Degradable Poly(ester amide)s for Biomedical Applications



Abstract
: Poly(ester amide)s are an emerging group of biodegradable polymers that may cover both commodity and speciality applications. These polymers have ester and amide groups on their chemical structure which are of a degradable character and provide good thermal and mechanical properties. In this sense, the strong hydrogen-bonding interactions between amide groups may counter some typical weaknesses of aliphatic polyesters like for example poly(ε-caprolactone). Poly(ester amide)s can be prepared from different monomers and following different synthetic methodologies which lead to polymers with random, blocky and ordered microstructures. Properties like hydrophilic/hydrophobic ratio and biodegradability can easily be tuned. During the last decade a great effort has been made to get functionalized poly(ester amide)s by incorporation of α-amino acids with hydroxyl, carboxyl and amine pendant groups and also by incorporation of carbon-carbon double bonds in both the polymer main chain and the side groups. Specific applications of these materials in the biomedical field are just being developed and are reviewed in this work (e.g., controlled drug delivery systems, hydrogels, tissue engineering and other uses like adhesives and smart materials) together with the main families of functionalized poly(ester amide)s that have been developed to date.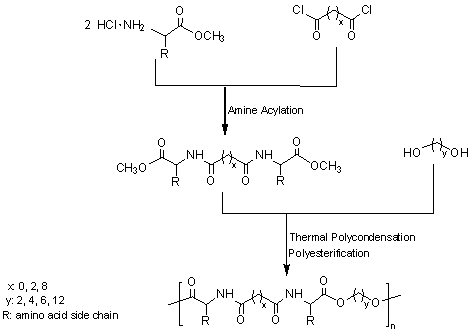
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Synthetic biodegradable polymers offer an alternative to nondegradable materials used in biomedical applications. Polymers can be synthesized with tailored mechanical and degradative properties by changing the composition and microstructure and consequently can be designed to fulfill specific requirements. Polymers can also be easily processed in different forms (i.e., multi- or monofilaments for surgical sutures, porous scaffolds with desired pore morphologic features conducive to tissue ingrowth or as micro/nanospheres for controlled drug delivery). Functionalized polymers are receiving great attention for these applications since they can link different kinds of drugs or modulate cellular function and induce tissue ingrowth [1,2].
Polyesters constitute nowadays the main family of synthetic biodegradable polymers used as commodity materials and even in the biomedical field. Most applications and studies concern polyglycolide, polylactide, poly(ε-caprolactone) and their copolymers. Considerable work has been performed with functionalized polyesters since they allow a subsequent chemical modification to achieve desirable properties including hydrophilicity, biodegradation rate or bioadhesion, to attach compounds with pharmacologic activity or in general targeting the subsequent medical treatments. Different strategies have been developed to synthesize functionalized polyesters with biologically relevant and compatible molecules. In this way, multihydroxy functional polyesters can be prepared by copolymerization of ε-caprolactone with 5-ethylenedioxy-ε-caprolactone and subsequent deprotection and reduction steps [3]. Polyesters functionalized with acid groups were easily prepared by reacting hydroxyl terminated oligo(ε-caprolactone)s with anhydrides like succinic, maleic and glutaric anhydrides [4]. This acid functionality could be enhanced by conversion to an acid chloride or an anhydride functionality by reaction with thionyl chloride or acetic anhydride, respectively. Polymerization of lactones containing protected functional groups (e.g., hydroxyl-, bis(hydroxyl)-, amino- and carboxyl-substituted) is also a suitable strategy when protecting groups could easily be removed [5].
Besides aliphatic polyesters, various types of functionalized synthetic polymers have been designed, tested and, in some cases, commercialized for biomedical applications, although in general are still in a premature stage. Poly(ester amide)s (PEAs) constitute a promising family of biodegradable materials since they combine a degradable character, afforded by hydrolizable ester groups (–COO–) placed in the backbone, with relatively good thermal and mechanical properties given by the strong intermolecular hydrogen bonding interactions that can be established between their amide groups (–NHCO–). Currently, a considerable variety of PEAs have been studied including the use of different monomers (e.g., α-amino acids, α,ω-aminoalcohols or carbohydrates) or different polymer microstructures (e.g., ordered, blocky or random monomer distributions) [6,7]. The presence of hydrolytically cleavable ester bonds in the backbone and the lowering of the crystallinity make also poly(ester amide)s promising materials for their use in medical fields. Note also that the incorporation of α-amino acid moieties may lead to biocompatible materials and, depending on their side groups, to functionalized polymers.
This review is focused on the synthesis and potential applications of functionalized PEAs. In this way, two synthetic routes will be discussed in Section 2: (a) Ring opening polymerization of functionalized lactones and (b) Polycondensation of functionalized monomers with reactive hydroxyl, amine or ester activated end groups. Section 3 is devoted to highlight relevant examples of functionalized PEAs, whereas main potential applications are discussed in Section 4.
2. Synthesis of Functionalized Poly(ester amide)s
2.1. Ring Opening Polymerization
2.1.1. Polydepsipeptides
Copolymers of α-hydroxy acids and α-amino acids, polydepsipeptides, are mainly prepared by the ring-opening polymerization of morpholine-2,5-dione derivatives. The use of single ring monomers logically led to poly(ester amide)s with a regular chemical structure, whereas random and even block copolymers were prepared according to the selected synthetic procedure (e.g., one step or two step synthesis) and using the appropriate combination of morpholine-2,5-diones.
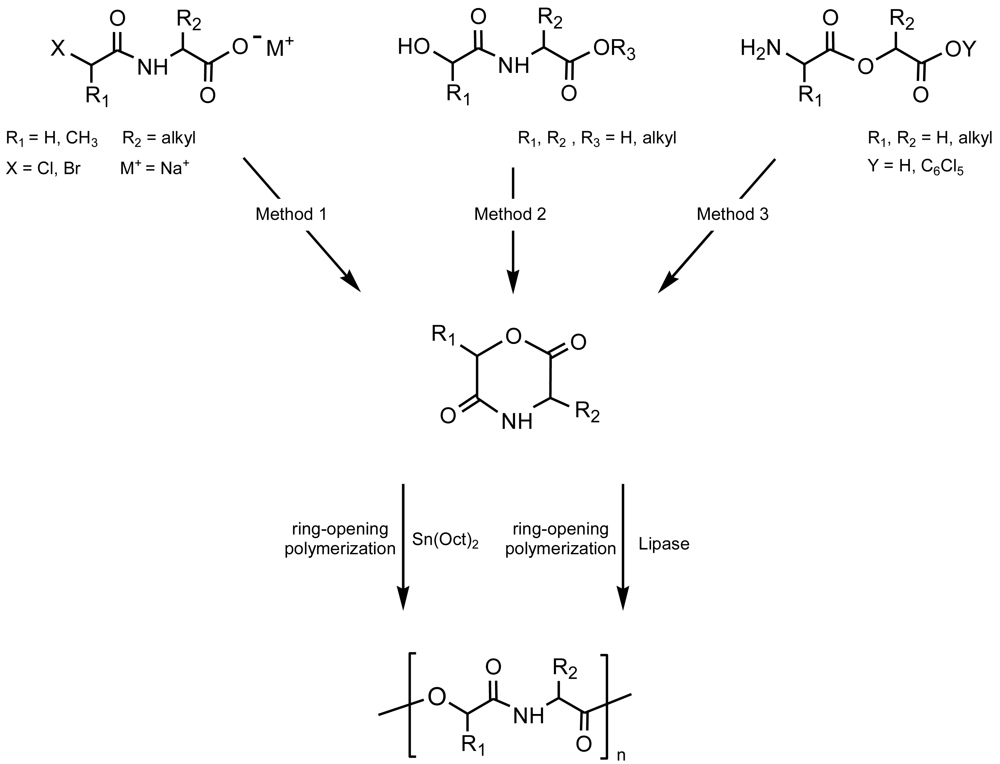
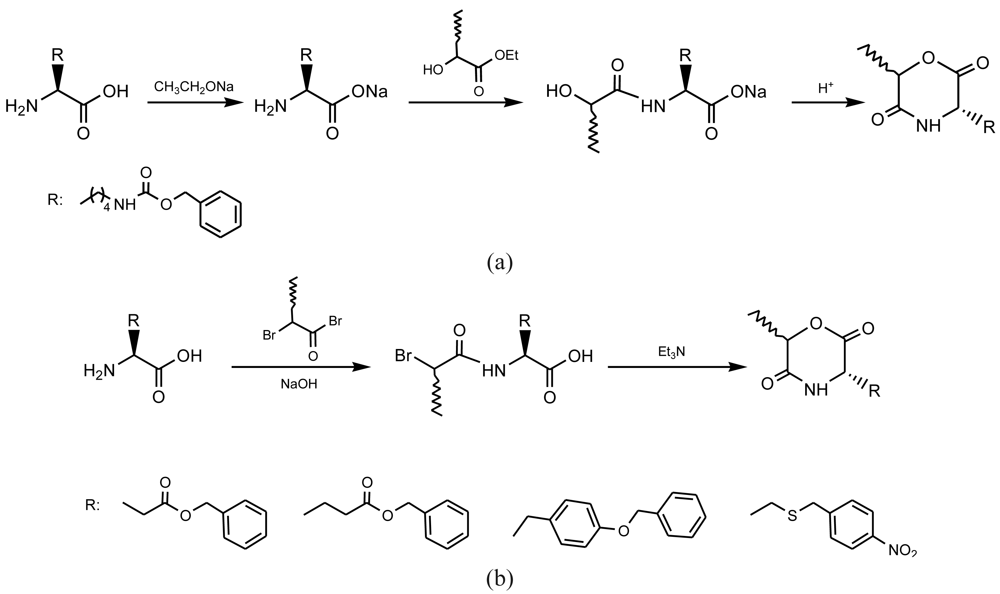
Derivatives of the 6-membered ring of morpholine-2,5-dione were synthesized by three main procedures [8] (Figure 1): (a) Cyclization of N-(α-haloacyl)-α-amino acid salts [9-13], (b) Intramolecular transesterification of N-(α-hydroxyacyl)-α-amino acid esters [14,15] and (c) Cyclization of O-(α-aminoacyl)-α-hydroxycarboxylic acids [16,17]. The first method probably gave the higher yields but conduced also to some degree of racemization when optically active monomers were employed. In this case, cyclization goes through an intramolecular SN reaction mechanism that may cause some racemization at the stereocenter of the hydroxy acid unit (C6). The third method based on an intramolecular amidation showed the lowest yields but some racemization at both C3 and C6 stereocenters.
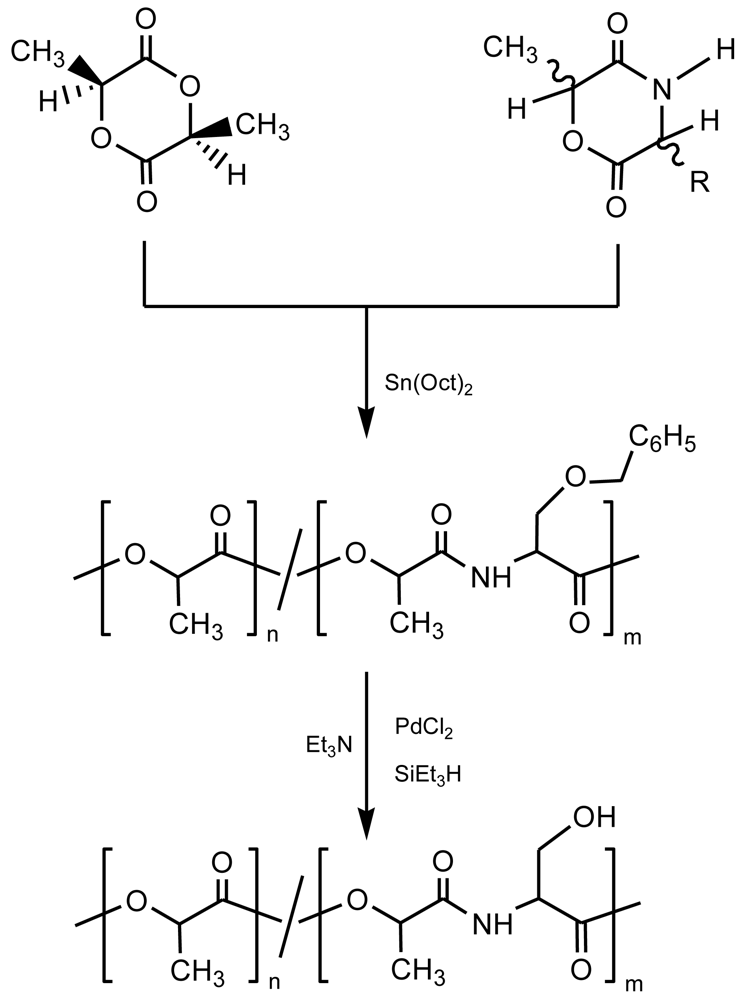
Polymerization of morpholine-2,5-diones were usually carried out in bulk using stannous octoate [Sn(Oct)2] as a catalyst, at reaction temperatures close to the melting temperature of the monomer, and adding water or alcohol molecules as initiators [18]. It was postulated that the ring opening reaction was initiated by a tin (II) hydroxyl or alkoxide group. Similarly, polymerizations were successfully assayed with several calcium alcoholates. Interestingly, the ring opening reaction didn't take place with N-alkyl substituted morpholine-2,5-diones, suggesting that the amido NH group should be involved in a reaction leading to a chelating intermediate when ring-opening occurred by attack at the ester group [19]. It is also assumed that an alkyl substitution at C3 decreased polymerizability in homopolymerization and even in copolymerization with other cyclic monomers.
