Effect of Cellulose Nanofibrils and TEMPO-mediated Oxidized Cellulose Nanofibrils on the Physical and Mechanical Properties of Poly(vinylidene fluoride)/Cellulose Nanofibril Composites

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Material and Film Preparation
2.3. Moprhological Characterization
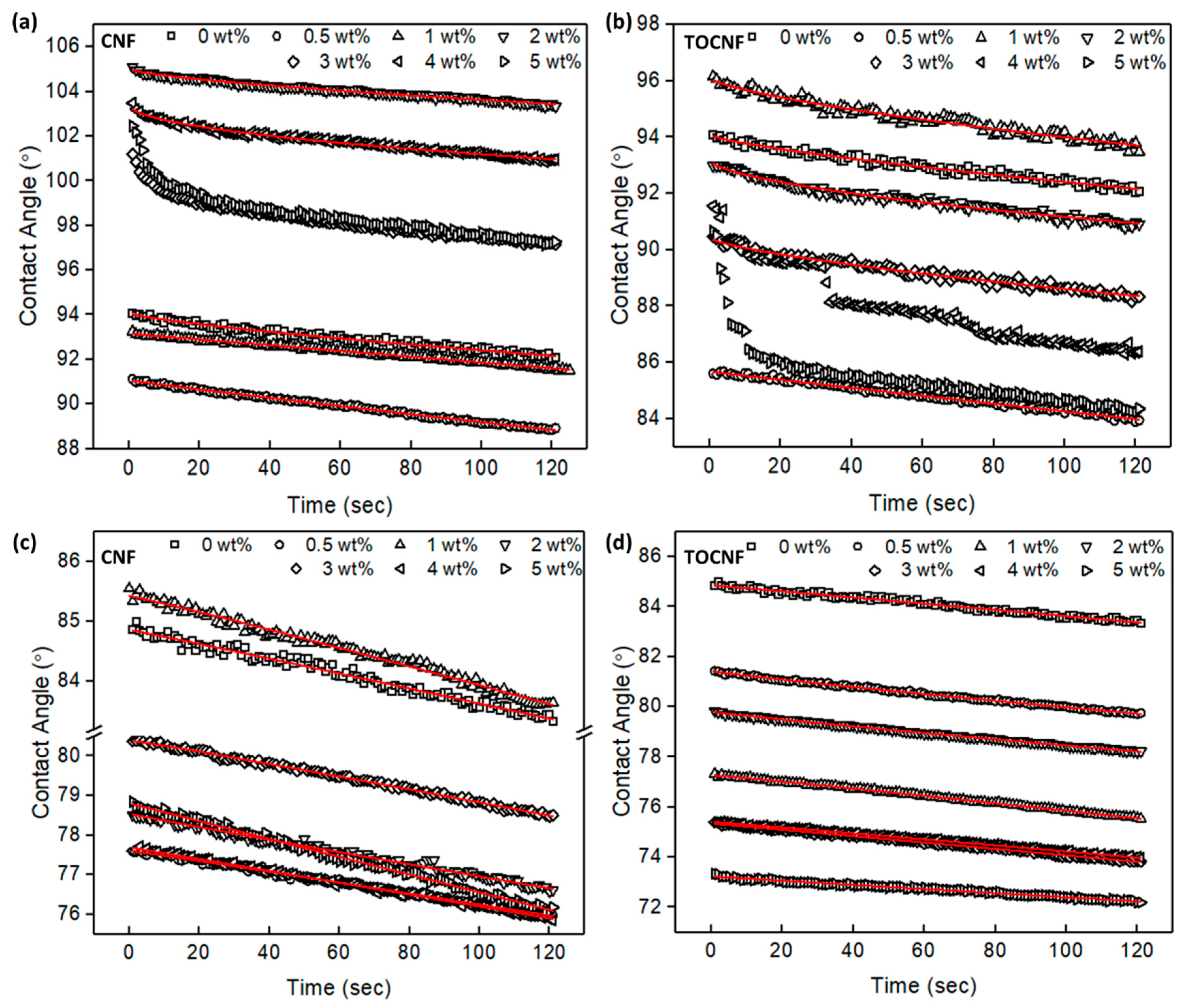
2.4. Contact Angle
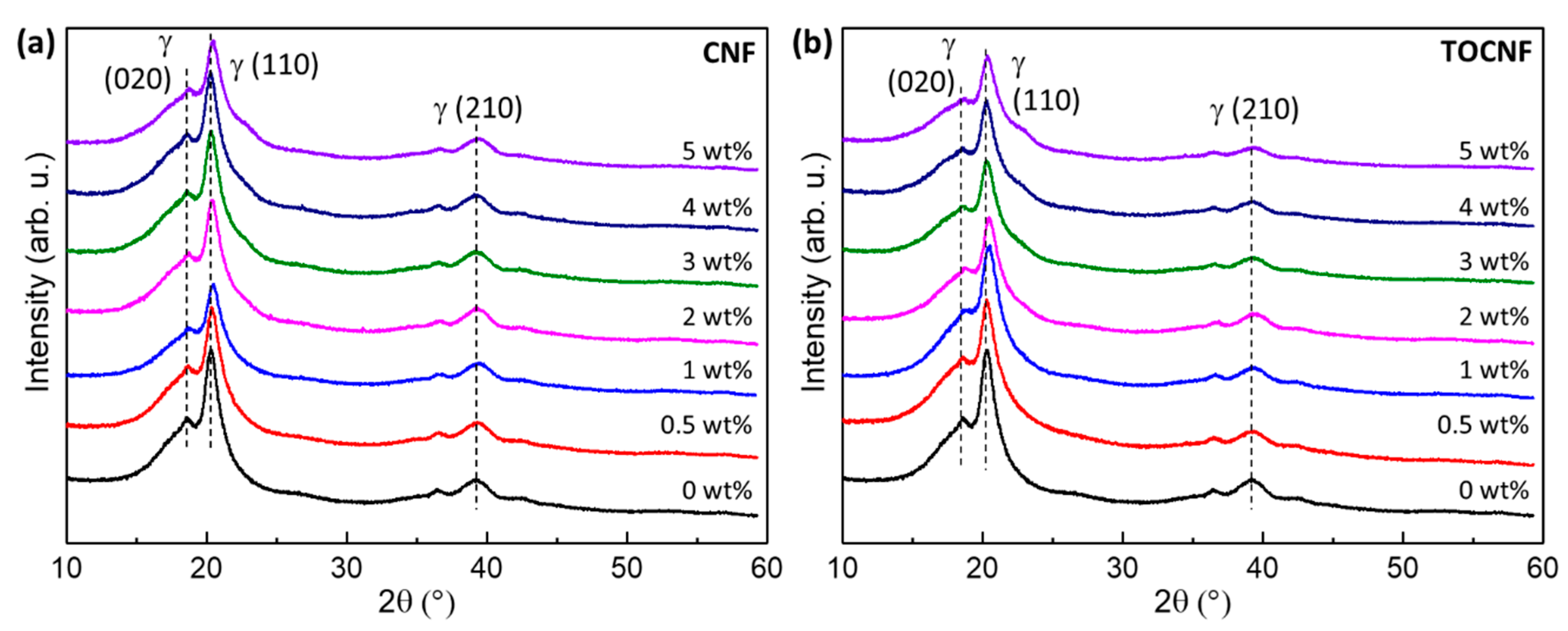
2.5. Crystalline Structure
2.6. Thermal and Mechanical Behavior
3. Results and Discussion
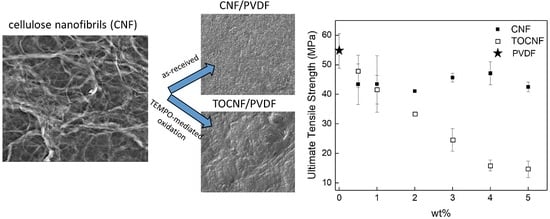
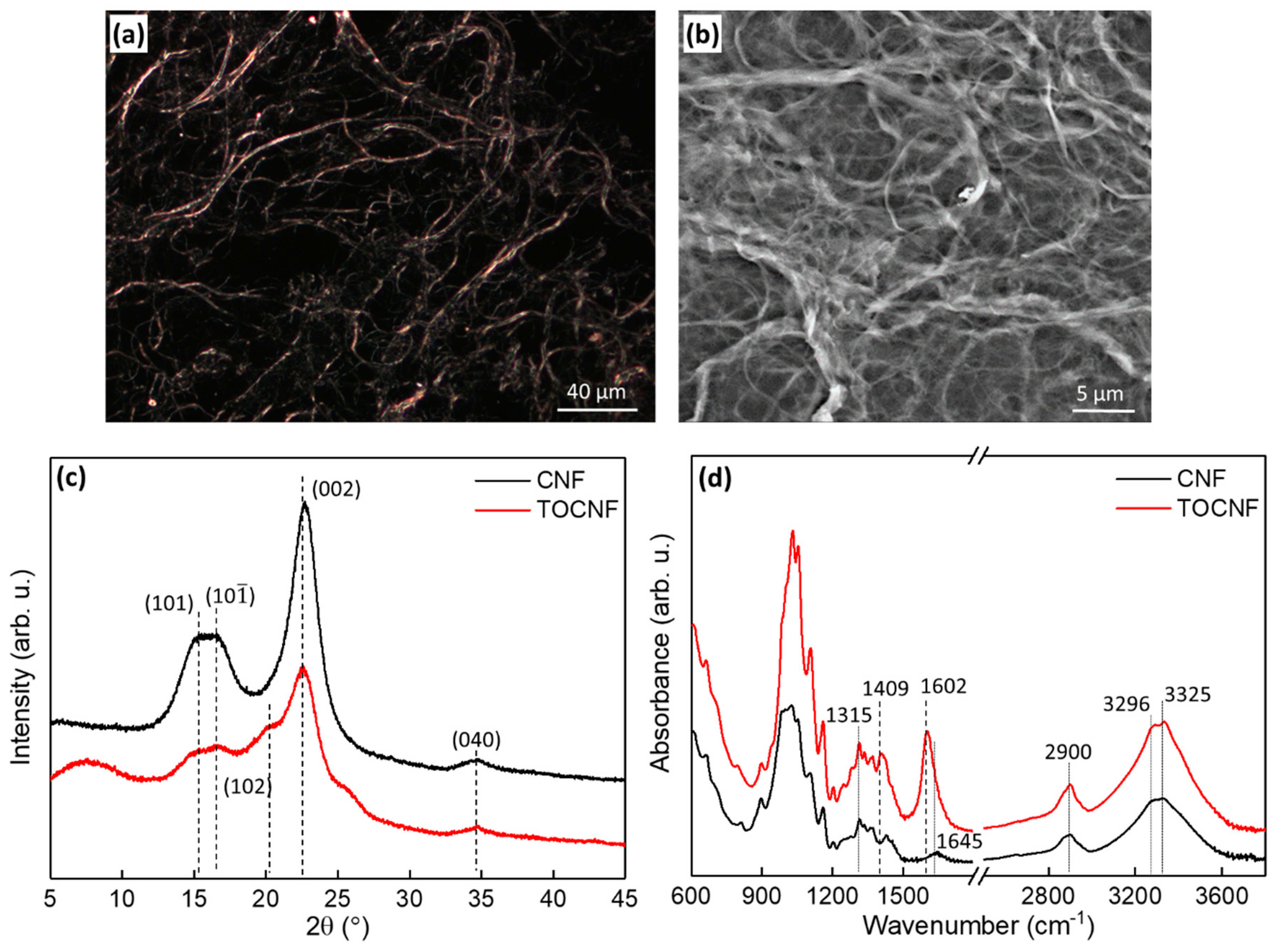
