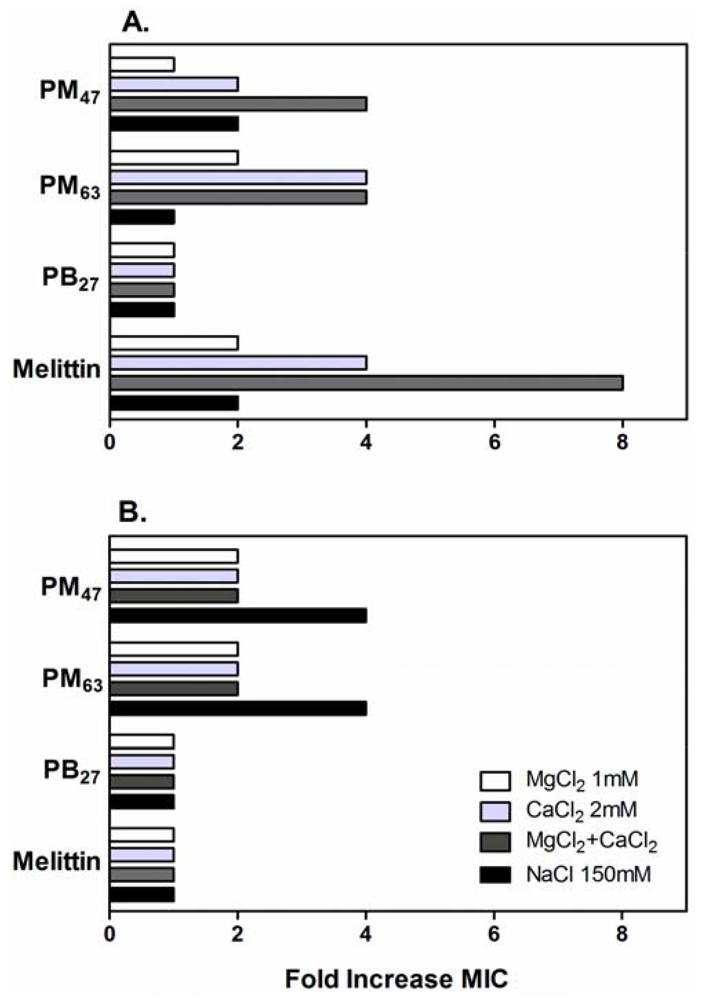
It has been reported that the activity of antimicrobial peptides was significantly reduced in high ionic strength solutions [60-62]. It has been postulated that the increased salt concentration screens the electrostatic attraction between the cationic peptides and anionic binding sites on the bacterial cell envelopes where potential polymer binding sites such as anionic phospholipids, lipopolysaccharides (LPS) in Gram-negative bacteria, and teichoic acids in Gram-positive bacteria are occupied by the salt cations [63,64]. This masking effect reduces the affinity of antimicrobial peptides for bacterial surface, resulting in reduction of antibacterial activity. Similarly, the physiological salts could impair the electrostatic binding of the cationic PMAs to bacteria. In addition, since the salt ions also curtain the cationic charges of polymer side chains, the polymers may have more compact conformations or undergo aggregation under these conditions, which could reduce the effective concentration of active polymer chains and result in lower activity. The antibacterial activity of PB27 was shown to be salt-resistant, indicating that the strong hydrophobicity of PB27 likely outweighs the contribution of electrostatic affinity to binding with anionic targets on bacteria cell membranes.
In order to investigate these possibilities, the effects of additional physiological salts in MH broth on the antibacterial activity of the selected PMAs were determined. The activity of these PMAs against E. coli and S. aureus was inhibited by the addition of physiological salts as evidenced by up to an eight-fold increase in the MIC values (Figure 5). The MICs of PM47 and PM63 against E. coli increased two- to four-fold by addition of CaCl2 and MgCl2 to the growth medium. These salt conditions had no effect on the activity of PB27, while the activity of melittin decreased four- to eight-fold. In contrast to CaCl2 and MgCl2, the addition of NaCl did not significantly affect the activity of PMAs against E. coli, even at a substantially higher NaCl concentration (150 mM) than those of CaCl2 (2 mM) or MgCl2 (1 mM). The MIC values of PMAs and melittin against S. aureus in the presence of CaCl2 and MgCl2 were equal to or two-fold greater than those in MH broth without salt additives. In addition, the MICs of PM47 and PM63 for S. aureus in the presence of NaCl increased four-fold.
In addition to potential effects on PMA binding to bacterial surfaces, the divalent cations Mg2+ and Ca2+ can neutralize and bridge the phosphate groups of LPS molecules on the outer membrane of Gram-negative strains, which is essential to maintaining the membrane integrity [65,66]. Hence, the inhibitory effect of Mg2+ and Ca2+ on the activity of PMAs against E. coli may indicate that the antibacterial mechanism involves polymer binding to LPS and subsequent disruption of E. coli outer membrane.