Enzymes such as lipases were another type of efficient catalyst [20-22] for polymerizations that, in this case, proceeded through ring-opening at the ester bond. Thus, the polymerization behavior was strongly influenced by the configuration of the hydroxyl acid moiety but not by the configuration of the α-amino acid moiety since steric effect of hydroxyl acid side chains decreased the activity of the cyclic monomer. In general, enzymatic polymerization gave rise to an enhanced racemization (mainly at the amino acid residue) than more conventional Sn(Oct)2-catalyzed polymerizations.
2.2. Polycondensation Methods
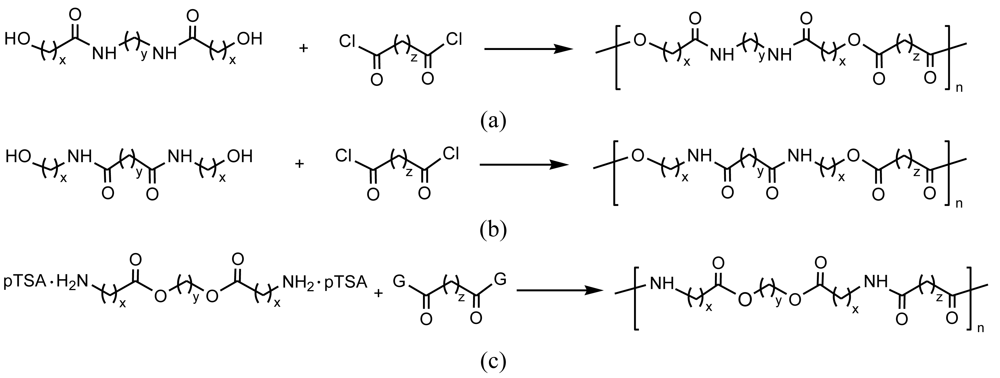
Polycondensations are usually applied to react diamide-diol, diester-diamine, ester-diamine or diamide-diester monomers with dicarboxylic acid derivatives or diols, as shown in Figure 2. Reaction of α,ω-aminoalcohols with acid anhydrides or dicarboxylic acid derivatives is another typical way to get PEAs from condensation reactions. It is easy to get functionalized PEAs when monomers incorporate α-amino acid or carbohydrate units, or have at least one unsaturated monomer which could be for example a simple dicarboxylic acid derivative.
2.2.1. Melt Polycondensation
This method is advantageous for industrial production because no post-treatment is necessary after the polymerization reaction. This is performed under reduced pressure and temperature to favor the elimination of condensation products and using transesterification catalysts. The synthesis is usually carried out in two temperature steps: The first one operates under milder conditions and gives rise to a prepolymer; in the second step the temperature is significantly raised to favor the condensation process and to get a high molecular weight sample.
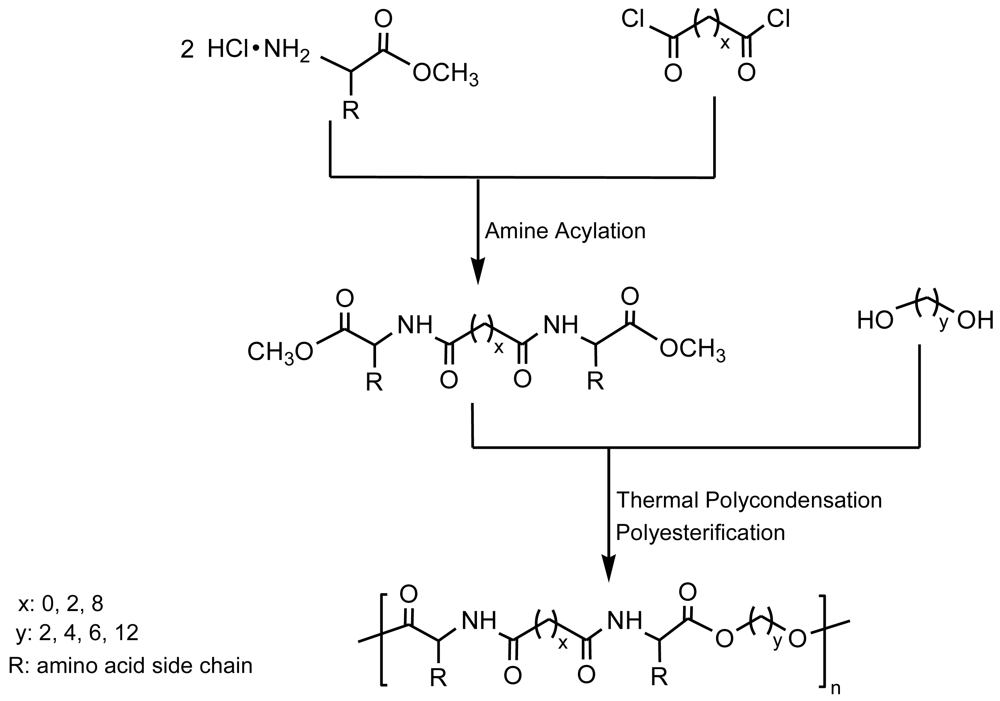
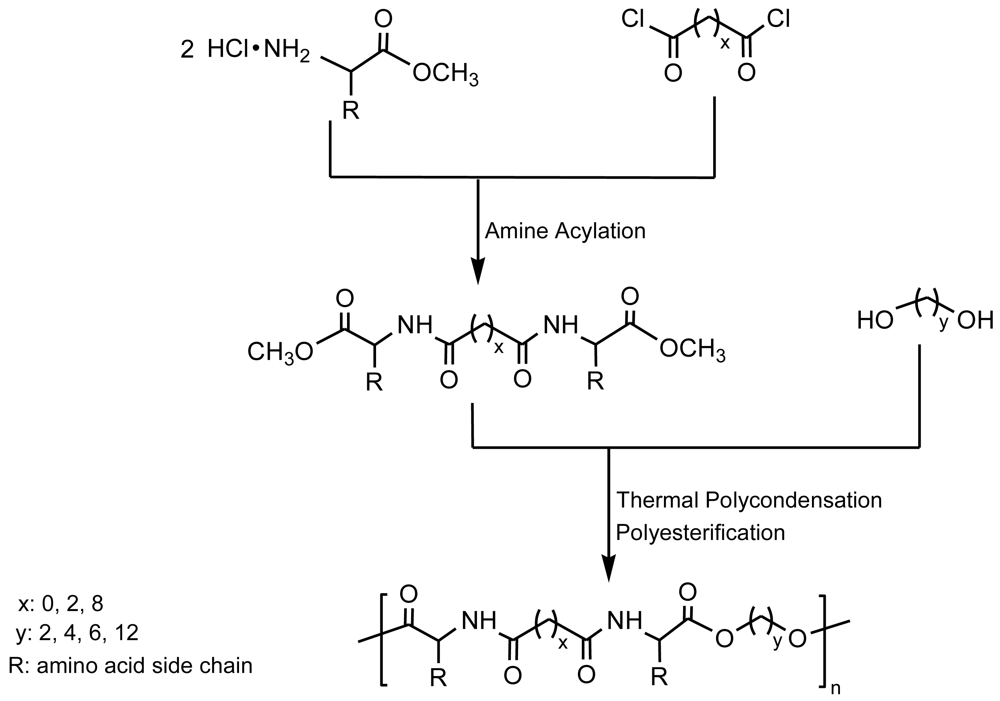
This procedure has been successfully applied to prepare poly(ester amide)s containing α-amino acid units by reaction of a diol with a diamide-diester previously obtained by condensation of a diacid chloride with an α-amino acid methyl ester [23,24] (Figure 3). Secondary reactions derived from the required high temperatures constitute the main disadvantage of this method that may limit the final molecular weight and cause problems when monomers have functional side groups highly susceptible to undertake these undesirable reactions.
2.2.2. Interfacial Polymerization
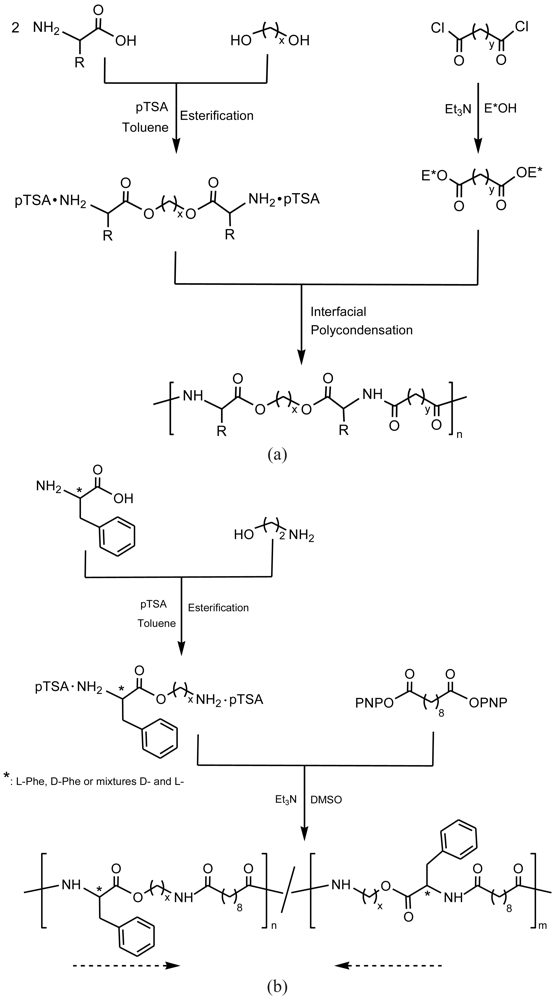
Interfacial polymerization can be carried out by reaction of a diacid chloride soluble in an organic solvent with a diamine or a diol soluble in a water medium. Reaction is strongly influenced by the solvent system, catalytic and surfactant additives. The choice of the organic solvent is important since it affects other polymerization factors, such as the potential partition of reactants between the two phases, the diffusion of the reactants, reaction rate, solubility and swelling or permeability of the growing polymer [25]. Surface-active agents can be added to increase interfacial area and contact between reactants. Phase-transfer catalysts, generally a small symmetric quaternary ammonium cation, may favor the nucleophilic displacement reaction characteristic of the interfacial polycondensation [26].
Interfacial polymerization reaction can be carried out at room temperature and consequently typical problems associated to the melt condensation process can be avoided. However, the low-temperature interfacial method has also several drawbacks, among which numerous side reactions that lead again to chain-termination and unit-heterogeneity should be noted. Interactions between aliphatic diacid chlorides and tertiary amines are especially noteworthy side reactions that prevent the formation of high molecular weight polymers.
Typically poly(ester amide)s containing α-amino acid units can be, in this case, prepared by reaction of a diacid chloride with a diester-diamine obtained by previous condensation of a diol with two α-amino acid units [27-30]. Since the diester-diamine is unstable as free base and tends to produce undesirable side reactions, the monomer is prepared as a stable salt of di-p-toluenesulfonic acid (Figure 4).
2.2.3. Solution Polycondensation
High polymerization rates, mild reaction conditions, high molecular weights and minimal side reactions are expected features of solution polycondensation reactions that make use of condensing agents or activation groups for the carboxylic acid in order to facilitate aminolysis reactions.
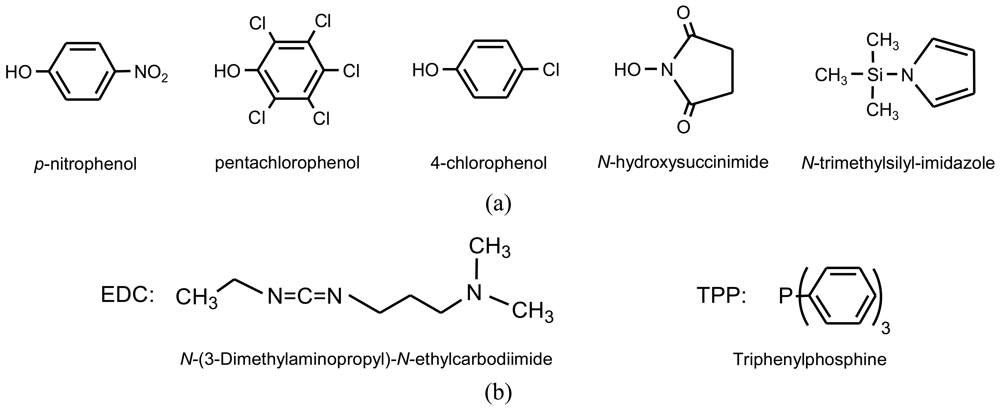
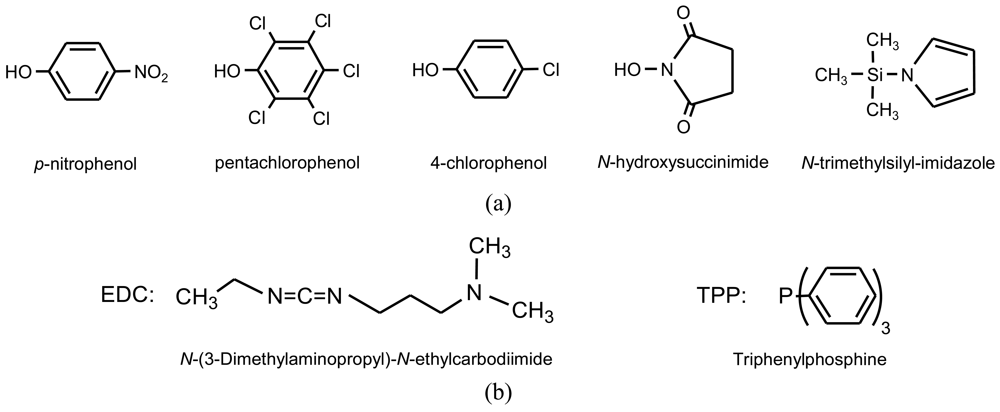
Activation of carboxylic groups is a well known method in peptide chemistry that is based on the activation of the carboxylic acid by leaving groups that form new ester or imide derivatives (Figure 5(a)). These leaving groups are liberated as low-molecular weight by-products after polycondensation. Figure 6 shows typical reactions to obtain α-amino acid derivatives by reaction of an activated dicarboxylic acid with a diester-diamine [31,32] (obtained by condensation of a diol with two α-amino acid units) and a ester-diamine [33] (obtained by condensation of an α,ω-aminoalcohol with an α-amino acid unit that gave rise to an aregic polymer).
