Synthesis of Amphiphilic Statistical Copolymers Bearing Methoxyethyl and Phosphorylcholine Groups and Their Self-Association Behavior in Water



Abstract
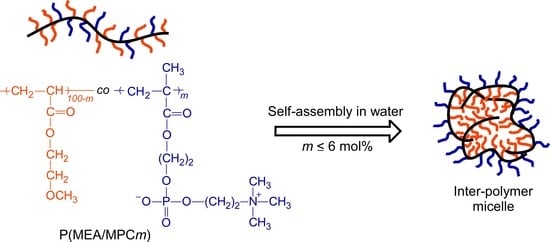
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Monomer Reactivity Ratio and Polymerization Kinetics
2.3. Preparation of the MEA Homopolymer
2.4. Preparation of the P(MEA/MPCm) Copolymer
2.5. Measurements
3. Results and Discussion
3.1. Determination of the Monomer Reactivity Ratio
3.2. Preparation of PMEA and P(MEA/MPCm)
3.3. Self-Association Behavior of P(MEA/MPCm) in Water
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Geng, Y.; Discher, D.E. Hydrolytic degradation of poly(ethylene oxide)-block-polycaprolactone worm micelles. J. Am. Chem. Soc. 2005, 127, 12780–12781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, R. The dawning era of polymer therapeutics. Nat. Rev. Drug Discov. 2003, 2, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Tong, R.; Cheng, J. Anticancer polymeric nanomedicines. Polym. Rev. 2007, 47, 345–381. [Google Scholar] [CrossRef]
- Choi, K.Y.; Liu, G.; Lee, S.; Chen, X. Theranostic nanoplatforms for simultaneous cancer imaging and therapy: Current approaches and future perspectives. Nanoscale 2012, 4, 330–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, S.; Yin, D.; Chen, Y.; Di, Y.; Chen, H.; Ma, Y.; Achilefu, S.; Gu, Y. In vivo targeted deep-tissue photodynamic therapy based on near-infrared light triggered upconversion nanoconstruct. ACS Nano 2013, 7, 676–688. [Google Scholar] [CrossRef]
- Lee, S.J.; Koo, H.; Jeong, H.; Huh, M.S.; Choi, Y.; Jeong, S.Y.; Byun, Y.; Choi, K.; Kim, K.; Kwon, I.C. Comparative study of photosensitizer loaded and conjugated glycol chitosan nanoparticles for cancer therapy. J. Control. Release 2011, 152, 21–29. [Google Scholar] [CrossRef]
- Williams, R.J.; Dove, A.P.; O’Reilly, R.K. Self-assembly of cyclic polymers. Polym. Chem. 2015, 6, 2998–3008. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Raghupathi, K.; Song, C.; Prasad, P.; Thayumanavan, S. Self-assembly of random copolymers. Chem. Commun. 2014, 50, 13417–13432. [Google Scholar] [CrossRef] [Green Version]
- Morishima, Y.; Nomura, S.; Ikeda, T.; Seki, M.; Kamachi, M. Characterization of unimolecular micelles of random copolymers of sodium 2-(acrylamido)-2-methylpropanesulfonate and methacrylamides bearing bulky hydrophobic substituents. Macromolecules 1995, 28, 2874–2881. [Google Scholar] [CrossRef]
- Yamamoto, H.; Morishima, Y. Effect of hydrophobe content on intra- and interpolymer self-associations of hydrophobically modified poly(sodium 2-(acrylamido)-2-methylpropanesulfonate) in water. Macromolecules 1999, 32, 7469–7475. [Google Scholar] [CrossRef]
- Terashima, T.; Sugita, T.; Fukae, K.; Sawamoto, M. Synthesis and single-chain folding of amphiphilic random copolymers in water. Macromolecules 2014, 47, 589–600. [Google Scholar] [CrossRef]
- Chang, Y.; McCormick, C.L. Water-soluble copolymers. 49. Effect of the distribution of the hydrophobic cationic monomer dimethyldodecyl(2-acrylamidoethyl)ammonium bromide on the solution behavior of associating acrylamide copolymers. Macromolecules 1993, 26, 6121–6126. [Google Scholar] [CrossRef]
- Yamamoto, H.; Hashidzume, A.; Morishima, Y. Micellization protocols for amphiphilic polyelectrolytes in water. How do polymers undergo intrapolymer associations? Polym. J. 2000, 32, 745–752. [Google Scholar] [CrossRef] [Green Version]
- Neal, T.J.; Beattie, D.L.; Byard, S.J.; Smith, G.N.; Murray, M.W.; Williams, N.S.J.; Emmett, S.N.; Armes, S.P.; Spain, S.G.; Mykhaylyk, O.O. Self-assembly of amphiphilic statistical copolymers and their aqueous rheological properties. Macromolecules 2018, 51, 1474–1487. [Google Scholar] [CrossRef] [Green Version]