3.5. Permeabilization of E. coli Membranes
In order to probe the antimicrobial mechanism of PMAs, we determined the effect of PMAs on the permeability of the outer (OM) and inner membranes (IM) of E. coli. The assays measure the ability of a chromogenic substrate to cross either the OM or the OM and IM to access a periplasmic or cytoplasmic enzyme, respectively. The OM permeability assay uses the periplasmic enzyme β-lactamase and the chromogenic substrate nitrocefin while the IM assay employs the cytoplasmic enzyme β-galactosidase and the chromogenic substrate ortho-nitrophenyl-β-galactoside (ONPG). These substrates show very low permeability across the membrane under control conditions, but the permeability is enhanced if a polymer, peptide, or other membrane disrupting agent is present [67]. As the substrates cross the membrane(s), the appropriate enzymes produce the chromogenic product which can be monitored by changes in the absorbance in the samples [68,69].
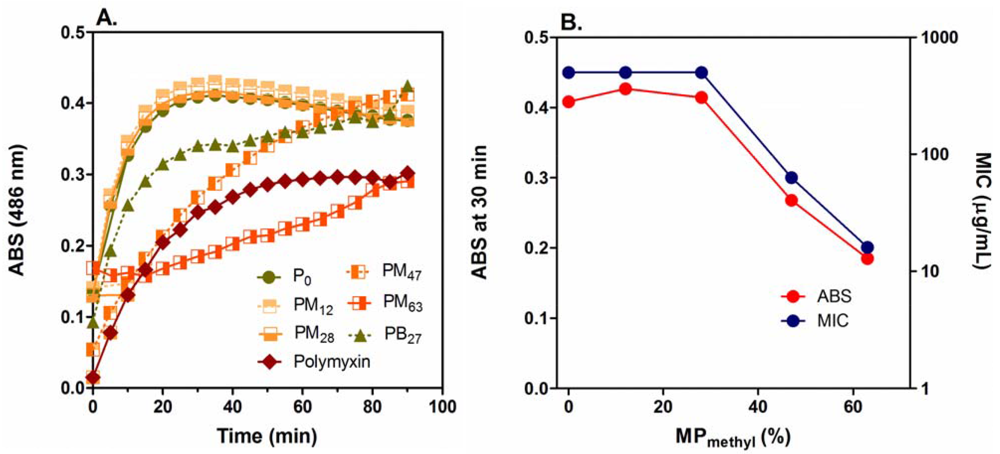
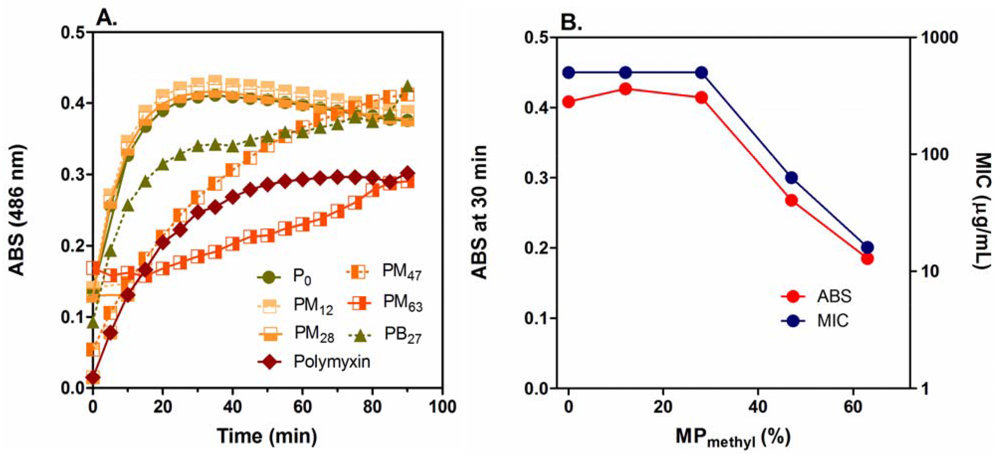
The PMAs did not cause any permeabilization of the IM up to their MIC concentrations (Figure S1 in Supplementary Data) although they disrupt the OM (Figure 6(A)) (discussed below) and model membranes (Figure S2 in Supplementary Data) at polymer concentrations lower than MICs. This indicates that the IM permeabilization may not be the primary factor for bacterial growth inhibition or killing by the PMAs, and other mechanisms may exert the antimicrobial effect against E. coli. On the other hand, the PMAs induced significant permeabilization of the OM at low μg/mL concentrations (Figure 6(A)) compared to the positive control of polymyxin B. Cationic antimicrobial peptides are known to permeabilize the E. coli OM, which promotes uptake of the peptides into the peptidoglycan layer (self-promoted uptake) [70]. The OM permeabilization by PMAs could also enhance their access to the peptidoglycan layers. The inhibition of PMA activity by CaCl2 and MgCl2 (Figure 5), which stabilizes the OM structure, may support the notion that the PMA antimicrobial mechanism involves OM permeabilization as uptake process to cell surfaces.