Esterification reactions between carboxylic and hydroxyl functions can also be favored by the use of condensing agents such as a carbodiimide (Figure 5(b)) that facilitate the elimination of water molecules under mild conditions. This procedure was applied to get functionalized PEAs from hydroxycarboxylic acids prepared from acid anhydrides (e.g., maleic anhydride) and aminoalcohols [34].
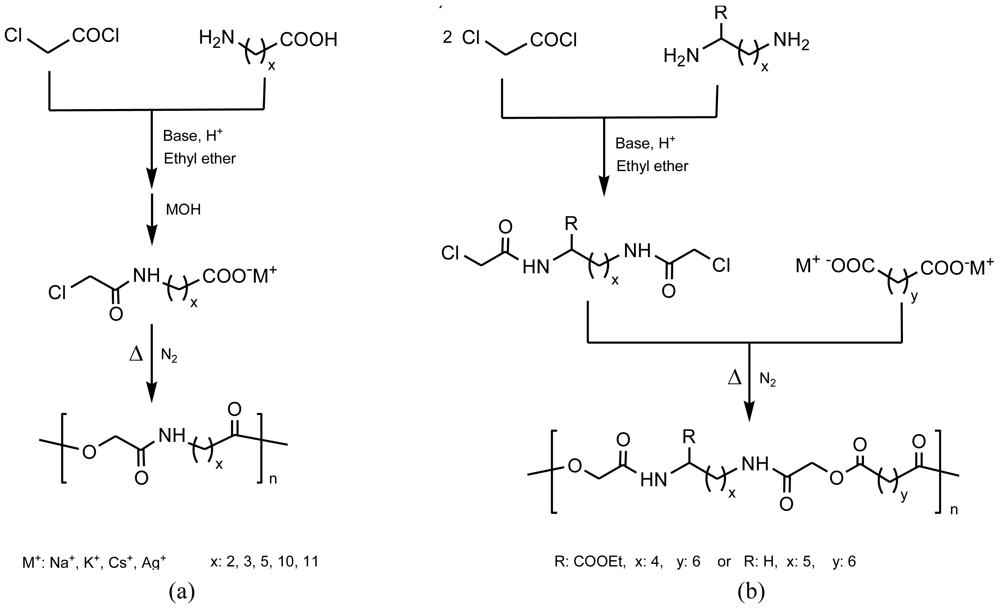
2.2.4. Solid/Liquefied State Polycondensation
A thermal polycondensation reaction based on the elimination of a metal halide as a driving force was also applied to prepare PEAs based on chloroacetate derivatives of an ω-amino acid [35,36] or a diamine [35,37] (Figure 7). Polymerizations can take place in the solid phase, the liquefied phase, or both phases, depending on the length of the monomers (i.e., the number of methylene groups) and the metal cation involved (e.g., cesium, potassium or sodium). In fact, these two conditioning factors have an influence on the melting point of the monomer, which can be either higher or lower than the required reaction temperature. This depends also on the cation and decreases in the order cesium < potassium < sodium salt. The method appears suitable to prepare functionalized PEAs using protected L-lysine as a diamine. Furthermore, the inorganic salt by-product can easily be removed by extensive washing with water, giving rise to a highly porous material of interest for some biomedical applications.
3. Main Families of Functionalized Poly(ester amide)s
3.1. Functionalized Poly(ester amide)s from Polymerization of Morpholine-2,5-dione Derivatives
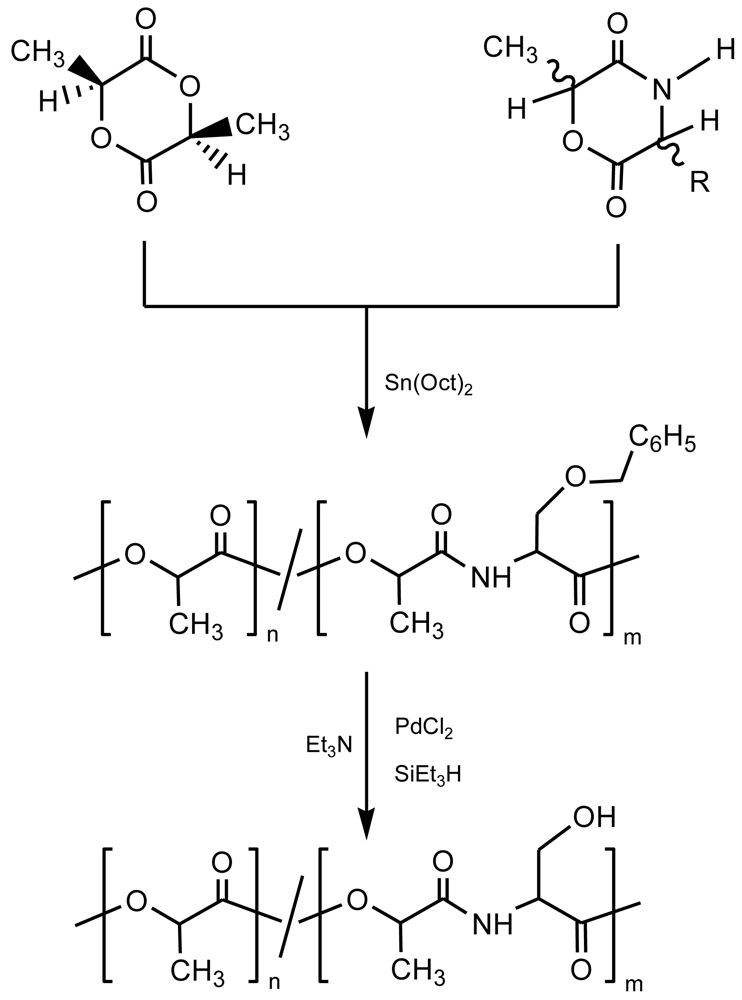
Functionalized PEAs can be obtained by ring opening polymerization of morpholine-2,5-diones derived from α-amino acids like L-aspartic acid, L-glutamic acid, L-lysine, L-serine (or L-tyrosine) and L-cysteine (Figure 8), which provide pendant carboxylic acid, amine, hydroxyl and thiol groups, respectively [38-43]. The synthesis of the six-membered ring required a previous protection of functional groups, which should be stable under the polymerization conditions [8]. Protective groups must be selectively removed without cleavage of ester and/or amide bonds of the polymer main chain once the polymerization step is finished.
The benzyl group was usually employed to protect carboxylic acid (e.g., aspartic or glutamic acids) and hydroxyl (i.e., tyrosine or serine) functions [14], respectively. The p-nitrobenzyl group appears adequate for thiol groups (e.g., cysteine), whereas the benzyloxycarbonyl group was employed for the ε-amino group of lysine. All these protective groups can easily be removed by catalytic hydrogenation without affecting the ester linkages of the polymer chain.
Polydepsipeptides derived from morpholine-2,5-diones containing glycolic acid and protected l-lysine, l-aspartic acid or l-glutamic acid [40,43] have been successfully synthesized although final molecular weights were low. In general, substituted morpholine-2,5-diones have a low reactivity and strategies based on the copolymerization with more reactive lactones (e.g., ε-caprolactone and D,L-lactide) were proposed [14,44] as well as the copolymerization of lactones with amino acid carboxyanhydrides [45].
Biodegradable copolymers having pendant reactive groups were obtained by ring-opening polymerization of L-lactide with morpholine-2,5-diones constituted by glycolic acid and aspartic acid or L-lysine, or L-lactic acid and L-lysine [39,40]. Interestingly, these polymers showed a faster enzymatic and hydrolytic degradation than polylactide
Gonsalves et al. [46] copolymerized the protected morpholine-2,5-dione derived from L-lactic acid and L-serine with different feed ratios (80 to 94 mol%) of L-lactide. After removal of the protective benzyl groups a copoly(ester amide) with pendant free hydroxy functional groups was obtained (Figure 9). These were also reacted with diisocyanate-terminated poly(ethylene glycol) to render polymers with a different degree of crosslinking (depending on the amount of free hydroxyl groups). The addition of poly(ethylene glycol) decreased the Tg (from 64 °C to 5 °C) and drastically increased the amorphous character.
Barrera et al. synthesised a copolymer of L-lactic acid and L-lysine containing about 2 mol% of L-lysine residues [39,47]. Further reaction of pendant lysine with Nε-(benzyloxycarbonyl)-L-lysine-N-carboxyanhydride provided up to a 35-fold increase in the lysine residue [48].
3.2. Poly(ester amide) s Prepared by Polycondensation Reactions and Containing Functionalized α-Amino Acids
The thermal polycondensation method based on the formation of metal halide salts as a driving force (Section 2.2.4.) was successfully applied to get potentially functionalized PEAs using the ethyl ester of L-lysine as a diamine unit. No secondary reactions such as transesterification occurred under the required polymerization conditions (Figure 10) [49].
Atkins et al. [50,51] used a simple and versatile strategy based on orthogonal protecting groups to get PEAs from polycondensation reactions of a diester-diamine and a diacid chloride. This method allows the incorporation of basic and acid functions using L-lysine or L-aspartic acid to prepare the diester-diamine monomer. Copolymerizations could also be performed using monomers based on different α-amino acids (Figure 11). Removal of the protective groups provides appropriate pendant amine or carboxylic acid functionalities to attach different compounds. For example, the carboxylic acid groups of a polymer containing L-aspartic acid units were converted to N-hydroxysuccinimidyl esters, which provided useful templates for further derivatization.
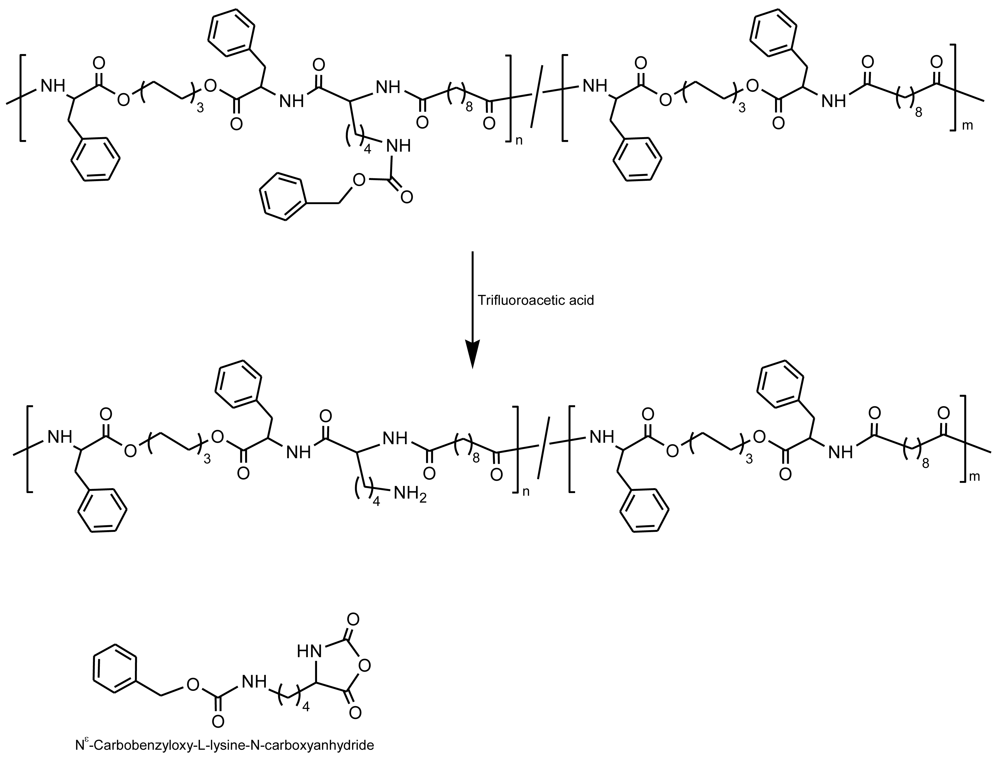
Chu et al. [52] have reported a method that allows the preparation of random PEAs having free amine groups (Figure 12). The procedure involved two steps: A ring-opening reaction of a protected amino acid derivative (e.g., Nε-(benzyloxycarbonyl)-L-lysine-N-carboxyanhydride (Z-LysNCA)) with di-p-toluenesulfonic acid salts of bis-L-phenylalanine hexane-1,6 diester (Phe-6)), followed by solution polycondensation of the above mixture of monomers with di-p-nitrophenyl sebacoylate. The pendant free amine groups on copoly(ester amide)s were easily regenerated by a subsequent deprotection under a simple acid treatment. The content of amine groups of the resulting functional PEA copolymers was controlled by adjusting the feed ratio of Phe-6 to Z-LysNCA.
All PEAs having L-lysine content were amorphous and exhibited Tgs ranging from 18 to 32 °C. The bulky pendant protective group in the L-lysine unit decreased the glass transition temperature due to its plasticizing effect. After deprotection, the recovered pendant amine groups strengthened intermolecular interactions among PEA chains via hydrogen bonds and increased the glass transition temperature. The preliminary data of cell proliferation and cytotoxicity of these new functional and positive charged PEAs show that they support bovine aortic endothelial cell proliferation without cytotoxicity.
Cytotoxicity, ability to support cell growth, inflammatory properties, and mechanical properties have been investigated for some amino and carboxylic acid functionalized PEAs. These were prepared by solution polycondensation of diamide-diols containing the α-amino acid units and the p-nitrophenyl ester of the appropriate dicarboxylic acid [53]. Results indicate that all forms of PEAs were noncytotoxic and noninflammatory in vitro. The amino-functionalized PEAs best supported endothelial cell adhesion, growth, and monolayer formation. Data suggested that PEAs could be a viable biomaterial for use in tissue engineering applications, particularly for use as a vascular graft.