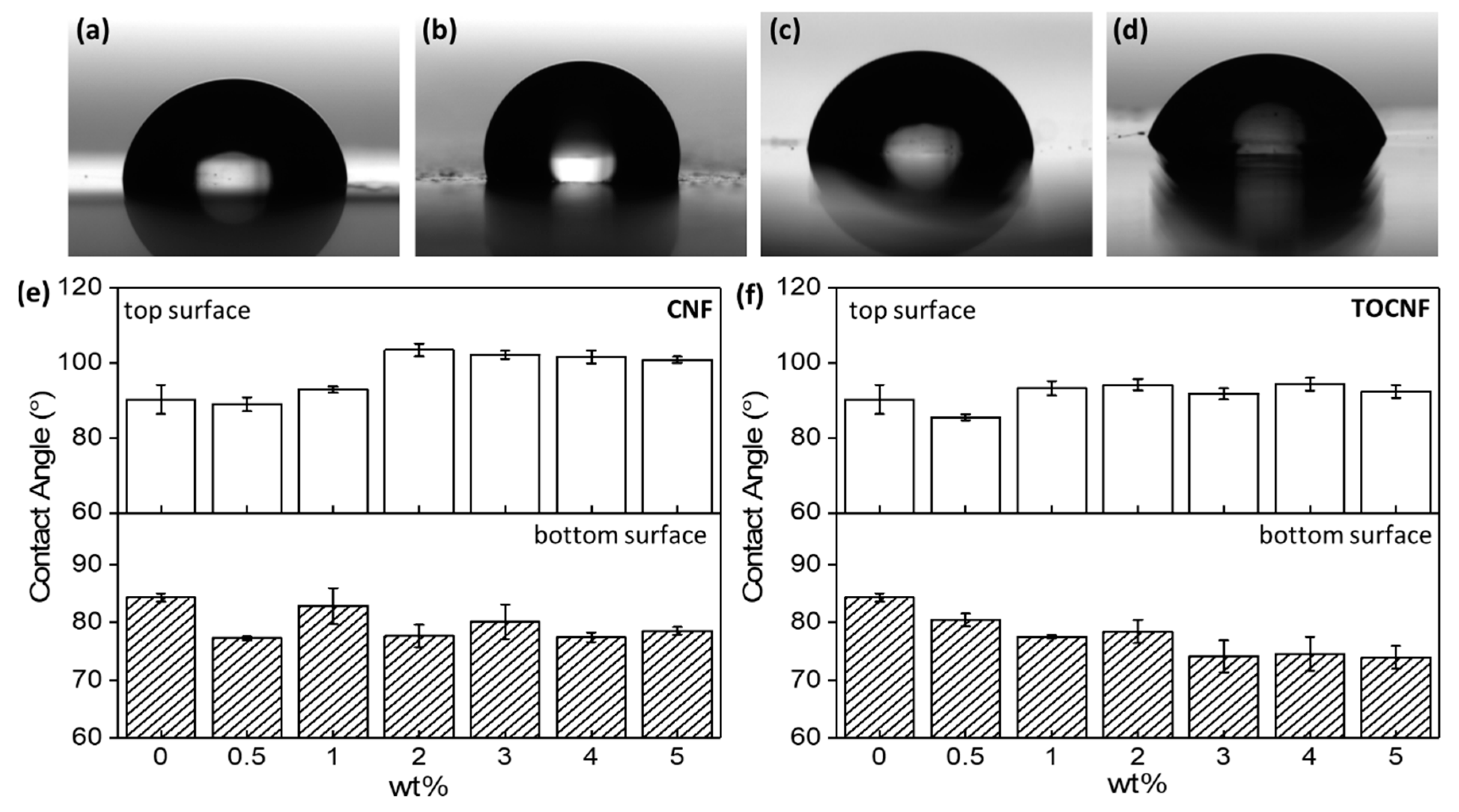
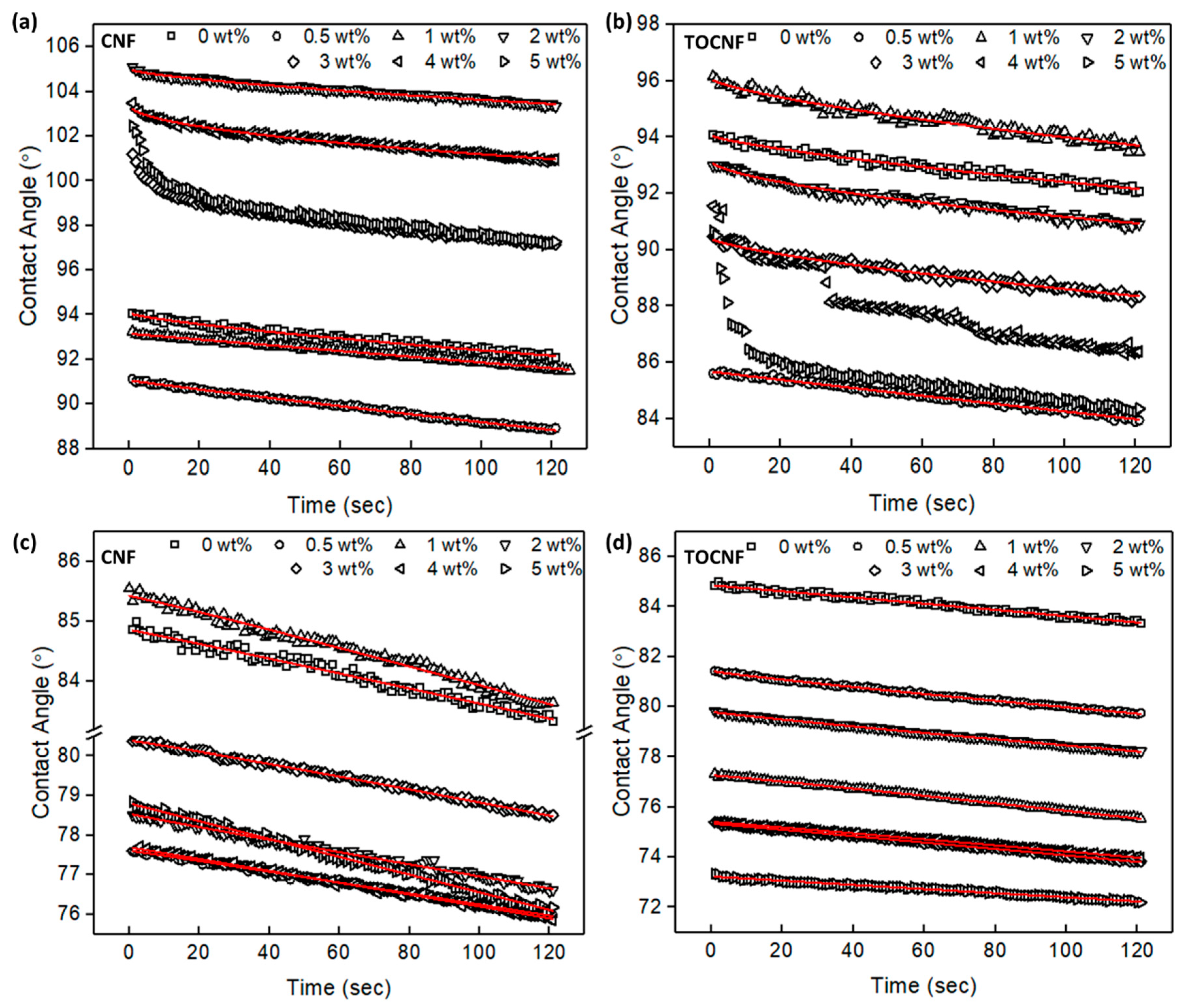
3.1. Surface Characterization and Morphology
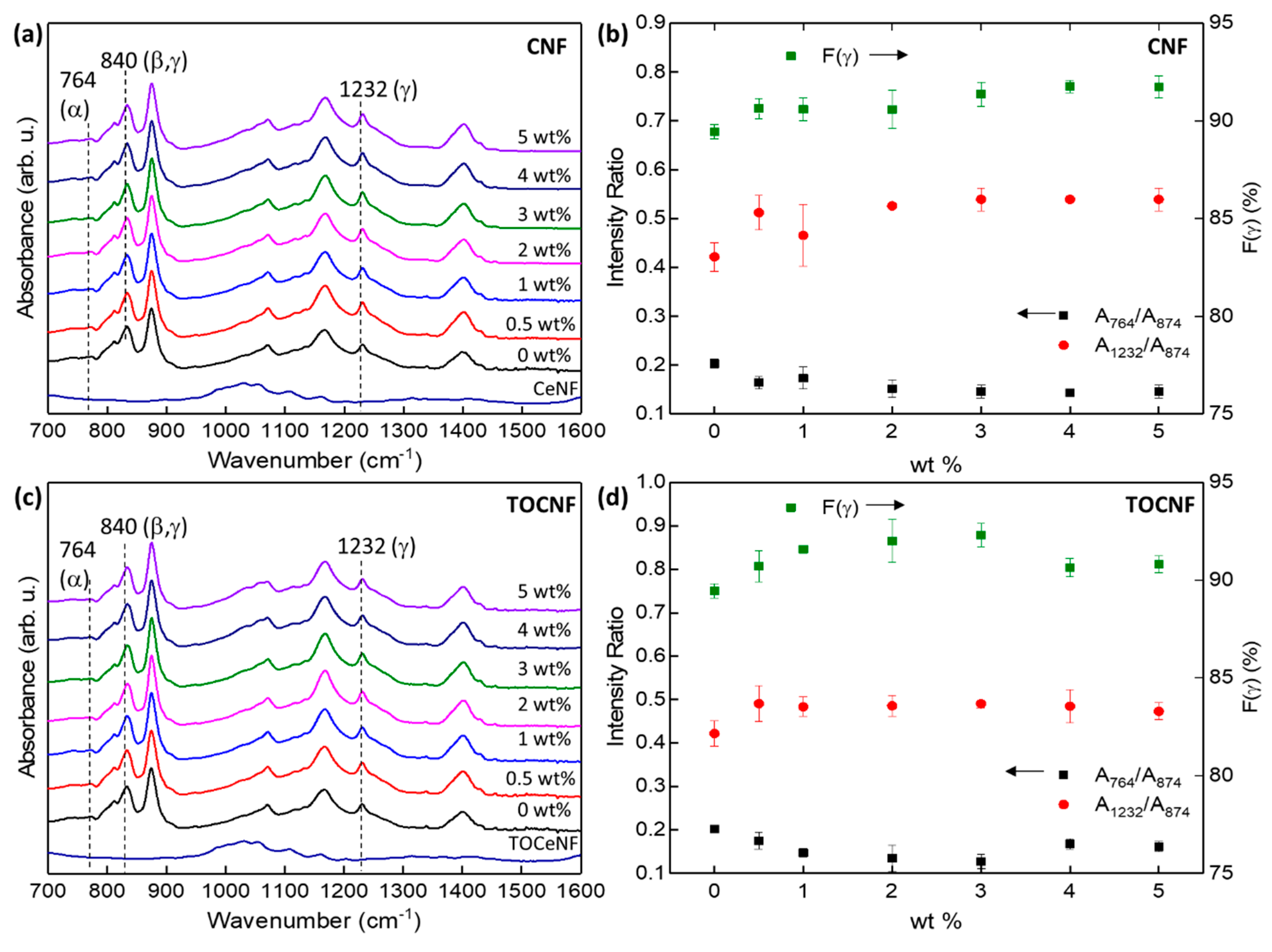
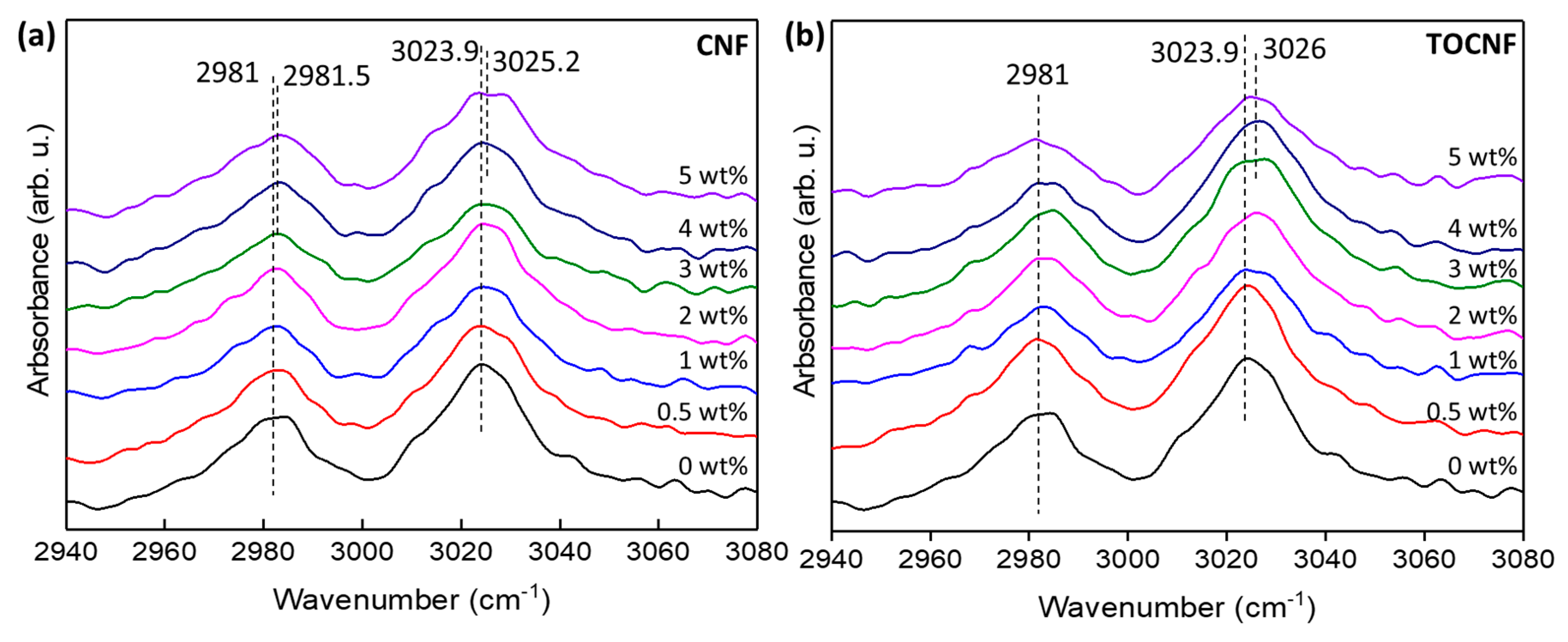
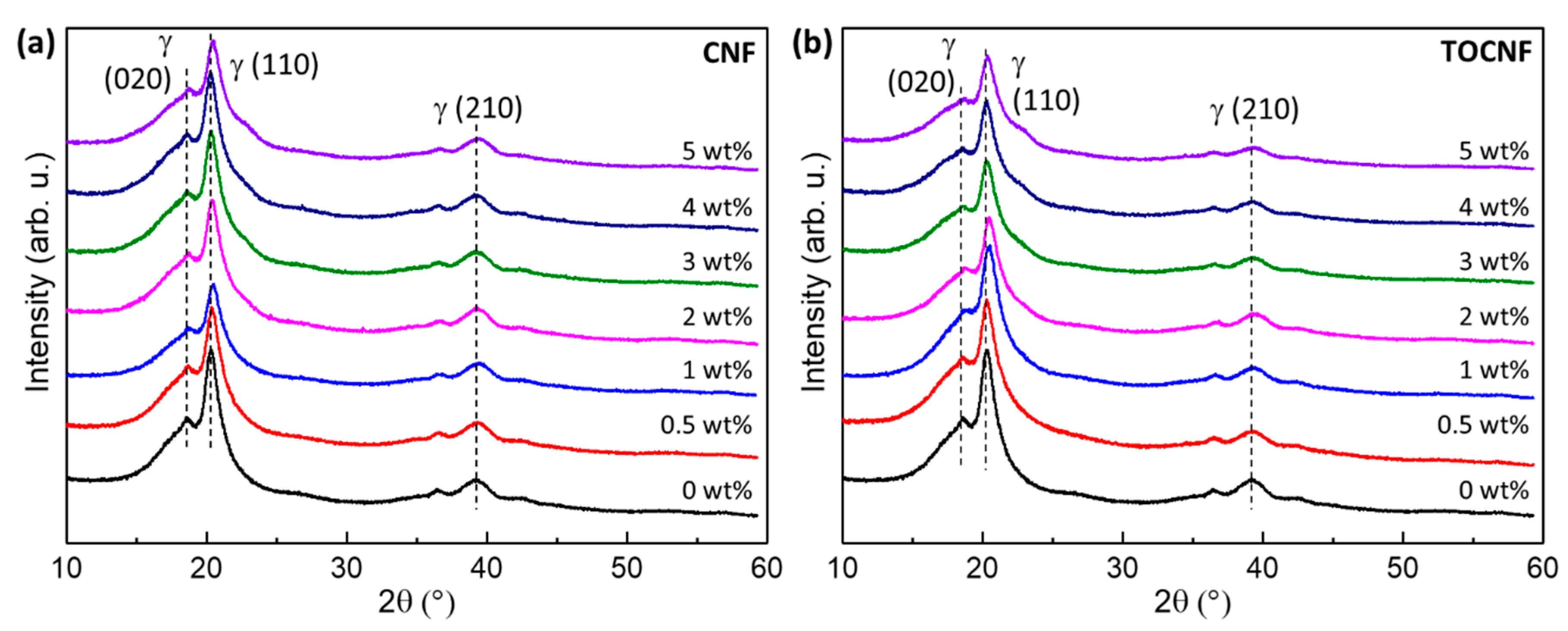
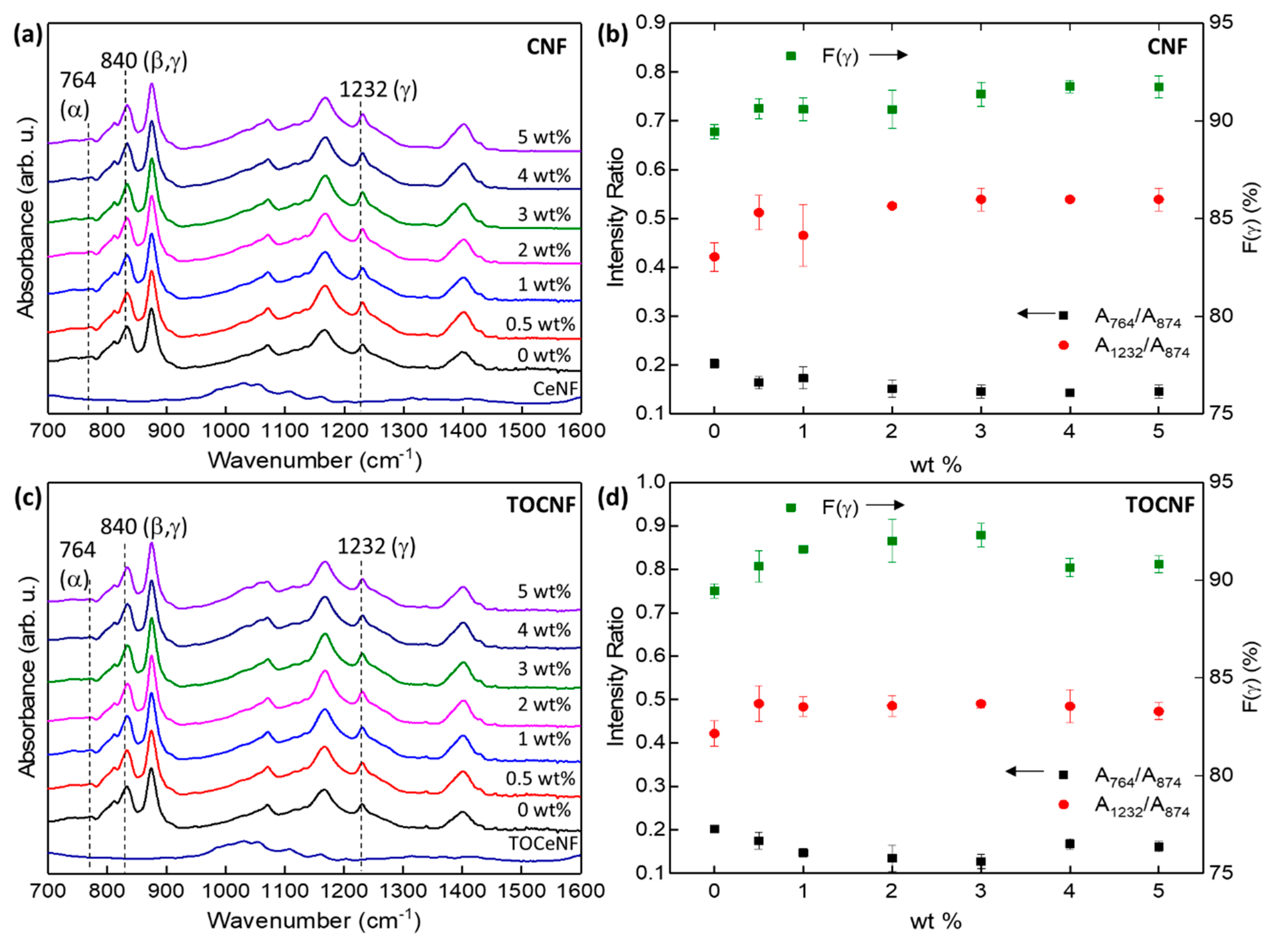
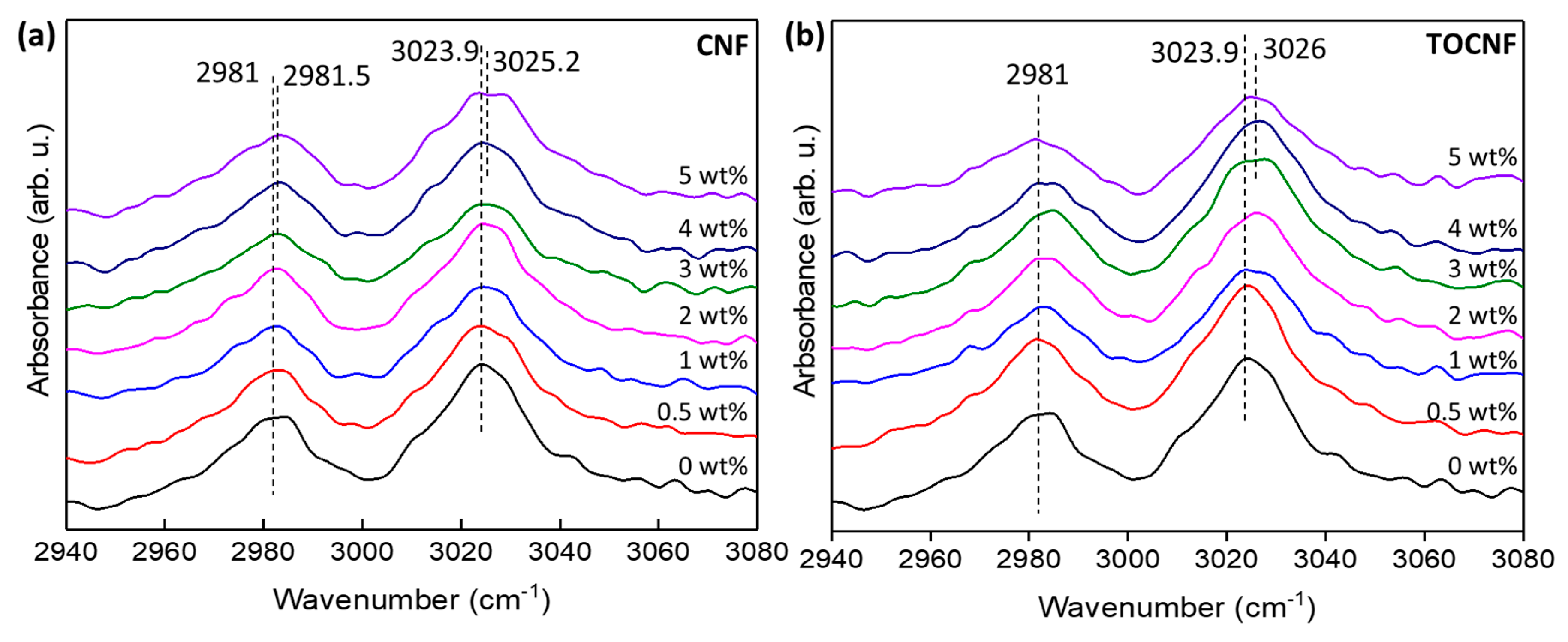
3.2. Crystalline Structure
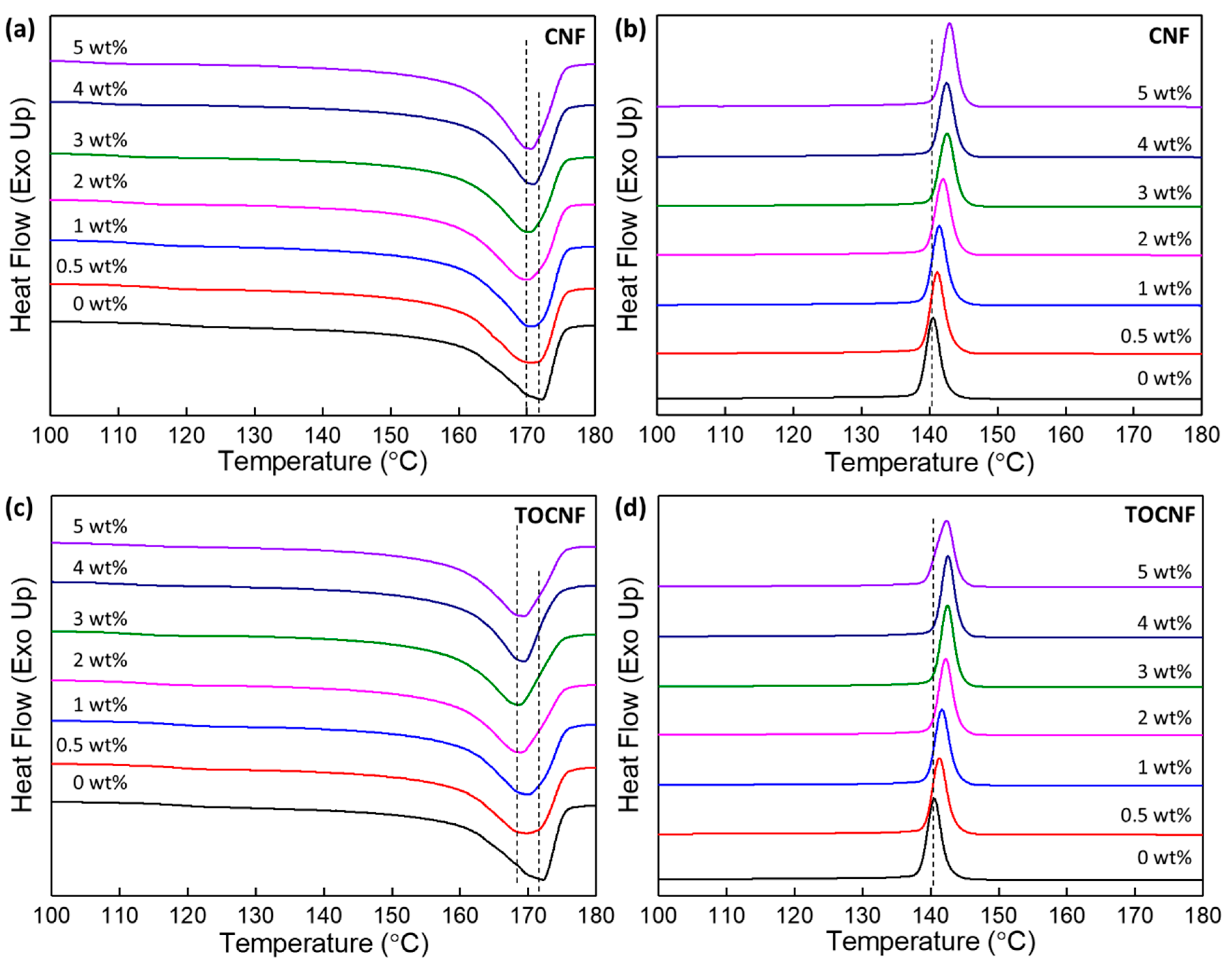
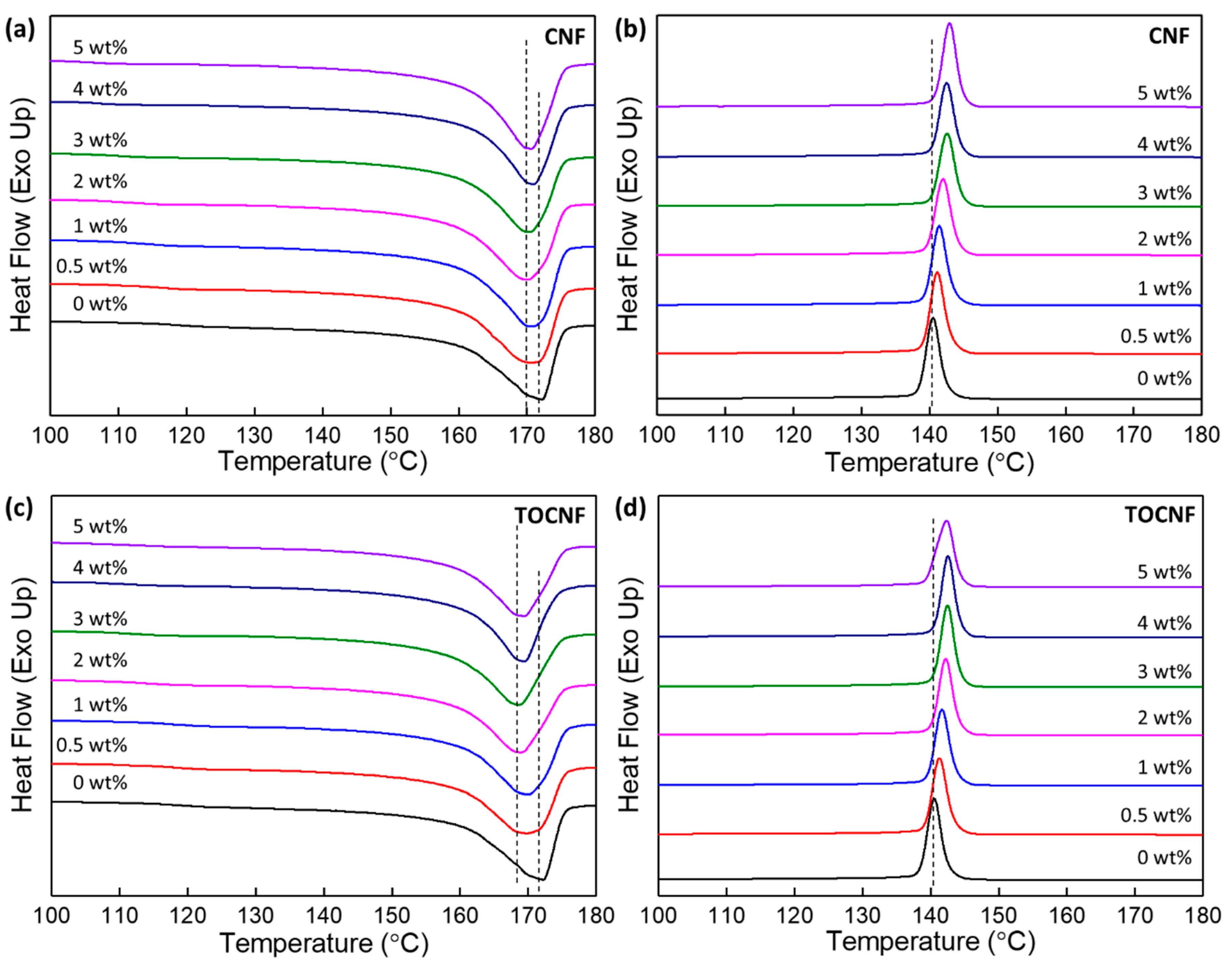
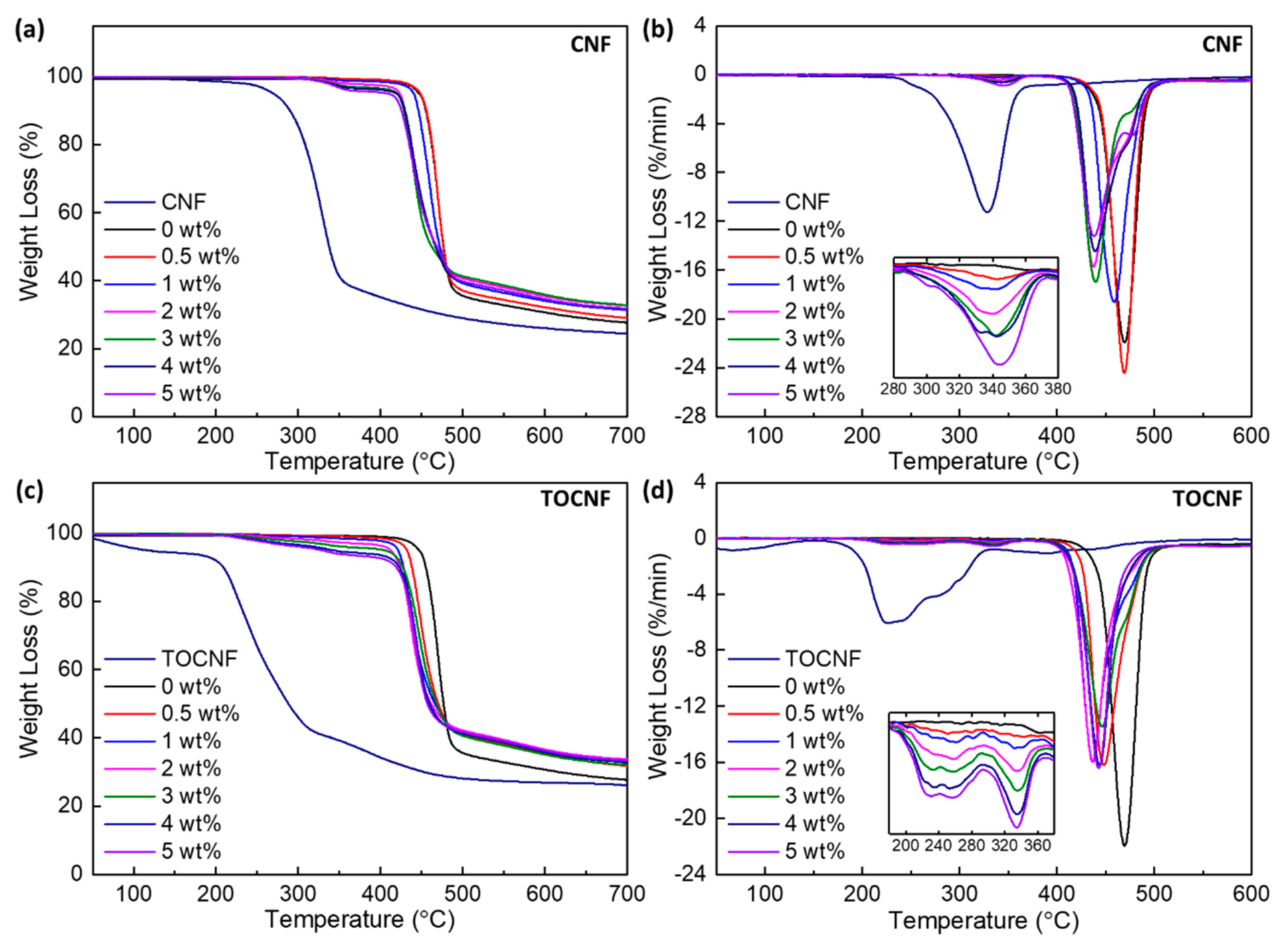
3.3. Thermal Stability and Crystallinity
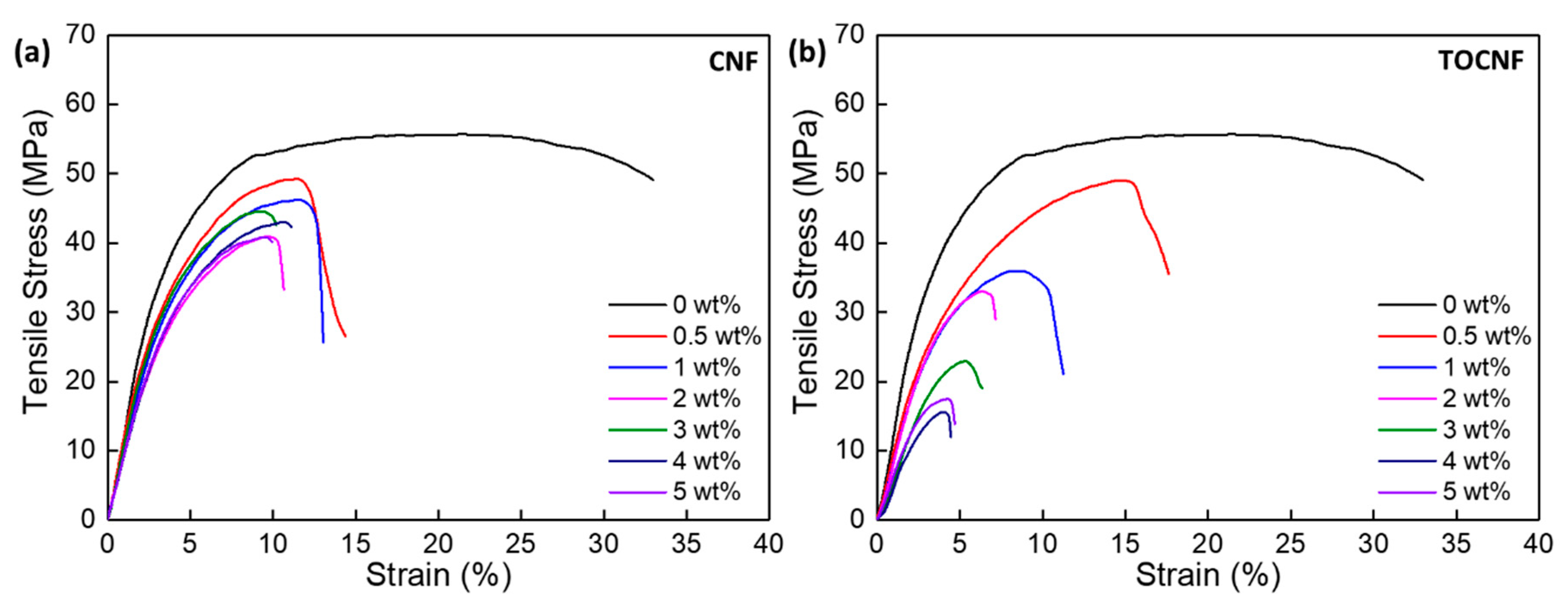