The homopolymer P0 and copolymers PM12 and PM28 displayed the same OM permeabilization effect at 16 μg/mL and relatively high production rates of products although these polymers are much less active against E. coli (MIC = 500 μg/mL) than other more active polymers. The MIC value and absorbance (product formation) at 30 min after incubation (Figure 6(C)) are constant up to 30% MPmethyl and decreased as the MPmethyl was further increased. This suggests that PMAs with more cationic groups are more advantageous in OM permeabilization, but they are less active against E. coli. This reverse relationship between the activity and OM permeabilization suggests that the ability of PMAs to permeabilize the E. coli OM does not directly reflect their antibacterial activity. This observed phenomenon may be a function of kinetically “trapping” the most cationic PMAs in the negatively charged lipopolysaccharide layer of the OM and peptidoglycan layer resulting in altered OM permeability but an inability to transit to the IM. In addition, since the PMAs could permeabilize model membranes at low μg/mL concentrations (Supplementary Data), the concentration of polymers which transient to the IM would be significantly lower than in bulk solution or the total bound to the bacterial cell. This could support the notion that the cationic PMAs are trapped in the cell wall structures before reaching to the IM.
It has been reported that some peptides and synthetic mimics exert their antimicrobial effect by mechanisms other than permeabilization of inner or cytoplasmic membranes. For example, the antimicrobial peptide buforin II penetrates through bacterial cell membranes and interacts with DNA/RNA leading to lethal action [71], while the peptide mersacidin has been shown to inhibit cell wall synthesis by inhibiting lipid II synthesis [2,72]. A synthetic polynorbornene modified with guanidine groups also showed antimicrobial activity without disrupting bacterial membranes [53]. Pleurocin-derived peptides inhibit macromolecular synthesis in E. coli at sub-lethal concentrations but did not induce IM permeabilization over the same concentration range [73]. Similarly, PMAs could diffuse into the peptidoglycan layers after the OM permeabilization and perturb peptidoglycan integrity and functions, which may result in inhibition of the cell wall synthesis or cellular uptake of metabolites, which has been postulated as a potential component of the mechanism of action of some peptides [74]. The PMAs might also penetrate the inner membranes without significant disruption and interact with intracellular targets. Although the molecular mechanisms and cellular targets remain unclear at this point, PMAs could have multiple targets, and the underlying individual mechanisms may not be mutually exclusive, but rather complementary or synergistic. Further investigation will provide insight into the mode and mechanism of antimicrobial action of these polymers.
4. Conclusions
In this report, we characterized the bactericidal action and hemocompatibility of PMAs in order to assess their potential for biomedical applications. The PMAs studied here displayed a broad spectrum of activity against clinically important bacterial pathogens, including the antibiotic-resistant S. aureus, with MIC values lower than that of the natural host-defense peptide magainin-2. The activity of PMAs showed bactericidal activity against E. coli and S. aureus subcultured from both exponential and stationary growth phases. In addition, PMAs showed low susceptibility to resistance development in E. coli. Furthermore, no cross-resistance with the traditional antibiotics CIP and NOR was detected after 21 passages in the presence of PMAs. The activity of PMAs was only slightly reduced in the presence of physiological salts. Since these polymers are not likely to form defined secondary structures such as an α-helix, the biological activity of amphiphilic PMAs does not rely on the polymer conformations, but rather the physical properties of cationic functionality and hydrophobicity in the polymer chain. Therefore, the design principles gleaned from studies on PMAs may be applicable to other synthetic polymer backbones. Taken together, these results demonstrate that PMAs are a new, versatile antimicrobial platform without any observed risk of resistance development in bacteria.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer b | R c | MPalkyl d(%) | DP e | MW f(kDa) |
|---|---|---|---|---|
| P0 | - | 0 | 14 | 2.4 |
| PM12 | methyl | 12 | 14 | 2.3 |
| PM28 | methyl | 28 | 15 | 2.3 |
| PM47 | methyl | 47 | 20 | 2.8 |
| PM63 | methyl | 63 | 17 | 2.2 |
| PB27 | butyl | 27 | 16 | 2.7 |
| Magainin-2 | - | - | 23 g | 2.5 |
| Melittin | - | - | 26g | 2.8 |
apreviously reported in Reference [46];bThe mole % of alkyl methacrylate is denoted in subscript x of PMx or PBx;cThe type of alkyl side chain;dMole percentage of alkyl side chains, determined by 1H NMR analysis;eDegree of polymerization, determined by 1H NMR analysis;fMolecular weight of polymers, calculated from the molecular weight of monomers and chain transfer agent, MPalkyl, and DP;gThe number of amino acid residues of magainin 2 [36] and melittin [37].