Random copoly(ester amide)s having pendant hydroxyl groups were prepared by a two-step polycondensation using dimethylolpropionic acid (DMPA), a diamine derived from glycine and 1,6-hexanediol, and sebacoyl dichloride. Among the three reactants, DMPA is a diol containing a pendant carboxyl group, which after incorporation into the molecular chain provides the possibility of further connecting drugs or other bioactive molecules to the polymer [54].
3.3. Unsaturated Poly(ester amide)s
Another approach to get functionalized PEAs is based on the incorporation of reactive carbon-carbon bonds which can be placed on either the side or the main chain.
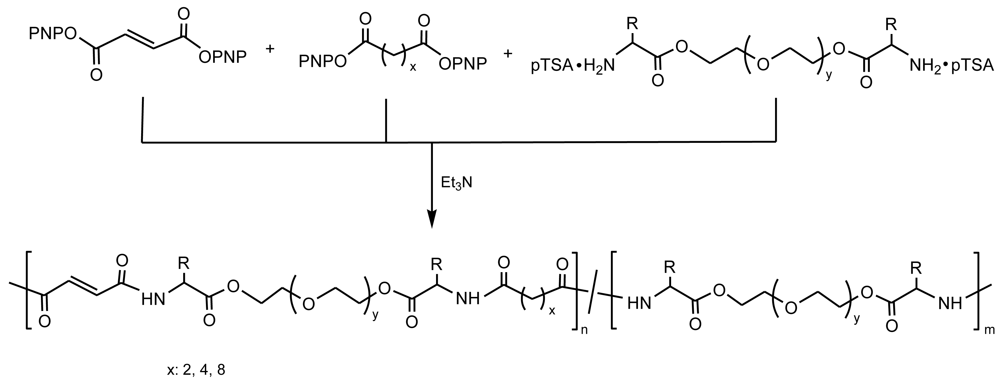
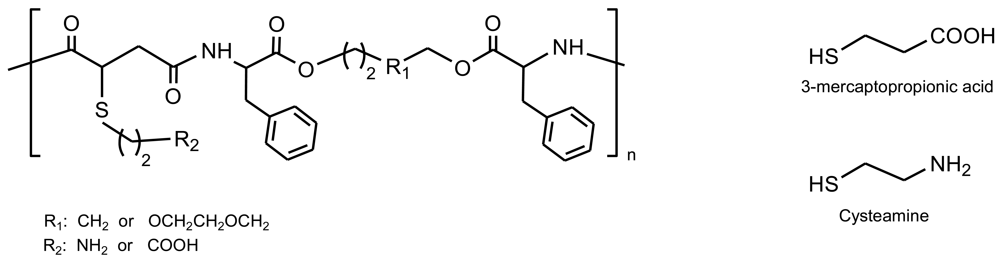
A series of biodegradable functional amino acid-based PEAs were designed and synthesized by the solution co-polycondensation of amino acid (L-phenylalanine and DL-2-allylglycine) based monomers and p-nitrophenyl esters of dicarboxylic acids [55] (Figure 13). Polymers incorporated pendant carbon-carbon double bonds through the DL-2-allylglycine units and consequently the content on these functional groups could be adjusted by tuning the feed ratio of L-phenylalanine to DL-2-allylglycine containing monomers. The glass transition temperature of PEAs decreased on increasing the methylene chain in both the amino acid and dicarboxilic acid segments. The reactivity and utility of the pendant double bonds was verified using a free-radical addition method [50,56,57], which converts them into carboxylic acid, amine, and sulfonate functionalities by reaction with 3-mercaptopropionic acid, 2-aminoethanethiol hydrochloride and sodium-3-mercapto-1-propanesulfonate, respectively. In this way, the incorporation of the functional pendant carbon-carbon double bonds along the polymer chains could significantly expand the biomedical applications via either their capability to conjugate bioactive agents or prepare additional useful functional derivatives.
A series of copoly(ester amide)s with positively charged guanidine side groups and pendant carbon-carbon double bonds were similarly prepared by co-polycondensation of amino acid (L-arginine and DL-2-allylglycine) based monomers and p-nitrophenyl esters of dicarboxylic acids [58]. All these cationic PEAs showed good solubility in polar solvents like water, alcohol, DMSO, and DMF. The cytotoxity studies indicated that the new PEAs were nontoxic to bovine aortic endothelial cells at the tested concentrations and exposure times.
The use of unsaturated diols and/or diacids could provide the PEA backbone of reactive double bonds [59-62]. These bonds are new reactive sites that could be used to make crosslinked hydrogel networks, which are highly interesting as drug carriers, as will be discussed in the next section [63].
Unsaturated PEAs containing α-amino acids have been synthesized by the conventional method, based on the condensation of bis(α-amino acid) α,ω-alkylene diesters and p-nitrophenyl esters of aliphatic dicarboxylic acids. Unsaturated or even mixtures of unsaturated and saturated dicarboxylic acids can be employed. Incorporation of the unsaturated units (e.g., by using bis-p-nitrophenyl fumarate) brings C=C double bonds in the backbone that increase rigidity and the glass transition temperature (respectively to saturated polymers with a similar chemical structure).
Chu et al. studied the polymerization of the p-toluenesulfonic acid salt of L-phenylalanine butane-1,4-diester with different mixtures of di-p-nitrophenyl esters of saturated (succinic, adipic or sebacic acids) and unsaturated (fumaric acid) dicarboxilic acids [59,64]. Copolymers could be obtained with rather high Mn molecular weights which varied between 14,600 and 36,900 g/mol. Copolymers having different unsaturation levels and properties were easily achieved by the adjustment of the feed ratio of the two dicarboxylic monomers. Thermal properties, solubility, and biodegradation rate was found to vary within the range defined by the characteristics of pure saturated and unsaturated polymers. In vitro biodegradation tests showed that these copolymers could be enzymatically hydrolyzed by α-chymotrypsin even at a low enzyme concentration, although their hydrolysis in a pure PBS buffer medium was slow. It was also found that the biodegradability of copolymers was affected by the length of the methylene groups in the saturated dicarboxylic units (the increase resulted in a lower hydrolyzable ester bond density and a slower biodegradation rate). The applied methodology seems highly interesting to tune properties in order to meet the requirements for a wide range of biomedical uses.
The range of application of amino acid based PEAs can be expanded by employing oligo(ethylene glycol)s instead of conventional diols in the solution polycondensation performed from diester-diamine monomers [65]. The obtained poly(ether ester amide)s have new blocks with ether linkages that may enhance hydrophilicity, flexibility and biodegradability. Depending on the type and concentration of monomers used, such as α-amino acid, saturated or unsaturated dicarboxylic acids, different dialcohols and oligo(ethylene glycol)s, materials with a wide range of thermal, mechanical, and biological properties could be attained in order to meet applications for pharmaceutical, biomedical, and tissue engineering applications.
A series of biodegradable random unsaturated/saturated poly(ether ester amide)s were synthesized by solution polycondensation of diamine salts of phenylalanine and triethylene glycol with the p-nitrophenyl active esters of mixtures between unsaturated (fumaric acid) and saturated dicarboxylic acids (succinic, adipic and sebacic acid) (Figure 14) [62]. These random copolymers were obtained with fairly good yields and Mn molecular weights ranging from 3,000 to 27,000 g/mol, and polydispersity indices between 1.52 and 2.13. Copolymers showed a high degradability in α-chymotrypsin enzyme solutions and the biodegradation rates decreased with the unsaturated content. Interestingly, it was concluded that upon adjusting monomers feed ratio, copolymers could have controlled chemical, physical, and biodegradation properties.
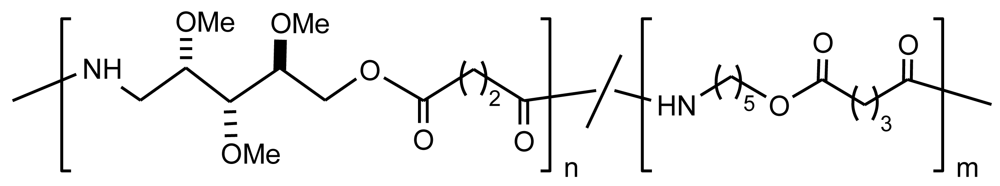
A series of unsaturated PEAs was synthesized using a mixture of diester-diamine monomers derived from oligo(ethylene glycol) and 2-butene-1,4-diol, and a mixture of p-nitrophenyl esters of saturated and unsaturated diacids [60,66] (Figure 15). Note that unsaturations were present in both diacid and diol moieties.
Unsaturated random copoly(ester amide)s were also synthesized by reaction of a mixture of phthalic and maleic anhydrides with ε-caprolactam and a mixture of ethylene and neopenthylene glycols [67]. The final oligomers (Mn 2,100–2,600 g/mol) were effectively crosslinked using vinyl acetate and benzoyl peroxide—ascorbic acid—as initiator-accelerant agents. The new materials showed a high compressive strength (104.0 MPa) and were hydrolytically degradable. Heat treatment conditions and crosslinker content played an important role to decrease the cumulative mass loss during the hydrolysis process. Measured properties suggested that these copolymers might be potentially used as a new type of bone fixation material. Copolymers with similar characteristics were prepared by the same procedure but changing ε-caprolactam with 1,6-hexanediamine [68] or glycine [69]. In the first case, preliminary biocompatibility in mice, skin was evaluated and the results were promising for their expected biomedical applications. Mechanical properties of composites constituted by the glycine derivative and calcium polyphosphate fibers were also evaluated. Results indicated that the mechanical strength increased very quickly as the fiber content raised. Flexural and compressive strength achieved maximal values when this content was close to 50–60 wt%.
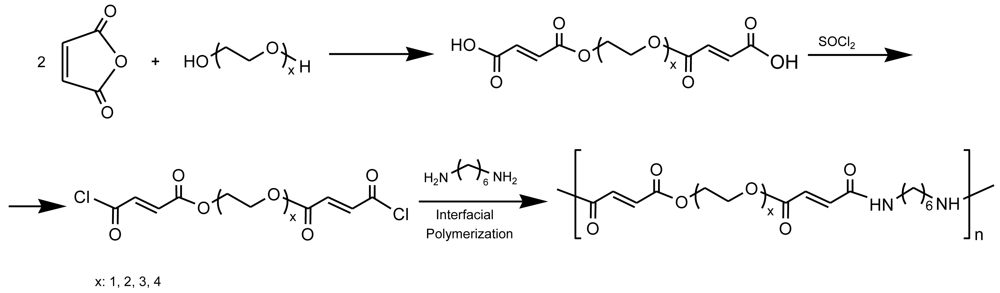
A series of biodegradable unsaturated PEAs containing also ethylene glycol moieties (from 1 to 4 units) was successfully prepared through interfacial polycondensation of 1,6-hexanediamine and the unsaturated diacylchloride derived from a previous reaction of maleic anhydride with oligo(ethylene glycol) (Figure 16) [70]. The obtained poly(ether ester amide)s were amorphous, and stable up to 300 °C under nitrogen, showed good hydrophilicity and improved solubility. Polymers were hydrolyzable in a rapid and steady way, increasing the rate of hydrolytic degradation by increasing the number of ether linkages per repeat unit.
An unsaturated dicarboxylic-terminated oligoester was prepared by reaction of ethylene glycol lactate diol with maleic anhydride [71]. The oligoester was then melt-polycondensed with toluene-2,4-diisocyanate to render a crosslinked resin with unsaturated double bonds (Figure 17). The new polymer showed a porous structure due to the formation of CO2 in the last synthesis step and was degradable. As a result of introducing isolated C=C double bonds, the polymer was flexible enough to exhibit shape-memory characteristics. It is worth mentioning that the glass transition temperature was close to human body temperature which meets one of the basic requirements for medical applications.
Chu et al. [72] reported an original way to get amino and carboxylic acid pendant groups from unsaturated poly(ester amide)s and poly(ether ester amide)s based on α-amino acids (i.e., L-phenylalanine). These functional groups were easily incorporated by the grafting of a thiol onto the unsaturated polymer via a thiol–ene reaction in the presence of a radical initiator (Figure 18). This one-step functionalization reaction was carried out with very high yields (close to 100%) under mild conditions and had clear advantages over synthesis based on selective protection and deprotection of functionalized α-amino acid derivatives.
3.4. Carbohydrate Derivatives
Polyesters derived from highly functionalized carbohydrates (e.g., hexitols such as sorbitol, iditol, glucitol, galacticol or mannitol and other alditols like xylitol, ribitol or arabitol) can be effectively synthesized by using lipases such as Novozyme 435, a lipase B from Candida antarctica immobilized on a resin [73-75]. The method is attractive since it appears to be regioselective, requiring mild reaction conditions, and the terminal hydroxyl groups of the hexitol or alditol units react faster. Thus, it is feasible to get polymers with different non reacted hydroxyl side groups. The procedure has not yet been applied to prepare PEAs, but hexitol and alditol derivatives have been synthesized by different methods, which seem interesting to review. In addition, the use of carbohydrates is also receiving greater attention since, in concert with the depletion of oil resources, the effective utilization of renewable resources (e.g., plant-biomass and carbohydrates) appear as an alternative that can be steadily supplied and used for polymer syntheses.
3.4.1. Derivatives of L-Arabinose and D-Xylose
L-Arabinose and D-xylose were transformed into 1-amino-1-deoxy-2,3,4-tri-O-methyl-5-O-[(pentachlorophenoxy)succinyl]-L-arabinitol and 1-amino-1-deoxy-2,3,4-tri-O-methyl-5-O-[(pentachlorophenoxy)succinyl]-D-xylitol hydrochlorides, respectively, in a seven step synthesis and then polymerized in solution using ethyldiisopropylamine as an acid acceptor (Figure 19) [76,77]. The regular PEA was derived from arabinose melted above 135 °C [140] whereas the polymer derived from xylose was amorphous indicating a great chain conformational flexibility that may contribute to the decreasing of cohesive forces between chains [78]. Degradation of this kind of polymers was found to occur by hydrolysis of the ester linkages at a rapid rate which depended on the crystallinity and hydrophilicity of the samples [79]. PEAs containing succinic acid units degraded faster according to a postulated mechanism where succinimide rings were formed [80].
