Mechanism Studies of LCP Synthesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
: The LCP (Liquid Crystal Polymer) known as Vectra is synthesized by acidolysis of 4-hydroxybenzoic acid with 6-hydroxy-2-naphthoic acid. The apparently simple acidolysis mechanism for LCP polycondensation is in fact a complex blend of mechanisms. Kinetics of model reactions and of actual polycondensations followed second-order kinetics and their rate constants were comparable. In the latter stages, ketene loss leads to phenolic ends, while decarboxylation provides phenyl ester ends. Accordingly, the mechanism changes to phenolysis. A quinone methide intermediate may also intervene, as revealed by kinetics studies and MALDI-TOF spectroscopy. Tailor-made matrices and synthesis of alternating well-defined oligomers assisted our studies. Nucleophilic aromatic substitutions may play a role, and we speculate on possible chain polycondensation. Esterolysis may be a useful alternative to LCP synthesis. Complications caused by ketene loss can be averted by the use of methoxycarbonyloxy monomers.1. Introduction
Thermotropic main chain LCP copolyesters are an important class of modern high tech polymers [1-3]. Their high mechanical strength and modulus, combined with stability to chemicals and heat lend them attractive properties. Their applications include electronic devices, fiber optic coupling discs, automotive components, distillation towers, and so on.
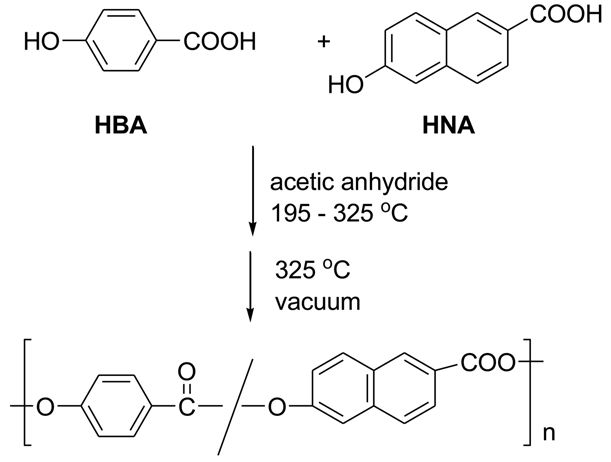
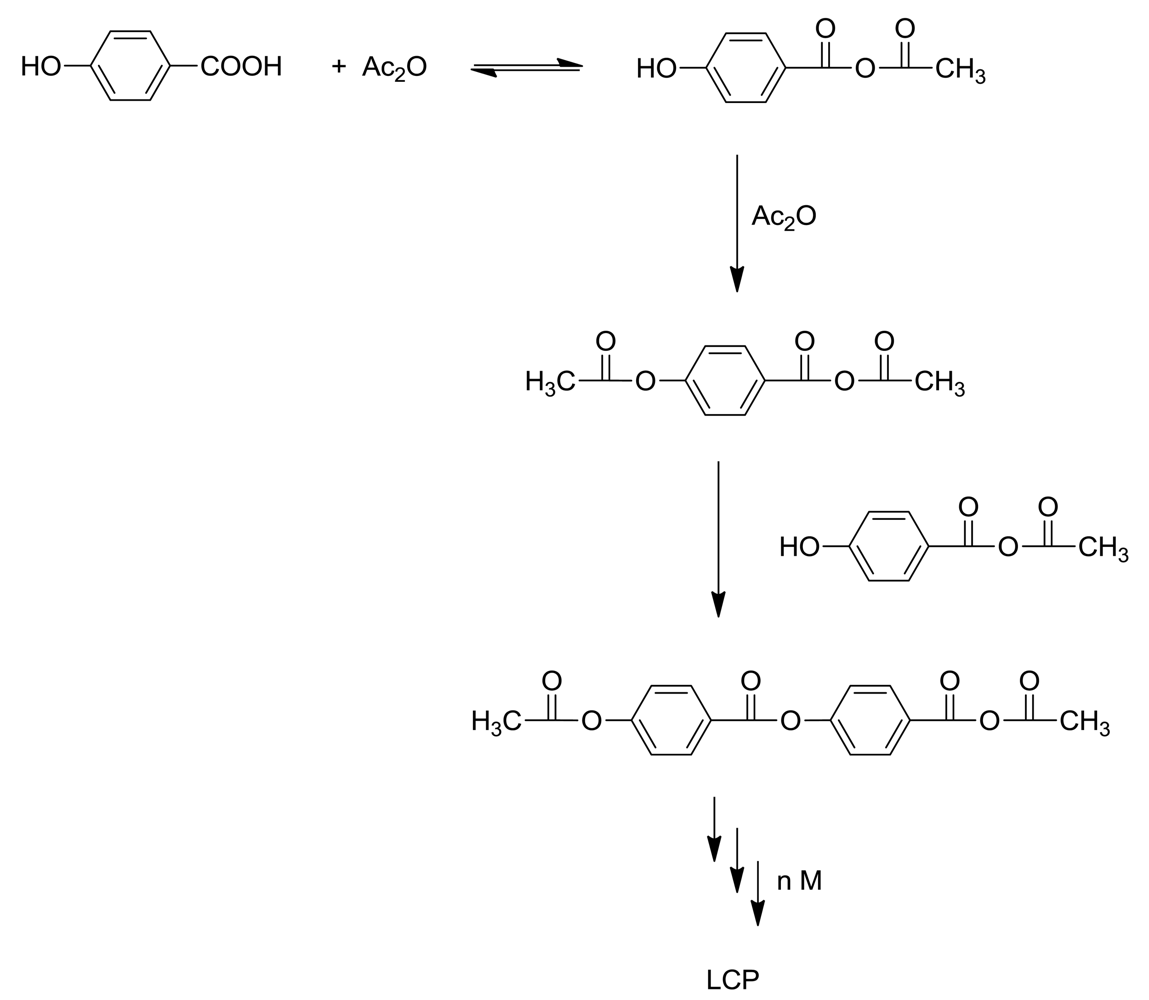
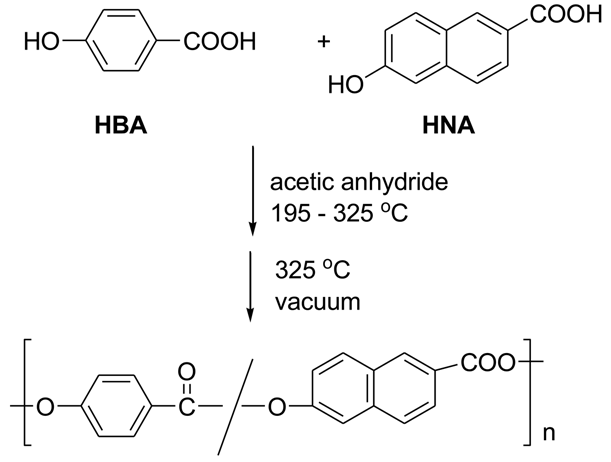
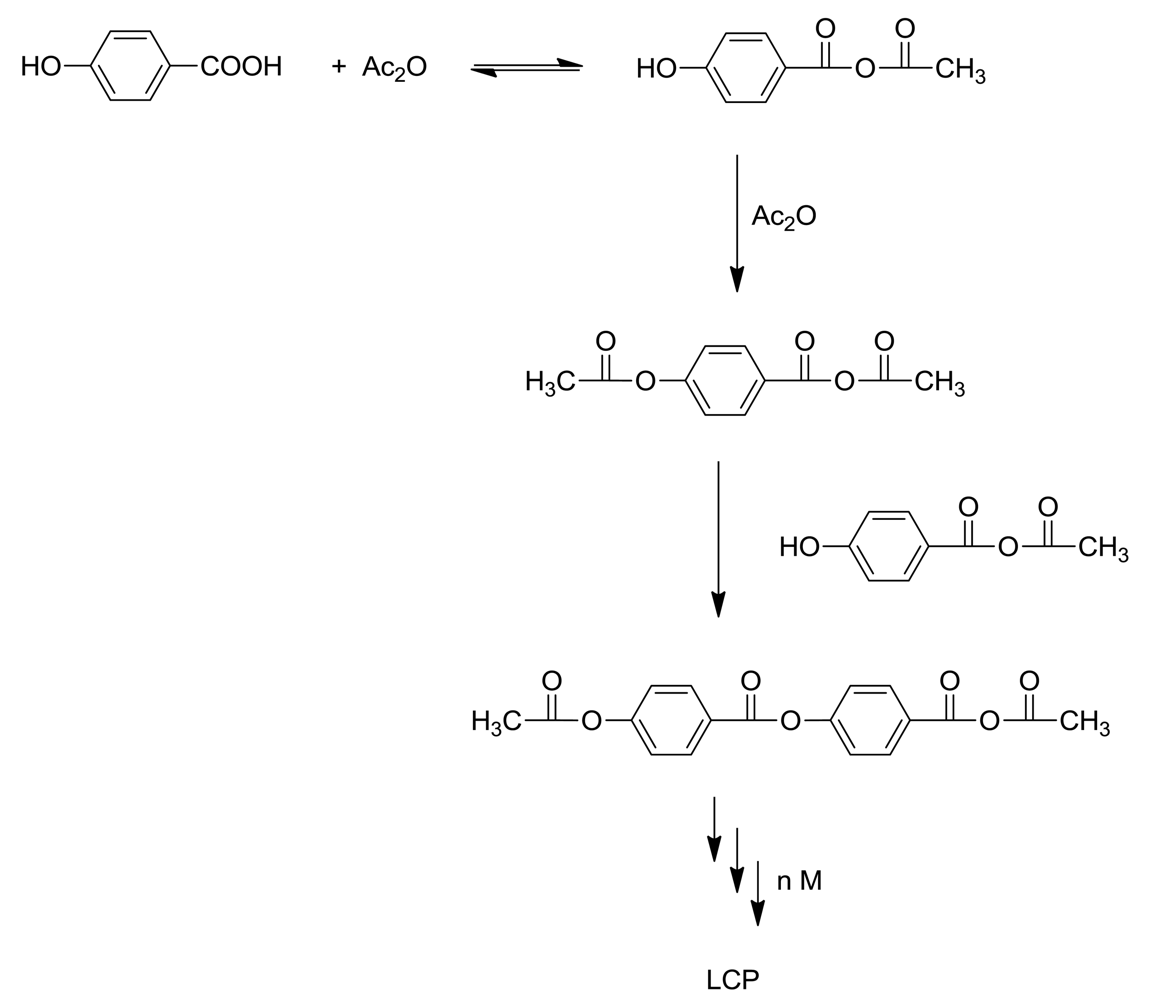