- Movassaghian, S.; Merkel, O.M.; Torchilin, V.P. Applications of polymer micelles for imaging and drug delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2015, 7, 691–707. [Google Scholar] [CrossRef]
- Hoshiba, T.; Otaki, T.; Nemoto, E.; Maruyama, H.; Tanaka, M. Blood-compatible polymer for hepatocyte culture with high hepatocyte-specific functions toward bioartificial liver development. ACS Appl. Mater. Interfaces 2015, 7, 18096–18103. [Google Scholar] [CrossRef]
- Murakami, D.; Kobayashi, S.; Tanaka, M. Interfacial structures and fibrinogen adsorption at blood-compatible polymer/water interfaces. ACS Biomater. Sci. Eng. 2016, 2, 2122–2126. [Google Scholar] [CrossRef]
- Tanaka, M.; Mochizuki, A.; Ishii, N.; Motomura, T.; Hatakeyama, T. Study of blood compatibility with poly(2-methoxyethyl acrylate). relationship between water structure and platelet compatibility in poly(2-methoxyethylacrylate-co-2-hydroxyethylmethacrylate). Biomacromolecules 2002, 3, 36–41. [Google Scholar] [CrossRef]
- Morita, S.; Tanaka, M.; Ozaki, Y. Time-resolved in situ ATR-IR observations of the process of sorption of water into a poly(2-methoxyethyl acrylate) film. Langmuir 2007, 23, 3750–3761. [Google Scholar] [CrossRef]
- Kobayashi, S.; Wakui, M.; Iwata, Y.; Tanaka, M. Poly(ω-methoxyalkyl acrylate)s: Nonthrombogenic polymer family with tunable protein adsorption. Biomacromolecules 2017, 18, 4214–4223. [Google Scholar] [CrossRef]
- Haraguchi, K.; Takehisa, T.; Mizuno, T.; Kubota, K. Antithrombogenic properties of amphiphilic block copolymer coatings: Evaluation of hemocompatibility using whole blood. ACS Biomater. Sci. Eng. 2015, 1, 352–362. [Google Scholar] [CrossRef]
- Kyomoto, M.; Ishihara, K. Self-initiated surface graft polymerization of 2-methacryloyloxyethyl phosphorylcholine on poly(ether ether ketone) by photoirradiation. ACS Appl. Mater. Interfaces 2009, 1, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Iwasaki, Y.; Nakabayashi, N. Polymeric lipid nanosphere consisting of water-soluble poly(2-methacryloyloxyethyl phosphorylcholine-co-n-butyl methacrylate). Polym. J. 1999, 31, 1231–1236. [Google Scholar] [CrossRef] [Green Version]
- Kojima, R.; Kasuya, M.C.Z.; Ishihara, K.; Hatanaka, K. Synthesis of amphiphilic copolymers by soap-free interface-mediated polymerization. Polym. J. 2009, 41, 370–373. [Google Scholar] [CrossRef]
- Kojima, R.; Kasuya, M.C.Z.; Ishihara, K.; Hatanaka, K. Physicochemical delivery of amphiphilic copolymers to specific organelles. Polym. J. 2011, 43, 718–722. [Google Scholar] [CrossRef]
- Ma, Y.; Tang, Y.; Billingham, N.C.; Armes, S.P.; Lewis, A.L.; Lloyd, A.W.; Salvage, J.P. Well-defined biocompatible block copolymers via atom transfer radical polymerization of 2-methacryloyloxyethyl phosphorylcholine in protic media. Macromolecules 2003, 36, 3475–3484. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Akiyoshi, K. Design of biodegradable amphiphilic polymers: Well-defined amphiphilic polyphosphates with hydrophilic graft chains via atrp. Macromolecules 2004, 37, 7637–7642. [Google Scholar] [CrossRef]
- Ishihara, K. Revolutionary advances in 2-methacryloyloxyethyl phosphorylcholine polymers as biomaterials. J. Biomed. Mater. Res. Part A 2019, 107, 933–943. [Google Scholar] [CrossRef]
- Chen, S.-H.; Chang, Y.; Ishihara, K. Reduced blood cell adhesion on polypropylene substrates through a simple surface zwitterionization. Langmuir 2017, 33, 611–621. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Ishihara, K. Cell membrane-inspired phospholipid polymers for developing medical devices with excellent biointerfaces. Sci. Technol. Adv. Mater. 2012, 13, 64101. [Google Scholar] [CrossRef]