Random copolymers were also prepared from appropriate mixtures of the above indicated arabinose-succinyl monomers and 5-amino-1-O-[(pentachlorophenoxy)glutaryl]pentanol hydrochloride (Figure 20) and their hydrolytic degradation studied [81]. Results demonstrated that the degradation rate could be enhanced by increasing the amount of the sugar-based monomer incorporated in the polymer chain. In this way, small amounts of this monomer were enough to produce a noticeable increase in polymer degradability.
3.4.2. Derivatives of Hydrophobic α-Amino Acids and Dianhydrohexitols
Dianhydrohexitols (e.g., 1,4:3,6-dianhydrosorbitol and 1,4:3,6-dianhydromannitol) were used as secondary diols to react with α-amino acids (L-phenylalanine, L-leucine, L-isoleucine, and L-methionine) in the presence of p-toluenesulfonic acid [82]. The obtained diester-diamines were then polycondensed in solution using the p-nitrophenyl active esters of even dicarboxylic acids with a number of methylene groups ranging from 4 to 10 (Figure 21). To our knowledge, PEAs derived from functionalized α-amino acids have not so far been prepared although their synthesis shows no additional problems regarding the above related PEAs.
All these polymers appear highly interesting since diols are available in industrial quantities, are derived entirely from renewable resources (starch) and are used in pharmacy (i.e., they are non-toxic). Problems associated with the low reactivity of the sterically hindered secondary hydroxyl groups were avoided with the preparation of O,O′-bis-α-aminoacyl derivatives which led to sterically unhindered (by the bicyclic fragments at least) and highly active functional amino groups that facilitated the polymer synthesis. In this way, polymers could be obtained with rather high molecular weights (e.g., 32,000 g/mol for the derivative of phenylalanine, sebacic acid and mannitol) and narrow polydispersities (1.0-1.7). Polymers had glass transition temperatures of up to 60–120 °C due to the presence of rigid cycles and higher than the related polymers derived from alkylenediols.
Degradation studies performed with α-chymotrypsin and lipase revealed highest tendency towards enzyme catalyzed hydrolysis for the PEAs based on L-phenylalanine, probably due to the highest hydrophobicity of the benzyl side groups. Hydrolysis decreased with the length of the diacid unit (i.e., with increasing hydrophobicity of the polymer backbone) since a competitive interaction between the hydrophobic acyl residue and the hydrophobic sites of the enzyme led to a non-productive binding and a decrease in the overall hydrolysis rate. In any case, degradation rates were comparable with those found for related α,ω-alkylenediol derivatives demonstrating that hydrolysis was not prevented by the rigid bicyclic fragments.
A series of similar PEAs, but derived from dianhydro-D-glucitol, were also synthesized by solution polycondensation. Again, relatively high number-average molecular weights were attained (i.e., up to 3.8 × 104 g/mol). These PEAs were, in general, degraded more slowly than the corresponding polyesters having the same aliphatic dicarboxylic acid units, both in composted soil and in an activated sludge. However enzymatic degradation tests using papain (an enzyme able to favor ester-bond and amide-bond cleavages) indicated a faster degradation for these new PEAs.
Intraocular polymer delivery systems based on biodegradable PEAs derived from bicyclic-fragments of 1,4:3,6-dianhydrohexitols such as D-glucitol, D-mannitol or L-iditol have been patented [83]. It has been claimed that the new systems released opthalmologic agents in a consistent and reliable manner into the exterior or interior of the eye by biodegradation of the polymer.
4. Biomedical Applications of Functionalized Poly(ester amide)s
Incorporation of functional pendant groups has expanded the applications of PEAs for two main reasons: (a) They allow tailoring properties such as mechanical, thermal, biodegradation rate and hydrophilicity; and (b) they allow further chemical conjugation with a wide variety of drugs, targeting groups, cell signaling molecules or other biological agents.
4.1. Drug Delivery Systems
Development of new synthetic degradable polymers for controlled release of drugs and proteins has been the focus of much attention since 1990s. First polymers were based on lactide and glycolide units due to their biodegradability and safe history as suture materials. Capability for processing into micro-and nanoparticles expanded their pharmaceutical applications for both oral and parenteral administration. The successful use of these polymers led to the evaluation of other aliphatic polyesters such as poly(ε-caprolactone). However, high crystallinity and hydrophobicity may cause an incomplete drug release that justifies the development of new polymers, copolymers and blends. In this way, a new series of materials with tailored properties can be achieved.
Random copolymers of lactide and morpholine-2,5-diones with reactive (hydrophilic) side-chain groups such as those derived from glycolic and aspartic acids or glycolic acid and lysine had been used to prepare microspheres with reactive surfaces [84-86]. These showed an efficient entrapment of ionic drugs and a slow drug release because of electrostatic interactions of the drug with the ionic side-chain groups of the polymer matrix. Amphiphilic block copolymers consisting of polylactide as hydrophobic segments and polydepsipeptides with amino or carboxylic acid groups as hydrophilic segments (Figure 22) were also considered. The microspheres of both random and block copolymers showed controlled release of growth factors to promote rapid growth of cells and regeneration of tissues.
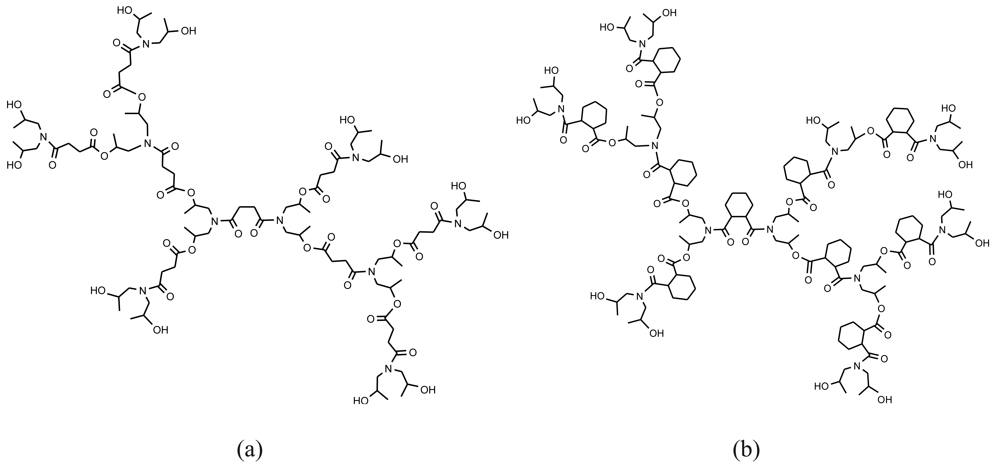
Some hyperbranched PEAs are now produced on an industrial scale and commercialized (Hybranes, Figure 23) at a very competitive cost. These PEAs were obtained from a monomer derived from a cyclic anhydride and a diisopropanol amine. Polycondensation was carried out via an oxazolinium intermediate in bulk, at relatively mild conditions, in the absence of catalyst [87]. By varying and combining anhydrides, and modification with several types of end groups, a large variety of structures with concomitant properties and industrial applications were obtained. In particular, compounds appear as promising candidates for pharmaceutical formulations as drug carriers (e.g., acetaminophen) [88]. It was also demonstrated that this kind of hyperbranched polymer with a large number of functional groups can act as solubilization enhancers for poorly water-soluble drugs, such as glimepiride, an antidiabetic drug [89]. Advantages are clear since the poor aqueous solubility and slow dissolution rate of glimepiride may lead to irreproducible clinical response or therapeutic failure due to subtherapeutic plasma drug levels. From an economical point of view, low oral bioavailability results in wasting a large portion of an oral dose and increases the cost of drug therapy, especially in the case of expensive drugs.
PEAs with elastomeric and bioabsorbable characteristics can also be used as coatings to which drugs can be covalently conjugated. Furthermore, composition and microstructure can be easily varied to allow a desired rate of degradation and drug release. Degradation rate of PEAs tends to increase with levels of inflammation, a feature that can be taken into account to control a treatment agent release rate (e.g., when the agent is directly linked to polymer side chains).
PEA coatings have been studied as efficient delivery systems of oxygen free radical scavengers (i.e., tempamine) which reduce tissue injury by neutralizing the toxic free radicals released during inflammation, and have therefore a positive effect on the vascular healing response. In particular, copoly(ester amide)s based on α-amino acids (e.g., L-leucine and L-lysine), diols and dicarboxylic acids were demonstrated to be biocompatible with the arterial walls [90,91]. Furthermore, PEAs coatings loaded with a 50% of tempamine had a tendency to decrease arterial injury as demonstrated by in vivo studies, in contrast with the severe inflammation observed when stents were directly loaded with the drug (i.e., without using the polymer coating) [92]. Several PEA coated stents have been patented for different medical procedures, such as treating occluded regions of blood vessels, thrombosis and restenosis [93,94]. Usually, a polymer solution which includes a dispersed therapeutic substance is applied to the stent. The solvent is allowed to evaporate, leaving a coating of the polymer on the stent surface and the therapeutic substance impregnated in the polymer.
A family of biodegradable copoly(ester amide)s based on naturally occurring α-amino acids (e.g., L-leucine and L-lysine derivatized by either benzyl alcohol or the nitroxide radical 4-amino-TEMPO as pendant group) has also been developed for applications ranging from biomedical device coatings to delivery of therapeutic biologics. An important feature of these coatings was their ability to support a natural healing response by attenuating the pro-inflammatory reaction to the implant and promoting growth of appropriate cells for repair of the tissue architecture. As a measure of pro-healing tissue compatibility for cardiovascular applications, endothelial cells adhered, spread, and proliferated on PEAs. Furthermore, new polymers were non-hemolytic and did not deplete platelets or leukocytes from whole blood [95].
4.2. Hydrogels
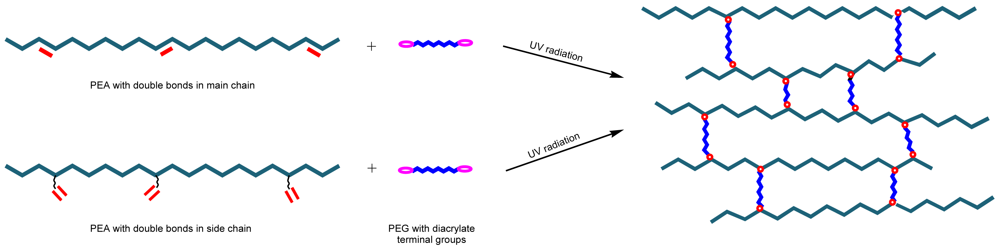
Hydrogels are of great interest for their use as medical implants, biosensors, bioseparators, and matrices for drug delivery and tissue engineering since they have high water content like body tissues and may be highly biocompatible. Biodegradable hydrogels are of interest as drug delivery systems for two main reasons: (a) A permanent foreign-body reaction is not produced after the materials serve their intended functions due to their biodegradability; and (b) biodegradable hydrogels could provide a drug release mechanism different to a traditional diffusion-based controlled release. The biodegradation-based mechanism is critical for complete release of large molecular weight drugs, especially pharmaceutically active proteins and peptides [96]. Hydrogels can be prepared by photocrosslinking PEAs with unsaturation on their main chain or in the side chain (Figure 24).
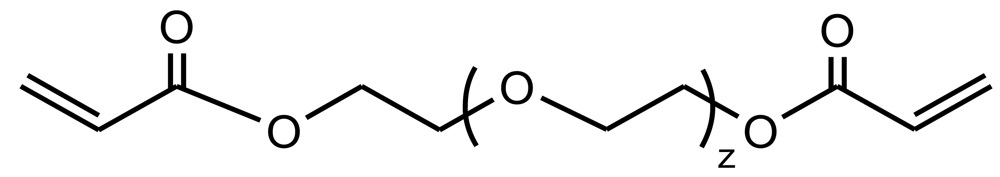
Hydrogels derived from copoly(ester amide)s with chemical structures displayed in Figure 15 (i.e., having carbon-carbon double bonds in their main chain) were prepared by photocrosslinking with poly(ethylene glycol) diacrylate (Figure 25). In fact, poly(ethylene glycol) is one of the frequently used hydrogel precursors since in vivo use is approved by the U.S. Food Drug Administration. Furthermore, elastic modulus, hydrophilicity and swelling behavior of these kind of hydrogels can be improved since the hydrophobic character of initial copoly(ester amide)s is balanced [97,98]. Based on the weight loss data, α-chymotrypsin had a much more profound effect on the hydrolysis (up to 32% weight loss on day 31) than PBS (less than 16%). The changes in elastic moduli and the interior morphology of the hydrogels were monitored during biodegradation and both the crosslinking density and the molecular weight between crosslinks determined. The differences in biodegradation rates showed that hydrogels could have controllable biodegradability by changing the concentration of α -chymotrypsin, the type of unsaturated precursor and the feed ratio (i.e., the ratio between the PEA precursor and the acrylate coupler).
New biodegradable hybrid hydrogels were also prepared by UV photocrosslinking with poly(ethylene glycol) diacrylate (Mn = 4,000 g/mol) or pluronic diacrylate (Mn = 7,000 g/mol) of pendant carbon-carbon double bonds in copoly(ester amide)s [99]. These were derived from L-phenylalanine and DL-2-allylglycine based monomers and p-nitrophenyl esters of dicarboxylic acids. The content of the pendant double bond in these unsaturated poly(ester amide)s was tunable by adjusting the feed ratio of allylglycine to another regular amino acid like L-phenylalanine. All these hybrid hydrogels showed three-dimensional porous structures. The gelation efficiency was lower when pluronic diacrylate was employed due its longer chain and consequently a looser network structure was formed. The hydrophobicity, crosslinking density and mechanical strength of the hybrid hydrogels increased with an increase in allylglycine content (i.e., double bond content) in the precursor polymer, but their swelling and pore size decreased. The biodegradation rate of these hybrid hydrogels in an enzyme (α-chymotrypsin) solution increased with an increase in the enzyme concentration and allylglycine content.