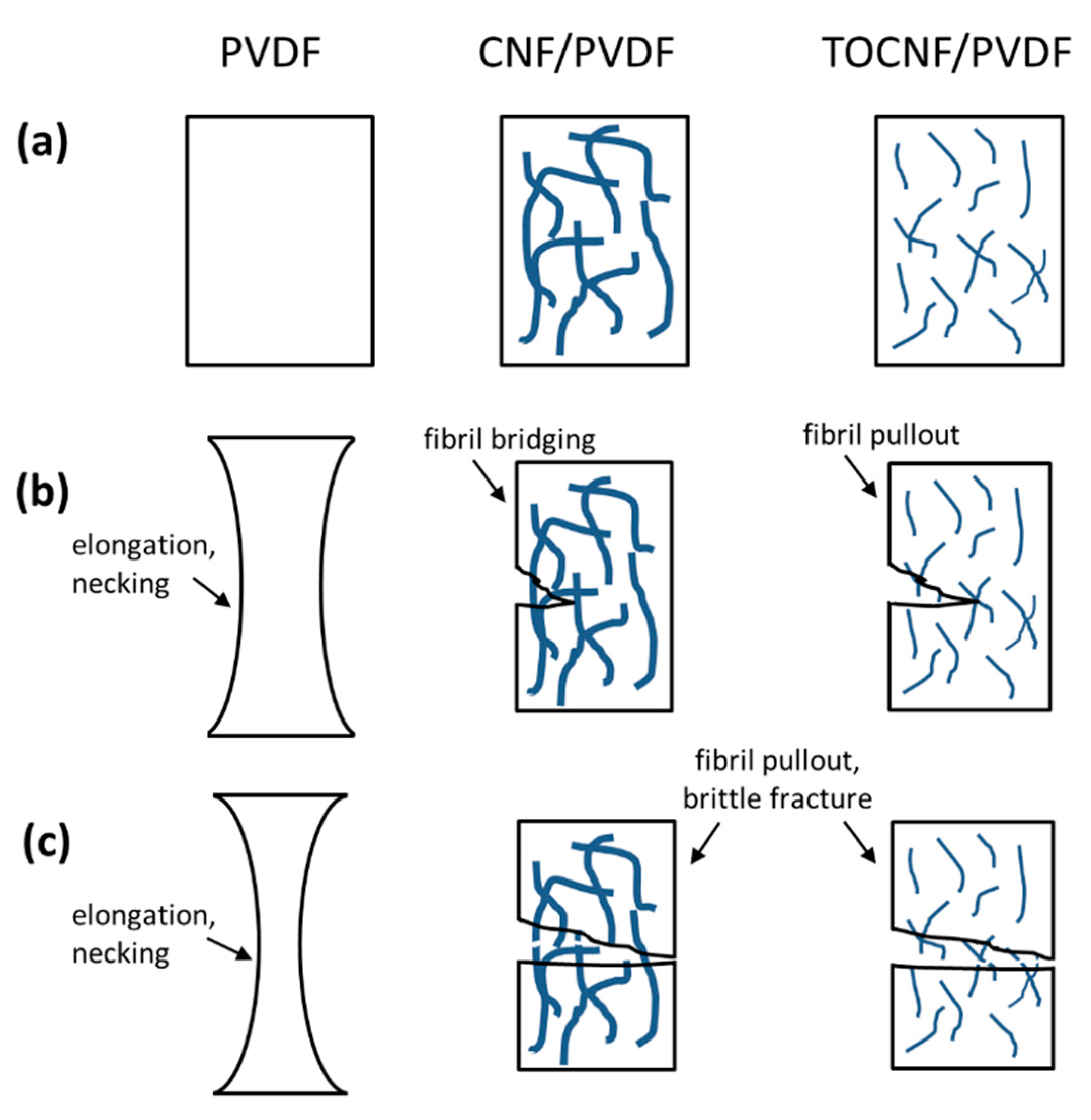
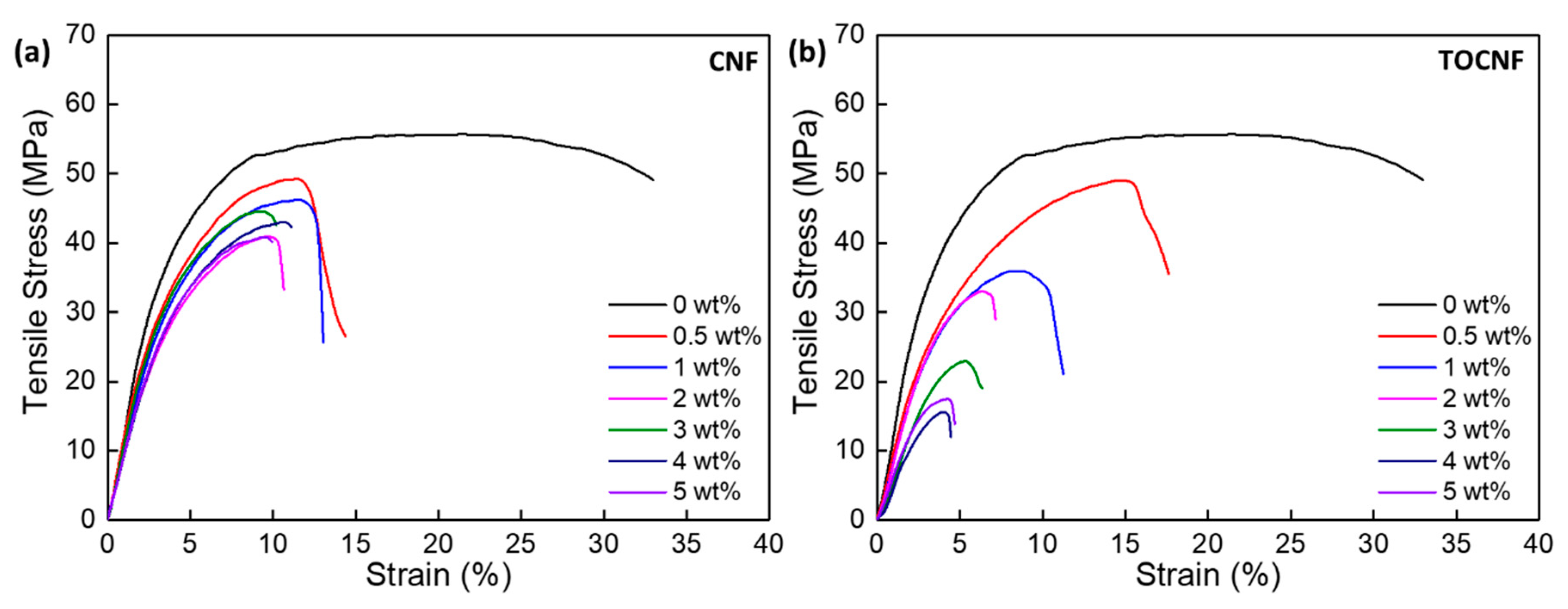
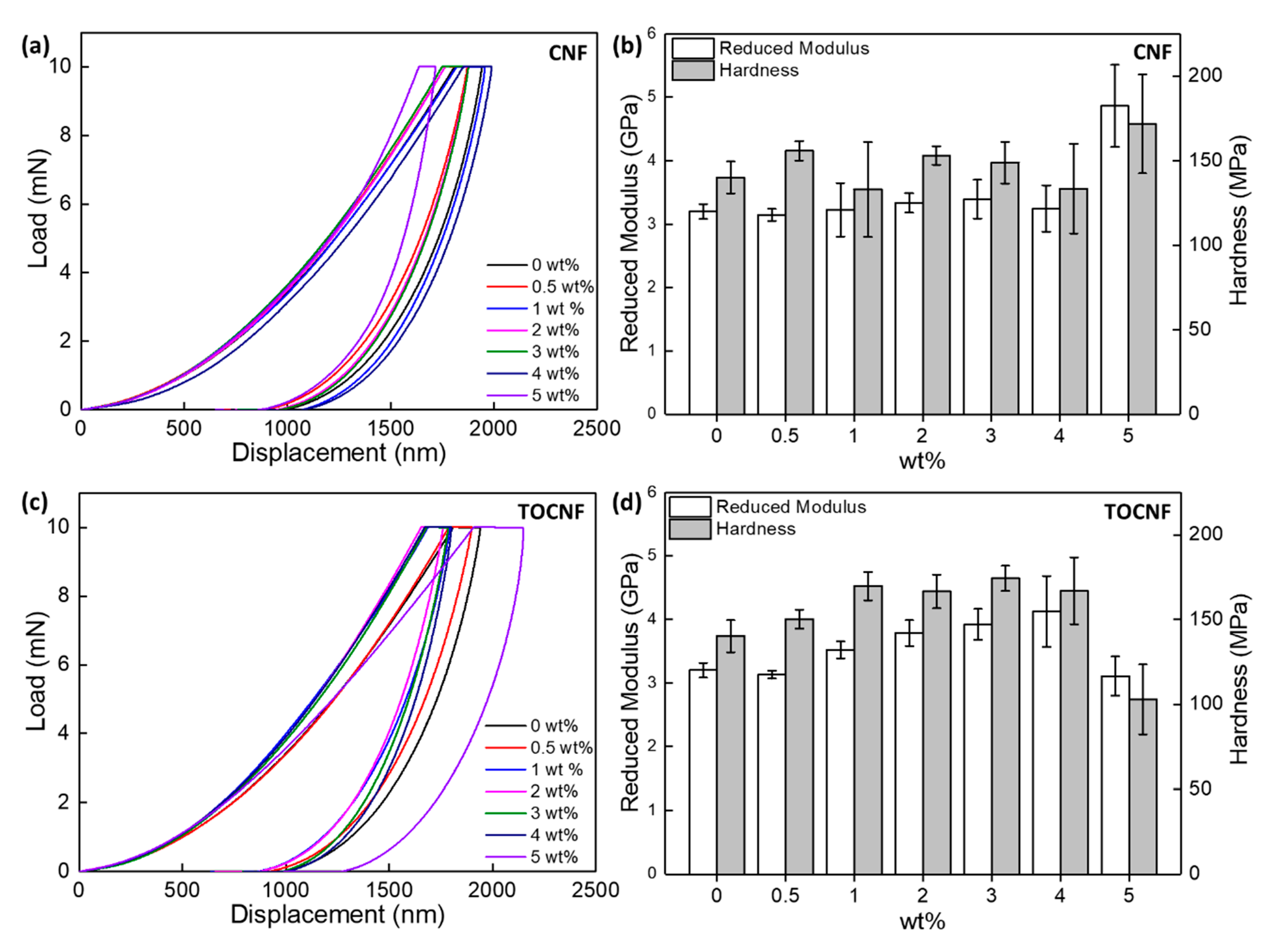
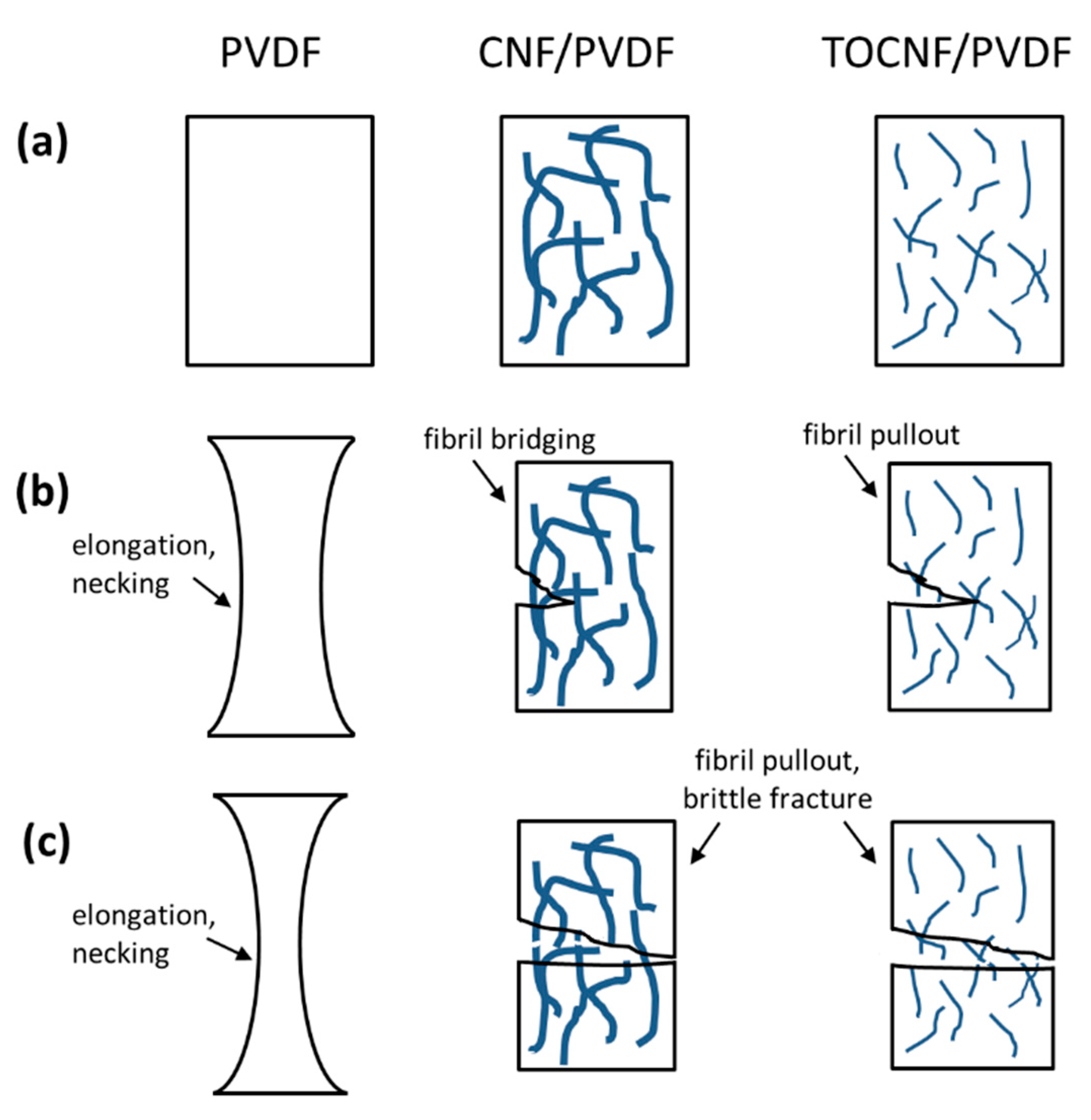
3.4. Mechanical Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Miao, C.W.; Hamad, W.Y. Cellulose reinforced polymer composites and nanocomposites: A critical review. Cellulose 2013, 20, 2221–2262. [Google Scholar] [CrossRef]
- Gabr, M.H.; Phong, N.T.; Abdelkareem, M.A.; Okubo, K.; Uzawa, K.; Kimpara, I.; Fujii, T. Mechanical, thermal, and moisture absorption properties of nano-clay reinforced