The main component of LCP is 4-hydroxybenzoic acid (HBA). This inexpensive ingredient is incorporated into linear aromatic polyesters and confers a rod-like structure to them. Unfortunately the homopolymer is quite intractable, melting at >500 °C and only slightly soluble in pentafluorophenol [4]. Accordingly, comonomers such as terephthalic acid and hydroquinone or 6-hydroxy-2-naphthoic acid (HNA) are added to make the polymers tractable and processable, so that commercial LCP's are always copolymers. Scheme 1 represents the basic polymerization reaction for the synthesis of Vectra®, a copolymer of 4-hydroxybenzoic acid and 6-hydroxy-2-naphthoic acid.
The industrial polymerization procedure consists of acetylating the components with acetic anhydride, followed by heating above 325 °C under a vacuum to eliminate acetic acid and form high polymer. Sodium or calcium acetate is present in small amounts. This apparently simple procedure is by no means the complete story. Kricheldorf [5] stated that the chemistry of 4-hydroxybenzoic acid polymerization above 300 °C is not simple, and we shall see that his statement is abundantly correct!
In this paper, the mechanism of the LCP polymerization is summarized, with an emphasis on our own research. The intractability of the polymer, even at low molecular weight, posed a constant problem in this study. For this reason, model studies were first carried out, followed by a study of the kinetics of the polymerization, and finally a study of the structure of these polymers. A better understanding of the mechanisms of these polymerizations will hopefully lead to improved processes and also improve the properties of the polymers.