The synthesis of poly (L-serine-alt-glycolic acid) and copolymers of poly (L-serine- co-glycolic acid-co-lactide) and poly (L-serine- co-glycolic acid- co-ε-caprolactone) has been performed from the appropriate morpholine-2,5-dione (Section 2.1.1) and lactide and ε-caprolactone rings. These biodegradable polymers contain a reactive primary hydroxy group on the side chain of serine and copolymers may show a variable degradation rate by changing the ratio of lactide and ε-caprolactone units. Polymers were also functionalized with the acrylate group through the reaction of the serine units with acryloyl chloride. Subsequent UV-photopolymerization of acrylate groups provided glassy and transparent networks with a gel content close to 90% that may be used for the purpose of encapsulating cells/drugs for injectable delivery [41].
4.3. Non-Viral Gene Carriers
Gene therapy has a great potential to provide effective treatments for many human diseases but its success depends on finding safe and effective delivery systems. Safety issues associated with current viral vectors, such as immune response, toxicity, chromosomal integration, and short shelf-life, may be overcome with non-viral delivery systems. Among different approaches, synthetic polymers appear attractive because of versatility in properties and types of modifications, low immunogenicity, unrestricted plasmid size, better shelf-life and the possibility of repeated administration. However, synthetic polymers are not able to deliver therapeutic DNA at a comparable efficiency than viral-based gene carriers.
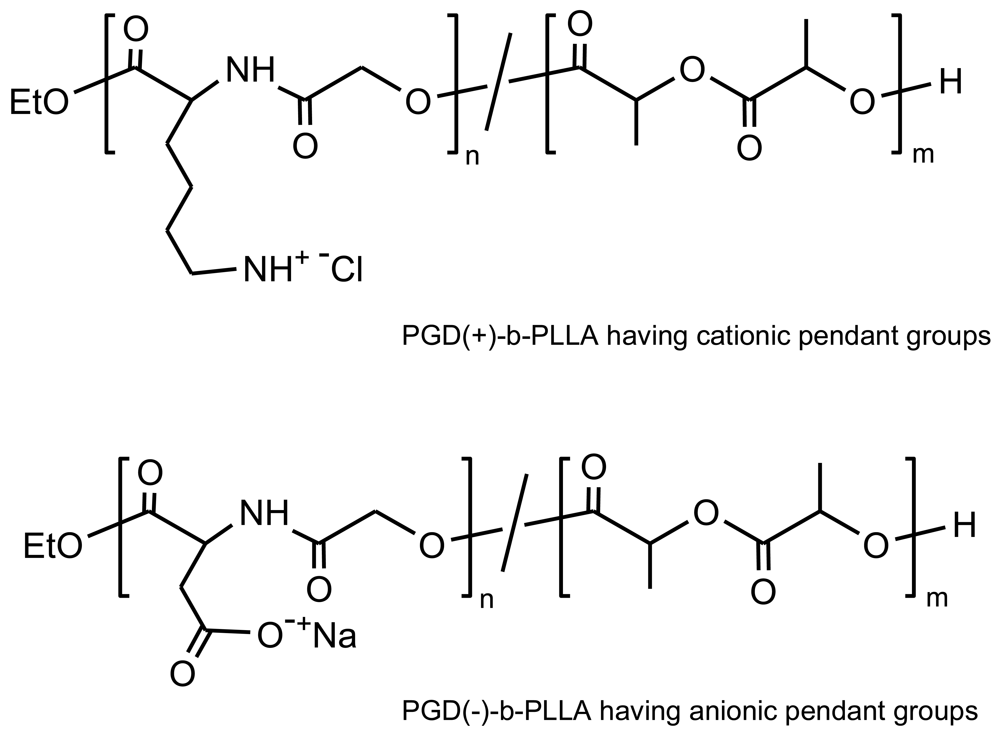
PEAs based on arginine units were prepared by the solution polycondensation method (from a diester-diamine and an activated dicarboxylic acid) and both their biosafety and capability to transfect rat vascular smooth muscle cells, a major cell type participating in vascular diseases, was demonstrated [100]. New PEAs showed high binding capacity toward plasmid DNA due to the strong basic guanidino group (pKa ∼ 12.5), which can condense negatively charged DNA. The binding activity was found to decrease with the increase of the number of methylene groups in the diol segment of PEAs. Furthermore, the use of arginine based PEAs as potential gene carriers was postulated to benefit from the fact that polyarginine enters cells more efficiently than other polycationic polymers that have been assayed as gene carriers.
4.4. Tissue Engineering
Materials to be used for tissue engineering applications and in vivo implantation should be characterized in terms of cytotoxicity, ability to support cell growth, inflammatory properties, or mechanical properties. Some of these features have been evaluated for new functionalized PEAs (Section 3.2) obtained by solution polycondensation and derived from amino-fuctionalized (L-lysine derivative), carboxylic acid-fuctionalized (L-aspartic acid derivative) and neutral (L-phenyalanine derivative) PEAs [53]. Results indicated that all forms of investigated PEAs were noncytotoxic and noninflammatory in vitro, although the amino-functionalized PEA best supported endothelial cell adhesion, growth, and monolayer formation. Mechanical testing indicated that the elastic moduli of these materials were strongly dependent on the charge formulation, but exhibited linearly elastic behavior at small strains (<10%). Data suggested that PEAs could be a viable biomaterial for use in tissue engineering applications and particularly for use as a vascular graft [53]. Over 600,000 coronary artery bypass grafts are implanted each year in the United States and Europe which justifies interest towards the development of effective synthetic vessel replacements as an alternative to autologous graft procedures, which may carry a substantial risk to the patient (i.e., a synthetic polymer, which closely mimics the mechanical properties of blood vessels, may minimize compliance mismatch and reduce the frequency of graft failure).
New PEAs based on the α-amino acids L-phenylalanine and L-methionine and a diol (1,4-butanediol or 1,6-hexanediol) and sebacic acid were synthesized with high molecular weight and narrow polydispersity indices. Porous 3D scaffolds prepared from these PEAs were found to have excellent porosities and appeared to be highly interesting for vascular tissue engineering [101].
New biodegradable elastomeric PEAs have also been synthesized for tissue engineering applications [102]. These new materials allow overcoming some limitations of conventional crosslinked aliphatic polyesters: (a) High crosslink densities, which result in exceedingly high stiffness; (b) rapid degradation upon implantation; or (c) limited chemical moieties for chemical modification. Poly(1,3-diamino-2-hydroxypropane-co-polyol sebacate)s formed crosslinked networks through the hydroxyl group and polyol (glycerol and/or D,L-threitol) units (Figure 26), and featured tensile Young's modulus on the order of 1 MPa and reversible elongations up to 92%. These polymers exhibited in vitro and in vivo biocompatibility and had projected degradation half-lives of up to 20 months in vivo.
Elastomeric biodegradable polymeric networks, and in particular semi-interpenetrating networks, can be formed using di- and poly-functional crosslinkers and functionalized poly(ester amide)s, based on α-amino acids and having double bonds in their main chain. It has been claimed that such materials have an appropriate combination of elasticity and toughness to be suitable for implantable biodegradable internal fixation devices. Components can be introduced in certain embodiments in vivo in a liquid state (i.e., prior to crosslinking) or implanted after being crosslinked ex-vivo [103].
4.5. Smart Materials
Stimuli-responsive polymers (e.g., thermosensitive polymers) are of great interest as materials for advances in biotechnology and the biomedical field due to their wide application as sensing, pollution control, drug delivery, biomimetic actuation, and catalysis. The thermosensitivity of any polymeric material can be regulated by controlling the hydrophobic–hydrophilic balance of the polymeric chains. Ohya et al. [104] induced thermosensitivity in the polydepsipeptide constituted by glycolic and aspartic acids by attaching the moderately hydrophobic isopropylamine group into its carboxylic side group. The new polymer was fully degradable in vitro at room temperature by cleavage of the ester bonds in the main chain. The cloud point at 29 °C (between room and body temperature) makes the polymer attractive for implants and other biomedical applications.
4.6. Composites and Adhesives
With the emergence of commercial hyperbranched and dendritic polymers, having a three-dimensional morphology with high peripheral functionality, new opportunities have been created for formulating dental adhesives and composites with enhanced mechanical and physical properties. Hybrane, a commercially available poly(ester amide), was effectively incorporated into acrylate-based dental composite and adhesive systems [105]. Addition of this hyperbranched polymer caused a higher cross-link density as a result of the large number of reactive end groups on the periphery that could react with the constituents of the polymer network. The enhanced cross-linking led to higher compressive strength (from 253 ± 20 MPa to 386 ± 20 MPa) and lower polymerization shrinkage (from 2.4 ± 0.2% to 1.5 ± 0.2 %) than a typical dental composite formulation. At concentrations higher than 0.5 wt%, the polymer acted as a plasticizer, reducing compressive strength and increasing shrinkage. It was also shown that the PEA added to the dental adhesive compositions increased the shear bond strength and enhanced the bond durability to a variety of dental surfaces.
5. Conclusions
Poly(ester amide)s constitute a peculiar family of biodegradable polymers, due to the presence of both ester and amide groups that guaranties degradability, and some improved properties with respect to related polyesters. A great effort has been made in the development of PEAs since the preparation of the first derivatives in the 1970s.
Synthesis methodologies are currently well established and allow attaining samples with appropriate molecular weights for most uses. Both ring opening polymerization of morpholine-2,5-dione derivatives and polycondensation reactions appear to be suitable procedures. A considerable body of literature refers to the preparation of PEAs with different architectures, which could lead to amorphous, semicrystalline and elastomeric materials. Furthermore, published data shows that a wide variety of monomers can be employed and that α-amino acid, carbohydrate or oligo(ethylene glycol) units can be easily incorporated. In this way, it is feasible to get a wide range of final properties.
Since the 1990s, research on poly(ester amide)s has mainly focused on the preparation of functionalized polymers. Strategies are based on the incorporation of amine, hydroxyl and carboxyl pendant groups, or even on the incorporation of carbon-carbon double bonds in either the chain backbone or the pendant groups. In this way, a great number of biomedical applications for these new polymers are currently being studied to cover topics such as controlled delivery systems, hydrogels and tissue engineering. Furthermore, several hyperbranched PEAs with interesting properties for their use in the biomedical field have been commercialized at competitive prices and even the number of patents related to the use of functionalized PEAs is steadily increasing. However, great efforts appear still necessary to validate the highly promising properties of new PEAs and also to improve their performance properties.









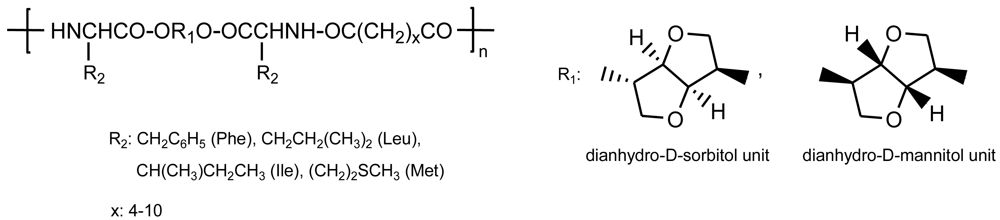

Acknowledgments
Authors want to indicate the support by CICYT and FEDER grants (MAT2009-11503) and by the Agència de Gestió d' Ajuts Universitaris i de Recerca (2009SGR-1208).