nano-cellulose biocomposites. Cellulose 2013, 20, 819–826. [Google Scholar] [CrossRef]
- Timmerman, J.F.; Hayes, B.S.; Seferis, J.C. Nanoclay reinforcement effects on the cryogenic microcracking of carbon fiber/epoxy composites. Compos. Sci. Technol. 2002, 62, 1249–1258. [Google Scholar] [CrossRef]
- Tibbetts, G.G.; Lake, M.L.; Strong, K.L.; Rice, B.P. A review of the fabrication and properties of vapor-grown carbon nanofiber/polymer composites. Compos. Sci. Technol. 2007, 67, 1709–1718. [Google Scholar] [CrossRef]
- Coleman, J.N.; Khan, U.; Gunko, Y.K. Mechanical reinforcement of polymers using carbon nanotubes. Adv. Mater. 2006, 18, 689–706. [Google Scholar] [CrossRef]
- Gacitua, W.E.; Ballerini, A.A.; Zhang, J. Polymer nanocomposites: Synthetic and natural fillers a review. Maderas Cienc. Tecnol. 2005, 7, 159–178. [Google Scholar] [CrossRef]
- Colvin, V.L. The potential environmental impact of engineered nanomaterials. Nat. Biotechnol. 2003, 21, 1166–1170. [Google Scholar] [CrossRef]
- National Institute for Occupational Safety and Health (NIOSH). Occupational exposure to carbon nanotubes and nanofibers. Current Intelligence Bulletin 65. Department of Health and Human Services (NIOSH) Publication No. 2013–145, April 2013. Available online: https://www.cdc.gov/niosh/docs/2013-145/default.html (accessed on 27 June 2019).
- Schulte, P.A.; Murashov, V.; Zumwalde, R.; Kuempel, E.D.; Geraci, C.L. Occupational exposure limits for nanomaterials: State of the art. J. Nanopart. Res. 2010, 12, 1971–1987. [Google Scholar] [CrossRef]
- Brandeli, A. Nanomaterials: Ecotoxicity, Safety, and Public Perception; Rai, M., Biswas, J.K., Eds.; Springer: Basel, Switzerland, Chapter 4; p. 69.