- Sugihara, S.; Blanazs, A.; Armes, S.P.; Ryan, A.J.; Lewis, A.L. Aqueous dispersion polymerization: A new paradigm for in situ block copolymer self-assembly in concentrated solution. J. Am. Chem. Soc. 2011, 133, 15707–15713. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Armes, S.P. Preparation of biocompatible zwitterionic block copolymer vesicles by direct dissolution in water and subsequent silicification within their membranes. Langmuir 2009, 25, 9564–9570. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Ueda, T.; Nakabayashi, N. Preparation of phospholipid polymers and their properties as polymer hydrogel membranes. Polym. J. 1990, 22, 355–360. [Google Scholar] [CrossRef]
- Fineman, M.; Ross, S.D. Linear method for determining monomer reactivity ratios in copolymerization. J. Polym. Sci. 1950, 5, 259–262. [Google Scholar] [CrossRef]
- Meiboom, S.; Gill, D. Modified spin-echo method for measuring nuclear relaxation times. Rev. Sci. Instrum. 1958, 29, 688–691. [Google Scholar] [CrossRef] [Green Version]
- Grassie, N.; Torrance, B.J.D.; Fortune, J.D.; Gemmell, J.D. Reactivity ratios for the copolymerization of acrylates and methacrylates by nuclear magnetic resonance spectroscopy. Polymer 1965, 6, 653–658. [Google Scholar] [CrossRef]
- Adolphi, U.; Kulicke, W.-M. Coil dimensions and conformation of macromolecules in aqueous media from flow field-flow fractionation/multi-angle laser light scattering illustrated by studies on pullulan. Polymer 1997, 38, 1513–1519. [Google Scholar] [CrossRef]
- Bloembergen, N.; Purcell, E.M.; Pound, R.V. Relaxation effects in nuclear magnetic resonance absorption. Phys. Rev. 1948, 73, 679–712. [Google Scholar] [CrossRef]
- Zhao, S.; Yuan, H.-Z.; Yu, J.-Y.; Du, Y.-R. Hydrocarbon chain packing in the micellar core of surfactants studied by 1H NMR relaxation. Colloid Polym. Sci. 1998, 276, 1125–1130. [Google Scholar] [CrossRef]
- Akcasu, A.Z.; Han, C.C. Molecular weight and temperature dependence of polymer dimensions in solution. Macromolecules 1979, 12, 276–280. [Google Scholar] [CrossRef]
- Bruns, W. The second osmotic virial coefficient of polymer solutions. Macromolecules 1996, 29, 2641–2643. [Google Scholar] [CrossRef]
- Matsuda, Y.; Kobayashi, M.; Annaka, M.; Ishihara, K.; Takahara, A. Dimensions of a free linear polymer and polymer immobilized on silica nanoparticles of a zwitterionic polymer in aqueous solutions with various ionic strengths. Langmuir 2008, 24, 8772–8778. [Google Scholar] [CrossRef]
- Kalyanasundaram, K.; Thomas, J.K. Environmental effects on vibronic band intensities in pyrene monomer fluorescence and their application in studies of micellar systems. J. Am. Chem. Soc. 1977, 99, 2039–2044. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| m a | Mn(SEC) b × 104 (g/mol) | Đ b | Mw(SLS) c × 105 (g/mol) | Rhd (nm) | Rgc (nm) | Rg/Rh |
|---|---|---|---|---|---|---|
| 6 | 1.41 | 2.61 | 2.36 | 9.6 | 15.5 | 1.6 |
| 12 | 1.46 | 2.24 | 1.53 | 10.0 | 14.8 | 1.5 |
| 46 | 2.69 | 2.04 | 0.73 | 9.2 | 12.8 | 1.4 |
| Sample | Mw(SLS) a × 10−7 (g/mol) | Rhb (nm) | Rga (nm) | Rg/Rh | RTEMc (nm) | Naggd | A2a × 105 (cm3 g−2 mol) | CAC e (g/L) |
|---|---|---|---|---|---|---|---|---|
| P(MEA/MPC6) | 3.37 | 96.9 | 92.1 | 0.95 | 103 | 143 | 2.5 | 0.0082 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.L.; Kawata, Y.; Ishihara, K.; Yusa, S.-i. Synthesis of Amphiphilic Statistical Copolymers Bearing Methoxyethyl and Phosphorylcholine Groups and Their Self-Association Behavior in Water. Polymers 2020, 12, 1808. https://doi.org/10.3390/polym12081808
Nguyen TL, Kawata Y, Ishihara K, Yusa S-i. Synthesis of Amphiphilic Statistical Copolymers Bearing Methoxyethyl and Phosphorylcholine Groups and Their Self-Association Behavior in Water. Polymers. 2020; 12(8):1808. https://doi.org/10.3390/polym12081808
Chicago/Turabian StyleNguyen, Thi Lien, Yuuki Kawata, Kazuhiko Ishihara, and Shin-ichi Yusa. 2020. "Synthesis of Amphiphilic Statistical Copolymers Bearing Methoxyethyl and Phosphorylcholine Groups and Their Self-Association Behavior in Water" Polymers 12, no. 8: 1808. https://doi.org/10.3390/polym12081808