References
- Akelah, A.; Moet, A. Functionalized Polymers and Their Applications; Chapman and Hall: London, UK, 1990. [Google Scholar]
- Kulshrestha, A.S.; Mahapatro, A. Polymers for Biomedical Applications. ACS Symp. Ser. 2008, 977, 1–7. [Google Scholar]
- Tian, D.; Dubois, P.; Grandfils, G.; Jérôme, J. Ring-Opening Polymerization of 1,4,8-Trioxaspiro[4.6]-9-undecanone: A New Route to Aliphatic Polyesters Bearing Functional Pendent Groups. Macromolecules 1997, 30, 406–409. [Google Scholar]
- Stevels, W.M.; Pintat, S.; Slaghek, T.M. Coupling Reactions of Activated Oligo(ε-caprolactone)s. Polym. Bull. 1999, 42, 257–264. [Google Scholar]
- Trollsas, M.; Lee, V.T.; Mercerreyes, D.; Löwenhielm, P.; Möller, M.; Millar, R.D.; Hedrick, J.L. Hydrophilic Aliphatic Polyesters: Design, Synthesis, and Ring-opening Polymerization of Functional Cyclic Esters. Macromolecules 2000, 33, 4619–4627. [Google Scholar]
- Okada, M. Chemical Synthesis of Biodegradable Polymers. Prog. Polym. Sci. 2002, 27, 87–133. [Google Scholar]
- Lips, P.A.M.; Dijkstra, P.J. Biodegradable Polymers for Industrials Applications; CRC Press: Boca Raton, FL, USA, 2005; Chapter 5; pp. 109–139. [Google Scholar]
- Feng, Y.; Guo, J. Biodegradable Polydepsipeptides. Int. J. Mol. Sci. Mol. Sci. 2009, 10, 589–615. [Google Scholar]
- Coin, I.; Dolling, R.; Krause, E.; Bienert, M.; Beyermann, M.; Sferdean, CD.; Carpino, L.A. Depsipeptide Methodology for Solid-phase Peptide Synthesis: Circumventing Side Reactions and Development of an Automated Technique via Depsidipeptide Units. J. Pept. Sci. 2010, 16, 223–230. [Google Scholar]
- Stawikowski, M.; Cudic, P. Depsipeptide Synthesis. In Peptide Characterization and Application Protocols; Fields, G.B., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2007; Volume 386, pp. 321–339. [Google Scholar]
- Chadwick, A.F.; Pascu, E. The Resolution and Rates of Hydrolysis of D,L-α-Bromopropionic Acid and Its Glycine Derivatives. J. Chem. Soc. 1943, 65, 392–402. [Google Scholar]
- Wang, D.; Feng, X.D. Copolymerization of ε-caprolactone with(3S)-3-[(benzyloxycarbonyl)methyl]morpholine-2,5-dione and the C-13 NMR SequenceAnalysis of the Copolymer. Macromolecules 1998, 31, 3824–3831. [Google Scholar]
- Zhang, G.D.; Wang, D.; Feng, X.D. Preliminary Study of Hydrogen Bonding in (3S)-3-[(benzyloxycarbonyl)methyl]morpholine-2,5-dione and Its Effect on Polymerization. Macromolecules 1998, 31, 6390–6092. [Google Scholar]
- In't Veld, P.J.A.; Dijkstra, P.J.; Feijen, J. Synthesis of Biodegradable Polyesteramides with Pendant Functional Groups. Macromol. Chem. Phys. 1992, 193, 2713–2730. [Google Scholar]
- Vinsova, J. Morpholine-2,5-diones. Their Preparation and Exploitation. Chem. Listy 2001, 95, 22–27. [Google Scholar]
- Hartwig, W.; Schoellkopf, U. Asymmetrische Synthesen über Heterocyclische Zwischenstufen, XVI. Enantioselektive Synthese von α-Alkyl-α-phenylglycinen durch Alkylieren von an C-6 Chiral Substituierten 3,6-Dihydro-3-phenyl-2H-1,4-oxazin-2-onen. Liebigs Ann. Chem. 1982, 1982, 1952–1970. [Google Scholar]
- Kardassis, G.; Brungs, P.; Nothhelfer, C.; Steckhan, E. Electrogenerated Chiral Cationic Glycine Equivalents—Parts 2: Chiral 3-Methoxy-2,5-morpholinediones from (S)-cz-Hydroxy Acids and Dimethyl Aminomalonate. Tetrahedron 1998, 54, 3479–3488. [Google Scholar]
- Nissen, D.; Gilon, C.; Goodmann, M. Polydepsipeptides. 4. Synthesis of the Alternating Polydepsipeptides Poly(Ala-Lac) and Poly(Val-Lac). Macromol. Chem. Supplement 1975, 1, 23–53. [Google Scholar]
- Chisholm, M.H.; Galucci, J.; Krempner, C.; Wiggenhorn, C. Comments on the Ring-opening Polymerization of Morpholine-2,5-dione Derivatives by Various Metal Catalysts and Characterization of the Products Formed in the Reactions Involving R(2)SnX(2), Where X = OPr(i) and NMe(2) and R = Bu(n), Ph and p-Me(2)NC(6)H(4). Dalton Trans. 2006, 6, 846–851. [Google Scholar]
- Samyn, C.; van Beylen, M. Polydepsipeptides: Ring-opening Polymerization of 3-Methyl-2,5-morpholinedione, 3,6-Dimethyl-2,5-morpholinedione and Copolymerization Thereof with D,L-Lactide. Macromol. Chem. Macromol. Symp. 1988, 19, 225–234. [Google Scholar]
- Feng, Y.; Klee, D.; Höcker, H. Biodegradable Block Copolymers with poly(ethylene oxide) and Poly(glycolic acid-valine) Blocks. J. Applied Polym. Sci. 2002, 86, 2916–2919. [Google Scholar]
- Feng, Y.; Knüfermann, J.; Klee, D.; Höcker, H. Enzyme-catalyzed Ring-opening Polymerization of 3(S)-isopropyl-morpholine-2,5-dione. Macromol. Rapid Comm. 1999, 20, 88–90. [Google Scholar]
- Montané, J.; Armelin, E.; Asín, L.; Rodríguez-Galán, A.; Puiggalí, J. Comparative Degradation Data of Polyesters and Related Poly(ester amide)s Derived from 1,4-butanediol, Sebacic Acid, and α-Amino Acids. J. Appl. Polym. Sci. 2002, 85, 1815–1824. [Google Scholar]
- Asín, L.; Armelin, E.; Montané, J.; Rodríguez-Galán, A.; Puiggalí, J. Sequential Poly(ester amide)s Based on Glycine, Diols, and Dicarboxylic Acids: Thermal Polyesterification versus Interfacial Polyamidation. Characterization of Polymers Containing Stiff Unit. J. Polym. Sci. A Polym. Chem. 2001, 39, 4283–4293. [Google Scholar]
- Morgan, P.W. Condensation Polymer by Interfacial and Solution Methods; John Wiley and Sons Inc.: New York, NY, USA, 1965. [Google Scholar]
- Starks, CM. Phase-transfer Catalysis. I. Heterogeneous Reactions Involving Anion Transfer by Quaternary Ammonium and Phosphonium Salts. J. Amer. Chem. Soc. 1971, 93, 195–199. [Google Scholar]
- Paredes, N.; Rodríguez-Galán, A.; Puiggalí, J. Synthesis and Characterization of a Family of Biodegradable Poly(ester amide)s Derived from Glycine. J. Polym. Sci. A Polym. Chem. 1998, 36, 1271–1282. [Google Scholar]
- Han, S.I.; Kim, B.S.; Kang, S.W.; Shirai, H.; Im, S.S. Cellular Interactions and Degradation of Aliphatic Poly(ester amide)s Derived from Glycine and/or 4-amino butyric acid. Biomaterials 2003, 24, 3453–3462. [Google Scholar]
- Paredes, N.; Rodríguez-Galán, A.; Puiggalí, J.; Peraire, C. Studies on the Biodegradation and Biocompatibility of a New Poly(ester amide) Derived from L-alanine. J. Appl. Polym. Sci. 1998, 69, 1537–1549. [Google Scholar]
- Nagata, M. Synthesis and Enzymatic Degradation of Poly(ester-amide) Stereocopolymers Derived from Alanine. Macromol. Chem. Phys. 1999, 200, 2059–2064. [Google Scholar]
- Katsarava, R.; Beridze, V.; Arabuli, N.; Kharadze, D.; Chu, C.C.; Won, C.Y. Amino Acid-Based Bioanalogous Polymers. Synthesis, and Study of Regular Poly(ester amide)s Based on Bis(α-Amino Acid) αω-Alkylene Diesters, and Aliphatic Dicarboxylic Acids. J. Polym. Sci. A Polym. Chem. 1999, 37, 391–407. [Google Scholar]
- Katsarava, R. Active Polycondensation: From Peptide Chemistry to Amino Acid Based Biodegradable Polymers. Macromol. Symp. 2003, 199, 419–429. [Google Scholar]
- Fan, Y.; Kobayashi, M.; Kise, H. Synthesis and Biodegradation of Poly(ester amide)s Containing Amino Acid Residues: The Effect of the Stereoisomeric Composition of L- and D-phenylalanines on the Enzymatic Degradation of the Polymers. J. Polym. Sci. A Polym. Chem. 2002, 40, 385–392. [Google Scholar]
- Koyama, E.; Sanda, F.; Endo, T. Polycondensations of Dicarboxylic Acids and Diols Derived from Optically Active Amino Alcohols. J. Polym. Sci. A Polym. Chem. 1997, 35, 2925–2934. [Google Scholar]
- Vera, M.; Rodríguez-Galán, A.; Puiggalí, J. New Method of Synthesis of Poly(ester amide)s Derived from the Incorporation of Glycolic Acid Residues into Aliphatic Polyamides. Macromol. Rapid Comm. 2004, 25, 812–817. [Google Scholar]
- Vera, M.; Franco, L.; Puiggalí, J. Synthesis and Characterization of Poly(glycolic acid-alt-6-aminohexanoic acid) and Poly(glycolic acid-alt-11-aminoundecanoic acid). Macromol. Chem. Phys. 2004, 205, 1782–1792. [Google Scholar]
- Vera, M.; Admetlla, M.; Rodríguez-Galán, A.; Puiggalí, J. Synthesis, Characterization and Degradation Studies of Sequential Poly(ester amide)s Derived from Glycolic Acid, 1,6-hexanediamine and Aliphatic Dicarboxylic Acids. Polym. Degrad. Stabil. 2005, 89, 21–32. [Google Scholar]
- Bettinger, C.J.; Bruggeman, J.P.; Borenstein, J.T.; Langer, R.S. Amino Alcohol-based Degradable Poly(ester amide) Elastomers. Biomaterials 2008, 29, 2315–2325. [Google Scholar]
- Barrera, D.A.; Zylstra, E.; Lansbury, P.T.; Langer, R. Synthesis and RGD Peptide Modification of a New Biodegradable Copolymer: Poly(lactic acid-co-lysine). J. Amer. Chem. Soc. 1993, 115, 11010–11011. [Google Scholar]
- Ouchi, T.; Nozaki, T.; Okamoto, Y.; Shiratani, M.; Ohya, Y. Synthesis and Enzymatic Hydrolysis of Polydepsipeptides with Functionalized Pendant Groups. Macromol. Chem. Phys. 1996, 197, 1823–1833. [Google Scholar]
- John, G.; Tsuda, S.; Morita, M. Synthesis and Modification of New Biodegradable Copolymers: Serine/Glycolic Acid Based Copolymers. J. Polym. Sci. A Polym. Chem. 1997, 35, 1901–1907. [Google Scholar]
- Wang, D.; Feng, X.D. Synthesis of Poly(glycolic acid-alt-L-aspartic acid) from a Morpholine-2,5-Dione Derivative. Macromolecules 1997, 30, 5688–5692. [Google Scholar]
- Ouchi, T.; Shiratani, M.; Jinno, M.; Hirao, M.; Ohya, Y. Synthesis of Poly[(glycolic acid)-alt-(L-aspartic acid)] and Its Biodegradation Behaviour in vitro. Macromol. Rapid Comm. 1993, 14, 825–831. [Google Scholar]
- Ouchi, T.; Nozaki, T.; Ishikawa, A.; Fujimoto, I.; Ohya, Y. Synthesis and Enzymatic Hydrolysis of Lactic Acid-Depsipeptide Copolymers with Functionalized Pendant Groups. J. Polym. Sci. A Polym. Chem. 1997, 35, 377–383. [Google Scholar]
- RypaÂcÏek, F.; SÏ tefko, I.; MachovaÂ, L.; Kubies, D.; Brus, J. Synthesis of Ester-amide Copolymers from Lactones and N-Carboxyanhydrides. Polym. Preprints 1998, 39, 126–127. [Google Scholar]
- Jin, J.; Gonsalves, K.E. Synthesis of Poly(L-lactide-co-serine) and Its Graft Copolymers with Poly(ethylene glycol). Polymer 1998, 39, 5155–5162. [Google Scholar]
- Barrera, D.A.; Zylstxa, E.; Lansbury, P.T.; Langer, R. Copolymerization and Degradation of Poly(lactic acid-co-lysine). Macromolecules 1995, 28, 425–432. [Google Scholar]
- Hrkach, J.S.; Ou, J.; Lotan, N.; Langer, R. Synthesis of Poly(L-lactic acid-co-L-lysine) Graft Copolymers. Macromolecules 1995, 28, 4736–4739. [Google Scholar]
- Vera, M.; Franco, L.; Puiggali, J. Synthesis of Poly(ester amide)s with Lateral Groups from a Bulk Polycondensation Reaction with Formation of Sodium Chloride Salts. J. Polym. Sci. A Polym. Chem. 2008, 46, 661–667. [Google Scholar]
- Atkins, K.M; Lopez, D.; Knight, D.K.; Mequanint, K.; Gillies, E.R. A Versatile Approach for the Syntheses of Poly(ester amide)s with Pendant Functional Groups. J. Polym. Sci. A Polym. Chem. 2009, 47, 3757–3772. [Google Scholar]
- de Wit, M.A.; Wang, Z.X.; Atkins, K.M.; Mequanint, K.; Gillies, E.R. Syntheses, Characterization, and Functionalization of Poly(ester amide)s with Pendant Amine Functional Groups. J. Polym. Sci. A: Polym. Chem. 2009, 47, 6376–6392. [Google Scholar]
- Deng, M.; Wu, J.; Reinhart-King, C.A.; Chu, C.C. Synthesis and Characterization of Biodegradable Poly(ester amide)s with Pendant Amine Functional Groups and in vitro Cellular Response. Biomacromolecules 2009, 10, 3037–3047. [Google Scholar]
- Horwitz, J.A.; Shum, K.M.; Bodle, J.C.; Deng, M.; Chu, C.C.; Reinhart-King, C.A. Biological Performance of Biodegradable Amino Acid-Based Poly(ester amide)s: Endothelial Cell Adhesion and Inflammation in vitro. J. Biomed. Mater. Res. A 2010, 95, 371–380. [Google Scholar]