2. Kinetics of Model Reactions of Monofunctional Compounds in AB Reaction
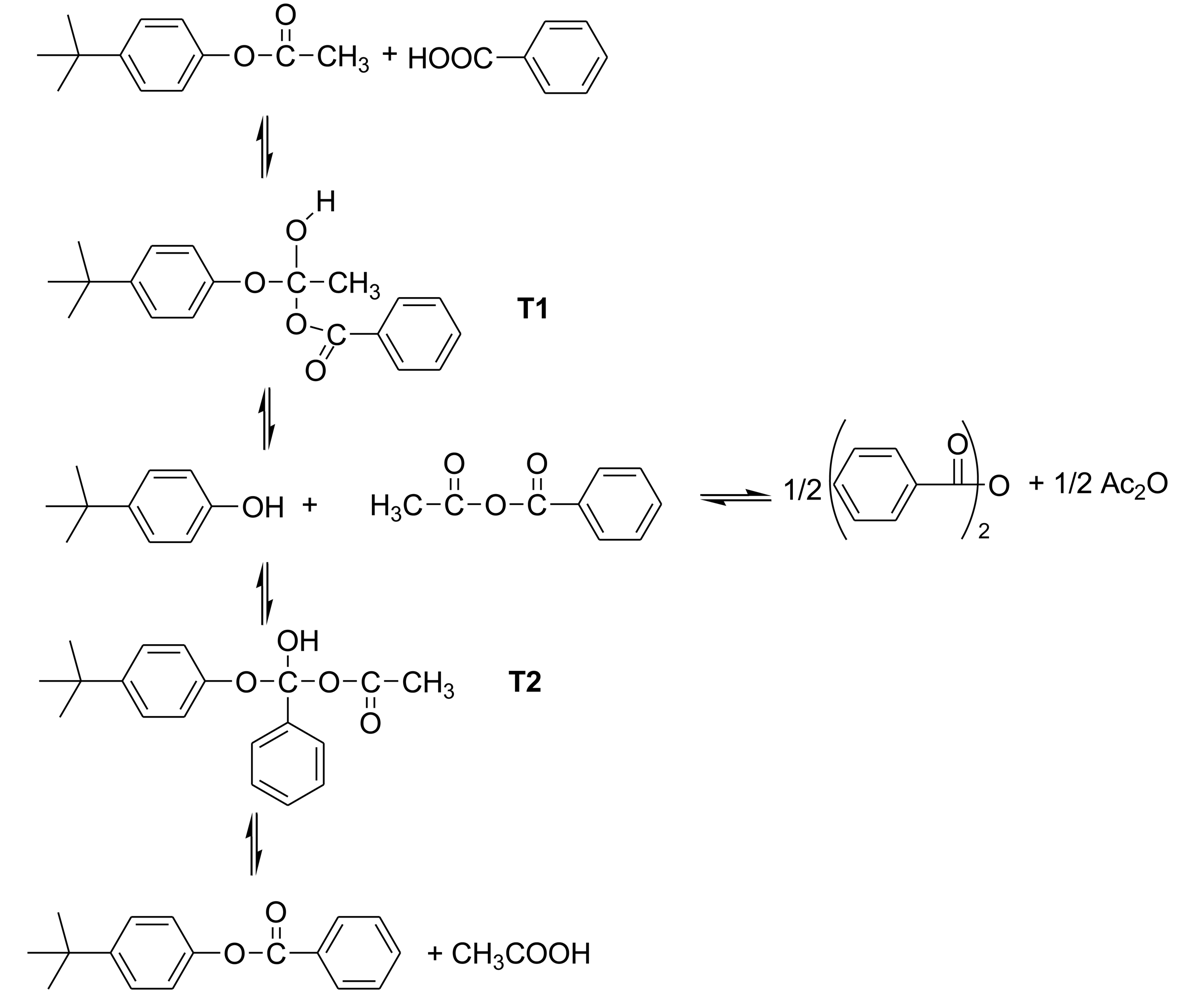
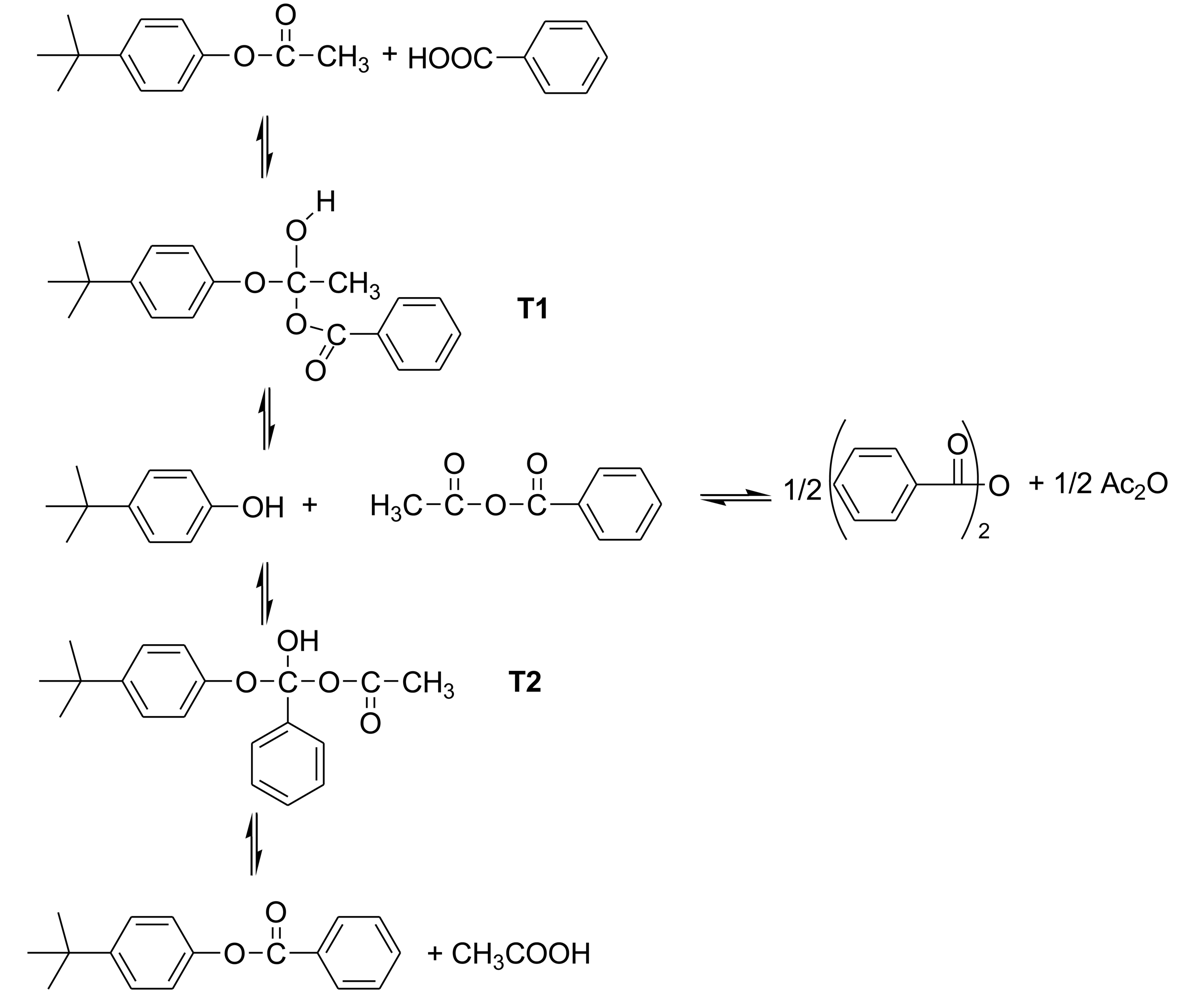
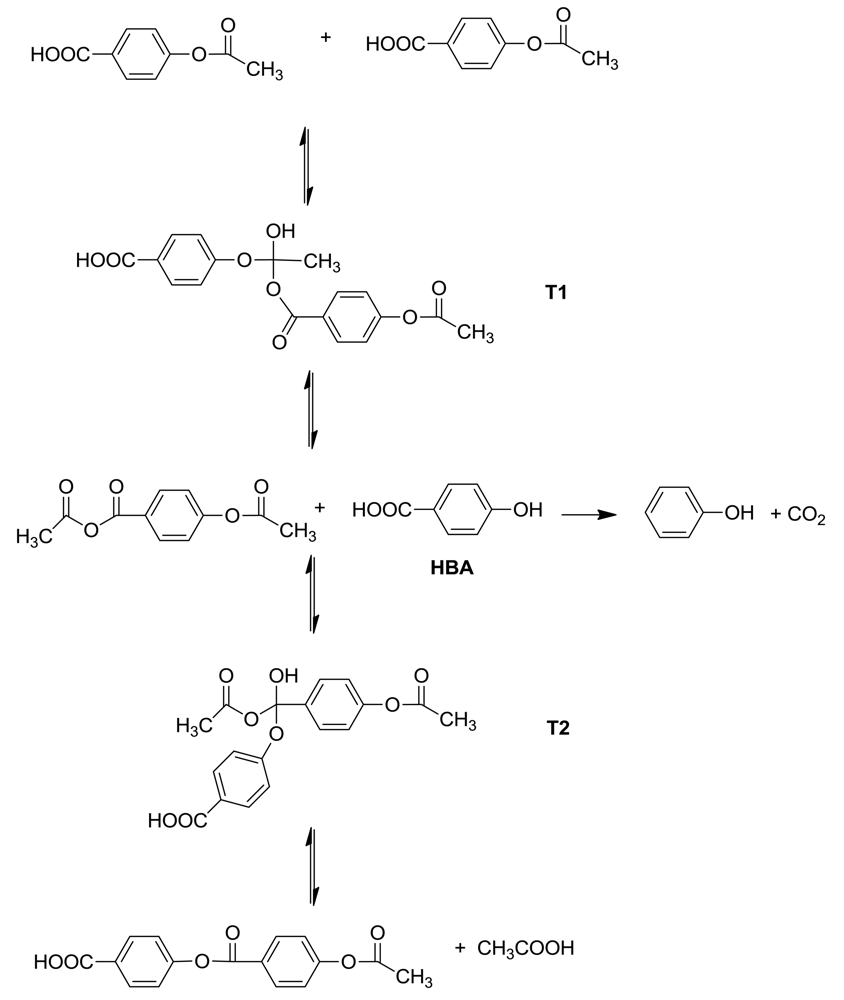
The starting point of this study was the investigation of the kinetics of the reaction of a model system for the acetolysis reaction. To simplify the kinetics investigations, the acetylated monomers are always used as the starting materials. The basic reaction in the polymerization is the formation of a new ester bond by the reaction of an aromatic acetate functionality with an aromatic carboxylic acid functionality, accompanied by the elimination of acetic acid. The model system was the reaction of p-t-butylphenyl acetate with benzoic acid in diphenyl ether solution at 200–250 °C to form p-t-butylphenyl benzoate and acetic acid [6]. Kinetics proved to be accurately second-order, first-order in each component; Arrhenius plots were linear and led to activation energies and entropies.

The proposed acidolysis mechanism involves two tetrahedral intermediates, as is customary for reactions with carbonyl groups, and agrees with the observed kinetics. (Scheme 2) In the first step, addition of benzoic acid to t-butylphenyl acetate leads to tetrahedral intermediate T1, which can revert to components or go forward to t-butylphenol and acetic benzoic anhydride. In turn these can recombine at the benzoyl carbonyl group giving tetrahedral intermediate T2, which again can revert back to components or proceed forward to t-butylphenyl benzoate and acetic acid. The asymmetric anhydride formed in this process can undergo metathesis to the symmetrical anhydrides.