| Bacteria | MIC or HC50a (μg/mL) | |||||||
|---|---|---|---|---|---|---|---|---|
| P0 | PM12 | PM28 | PM47 | PM63 | PB27 | Mag b | Mel c | |
| Gram negative | ||||||||
| E. coli | 500 | 500 | 500 | 63 | 16 | 16 | 125 | 13 |
| P. aeruginosa | 1,000 | 500 | 500 | 63 | 31 | 31 | 500 | 100 |
| S. enterica | >1,000 | >1,000 | >500 | 125 | 63 | 31 | 500 | 50 |
| Gram positive | ||||||||
| S. aureus | 125 | 125 | 250 | 125 | 125 | 16 | >500 | 6 |
| S. aureus (MRSA e) | 125 | 125 | 250 | 125 | 125 | 16 | >500 | 3 |
| E. faecalis | >1,000 | >1,000 | 1,000 | 250 | 63 | 16 | >500 | 6 |
| B. subtilis | 16 | 16 | 8 | 8 | 4 | 4 | 63 | 2 |
| P. acnes | n.d. | n.d. | n.d. | ≤2 | 2 | ≤0.5 | n.d. | n.d. |
| Human red blood | >2,000 | >2,000 | >2,00 | >2,000 | 114 | 13 | >250 | 2 |
| cells (HC50) d | (12%) | (35 percnt;) | (3 percnt;) | (7 percnt;) | (9 percnt;) | |||
aHC50: Polymer concentration for 50% hemolysis, previously reported in reference [46];bMagainin-2;cMelittin;dThe hemolysis percentage at the highest polymer concentration (2,000 μg/mL) is given in the parenthesis;eMethicillin-resistant S. aureus.
| Polymer | Selectivity index (HC50/MIC) | |
|---|---|---|
| E. coli | S. aureus | |
| P0 | >4 | >16 |
| PM12 | >4 | >16 |
| PM28 | >4 | >8 |
| PM47 | >32 | >16 |
| PM63 | 7.1 | 0.91 |
| PB | 0.81 | 0.81 |
| Magainin-2 | >2 | - |
| Melittin | 0.15 | 0.33 |
Acknowledgments
This research was supported by the Department of Biologic and Materials Sciences, University of Michigan School of Dentistry. A part of research was supported by NSF Career Award (DMR-0845592) to KK. GC acknowledges support from the Rowan University NSFSG program for support of a portion of this work. We thank Robert Davenport at the University of Michigan Hospital for supplying the red blood cells. K. Kuroda is a coinventor on a patent application filed by the University of Pennsylvania covering “Antimicrobial Copolymers and Uses Thereof”. The patent application has been licensed to PolyMedix Inc. (Radnor, PA). PolyMedix did not play a role in the design and conduct of this study, in the collection, analysis, or interpretation of the data, or in the preparation, review, or approval of the article.
References
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar]
- Dathe, M.; Nikolenko, H.; Meyer, J.; Beyermann, M.; Bienert, M. Optimization of the antimicrobial activity of magainin peptides by modification of charge. FEBS Lett. 2001, 501, 146–150. [Google Scholar]
- Dathe, M.; Wieprecht, T.; Nikolenko, H.; Handel, L.; Maloy, W.L.; MacDonald, D.L.; Beyermann, M.; Bienert, M. Hydrophobicity, hydrophobic moment and angle subtended by charged residues modulate antibacterial and haemolytic activity of amphipathic helical peptides. FEBS Lett. 1997, 403, 208–212. [Google Scholar]
- Jiang, Z.Q.; Vasil, A.I.; Hale, J.D.; Hancock, R.E.W.; Vasil, M.L.; Hodges, R.S. Effects of net charge and the number of positively charged residues on the biological activity of amphipathic alpha-helical cationic antimicrobial peptides. Biopolymers 2008, 90, 369–383. [Google Scholar]
- Matsuzaki, K.; Nakamura, A.; Murase, O.; Sugishita, K.; Fujii, N.; Miyajima, K. Modulation of magainin 2-lipid bilayer interactions by peptide charge. Biochemistry 1997, 36, 2104–2111. [Google Scholar]
- Krishnakumari, V.; Singh, S.; Nagaraj, R. Antibacterial activities of synthetic peptides corresponding to the carboxy-terminal region of human beta-defensins 1-3. Peptides 2006, 27, 2607–2613. [Google Scholar]
- Leippe, M.; Andra, J.; Mullereberhard, H.J. Cytolytic and antibacterial activity of synthetic peptides derived from amoebapore, the pore-forming peptide of entamoeba-histolytica. Proc. Natl. Acad. Sci. USA 1994, 91, 2602–2606. [Google Scholar]