- Moon, R.J.; Martini, A.; Nairn, J.; Simonsen, J.; Youngblood, J. Cellulose nanomaterials review: Structure, properties and nanocomposites. Chem. Soc. Rev. 2011, 40, 3941–3994. [Google Scholar] [CrossRef]
- Zhou, C.; Chu, R.; Wu, R.; Wu, Q. Electrospun polyethylene oxide/cellulose nanocrystal composite nanofibrous mats with homogeneous and heterogeneous microstructures. Biomacromolecules 2011, 12, 2617–2625. [Google Scholar] [CrossRef]
- Xu, X.; Liu, F.; Jiang, L.; Zhu, J.Y.; Haagenson, D.; Wiesenborn, D.P. Cellulose nanocrystals vs. cellulose nanofibrils: A comparative study on their microstructures and effects as polymer reinforcing agents. ACS Appl. Mater. Interfaces 2013, 5, 2999–3009. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Sliozberg, H.R.; Snyder, J.F.; Steele, J.; Chantawansri, T.L.; Orlicki, J.A.; Walck, S.D.; Reiner, R.S.; Rudie, A.W. Highly transparent and toughened poly (methyl methacrylate) nanocomposite films containing networks of cellulose nanofibrils. ACS Appl. Mater. Interfaces 2015, 7, 25464–25472. [Google Scholar] [CrossRef] [PubMed]
- Martins, P.; Lopes, A.C.; Lanceros-Mendez, S. Electroactive phases of poly (vinylidene fluoride): Determination, processing and applications. Prog. Polym. Sci. 2014, 39, 683–706. [Google Scholar] [CrossRef]
- Zhao, X.Z.; Bharti, V.; Zhang, Q.M.; Romotowski, T.; Tito, F.; Ting, R. Electromechanical properties of electrostrictive poly (vinylidene fluoride–trifluoroethylene) copolymer. Appl. Phys. Lett. 1998, 73, 2054–2056. [Google Scholar] [CrossRef]
- Perez, R.; Kral, M.; Bleuler, H. Study of polyvinylidene fluoride (PVDF) based bimorph actuators for laser scanning actuation at kHz frequency range. Sens. Actuators A 2012, 183, 84–94. [Google Scholar] [CrossRef]
- Bar-Cohen, Y.; Zhang, Q. Electroactive polymer actuators and sensors. MRS Bulletin 2008, 33, 173–181. [Google Scholar] [CrossRef]
- Sirohi, J.; Chopra, I. Fundamental understanding of piezoelectric strain sensors. J. Intell. Material. Syst. Struct. 2000, 11, 246–257. [Google Scholar] [CrossRef]
- Ying, L.; Kang, E.T.; Neoh, K.G. Covalent immobilization of glucose oxidase on microporous membranes prepared from poly(vinylidene fluoride) with grafted poly(acrylic acid) side chains. J. Membr. Sci. 2002, 208, 361–374. [Google Scholar] [CrossRef]
- Ouyang, J.Y.; Chu, C.W.; Szmanda, C.R.; Ma, L.P.; Yang, Y. Programmable polymer thin film and non-volatile memory device. Nat. Mater. 2004, 3, 918–922. [Google Scholar] [CrossRef]
- Scott, J.C.; Bozano, L.D. Nonvolatile memory elements based on organic materials. Adv. Mater. 2007, 19, 1452–1463. [Google Scholar] [CrossRef]
- Chang, C.; Tran, V.H.; Wang, J.; Fuh, Y.K.; Lin, L. Direct-write piezoelectric polymeric nanogenerator with high energy conversion efficiency. Nano Lett. 2010, 10, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Beeby, S.P.; Tudor, M.J.; White, N.M. Energy harvesting vibration sources for microsystems applications. Meas. Sci. Technol. 2006, 17, R175–R195. [Google Scholar] [CrossRef]
- Beeby, S.P.; Torah, R.N.; Tudor, M.J.; Glynne-Jones, P.; O’Donnell, T.; Saha, C.R.; Roy, S. A micro electromagnetic generator for vibration energy harvesting. J. Micromech. Microeng. 2007, 17, 1257–1265. [Google Scholar] [CrossRef]
- Lee, M.; Chen, C.Y.; Wang, S.; Cha, S.N.; Park, Y.J.; Kim, J.M.; Chou, L.J.; Wang, Z.L. A hybrid piezoelectric structure for wearable nanogenerators. Adv. Mater. 2012, 24, 1759–1764. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, S.; Litt, M.H.; Lando, J.B. The crystal structure of the γ phase of poly (vinylidene fluoride). Macromolecules 1980, 3, 1178–1183. [Google Scholar] [CrossRef]
- Lovinger, A.J. Unit cell of the γ phase of poly (vinylidene fluoride). Macromolecules 1981, 4, 322–325. [Google Scholar] [CrossRef]
- Bodkhe, S.; Rajesh, P.S.M.; Kamle, S.; Verma, V. Beta-phase enhancement in polyvinylidene fluoride through filler addition: Comparing cellulose with carbon nanotubes and clay. J. Polym. Res. 2014, 21, 434. [Google Scholar] [CrossRef]
- Rajesh, P.S.M.; Bodkhe, S.; Kamle, S.; Verma, V. Enhancing beta-phase in PVDF through physicochemical modification of cellulose. Electron. Mater. Lett. 2014, 10, 315–319. [Google Scholar] [CrossRef]
- Zhang, Z.; Wu, Q.; Song, K.; Lei, T.; Wu, Y. Poly(vinylidene fluoride)/cellulose nanocrystals composites: Rheological, hydrophilicity, thermal and mechanical properties. Cellulose 2015, 22, 2431–2441. [Google Scholar] [CrossRef]
- Fu, R.; Chen, S.; Lin, Y.; Zhang, S.; Jiang, J.; Li, Q.; Gu, Y. Improved piezoelectric properties of electrospun poly (vinylidene fluoride) fibers blended with cellulose nanocrystals. Mater. Lett. 2017, 187, 86–88. [Google Scholar] [CrossRef]
- Rak, K.; Phakdeepataraphan, E.; Bunnak, N.; Ummartyotin, S.; Sain, M.; Manuspiya, H. Development of bacterial cellulose and poly(vinylidene fluoride) binary blend system: Structure and properties. Chem. Eng. J. 2014, 237, 396–402. [Google Scholar] [CrossRef]
- Vidhate, S.; Shaito, A.; Chung, J.; D’Souza, N.A. Crystallization, mechanical, and rheological behavior of.polyvinylidene fluoride/carbon nanofiber composites. J. Appl. Polym. Sci. 2009, 112, 254–260. [Google Scholar] [CrossRef]
- Shamsuddin, S.R.; Hodgkinson, J.; Asp, L.; Langstrom, R.; Bismarck, A. Carbon fibre reinforced PVDF composites. In Proceedings of the ECCM15–15th European Conference on Composite Materials, Venice, Italy, 24–28 June 2012. [Google Scholar]
- Wu, Q.; Xie, D.-J.; Zhang, Y.-D.; Jia, Z.-M.; Zhang, H.-Z. Mechanical properties and simulation of nanographene/polyvinylidene fluoride composite films. Composites Part B 2019, 156, 148–155. [Google Scholar] [CrossRef]
- Al-Saygh, A.; Ponnamma, D.; AlMaadeed, M.A.; Vijayan, P.; Karim, A.; Hassan, M.K. Flexible Pressure sensor based on PVDF nanocomposites containing reduced graphene oxide-titania hybrid nanolayers. Polymers 2017, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Eggedi, O.; Valiyaneerilakkal, U.; Darla, M.R.; Varghese, S. Nanoindentation and thermal characterization of poly (vinylidenefluoride)/MWCNT nanocomposites. AIP Adv. 2014, 4, 047102. [Google Scholar] [CrossRef] [Green Version]
- Sacui, I.A.; Nieuwendaal, R.C.; Burnett, D.J.; Stranick, S.J.; Jorfi, M.; Weder, C.; Johan Foster, E.; Olsson, R.T.; Gilman, J.W. Comparison of the properties of cellulose nanocrystals and cellulose nanofibrils isolated from bacteria, tunicate, and wood processed using acid, enzymatic, mechanical, and oxidative methods. ACS Appl. Mater. Interfaces 2014, 6, 6127–6138. [Google Scholar] [CrossRef]
- Necas, D.; Klapetek, P. Gwyddion: An open-source software for SPM data analysis. Open Phys. 2012, 10, 181–188. [Google Scholar] [CrossRef]
- Cataldo, V.A.; Cavallaro, G.; Lazzara, G.; Milioto, S.; Parisi, F. Coffee grounds as filler for pectin: Green composites with competitive performances dependent on the UV irradiation. Carbohydr. Polym. 2017, 170, 198–205. [Google Scholar] [CrossRef]
- Bertolino, V.; Cavallaro, G.; Lazzara, G.; Milioto, S.; Parisi, F. Halloysite nanotubes sandwiched between chitosan layers: Novel bionanocomposites with multilayer structures. New J. Chem. 2018, 42, 8384–8390. [Google Scholar] [CrossRef]
- Segal, L.; Creely, J.J.; Martin, A.E.; Conrad, C.M. An empirical method for estimating the degree of crystallinity of native cellulose using the X-ray diffractometer. Tex. Res. J. 1959, 29, 786–794. [Google Scholar] [CrossRef]
- Wang, L.; Chen, S. Crystallization behaviors of poly (vinylidene fluoride) and poly (methyl methacrylate)-block-poly (2-vinyl pyridine) block copolymer blends. J. Therm. Anal. Calorim. 2016, 125, 215–230. [Google Scholar] [CrossRef]
- Lee, K.Y.; Tammelin, T.; Schulfter, K.; Kiiskinen, H.; Samela, J.; Bismarck, A. High performance cellulose nanocomposites: Comparing the reinforcing ability of bacterial cellulose and nanofibrillated cellulose. ACS Appl. Mater. Interfaces 2012, 4, 4078–4086. [Google Scholar] [CrossRef] [PubMed]
- Oudiani, A.E.; Chaabouni, Y.; Msahli, S.; Sakli, F. Crystal transition from cellulose I to cellulose II in NaOH treated Agave americana L. fibre. Carbohyd. Polym. 2011, 86, 1221–1229. [Google Scholar] [CrossRef]
- Han, J.; Zhou, C.; Wu, Y.; Liu, F.; Wu, Q. Self-assembling behavior of cellulose nanoparticles during freeze-drying: Effect of suspension concentration, particle size, crystal structure, and surface charge. Biomacromolecules 2013, 14, 1529–1540. [Google Scholar] [CrossRef] [PubMed]
- Johar, N.; Ahmad, I.; Dufresne, A. Extraction, preparation and characterization of cellulose fibres and nanocrystals from rice husk. Ind. Crop. Prod. 2012, 37, 93–99. [Google Scholar] [CrossRef]
- Cheng, D.; Wen, Y.; An, X.; Zhu, X.; Ni, Y. TEMPO-oxidized cellulose nanofibers (TOCNs) as a green reinforcement for waterborne polyurethane coating (WPU) on wood. Carbohyd. Polym. 2016, 151, 326–334. [Google Scholar] [CrossRef]
- Gregorio Jr., R.; Sousa Borges, D. Effect of crystallization rate on the formation of the polymorphs of solution cast poly (vinylidene fluoride). Polymer 2008, 49, 4009–4016. [Google Scholar] [CrossRef]
- Li, M.; Katsouras, I.; Piliego, C.; Glasser, G.; Lieberwirth, I.; Blomb, P.W.M.; de Leeuw, D.M. Controlling the microstructure of poly (vinylidene-fluoride)(PVDF) thin films for microelectronics. J. Mater. Chem. C 2013, 1, 7695–7702. [Google Scholar] [CrossRef]
- Okita, Y.; Fujisawa, S.; Saito, T.; Isogai, A. TEMPO-oxidized cellulose nanofibrils dispersed in organic solvents. Biomacromolecules 2011, 12, 518–522. [Google Scholar] [CrossRef]