3. Kinetics of AB Acetolysis Polymerizations
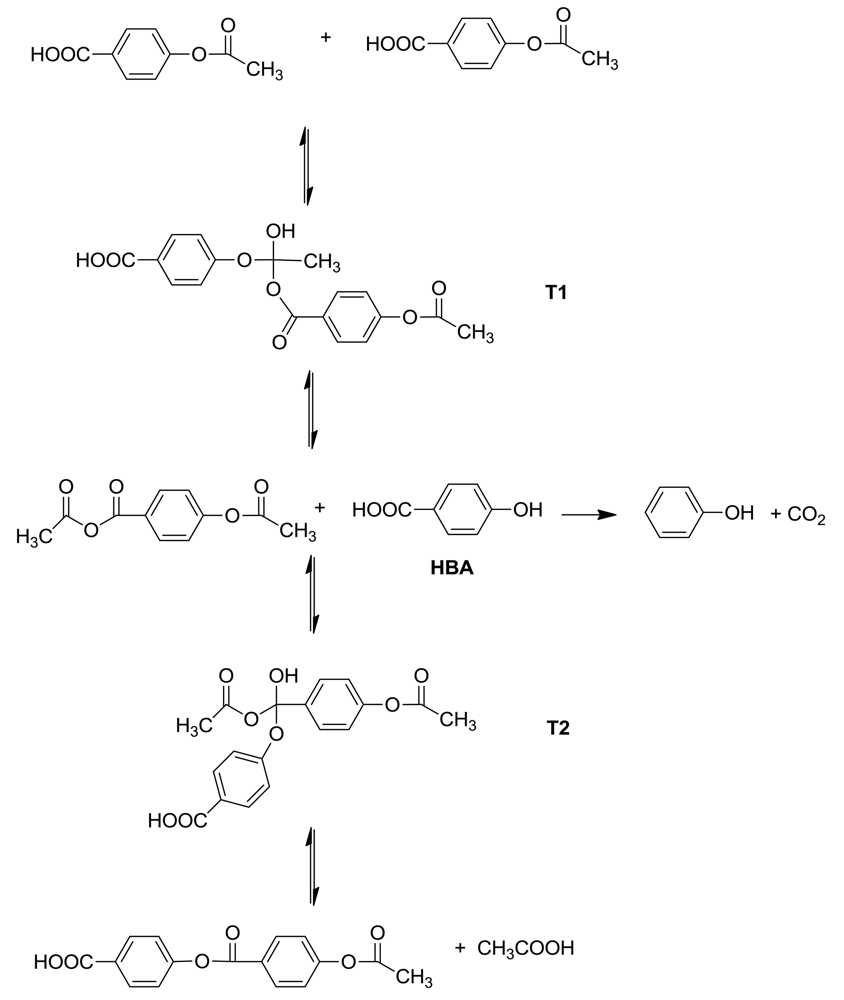
To investigate the kinetics of the Vectra® polymerization, 4-acetoxybenzoic acid and 6-acetoxy-2- naphthoic acid were allowed to react at different temperatures and the evolved acetic acid was measured [8]. All co- and homopolymerizations were second order kinetics up to 70% conversion. Two kinetic regions were observed, with the rate of the initial reaction being larger than the later reactions; the time of rate change corresponded to a change from homogeneous to heterogeneous reaction conditions.
The polymerization mechanism follows the same pattern as the ester formation in the model reaction and is shown in Scheme 3. Two tetrahedral intermediates lead to the formation of a new ester link during the addition of a new monomer to the chain. Reversion of the second tetrahedral intermediate T2 restores p-hydroxybenzoic acid as a transitory intermediate. This is a crucial point, because at 150 °C, HBA can decarboxylate in a competitive side reaction to form phenol [9]. The consequences of its presence are discussed below.
4. Study of the Small Molecule Products
In a second approach we examined the small molecules formed as the polymerization proceeds [7]. The polymerization reactor for the copolymerization of HBA and HNA was therefore connected to a series of three traps: the first trap was held at −10 °C for the collection of acetic acid, phenyl acetate, acetic anhydride, phenol, and the starting monomers HBA and HNA; the second trap contained a solution of aniline in tetrahydrofuran for the collection and detection of ketene as acetanilide, and the third trap contained a solution of barium hydroxide and water for the detection of carbon dioxide.
The content of the first trap at −10 °C was analyzed by HPLC, except for the acetic acid, which was titrated for the kinetics study described above. The most interesting compound for this study was phenol, which was not formed at first, but the amounts of which became substantial in the latter stages of polymerization.
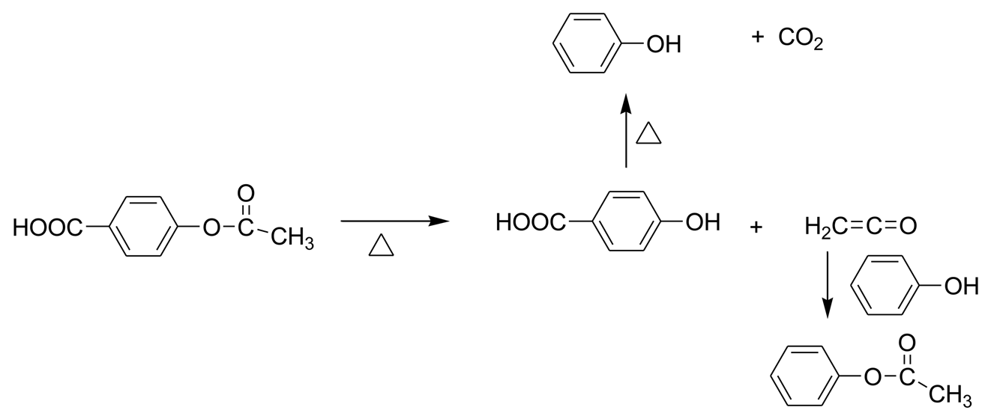
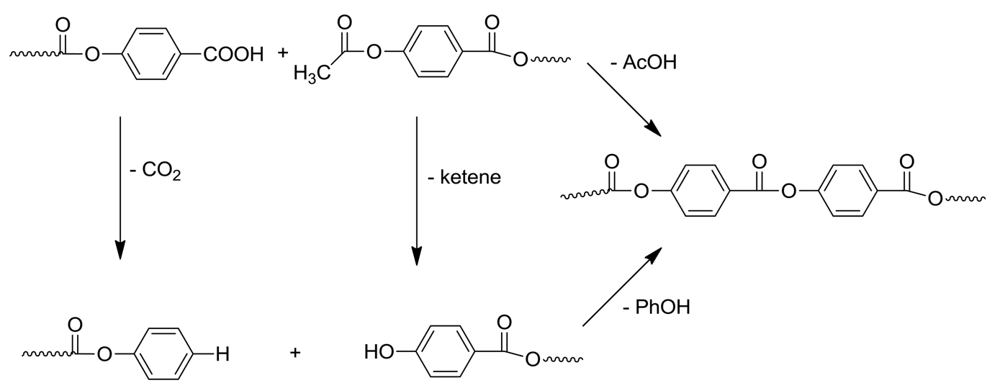
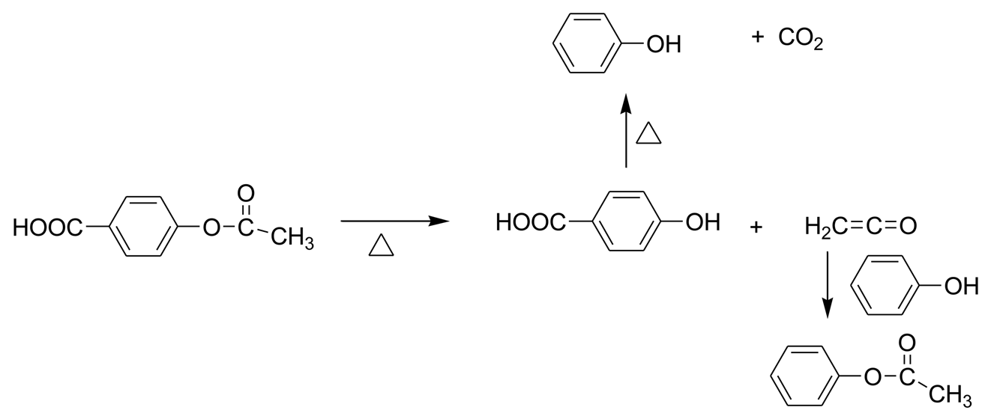
The second trap was designed to monitor the formation of ketene, which has been implicated in the unwanted color formation of the LCP's and side reactions [10]. Ketene was trapped with aniline, and the amount of formed acetanilide was determined by GC/MS. The elimination of ketene from 4-acetoxyarenes at high temperature has been previously described in the literature [10], leading to the conclusion that the terminal aromatic acetoxy groups are not stable. Recently, we presented evidence for the chain free radical mechanism for this ketene loss [11]. Under these polymerization conditions, ketene could also form from acetic anhydride or a mixed anhydride. Finally, ketene loss from p-acetoxybenzoic acid could give p-hydroxybenzoic acid, leading to phenol and carbon dioxide. (Scheme 4).