- Nakajima, Y.; AlvarezBravo, J.; Cho, J.H.; Homma, K.; Kanegasaki, S.; Natori, S. Chemotherapeutic activity of synthetic antimicrobial peptides: Correlation between chemotherapeutic activity and neutrophil-activating activity. FEBS Lett. 1997, 415, 64–66. [Google Scholar]
- Hamuro, Y.; Schneider, J.P.; DeGrado, W.F. De novo design of antibacterial beta-peptides. J. Am. Chem. Soc. 1999, 121, 12200–12201. [Google Scholar]
- Liu, D.H.; DeGrado, W.F. De novo design, synthesis, and characterization of antimicrobial beta-peptides. J. Am. Chem. Soc. 2001, 123, 7553–7559. [Google Scholar]
- Porter, E.A.; Weisblum, B.; Gellman, S.H. Mimicry of host-defense peptides by unnatural oligomers: Antimicrobial beta-peptides. J. Am. Chem. Soc. 2002, 124, 7324–7330. [Google Scholar]
- Ivankin, A.; Livne, L.; Mor, A.; Caputo, G.A.; DeGrado, W.F.; Meron, M.; Lin, B.; Gidalevitz, D. Role of the conformational rigidity in the design of biomimetic antimicrobial compounds. Angew. Chem. Int. Ed. 2010, 49, 8462–8465. [Google Scholar]
- Chongsiriwatana, N.P.; Patch, J.A.; Czyzewski, A.M.; Dohm, M.T.; Ivankin, A.; Gidalevitz, D.; Zuckermann, R.N.; Barron, A.E. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc. Natl. Acad. Sci. USA 2008, 105, 2794–2799. [Google Scholar]
- Patch, J.A.; Barron, A.E. Helical peptoid mimics of magainin-2 amide. J. Am. Chem. Soc. 2003, 125, 12092–12093. [Google Scholar]
- Chen, C.Z.S.; Beck-Tan, N.C.; Dhurjati, P.; van Dyk, T.K.; LaRossa, R.A.; Cooper, S.L. Quaternary ammonium functionalized poly(propylene imine) dendrimers as effective antimicrobials: Structure-activity studies. Biomacromolecules 2000, 1, 473–480. [Google Scholar]
- Dizman, B.; Elasri, M.O.; Mathias, L.J. Synthesis and antibacterial activities of water-soluble methacrylate polymers containing quaternary ammonium compounds. J. Polym. Sci. Part A: Polym. Chem. 2006, 44, 5965–5973. [Google Scholar]
- Ikeda, T.; Hirayama, H.; Yamaguchi, H.; Tazuke, S.; Watanabe, M. polycationic biocides with pendant active groups—molecular-weight dependence of antibacterial activity. Antimicrob. Agents Chemother. 1986, 30, 132–136. [Google Scholar]
- Kenawy, E.R.; Worley, S.D.; Broughton, R. The chemistry and applications of antimicrobial polymers: A state-of-the-art review. Biomacromolecules 2007, 8, 1359–1384. [Google Scholar]
- Tashiro, T. Antibacterial and bacterium adsorbing macromolecules. Macromol. Mater. Eng. 2001, 286, 63–87. [Google Scholar]
- Kugler, R.; Bouloussa, O.; Rondelez, F. Evidence of a charge-density threshold for optimum efficiency of biocidal cationic surfaces. Microbiology 2005, 151, 1341–1348. [Google Scholar]
- Lee, S.B.; Koepsel, R.R.; Morley, S.W.; Matyjaszewski, K.; Sun, Y.J.; Russell, A.J. Permanent, nonleaching antibacterial surfaces. 1. Synthesis by atom transfer radical polymerization. Biomacromolecules 2004, 5, 877–882. [Google Scholar]
- Milovic, N.M.; Wang, J.; Lewis, K.; Klibanov, A.M. Immobilized N-alkylated polyethylenimine avidly kills bacteria by rupturing cell membranes with no resistance developed. Biotechnol. Bioeng. 2005, 90, 715–722. [Google Scholar]
- Murata, H.; Koepsel, R.R.; Matyjaszewski, K.; Russell, A.J. Permanent, non-leaching antibacterial surfaces-2: How high density cationic surfaces kill bacterial cells. Biomaterials 2007, 28, 4870–4879. [Google Scholar]
- Tiller, J.C.; Liao, C.J.; Lewis, K.; Klibanov, A.M. Designing surfaces that kill bacteria on contact. Proc. Natl. Acad. Sci. USA 2001, 98, 5981–5985. [Google Scholar]