- Tsuguyuki, S.; Satoshi, K.; Yoshiharu, N.; Akira, I. Cellulose nanofibers prepared by TEMPO-mediated oxidation of native cellulose. Biomacromolecules 2007, 8, 2485–2491. [Google Scholar] [CrossRef]
- Ma, W.; Zhang, J.; Wang, X.; Wang, S. Effect of PMMA on crystallization behavior and hydrophilicity of poly (vinylidene fluoride)/poly (methyl methacrylate) blend prepared in semi-dilute solutions. Appl. Surf. Sci. 2007, 253, 8377–8388. [Google Scholar] [CrossRef]
- Cassie, B.D.; Baxter, S. Wettability of porous surfaces. Trans. Faraday Soc. 1944, 40, 546–551. [Google Scholar] [CrossRef]
- Lopes, A.C.; Costa, C.M.; Tavares, C.J.; Neves, I.C.; Lanceros-Méndez, S. Nucleation of the electroactive γ phase and enhancement of the optical transparency in low filler content poly (vinylidene)/clay nanocomposites. J. Phys. Chem. C 2011, 115, 18076–18082. [Google Scholar] [CrossRef]
- Benz, M.; Euler, W.B.; Gregory, O.J. The role of solution phase water on the deposition of thin films of poly (vinylidene fluoride). Macromolecules 2002, 35, 2682–2688. [Google Scholar] [CrossRef]
- Tamang, A.; Kumar Ghosh, S.K.; Garain, S.; Alam, M.M.; Haeberle, J.; Henkel, K.; Schmeisser, D.; Mandal, D. DNA-Assisted β-phase nucleation and alignment of molecular dipoles in PVDF film: A realization of self-poled bioinspired flexible polymer nanogenerator for portable electronic devices. ACS Appl. Mater. Interfaces 2015, 7, 16143–16147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wu, Q.; Song, K.; Ren, S.; Lei, T.; Zhang, Q. Using cellulose nanocrystals as a sustainable additive to enhance hydrophilicity, mechanical and thermal properties of poly(vinylidene fluoride)/poly(methyl methacrylate) blend. ACS Sustain. Chem. Eng. 2015, 3, 574–582. [Google Scholar] [CrossRef]
- Issa, A.A.; Al-Maadeed, M.; Luyt, A.S.; Mrlik, M.; Hassan, M.K. Investigation of the physico-mechanical properties of electrospun PVDF/cellulose (nano) fibers. J. Appl. Polym. Sci. 2016, 43594. [Google Scholar] [CrossRef]
- Martins, P.; Cparros, C.; Goncalves, R.; Martins, P.M.; Benelmekki, M.; Botelho, G.; Lanceros-Mendez, S.J. Role of nanoparticle surface charge on the nucleation of the electroactive β-poly (vinylidene fluoride) nanocomposites for sensor and actuator applications. J. Phys. Chem. C 2012, 116, 15790–15794. [Google Scholar] [CrossRef]
- Imran-ul-haq, M.; Tiersch, B.; Beuermann, S. Influence of polymer end groups on crystallization and morphology of poly (vinylidene fluoride) synthesized in homogeneous phase with supercritical carbon dioxide. Macromolecules 2008, 41, 7453–7462. [Google Scholar] [CrossRef]
- Saito, T.; Hirota, M.; Tamura, N.; Kimura, S.; Fukuzumi, H.; Heux, L.; Isogai, A. Individualization of nano-sized plant cellulose fibrils by direct surface carboxylation using TEMPO catalyst under neutral conditions. Biomacromolecules 2009, 10, 1992–1996. [Google Scholar] [CrossRef]
- Sharma, M.; Sharma, K.; Bose, S. Segmental relaxations and crystallization-induced phase separation in PVDF/PMMA blends in the presence of surface-functionalized multiwall carbon nanotubes. J. Phys. Chem. B 2013, 117, 8589–8602. [Google Scholar] [CrossRef]
- Ghosh, S.K.; Alam, M.M.; Mandal, D. The in situ formation of platinum nanoparticles and their catalytic role in electroactive phase formation in poly(vinylidene fluoride): A simple preparation of multifunctional poly(vinylidene fluoride) films doped with platinum nanoparticles. RSC Adv. 2014, 4, 41886–41894. [Google Scholar] [CrossRef]
- Kumar, A.; Negi, Y.S.; Choudhary, V.; Bhardwaj, N.K. Characterization of cellulose nanocrystals produced by acid-hydrolysis from sugarcane bagasse as agro-waste. J. Mater. Phys. Chem. 2014, 2, 1–8. [Google Scholar] [CrossRef]
- Fukuzumi, H.; Saito, T.; Okita, Y.; Isogai, A. Thermal stabilization of TEMPO-oxidized cellulose. Polym. Degrad. Stab. 2010, 95, 1502–1508. [Google Scholar] [CrossRef]
- Liu, D.G.; Sun, X.; Tian, H.F.; Maiti, S.; Ma, Z.S. Effects of cellulose nanofibrils on the structure and properties on PVA nanocomposites. Cellulose 2013, 20, 2981–2989. [Google Scholar] [CrossRef]
- Park, K.H.; Shin, C.; Song, Y.-S.; Lee, H.-J.; Shin, C.; Kim, Y. Recyclable and mendable cellulose-reinforced composites crosslinked with Diels–Alder adducts. Polymers 2019, 11, 117. [Google Scholar] [CrossRef]
- Ten, E.; Jiang, L.; Wolcott, M.P. Crystallization kinetics of poly (3-hydroxybutyrate-co-3-hydroxyvalerate)/cellulose nanowhiskers composites. Carbohyd. Polym. 2012, 90, 541–550. [Google Scholar] [CrossRef]
- Wang, H.; Li, L.; Wu, T.; Wang, X.; Cheng, X.; Li, D. A Comparative study on the characterization of nanofibers with cellulose I, I/II, and II polymorphs from wood. Polymers 2019, 11, 153. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| top surface | bottom surface | |||||
|---|---|---|---|---|---|---|
| wt % | θi | k (×10−4 s−1) | n | θi | k (×10−4 s−1) | n |
| 0 | 94.08 | 5.6 | 0.75 | 84.87 | 1.4 | 1.00 |
| CNF | ||||||
| 0.5 | 91.07 | 2.6 | 0.95 | 77.63 | 1.8 | 1.00 |
| 1 | 93.16 | 1.3 | 1.00 | 85.48 | 1.8 | 1.00 |
| 2 | 105.20 | 5.3 | 0.70 | 78.54 | 2.4 | 0.97 |
| 3 | - | - | - | 80.43 | 2.0 | 1.00 |
| 4 | 103.42 | 2.1 | 0.51 | 77.70 | 1.9 | 1.00 |
| 5 | - | - | - | 78.80 | 2.8 | 1.00 |
| TOCNF | ||||||
| 0.5 | 85.68 | 1.9 | 0.98 | 81.43 | 2.9 | 0.89 |
| 1 | 96.10 | 8.9 | 0.70 | 77.32 | 1.8 | 1.00 |
| 2 | 93.20 | 14.9 | 0.58 | 79.80 | 2.5 | 0.91 |
| 3 | 90.43 | 7.8 | 0.71 | 75.36 | 1.7 | 1.00 |
| 4 | - | - | - | 75.43 | 1.6 | 1.00 |
| 5 | - | - | - | 73.23 | 1.2 | 0.99 |
| wt% | Tm (°C) | Tcryst (°C) | ΔHf (J/g) | Xc (%) | T10% (°C) | T50% (°C) | Tp1 (°C) | Tp2 (°C) | Tp3 (°C) |
|---|---|---|---|---|---|---|---|---|---|
| 0 | 172.2 | 140.6 | 52.1 | 49.8 | 455.0 | 476.6 | - | 469.5 | - |
| CNF | - | - | - | - | 289.9 | 337.5 | 328.5 | - | - |
| 0.5 | 170.6 | 141.1 | 54.7 | 52.6 | 455.7 | 476.1 | - | 467.8 | - |
| 1 | 170.8 | 141.4 | 55.0 | 53.1 | 446 | 472.1 | 341.2 | 456.8 | - |
| 2 | 170.1 | 142.0 | 52.8 | 51.5 | 429.7 | 467.5 | 341.4 | 436.1 | - |
| 3 | 170.4 | 142.6 | 48.2 | 47.5 | 428.5 | 462.4 | 341 | 443.2 | - |
| 4 | 170.9 | 142.5 | 50.4 | 50.2 | 429.5 | 466.9 | 338.5 | 439.4 | - |
| 5 | 170.5 | 142.9 | 54.3 | 54.6 | 425.5 | 468.5 | 341.2 | 435.1 | 476.1 |
| TOCNF | - | - | - | - | 209.1 | 288 | 224.1 | - | - |
| 0.5 | 169.8 | 141.3 | 51.0 | 49.0 | 438.2 | 470.1 | - | 449.7 | - |
| 1 | 169.9 | 141.6 | 50.7 | 49.0 | 427.9 | 466.2 | 331.8 | 436.5 | - |
| 2 | 168.9 | 142.2 | 48.6 | 47.4 | 423.7 | 461.6 | 332.9 | 437.3 | - |
| 3 | 168.6 | 142.5 | 49.5 | 48.8 | 427.1 | 468.3 | 333 | 446.6 | - |
| 4 | 169.4 | 142.5 | 46.2 | 46.0 | 422.9 | 459.7 | 332.8 | 444.6 | - |
| 5 | 169.4 | 142.3 | 49.0 | 49.0 | 419.6 | 457 | 333.4 | 442.7 | - |
| wt% | Tensile Test | Nanoindentation | |||||
|---|---|---|---|---|---|---|---|
| Ultimate Tensile Strength (MPa) | Tensile Modulus (GPa) | Elong. at Break (%) | Yield Stress (MPa) | Tensile Toughness (103 kJ/m3) | Reduced Modulus (GPa) | Hardness (MPa) | |
| 0 | 54.7 ± 5.9 | 1.12 ± 0.03 | 37.7 ± 4.2 | 29.2 ± 1.4 | 17.6 ± 3.1 | 3.20 ± 0.12 | 140.0 ± 9.5 |
| CNF | |||||||
| 0.5 | 43.4 ± 6.8 | 1.22 ± 0.18 | 14.1 ± 1.1 | 21.1 ± 3.2 | 4.5 ± 1.1 | 3.15 ± 0.10 | 155.8 ± 5.8 |
| 1 | 43.5 ± 9.5 | 1.08 ± 0.28 | 11.7 ± 2.9 | 20.4 ± 1.7 | 3.6 ± 1.5 | 3.22 ± 0.42 | 133.0 ± 28 |
| 2 | 41.1 ± 0.5 | 0.75 ± 0.04 | 19.0 ± 7.6 | 21.7 ± 1.9 | 6.5 ± 3.0 | 3.33 ± 0.15 | 153.1 ± 5.5 |
| 3 | 45.6 ± 1.4 | 0.94 ± 0.04 | 10.2 ± 1.0 | 21.6 ± 1.2 | 3.4 ± 0.1 | 3.40 ± 0.31 | 148.9 ± 12.3 |
| 4 | 47.1 ± 3.9 | 1.09 ± 0.01 | 10.8 ± 0.3 | 25.1 ± 0.4 | 3.7 ± 0.3 | 3.25 ± 0.13 | 133.5 ± 26.5 |
| 5 | 42.5 ± 1.6 | 1.05 ± 0.08 | 11.7 ± 2.1 | 21.5 ± 4.8 | 3.4 ± 0.7 | 4.86 ± 0.65 | 171.7 ± 29.2 |
| TOCNF | |||||||
| 0.5 | 47.8 ± 5.4 | 1.13 ± 0.06 | 18.1 ± 5.7 | 21.1 ± 3.1 | 6.6 ± 2.5 | 3.13 ± 0.06 | 150.1 ± 5.6 |
| 1 | 41.5 ± 4.9 | 1.33 ± 0.05 | 12.2 ± 0.9 | 23.8 ± 2.6 | 3.8 ± 0.7 | 3.52 ± 0.14 | 169.6 ± 8.5 |
| 2 | 33.3 ± 0.6 | 0.75 ± 0.06 | 8.1 ± 0.3 | 20.7 ± 0.6 | 1.8 ± 0.1 | 3.78 ± 0.20 | 166.5 ± 9.9 |
| 3 | 24.6 ± 3.8 | 0.65 ± 0.12 | 6.6 ± 0.6 | 17.9 ± 1.2 | 1.0 ± 0.2 | 3.92 ± 0.24 | 174.3 ± 7.4 |
| 4 | 15.8 ± 1.8 | 0.20 ± 0.02 | 5.1 ± 0.6 | 11.4 ± 1.1 | 0.4 ± 0.1 | 4.13 ± 0.56 | 166.9 ± 19.8 |
| 5 | 14.6 ± 2.7 | 0.48 ± 0.04 | 4.6 ± 0.9 | 11.4 ± 0.7 | 0.3 ± 0.2 | 3.11 ± 0.31 | 102.8 ± 20.6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barnes, E.; Jefcoat, J.A.; Alberts, E.M.; McKechnie, M.A.; Peel, H.R.; Buchanan, J.P.; Weiss Jr., C.A.; Klaus, K.L.; Mimun, L.C.; Warner, C.M. Effect of Cellulose Nanofibrils and TEMPO-mediated Oxidized Cellulose Nanofibrils on the Physical and Mechanical Properties of Poly(vinylidene fluoride)/Cellulose Nanofibril Composites. Polymers 2019, 11, 1091. https://doi.org/10.3390/polym11071091
Barnes E, Jefcoat JA, Alberts EM, McKechnie MA, Peel HR, Buchanan JP, Weiss Jr. CA, Klaus KL, Mimun LC, Warner CM. Effect of Cellulose Nanofibrils and TEMPO-mediated Oxidized Cellulose Nanofibrils on the Physical and Mechanical Properties of Poly(vinylidene fluoride)/Cellulose Nanofibril Composites. Polymers. 2019; 11(7):1091. https://doi.org/10.3390/polym11071091
Chicago/Turabian StyleBarnes, Eftihia, Jennifer A. Jefcoat, Erik M. Alberts, Mason A. McKechnie, Hannah R. Peel, J. Paige Buchanan, Charles A. Weiss Jr., Kyle L. Klaus, L. Christopher Mimun, and Christopher M. Warner. 2019. "Effect of Cellulose Nanofibrils and TEMPO-mediated Oxidized Cellulose Nanofibrils on the Physical and Mechanical Properties of Poly(vinylidene fluoride)/Cellulose Nanofibril Composites" Polymers 11, no. 7: 1091. https://doi.org/10.3390/polym11071091