In the copolymerization of HBA and HNA in the presence of acetic anhydride, loss of ketene leads to a terminal phenolic end group. Model reaction studies showed that attempted condensation reactions of such a terminal phenolic group with a p-oxybenzoic acid with loss of water do not occur. This explains the fact that aromatic p-hydroxyacids do not lead to polymer upon heating; the acetic anhydride is a necessary ingredient for the polymerization. Accordingly this polymerization would stop were it not for the intervention of phenol (see below).
The other byproduct is CO2, rate of formation of which is determined by weighing the formed barium carbonate in the third trap. Our investigations showed that carbon dioxide formed during the initial stages of polymerization, and became more substantial as the polymerization proceeded. Decarboxylation of chain ends can also occur and leads to terminal phenyl esters.
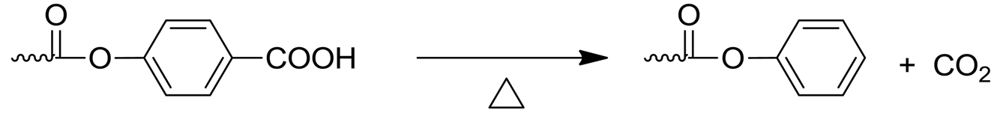
5. Phenolysis Mechanism
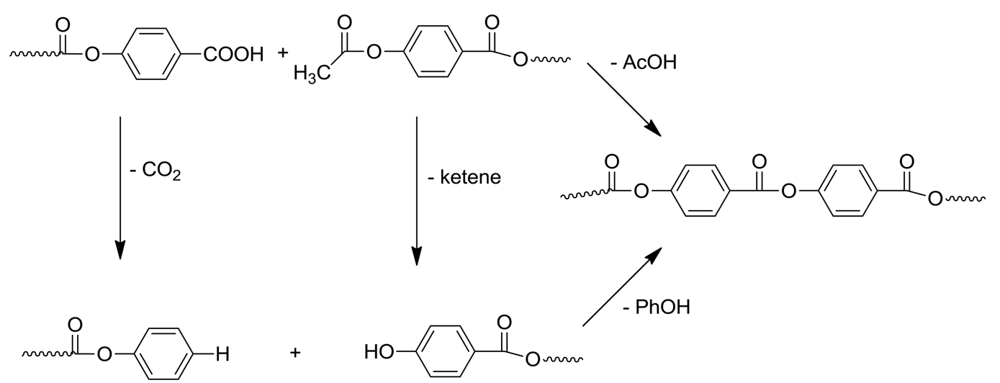
Through the study of model reactions and the small molecules formed during the polymerization, we have shown that the end groups of the growing polymer chains change as the polymerization proceeds. (Scheme 5) The benzoic acid end groups can decarboxylate, leading to phenyl esters, while the acetoxybenzene end groups lose ketene, resulting in phenolic end groups. The fascinating thing about the LCP polymerization is that the growth of molecular weight proceeds even though the end groups have changed. The polymerization switches mechanism from acetolysis to phenolysis [7], i.e., phenol evolves as new ester bonds form, resulting in higher molecular weights. It is in the phenolysis reaction that the reported catalysis by sodium acetate, can occur; it does not catalyze acidolysis [12], but is known to catalyze phenyl ester interchange [13,14].
The question of which comes first, ketene loss to form hydroxyl ends or decarboxylation to form phenyl ester ends, has to be asked. We found that ketene loss occurs before phenyl ester formation, as demonstrated by polymer end group analysis and molecular weight determination. The latter was determined in two ways: the polymer was depolymerized by sodium deuteroxide in D2O; the acetic acid peak, which results from the acetoxy end group, was measured in NMR; and the molecular weight was determined by light scattering. Comparison of these two values revealed a discrepancy of about 50%, reflecting a loss of acetyl groups. No phenol was detected, so the phenyl ester had not yet formed [7].
6. The Role of Phenol
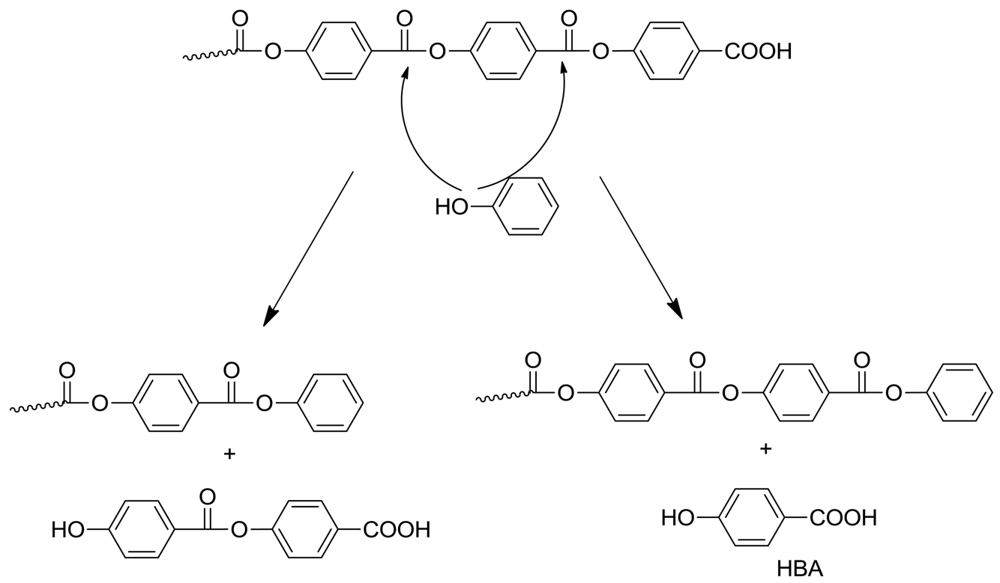
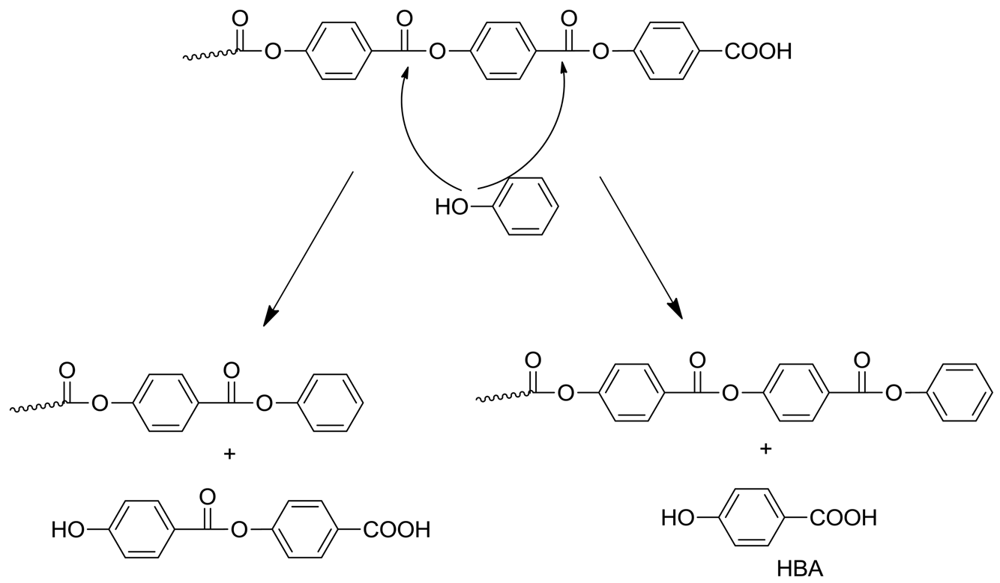
The presence of phenol toward the end of polymerization is extremely significant. Phenol arises from several mechanisms. One source is the facile decarboxylation of HBA, which is an intermediate compound formed from the tetrahedral intermediate, as shown in Scheme 3. Indian workers [9] showed that HBA decomposition is rapid at the temperatures involved. However the main source of phenol is the secondary polymerization, after the switch to the phenolysis mechanism.