- Ikeda, T.; Tazuke, S.; Suzuki, Y. Biologically-active polycations, 4. Synthesis and antimicrobial activity of poly(trialkylvinylbenzylammonium chloride)s. Makromol. Chem. 1984, 185, 869–876. [Google Scholar]
- Mowery, B.P.; Lee, S.E.; Kissounko, D.A.; Epand, R.F.; Epand, R.M.; Weisblum, B.; Stahl, S.S.; Gellman, S.H. Mimicry of antimicrobial host-defense peptides by random copolymers. J. Am. Chem. Soc. 2007, 129, 15474–15476. [Google Scholar]
- Ilker, M.F.; Schule, H.; Coughlin, E.B. Modular norbornene derivatives for the preparation of well-defined amphiphilic polymers: Study of the lipid membrane disruption activities. Macromolecules 2004, 37, 694–700. [Google Scholar]
- Kenawy, E.R.; Abdel-Hay, F.I.; El-Raheem, A.; El-Shanshoury, R.; El-Newehy, M.H. Biologically active polymers: Synthesis and antimicrobial activity of modified glycidyl methacrylate polymers having a quaternary ammonium and phosphonium groups. J. Controlled Release 1998, 50, 145–152. [Google Scholar]
- Kuroda, K.; DeGrado, W.F. Amphiphilic polymethacrylate derivatives as antimicrobial agents. J. Am. Chem. Soc. 2005, 127, 4128–4129. [Google Scholar]
- Gelman, M.A.; Weisblum, B.; Lynn, D.M.; Gellman, S.H. Biocidal activity of polystyrenes that are cationic by virtue of protonation. Org. Lett. 2004, 6, 557–560. [Google Scholar]
- Eren, T.; Som, A.; Rennie, J.R.; Nelson, C.F.; Urgina, Y.; Nusslein, K.; Coughlin, E.B.; Tew, G.N. Antibacterial and hemolytic activities of quaternary pyridinium functionalized polynorbornenes. Macromol. Chem. Phys. 2008, 209, 516–524. [Google Scholar]
- Ishitsuka, Y.; Arnt, L.; Majewski, J.; Frey, S.; Ratajczek, M.; Kjaer, K.; Tew, G.N.; Lee, K.Y.C. Amphiphilic poly(phenyleneethynylene)s can mimic antimicrobial peptide membrane disordering effect by membrane insertion. J. Am. Chem. Soc. 2006, 128, 13123–13129. [Google Scholar]
- Liu, D.H.; Choi, S.; Chen, B.; Doerksen, R.J.; Clements, D.J.; Winkler, J.D.; Klein, M.L.; DeGrado, W.F. Nontoxic membrane-active antimicrobial arylamide oligomers. Angew. Chem. Int. Ed. 2004, 43, 1158–1162. [Google Scholar]
- Tew, G.N.; Liu, D.H.; Chen, B.; Doerksen, R.J.; Kaplan, J.; Carroll, P.J.; Klein, M.L.; DeGrado, W.F. De novo design of biomimetic antimicrobial polymers. Proc. Natl. Acad. Sci. USA 2002, 99, 5110–5114. [Google Scholar]
- Correia, V.G.; Bonifácio, V.D.B.; Raje, V.P.; Casimiro, T.; Moutinho, G.; da Silva, C.L.; Pinho, M.G.; Aguiar-Ricardo, A. Oxazoline-based antimicrobial oligomers: Synthesis by CROP using supercritical CO2. Macromol. Biosci. 2011, 11, 1128–1137. [Google Scholar]
- Stratton, T.R.; Howarter, J.A.; Allison, B.C.; Applegate, B.M.; Youngblood, J.P. Structure-activity relationships of antibacterial and biocompatible copolymers. Biomacromolecules 2010, 11, 1286–1290. [Google Scholar]
- Chakrabarty, S.; King, A.; Kurt, P.; Zhang, W.; Ohman, D.E.; Wood, L.F.; Lovelace, C.; Rao, R.; Wynne, K.J. Highly effective, water-soluble, hemocompatible 1,3-propylene oxide-based antimicrobials: poly[(3,3-quaternary/PEG)-copolyoxetanes]. Biomacromolecules 2011, 12, 757–769. [Google Scholar]
- Kuroda, K.; Caputo, G.A.; DeGrado, W.F. The role of hydrophobicity in the antimicrobial and hemolytic activities of polymethacrylate derivatives. Chem. Eur. J. 2009, 15, 1123–1133. [Google Scholar]
- Palermo, E.F.; Kuroda, K. Chemical structure of cationic groups in amphiphilic polymethacrylates modulates the antimicrobial and hemolytic activities. Biomacromolecules 2009, 10, 1416–1428. [Google Scholar]
- Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; NCCLS Approved standards M7-A3; NCCLS: Wayne, PA, USA, 2003.