- Guan, H.L.; Deng, C.; Xu, X.Y.; Liang, Q.Z.; Chen, X.S.; Jing, X.B. Synthesis of Biodegradable Poly(ester amide)s Containing Functional Groups. J. Polym. Sci. A Polym. Chem. 2005, 43, 1144–1149. [Google Scholar]
- Pang, X.; Chu, C.C. Synthesis, Characterization and Biodegradation of Functionalized Amino Acid-Based Poly(ester amide)s. Biomaterials 2010, 31, 3745–3754. [Google Scholar]
- Hoyle, C.E.; Lee, T.Y.; Roper, T. Thiol-enes: Chemistry of the Past with Promise for the Future. J. Polym. Sci. A Polym. Chem. 2004, 42, 5301–5338. [Google Scholar]
- Bantchev Grigor, B.; Kenar James, A.; Biresaw, G.; Han Moon, G. Free Radical Addition of Butanethiol to Vegetable Oil Double Bonds. J. Agricul. Food Chem. 2009, 57, 1282–1290. [Google Scholar]
- Pang, X.; Wu, J.; Reinhart-King, C.; Chu, C.C. Synthesis and Characterization of Functionalized Water Soluble Cationic Poly(ester amide)s. J. Polym. Sci. A Polym. Chem. 2010, 48, 3758–3766. [Google Scholar]
- Guo, K.; Chu, C.C. Synthesis, Characterization, and Biodegradation of Copolymers of Unsaturated and Saturated Poly(ester amide)s. J. Polym. Sci. A Polym. Chem. 2007, 45, 1595–1606. [Google Scholar]
- Guo, K.; Chu, C.C. Biodegradation of Unsaturated Poly(ester-amide)s and Their Hydrogels. Biomaterials 2007, 28, 3284–3294. [Google Scholar]
- Chu, C.C.; Katsarava, R. Elastomeric Functional Biodegradable Copolyester Amides and Copolyester Urethanes. U.S. Patent 6,503,538, 7 January 2003. [Google Scholar]
- Guo, K.; Chu, C.C. Copolymers of Unsaturated and Saturated Poly(ether ester amide)s: Synthesis, Characterization, and Biodegradation. J. Appl. Polym. Sci. 2008, 110, 1858–1869. [Google Scholar]
- Guo, K.; Chu, C.C. Controlled Release of Paclitaxel from Biodegradable Unsaturated Poly(ester amide)s/Poly(ethylene glycol) Diacrylate Hydrogels. J. Biomater. Sci. Polym. Ed. 2007, 18, 489–504. [Google Scholar]
- Guo, K.; Chu, C.C.; Chkhaidze, E.; Katsarava, R. Synthesis and Characterization of Novel Biodegradable Unsaturated Poly(ester amide)s. J. Polym. Sci. A Polym. Chem. 2005, 43, 1463–1477. [Google Scholar]
- Guo, K.; Chu, C.C. Synthesis, Characterization, and Biodegradation of Novel Poly(ether ester amide)s Based on L-phenylalanine and Oligoethylene Glycol. Biomacromolecules 2007, 8, 2851–2861. [Google Scholar]
- Guo, K.; Chu, C.C. Synthesis and Characterization of Novel Biodegradable Unsaturated Poly(ester amide)/Poly(ethylene glycol) Diacrylate Hydrogel. J. Polym. Sci. A Polym. Chem. 2005, 43, 3932–3944. [Google Scholar]
- Ai, Y.; Shi, Z.; Guo, W. A New Type of Unsaturated Poly(ester-amide): Synthesis and Compressive Strength. Mater. Design 2009, 30, 892–895. [Google Scholar]
- Ai, Y.; Shi, Z.; Guo, W.; Xie, S. Synthesis and Characterization of a Potential Material as Internal Fixation of Bone Fracture. Mater. Sci. Eng. 2009, 29, 1001–1005. [Google Scholar]
- Ai, Y.; Shi, Z.; Guo, W. Calcium Polyphosphate Fibers/Unsaturated Poly(ester-amide) Composites for Bone-Fixation Materials. Polym. Composite 2009, 30, 1119–1124. [Google Scholar]
- Wang, L.; Wang, Y.; Cao, D. Synthesis and Characterization of new Unsaturated Degradable Poly(ether ester amide)s Containing Ethylene Oxide Moieties. J. Macromol. Sci. Pure 2009, 46, 282–289. [Google Scholar]
- Xiao, C.; He, Y. Tailor-Made Unsaturated Poly(esteramide) Network that Contains Monomeric Lactate Sequences. Polym. Int. 2007, 56, 816–819. [Google Scholar]
- Guo, K.; Chu, C.C. Synthesis of Biodegradable Amino-Acid-Based Poly(ester amide)s and Poly(ether ester amide)s with Pendant Functional Groups. J. Appl. Polym. Sci. 2010, 117, 3386–3394. [Google Scholar]
- Williams, C.K. Synthesis of Functionalized Biodegradable Polyesters. Chem. Soc. Rev. 2007, 36, 1573–1580. [Google Scholar]
- Kim, D.Y.; Dordick, J.S. Combinatorial Array-Based Enzymatic Polyester Synthesis. Biotechnol. Bioeng. 2001, 76, 200–206. [Google Scholar]
- Kumar, R.; Kulshrestha, A.; Gao, W.; Gross, R.A. Versatile Route to Polyol Polyesters by Lipase Catalysis. Macromolecules 2003, 36, 8219–8221. [Google Scholar]
- Molina Pinilla, I.; Bueno Martinez, M.; Galbis Pérez, J.A. Synthesis of a Stereoregular Poly(ester amide) Derived from L-Arabinose. Macromolecules 1995, 28, 3766–3770. [Google Scholar]
- Molina Pinilla, I.; Bueno Martínez, M.; Zamora Mata, F.; Galbis, J.A. Synthesis and Properties of Stereoregular Poly(ester amides) Derived from Carbohydrates. J. Polym. Sci. A Polym. Chem. 1998, 36, 67–77. [Google Scholar]
- Pérez-Rodríguez, A.; Alla, A.; Fernández-Santín, J.M.; Muñoz-Guerra, S. Poly(ester amide)s Derived from Tartaric and Succinic Acids: Changes in Structure and Properties upon Hydrolytic Degradation. J. Appl. Polym. Sci. 2000, 78, 486–494. [Google Scholar]
- Molina Pinilla, I.; Bueno Martínez, M.; Zamora Mata, F.; Galbis, J.A. Hydrolytic Degradation of Poly(ester amides) Derived from Carbohydrates. Macromolecules 1997, 30, 3197–3203. [Google Scholar]
- Villuendas, I.; Molina, I.; Regaño, C.; Bueno, M.; Martínez de Ilarduya, A.; Galbis, J.A.; Muñoz-Guerra, S. Hydrolytic Degradation of Poly(ester amide)s Made from Tartaric and Succinic Acids: Influence of the Chemical Structure and Microstructure on Degradation Rate. Macromolecules 1999, 32, 8033–80. [Google Scholar]
- Molina Pinilla, I.; Bueno Martínez, M.; Zamora Mata, F.; Galbis, J.A. Carbohydrate-Based Copolymers. Synthesis and Characterization of Copoly(ester amide)s Containing L-Arabinose Units. Macromolecules 2002, 35, 2977–2984. [Google Scholar]
- Gomurashvili, Z.; Kricheldorf, H.R.; Katsarava, R. Amino Acid Based Bioanalogous Polymers. Synthesis and Study of New Poly(ester amide)s Composed of Hydrophobic α-amino Acids and Dianhydrohexitoles. J. Macromol. Sci. Pure 2000, 37, 215–227. [Google Scholar]
- Landis, G.C.; Turnell, W.G.; Yumin, Y. Delivery of Ophthalmologic Agents to the Exterior or Interior of the Eye. WO 2007/130477A2, 19 November 2007. [Google Scholar]
- Ouchi, T.; Hamada, A.; Ohya, Y. Biodegradable Microspheres Having Reactive Groups Prepared From L-Lactic Acid-Depsipeptide Copolymers. Macromol. Chem. Phys. 1999, 200, 436–441. [Google Scholar]
- Ouchi, T.; Ohya, Y. Design of Lactide Copolymers as Biomaterials. J. Polym. Sci. A Polym. Chem. 2004, 42, 453–462. [Google Scholar]
- Ouchi, T.; Toyohara, M.; Arimura, H.; Ohya, Y. Preparation of Poly(L-lactide)-Based Microspheres Having a Cationic or Anionic Surface Using Biodegradable Surfactants. Biomacromolecules 2002, 3, 885–888. [Google Scholar]
- Froehling, P. Development of DSM' s Hybrane—Hyperbranched Polyesteramides. J. Polym. Sci. A Polym. Chem. 2004, 42, 3110–3115. [Google Scholar]
- Suttiriuengwong, S.; Rolker, J.; Smirnova, I.; Arit, W.; Seiler, M.; Lüderitz, L.; Perez de Diego, Y.; Jansens, P.J. Hyperbranched Polymers as Drug Carriers: Microencapsulation and Release Kinetics. Pharm. Dev. Technol. 2006, 11, 55–70. [Google Scholar]
- Sebastjan Reven, S.; Grdadolnik, J.; Kristl, J.; Zagar, E. Hyperbranched Poly(esteramides) as Solubility Enhancers for Poorly Water-Soluble Drug Glimepiride. Int. J. Pharm. 2010, 396, 119–126. [Google Scholar]
- Lee, S.H.; Szinai, I.; Carpenter, K.; Katsarava, R.; Jokhadze, G.; Chu, C.C.; Huang, Y.; Verbeken, E.; Bramwel, O.; De Scheerder, I.; Hong, M.K. In-vivo Biocompatibility Evaluation of Stents Coated with a New Biodegradable Elastomeric and Functional Polymer. Coronary Artery Dis. 2002, 13, 237–241. [Google Scholar]
- Desnoyer, J.R.; Pacetti, D.P.; Hossainy, S.F.A.; Kleiner, L.; Tang, Y.; Zhang, G. Poly(ester amide) Filler Blends for Modulation of Coating Properties. U.S. 7,749,263 B2, 06 July 2010. [Google Scholar]
- Huang, Y.; Wang, L.; Li, S.; Liu, X.; Lee, K.; Verbeken, E.; van de Werf, F.; de Scheerder, I. Stent-Based Tempamine Delivery on Neointimal Formation in a Porcine Coronary Model. Acute Cardiac Care. 2006, 8, 210–216. [Google Scholar]
- Pacetti, S; Desnoyer, J.R. Biologically Absorbable Coatings for Implantable Devices Based on Poly(ester amides) and Methods for Fabricating the Same. U.S. 7,220,816, 22 May 2007. [Google Scholar]
- Trollsas, M.O.; Lim, F.; Ngo, M.H.; Hossainy, S.F.A. Biodegradable Poly(ester-amide) and Poly(amide) Coatings for Implantable Medical Devices with Enhanced Bioabsorption Times. Patent application number 20100047319.
- DeFife, K.M.; Grako, K.; Cruz-Aranda, G.; Price, S.; Chantung, R.; Macpherson, K.; Khoshabeh, R.; Gopalan, S.; Turnell, A.G. Poly(ester amide) Copolymers Promote Blood and Tissue Compatibility. J. Biomater. Sci. Ed. 2009, 43, 1495–1511. [Google Scholar]
- Park, K.; Shalaby, W.S.W.; Park, H. Biodegradable Hydrogels for Drug Delivery; Technomic: Lancaster, PA, USA, 1993. [Google Scholar]
- Wu, D.; Zhang, X.; Chu, C.C. Synthesis, Characterization and Drug Release From Three-Arm Poly(ε-caprolactone) Maleic Acid/Poly(ethylene glycol) Diacrylate Hydrogels. J. Biomater. Sci. Polym. Ed. 2003, 14, 777–802. [Google Scholar]
- Park, Y.D.; Tirelli, N.; Hubbell, J.A. Photopolymerized Hyaluronic Acid-Based Hydrogels and Interpenetrating Networks. Biomaterials 2003, 24, 893–900. [Google Scholar]
- Pang, X.; Chu, C.C. Synthesis, Characterization and Biodegradation of Poly(ester amide)s Based Hydrogels. Polymer 2010, 51, 4200–4210. [Google Scholar]
- Dai Yamanouchi, D.; Wu, J.; Lazar, A.N.; Kent, K.C.; Chu, C.C.; Liu, B. Biodegradable Arginine-Based Poly(ester-amide)s as Non-Viral Gene Delivery Reagents. Biomaterials 2008, 29, 3269–3277. [Google Scholar]
- Karimi, P.; Rizkalla, A.S.; Mequanint, K. Versatile Biodegradable Poly(ester amide)s Derived from α-Amino Acids for Vascular Tissue Engineering. Materials 2010, 3, 2346–2368. [Google Scholar]
- Bettinger, C.J.; Bruggeman, J.P.; Borenstein, J.T.; Langer, R.S. Amino Alcohol-Based Degradable Poly(ester amide) Elastomers. Biomaterials 2008, 29, 2315–2325. [Google Scholar]
- Gomurashvili, Z.D.; Katsarava, R.; Chumburdze, G.; Mumladze, N.; Tugushi, D. Bioabsorbable Elastomeric Polymer Networks, Cross-Linkers and Methods of Use. U.S. 2009/0253809 A, 8 October 2009. [Google Scholar]
- Ohya, Y.; Toyohara, M.; Sasakawa, M.; Arimura, H.; Ouchi, T. Thermosensitive Biodegradable Polydepsipeptide. Macromol. Biosci. 2005, 5, 273–276. [Google Scholar]
- Dodiuk-Kenig, H.; Lizenboim, K.; Eppelbaum, I.; Zalsman, B.; Kenig, S. The Effect of Hyper-branched Polymers on the Properties of Dental Composites and Adhesives. J. Adhes. Sci. Technol. 2004, 18, 1723–1737. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rodriguez-Galan, A.; Franco, L.; Puiggali, J. Degradable Poly(ester amide)s for Biomedical Applications. Polymers 2011, 3, 65-99. https://doi.org/10.3390/polym3010065
Rodriguez-Galan A, Franco L, Puiggali J. Degradable Poly(ester amide)s for Biomedical Applications. Polymers. 2011; 3(1):65-99. https://doi.org/10.3390/polym3010065
Chicago/Turabian StyleRodriguez-Galan, Alfonso, Lourdes Franco, and Jordi Puiggali. 2011. "Degradable Poly(ester amide)s for Biomedical Applications" Polymers 3, no. 1: 65-99. https://doi.org/10.3390/polym3010065
APA StyleRodriguez-Galan, A., Franco, L., & Puiggali, J. (2011). Degradable Poly(ester amide)s for Biomedical Applications. Polymers, 3(1), 65-99. https://doi.org/10.3390/polym3010065