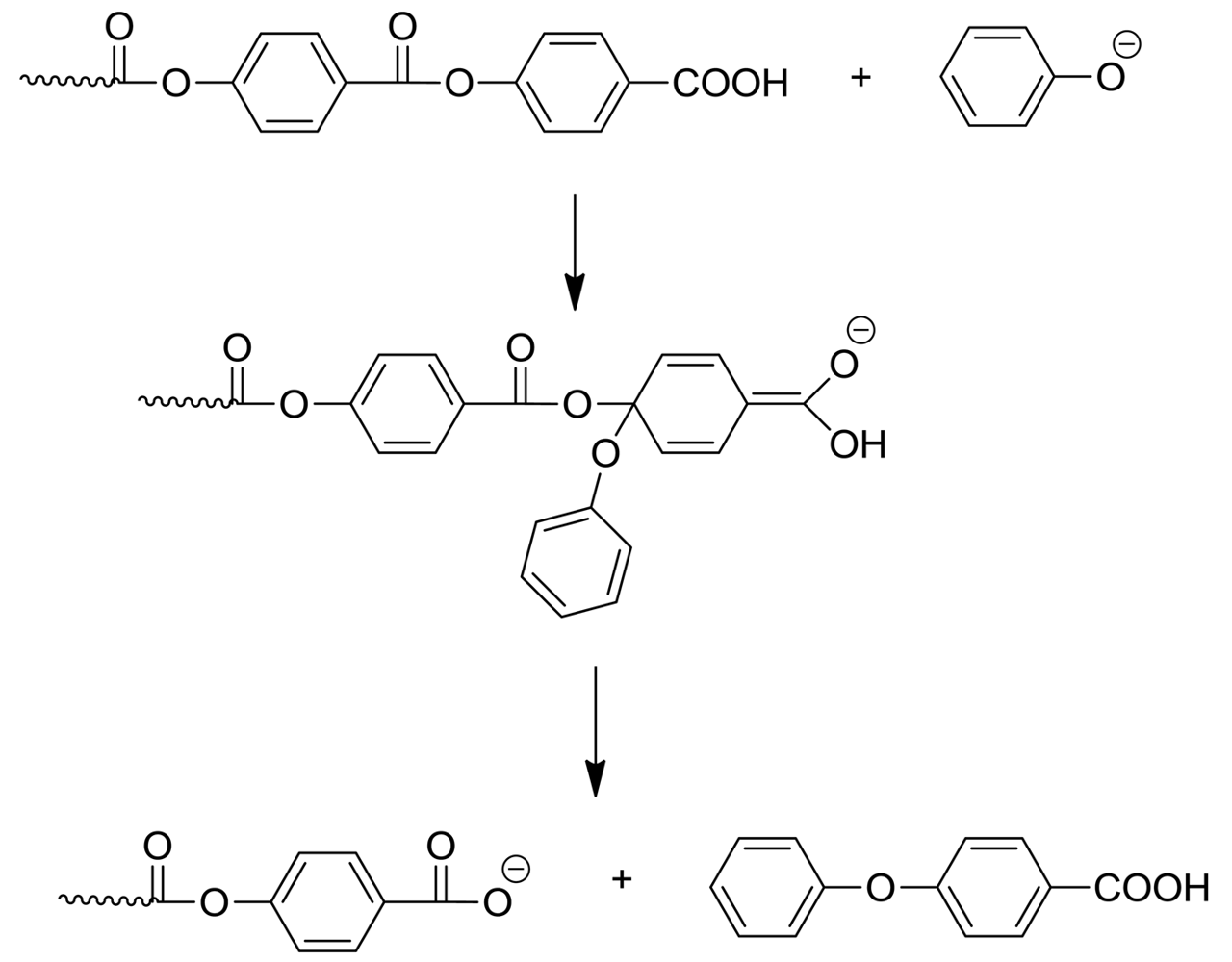
Once formed, the phenol can play several roles. It can take part in phenyl ester interchange; the attack of phenol at the ultimate or penultimate ester group is particularly interesting (Scheme 6). The former reaction forms a polymer with phenyl ester end group and expels HBA; decarboxylation of the latter would in turn form more phenol, so a chain process can take place. Attack at the penultimate ester group also leads to a terminal phenyl ester and the dimer of HBA. If phenol participates in mid-chain cleavage, this will still give a phenyl ester-tipped polymer. Of course mass transfer must also be considered at this stage: depending on the configuration of the polymer reactor, phenol may be removed to a greater or lesser extent and accordingly phenyl ester formation will also be variable. In future work it may be of interest to deliberately add a high-boiling phenol at the beginning in order to facilitate this change.
7. Kinetics of AA/BB Polymerizations
The studies of the AB polymerizations were extended to the AA/BB polymerizations of aromatic diacids with diacetates [15]. The model A/BB reaction of hydroquinone diacetate with p-t-butylbenzoic acid also followed second-order kinetics in homogeneous conditions.
The diacids used in AA/BB polymerization systems are terephthalic acid (TA) and 2,6-naph-thalene-dicarboxylic acid (NDA), which are highly insoluble, leading to heterogeneous reactions from the beginning of the polymerization; the BB-mononers were hydroquinone diacetate and 2-phenylhydroquinone diacetate. Again, the amount of evolved acetic acid during the polymerization was measured. The kinetics were strictly second-order, with rate constants comparable to those measured in the corresponding model study.
In contrast with the AB polymerizations described above, the AA/BB polymerization continues even after ketene loss, because in this case the dehydration reaction is possible. The explanation for the difference is in the greater electrophilicity of the carboxylic acid functionality of aromatic diacid, such as TA, over HBA, and the greater nucleophilicity of the aromatic hydroxyl group of hydroquinone over that of HBA.
8. LCP Structure Characterization by MALDI-TOF Spectroscopy
The LCP's intractability makes conventional spectroscopic methods, such as NMR or IR, unsuitable for structure determination. So we resorted to MALDI-TOF spectroscopy. However, it must be noted that major limitations also exist in this case, namely molecular weight limitations. Even though the structure of monodisperse biopolymers with molecular weights of up to 1 million dalton has been determined with this technique, synthetic polycondensation polymers generally give spectra only up to 5–10 KD. Even conventional polyethylene terephthalate and nylon have not been studied at molecular weights above 10 KD. The reason for these difficulties is not entirely clear. However it is unsatisfactory in that the mass spectrum of polymers of high molecular weight, which confer useful properties above the entanglement length, cannot readily be determined.
Despite this limitation, MALDI-TOF spectroscopy can be quite fruitful. To begin, we synthesized a soluble low molecular weight copolymer of 3-HBA with 2,6-HNA by the conventional acidolysis process [16]. MALDI spectra of this polymer were entirely consistent with expectations, in particular regarding the end groups which we observed. We concluded that we had detected the actual polymer molecules, with a terminal acylium ion (∼COO-Ar-CO+).
Next, we examined a low molecular weight copolymer of 4-HBA with the same 2,6-HNA [16]. A complex situation then presented itself. It appeared that we were not observing the signals for the molecules present in the polymerizing mixture, but rather truncated forms of these, and possibly with rearranged monomer units. Several series of fragments with various combinations of end groups were observed, but no phenyl ester-tipped polymers were seen. Moreover, series of fragments with different terminal groups were observed, such as ∼COO-Ar-CO+, ∼COO-Ar-COOH, Na+, and among others ∼COO-Ar-COOH, H+.