- Hancock, R.E.W. Hancock Laboratory Methods; Department of Microbiology and Immunology, University of British Columbia: British Columbia, Canada, 2001. Avalable online: http://www.cmdr.ubc.ca/bobh/methods.htm (accessed on 29 Auguest 2011).
- Giacometti, A.; Cirioni, O.; Barchiesi, F.; Del Prete, M.S.; Fortuna, M.; Caselli, F.; Scalise, G. In vitro susceptibility tests for cationic peptides: Comparison of broth microdilution methods for bacteria that grow aerobically. Antimicrob. Agents Chemother. 2000, 44, 1694–1696. [Google Scholar]
- Turner, J.; Cho, Y.; Dinh, N.N.; Waring, A.J.; Lehrer, R.I. Activities of LL-37, a cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob. Agents Chemother. 1998, 42, 2206–2214. [Google Scholar]
- NCCLS. Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria, 5th ed.; Approved Standard M11-A6; NCCLS: Wayne, PA, USA, 2000. [Google Scholar]
- Sovadinova, I.; Palermo, E.F.; Huang, R.; Thoma, L.M.; Kuroda, K. Mechanism of polymer-induced hemolysis: Nanosized pore formation and osmotic lysis. Biomacromolecules 2011, 12, 260–268. [Google Scholar]
- Lienkamp, K.; Madkour, A.E.; Musante, A.; Nelson, C.F.; Nusslein, K.; Tew, G.N. Antimicrobial polymers prepared by ROMP with unprecedented selectivity: A molecular construction kit approach. J. Am. Chem. Soc. 2008, 130, 9836–9843. [Google Scholar]
- Lienkamp, K.; Kumar, K.N.; Som, A.; Nusslein, K.; Tew, G.N. “Doubly selective” antimicrobial polymers: How do they differentiate between bacteria? Chem. Eur. J. 2009, 15, 11710–11714. [Google Scholar]
- Epand, R.F.; Sarig, H.; Mor, A.; Epand, R.M. Cell-wall interactions and the selective bacteriostatic activity of a miniature oligo-acyl-lysyl. Biophys. J. 2009, 97, 2250–2257. [Google Scholar]
- Eady, E.A.; Gloor, M.; Leyden, J.J. Propionibacterium acnes resistance: A worldwide problem. Dermatology 2003, 206, 54–56. [Google Scholar]
- Ross, J.I.; Snelling, A.M.; Carnegie, E.; Coates, P.; Cunliffe, W.J.; Bettoli, V.; Tosti, G.; Katsambas, A.; Del Pulgar, J.; Rollman, O.; et al. Antibiotic-resistant acne: Lessons from Europe. Br. J. Dermatol. 2003, 148, 467–478. [Google Scholar]
- Mowery, B.P.; Lindner, A.H.; Weisblum, B.; Stahl, S.S.; Gellman, S.H. Structure-activity relationships among random nylon-3 copolymers that mimic antibacterial host-defense peptides. J. Am. Chem. Soc. 2009, 131, 9735–9745. [Google Scholar]
- Gabriel, G.J.; Madkour, A.E.; Dabkowski, J.M.; Nelson, C.F.; Nusslein, K.; Tew, G.N. Synthetic mimic of antimicrobial peptide with nonmembrane-disrupting antibacterial properties. Biomacromolecules 2008, 9, 2980–2983. [Google Scholar]
- Rawlinson, L.A.B.; Ryan, S.M.; Mantovani, G.; Syrett, J.A.; Haddleton, D.M.; Brayden, D.J. Antibacterial effects of poly(2-(dimethylamino ethyl)methacrylate) against selected gram-positive and gram-negative bacteria. Biomacromolecules 2010, 11, 443–453. [Google Scholar]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar]
- Hirai, K.; Aoyama, H.; Suzue, S.; Irikura, T.; Iyobe, S.; Mitsuhashi, S. Isolation and characterization of norfloxacin-resistant mutants of escherichia-coli K-12. Antimicrob. Agents Chemother. 1986, 30, 248–253. [Google Scholar]
- Hooper, D.C. Emerging mechanisms of fluoroquinolone resistance. Emerg. Infect. Dis. 2001, 7, 337–341. [Google Scholar]
- Tew, G.N.; Clements, D.; Tang, H.Z.; Arnt, L.; Scott, R.W. Antimicrobial activity of an abiotic host defense peptide mimic. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1387–1392. [Google Scholar]
- Choi, S.; Isaacs, A.; Clements, D.; Liu, D.H.; Kim, H.; Scott, R.W.; Winkler, J.D.; DeGrado, W.F. De novo design and in vivo activity of conformationally restrained antimicrobial arylamide foldamers. Proc. Natl. Acad. Sci. USA 2009, 106, 6968–6973. [Google Scholar]