To investigate this topic further we synthesized a variety of well-defined alternating tetramers of 4-HBA with 2,6-HNA [17]. These were synthesized by the use of protected intermediates, stepwise couplings and deprotection. MALDI-TOF spectra of these alternating oligomers also gave complex MALDI-TOF spectra. The appearance of peaks containing up to 5 HBA units linked together was particularly noteworthy, even though the oligomers were strictly alternating. For example, the following peaks were detected in the MALDI-TOF spectra: mass 361 corresponding to [HB3]+, 533 [AcB3OBn]Na+, 653 [Ac-B4OBn]Na+, and 773 [AcB5OBn]Na+, with B = p-oxybenzoate and Bn = benzyl, clearly indicating extensive reshuffling among the monomer units in the tetramer, and therefore also in the polymer.
We postulate that depolymerization of HBA units occurs from the acylium ion series, which would involve loss of a quinone methide intermediate. This is followed by oligomerization of the quinone methide intermediates; hence the monomer groups are rearranged in the detected cations.

Italian and British workers have encountered exactly this intermediate in the saponification of the dinitrophenyl ester of 4-hydroxybenzoic acid [18].
If the MALDI-TOF results can be extrapolated to molten polymer, this cleavage possibility may make LCP's “fragile” polymers, which undergo homolysis with concomitant quinone methide formation during molding, and consequent unit reshuffling during recombination.
9. Tailor-Made MALDI-TOF Matrices for LCP polymers
We wondered whether some of the problems involved in MALDI spectroscopy of 4-HBA copolymers might be due to the incompatibility of the polymer with the matrix, which would make the aggregated polymer difficult to vaporize. To address this problem we recalled that one of the very few solvents for the LCPs is pentafluorophenol. Accordingly we synthesized a number of octafluoroazobenzene derivatives containing phenolic and/or carboxylic acid groups which should solvate and compatibilize the polymer with the matrix [19]. 4,4′-Dihydroxyoctafluoroazobenzene was the most successful matrix, which indeed gave superior spectra to those obtained using conventional matrices such as dithranol or HABA. With this new matrix, we were able to obtain much better signal to noise ratio and also detect higher molecular weight signals for a series of polyesters. However, the same complexity of behavior and contrast between 4-HBA copolymers, which can form the quinone methide intermediate (see above), and the 3-HBA copolymers, which cannot, was found with the new matrices.
10. Other Possible Side Reactions during the LCP Polymerization
In the literature, both Kricheldorf [5] and Economy [4] mention 4-phenoxybenzoic acid as a byproduct in LCP preparation; it could be formed through nucleophilic aromatic substitution (Scheme 7). This would correspond to nucleophilic aromatic displacement, a reaction for which the polymer structure is very well adapted. It is quite analogous to polysulfones where this phenomenon is well known. This reaction would consume phenoxide anion, which would have to be replaced by further decarboxylation to allow the polymerization to proceed via the phenolysis mechanism.
In Japan, Yokozawa [20] has demonstrated chain polycondensation: an all-para aromatic LCP polyester could be obtained with a molecular weight distribution close to 1 rather than 2, as in stepwise polycondensation. Yokozawa used a 3-octyl-4-hydroxybenzoic acid activated derivative for solubility: the very specific leaving group benzothiazole was required. The leaving group had to be specifically chosen, as some of the leaving groups may give rise to the quinone methide intermediate.
Speculatively, this chain polycondensation mechanism might be applicable to ordinary LCP polycondensation. HBA reacts twice with acetic anhydride to yield the acetoxy mixed anhydride shown in Scheme 8. This doubly acetylated HBA acts as an electrophilic active initiator in which the carbonyl of the mixed anhydride is the more electrophilic carbonyl. This initiator reacts with the mixed anhydride of HBA and acetic anhydride, which acts as the “nucleophilic” monomer through the more nucleophilic hydroxyl. If acetylation of 4-HBA is followed by a chain condensation reaction, this could speculatively explain so-called runaway polymerizations encountered when excess acetic anhydride is used [21].
11. Other Reaction Pathways to LCPs
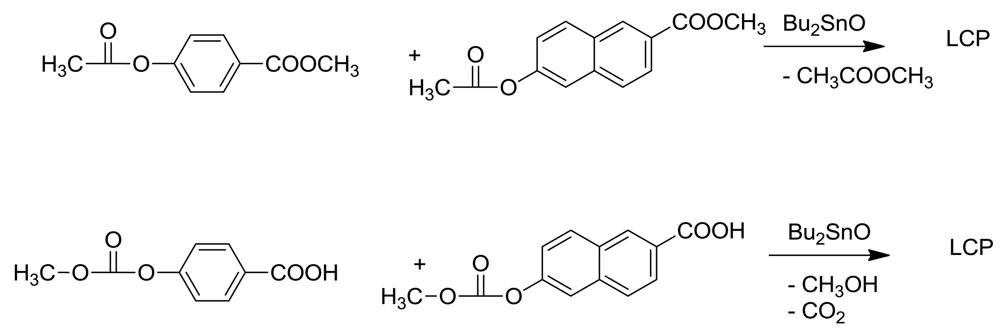
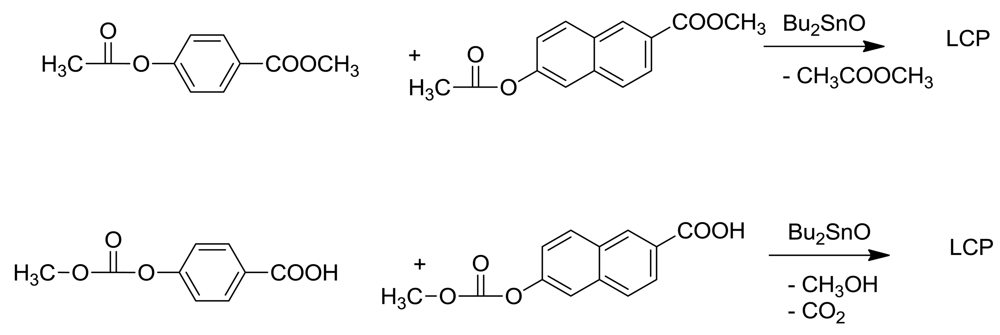
An alternative route to LCPs which may avoid ketene loss lies in an ester interchange reaction (esterolysis), catalyzed by tin or titanium catalysts. We had previously utilized this route to synthesize polyarylates by reacting a mixture of dimethyl isophthalate and dimethyl 2,6-naphthalenedicarboxylate with bis-acetoxy bisphenol A by gradually heating this mixture in diphenyl ether in the presence of dibutyltin oxide for three days up to 340 °C [13,14]. A polymer with inherent viscosity of 0.48 dl.g−1 was obtained in laboratory conditions. In that study, esterolysis proved to be much superior to alcoholysis of dimethyl isophthalate with bisphenol A, which gave substantial discoloration, probably owing to the thermal instability of free bisphenol A.
This route was successfully applied to the synthesis of LCPs by copolymerizing methyl p-acetoxybenzoate with methyl 6-acetoxynaphthoate in presence of dibutyltin oxide [22]. In exploratory studies, the melt viscosities of these polymers were a little lower than those prepared by acidolysis. (Scheme 9) The only drawback was the apparent requirement for a rather high concentration of stannous octanoate in these exploratory experiments. However, a variety of Sn(II) catalysts are available and large differences in their catalytic activity can be anticipated. Whether ketene loss can be minimized by this route remains to be seen.
The plethora of reaction mechanisms arising from ketene loss prompted us to seek other monomers not susceptible to ketene loss. We chose p-methoxycarbonyloxy aromatic acids, which are readily available by reaction of methyl chloroformate with hydroxyacid in presence of base [23]. Thermal copolymerization of a 73:27 mixture of the methoxycarbonyloxy-benzoic and -napththoic acid proceeded smoothly with the evolution of methanol and carbon dioxide. At a lower temperature (230 °C) than that used for the classical LCP polymerization (starting at 250 °C), the rate of this polymerization was faster than for the classical acetoxy route described in this review, and the color was substantially improved. (Scheme 9) Polyarylate forms from bisphenol-A-bis-methylcarbonate and dimethyl terephthalate with evolution of dimethyl carbonate [24]; in these polymerizations ketene loss is not possible. A tin catalyzed ester interchange of methyl p-hydroxybenzoate, or HBA itself, with dimethyl carbonate is therefore a possible practical synthesis.
12. Conclusions
The apparently simple acidolysis mechanism for LCP polycondensation is in fact a complex blend of mechanisms. The kinetics of model reactions and of actual polycondensations followed second-order kinetics and their rate constants were comparable. In the latter stages, ketene loss leads to phenyl ester ends and the mechanism changes to phenolysis. A quinone methide intermediate may also intervene, as revealed by kinetics studies and MALDI-TOF results. Tailor-made matrices and synthesis of alternating well-defined oligomers assisted our studies. Nucleophilic aromatic substitutions may play a role, and we speculate on possible chain polycondensation. Esterolysis may be a useful alternative to LCP synthesis. Complication caused by ketene loss can be averted by the use of methoxycarbonyloxy monomers.

Acknowledgments
The authors acknowledge the contributions of Hoechst Ticona Corporation colleagues whose names appear on the publications cited, in particular H. Clay Linstid (deceased) of the Ticona Corporation, and of our own colleagues.
Dedicated to the memory of H.C. Linstid, Research Laboratories, Ticona Corp., Florence, Kentucky, who passed away unexpectedly on June 28, 2008. He made many contributions to this work and is greatly missed by his family, colleagues, and friends.
References
- Jin, J.I.; Kang, C.S. Thermotropic main chain polyesters. Progr. Polym. Sci. 1997, 22, 937–973. [Google Scholar]
- Han, H.; Bhowmik, P.K. Wholly aromatic liquid-crystalline polymers. Progr. Polym. Sci. 1997, 22, 1431–1502. [Google Scholar]
- Negi, Y.S.; Goyal, R.K. Development of thermotropic liquid crystal polymers. Int. J. Plastics Technol. 2003, 7, 99–118. [Google Scholar]
- Economy, J.; Storm, R.S.; Matkovick, M.I.; Cottis, S.G.; Novak, B.E. Synthesis and structure of the p-hydroxybenzoic acid polymer. J. Polym. Sci. Polym. Chem. Ed. 1976, 14, 2207. [Google Scholar]
- Kricheldorf, H.; Schwarz, G. New polymer syntheses, 7. Synthesis and characterization of oligomeric and high molecular weight poly(4-hydroxybenzoate). Makromol. Chem. 1983, 184, 475–496. [Google Scholar]
- Huang, J.; Leblanc, J.P.; Hall, H.K., Jr. Model studies on the kinetics and mechanism of polyarylate synthesis by acidolysis. J. Polym. Sci. Polym. Chem. Ed. 1992, 30, 345–354. [Google Scholar]
- Han, X.; Williams, P.A.; Padias, A.B.; Hall, H.K., Jr.; Sung, H.N.; Linstid, H.C.; Lee, C. A change in mechanism from acidolysis to phenolysis in the bulk copolymerization of 4-acetoxybenzoic acid and 6-acetoxy-2-naphthoic acid. Macromolecules 1996, 29, 8313. [Google Scholar]
- Williams, P.A.; Han, X.; Padias, A.B.; Hall, H.K., Jr. Kinetics of the polymerization of 4-acetoxybenzoic acid and 6-acetoxynaphthoic acid. Macromolecules 1996, 29, 1874. [Google Scholar]
- Sitamanikyam, D.; Sundaram, E.V. Kinetics of thermal decarboxylation of p-substituted benzoic acids in glycerol. Ind. J. Chem. 1972, 10, 184. [Google Scholar]
- Leblanc, J.P.; Huang, J.; Padias, A.B.; Hall, H.K., Jr. Thermolysis of polyarylate model compounds. J. Polym. Sci. Polym. Chem. Ed. 1992, 30, 2321. [Google Scholar]
- Stuparu, M.C.; Xu, J.; Hall, H.K., Jr. Free radical generation of ketene during the synthesis of LCPs. Tetrah. Lett. 2009, 50, 6743–6744. [Google Scholar]
- Han, X. These Laboratories. Unpublished data. 2000.
- Oishi, T.; Hall, H.K., Jr. Model studies and a new melt polycondensation route to poly-bisphenol A-Iso/terephthalate (Polyarylate). J. Polym. Sci. Polym. Chem. Ed. 1992, 30, 83. [Google Scholar]
- Han, X.; Padias, A.B.; Hall, H.K., Jr. Syntheses of polyarylates by alcoholysis and esterolysis. J. Polym. Chem. Polym. Chem. 1999, 37, 2891. [Google Scholar]
- Han, X.; Padias, A.B.; Hall, H.K., Jr. Kinetics of the bulk polymerization of aromatic diacids and diacetates. Macromolecules 1997, 30, 8205. [Google Scholar]
- Somogyi, A.; Bojkova, N.; Padias, A.B.; Hall, H.K., Jr. Analysis of aromatic polyesters by matrix-assisted laser desorption/ionization time-time-of-flight mass spectrometry. Macromolecules 2005, 38, 4067–4071. [Google Scholar]
- Elandaloussi, E.H.; Somogyi, A.; Padias, A.B.; Bates, R.B.; Hall, H.K., Jr. Resequencing of comonomer units of well-defined vectro oligomers during MALDI-TOF mass spectral measurements. Macromolecules 2006, 39, 6913–6923. [Google Scholar]
- Cevasco, G.; Gaunti, G.; Hopkins, A.R.; Thea, S.; Williams, A. A novel dissociative mechanism. J. Org. Chem. 1985, 50, 479–484. [Google Scholar]
- Somogyi, A.; Elandaloussi, E.H.; Hall, D.E.; Padias, A.B.; Bates, R.B.; Hall, H.K., Jr. Powerfully solvating matrices for MALDI-TOD spectroscopy of aromatic polyesters. Macromolecules 2007, 40, 5311–5321. [Google Scholar]
- Yokoyama, A.; Iwashita, K.I.; Hirabayashi, K.; Alyama, K.; Yokozawa, T. Investigation of aromatic polyester synthesis by the chain-growth polycondensation synthesis. Macromolecules 2003, 36, 4328–4336. [Google Scholar]
- Linstid, H.C. Ticona Corp. Personal Communication. 2006.
- Choi, W.S.; Padias, A.B.; Hall, H.K., Jr. LCP aromatic polyesters by esterolysis melt polymerization. J. Polym. Sci. A Polym. Chem. 2000, 38, 3586–3595. [Google Scholar]
- Han, X.; Padias, A.B.; Hall, H.K., Jr. Synthesis of thermotropic LCP's using p-methoxycarbonyloxy aromatic acids. J. Polym. Chem. Polym. Chem. Ed. 1999, 37, 1703. [Google Scholar]
- Berti, C.; Bonora, V.; Pilati, F.; Fiorini, M. Aromatic polyesters based on bisphenol A. Macromolecules 1991, 24, 5269–5272. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Buyle Padias, A.; Hall, Jr., H.K. Mechanism Studies of LCP Synthesis. Polymers 2011, 3, 833-845. https://doi.org/10.3390/polym3020833
Buyle Padias A, Hall, Jr. HK. Mechanism Studies of LCP Synthesis. Polymers. 2011; 3(2):833-845. https://doi.org/10.3390/polym3020833
Chicago/Turabian StyleBuyle Padias, Anne, and Henry K. Hall, Jr. 2011. "Mechanism Studies of LCP Synthesis" Polymers 3, no. 2: 833-845. https://doi.org/10.3390/polym3020833
APA StyleBuyle Padias, A., & Hall, Jr., H. K. (2011). Mechanism Studies of LCP Synthesis. Polymers, 3(2), 833-845. https://doi.org/10.3390/polym3020833