- Marr, A.K.; Gooderham, W.J.; Hancock, R.E.W. Antibacterial peptides for therapeutic use: Obstacles and realistic outlook. Curr. Opin. Pharmacol. 2006, 6, 468–472. [Google Scholar]
- Goldman, M.J.; Anderson, G.M.; Stolzenberg, E.D.; Kari, U.P.; Zasloff, M.; Wilson, J.M. Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 1997, 88, 553–560. [Google Scholar]
- Maisetta, G.; di Luca, M.; Esin, S.; Florio, W.; Brancatisano, F.L.; Bottai, D.; Campa, M.; Batoni, G. Evaluation of the inhibitory effects of human serum components on bactericidal activity of human beta defensin 3. Peptides 2008, 29, 1–6. [Google Scholar]
- Deslouches, B.; Islam, K.; Craigo, J.K.; Paranjape, S.M.; Montelaro, R.C.; Mietzner, T.A. Activity of the de novo engineered antimicrobial peptide WLBU2 against pseudomonas aeruginosa in human serum and whole blood: Implications for systemic applications. Antimicrob. Agents Chemother. 2005, 49, 3208–3216. [Google Scholar]
- Matsuzaki, K. Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta Biomembr. 1999, 1462, 1–10. [Google Scholar]
- Coughlin, R.T.; Tonsager, S.; McGroarty, E.J. Quantitation of metal-cations bound to membranes and extracted lipopolysaccharide of escherichia-coli. Biochemistry 1983, 22, 2002–2007. [Google Scholar]
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar]
- Mensa, B.; Kim, Y.; Choi, S.; Scott, R.; Caputo, G.; DeGrado, W. Antibacterial mechanism of action of arylamide foldamers. Antimicrob. Agents Chemother. 2011. [Google Scholar] [CrossRef]
- Epand, R.F.; Schmitt, M.A.; Gellman, S.H.; Epand, R.M. Role of membrane lipids in the mechanism of bacterial species selective toxicity by two alpha/beta-antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1343–1350. [Google Scholar]
- Hancock, R.E.W.; Wong, P.G.W. Compounds which increase the permeability of the pseudomonas-aeruginosa outer-membrane. Antimicrob. Agents Chemother. 1984, 26, 48–52. [Google Scholar]
- Hancock, R.E.W. Peptide antibiotics. Lancet 1997, 349, 418–422. [Google Scholar]
- Park, C.B.; Kim, H.S.; Kim, S.C. Mechanism of action of the antimicrobial peptide buforin II: Buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun. 1998, 244, 253–257. [Google Scholar]
- Brotz, H.; Bierbaum, G.; Leopold, K.; Reynolds, P.E.; Sahl, H.G. The lantibiotic mersacidin inhibits peptidoglycan synthesis by targeting lipid II. Antimicrob. Agents Chemother. 1998, 42, 154–160. [Google Scholar]
- Patrzykat, A.; Friedrich, C.L.; Zhang, L.J.; Mendoza, V.; Hancock, R.E.W. Sublethal concentrations of pleurocidin-derived antimicrobial peptides inhibit macromolecular synthesis in Escherichia coli. Antimicrob. Agents Chemother. 2002, 46, 605–614. [Google Scholar]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sovadinova, I.; Palermo, E.F.; Urban, M.; Mpiga, P.; Caputo, G.A.; Kuroda, K. Activity and Mechanism of Antimicrobial Peptide-Mimetic Amphiphilic Polymethacrylate Derivatives. Polymers 2011, 3, 1512-1532. https://doi.org/10.3390/polym3031512
Sovadinova I, Palermo EF, Urban M, Mpiga P, Caputo GA, Kuroda K. Activity and Mechanism of Antimicrobial Peptide-Mimetic Amphiphilic Polymethacrylate Derivatives. Polymers. 2011; 3(3):1512-1532. https://doi.org/10.3390/polym3031512
Chicago/Turabian StyleSovadinova, Iva, Edmund F. Palermo, Michael Urban, Philomene Mpiga, Gregory A. Caputo, and Kenichi Kuroda. 2011. "Activity and Mechanism of Antimicrobial Peptide-Mimetic Amphiphilic Polymethacrylate Derivatives" Polymers 3, no. 3: 1512-1532. https://doi.org/10.3390/polym3031512
APA StyleSovadinova, I., Palermo, E. F., Urban, M., Mpiga, P., Caputo, G. A., & Kuroda, K. (2011). Activity and Mechanism of Antimicrobial Peptide-Mimetic Amphiphilic Polymethacrylate Derivatives. Polymers, 3(3), 1512-1532. https://doi.org/10.3390/polym3031512