Fabrication and Performance of Composite Microencapsulated Phase Change Materials with Palmitic Acid Ethyl Ester as Core

Abstract
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Microencapsulation Process
2.2.1. Fabrication of SiO2–Polyurea Microcapsules
2.2.2. Preparation of Polyurea–Polyurethane–SiO2 Microcapsules
2.3. Characterization
2.3.1. Fourier Transform Infrared Spectroscopy (FT-IR)
2.3.2. Field Emission-Scanning Electron Microscopy (FE-SEM)
2.3.3. The Phase Change Properties Analysis
2.3.4. Thermal Gravimetric Analysis (TGA)
2.4. Compactness Test
3. Results and Discussion
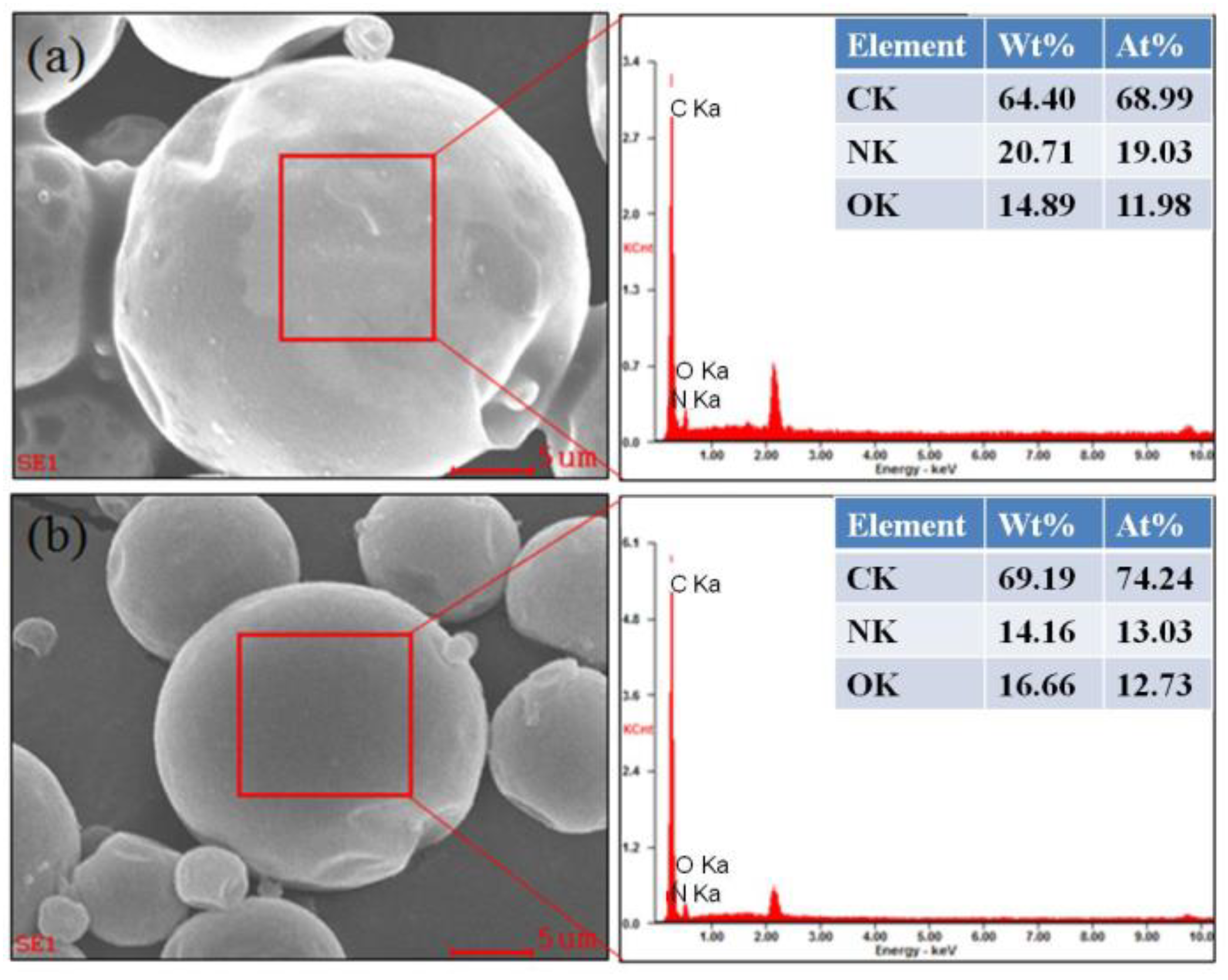
3.1. Morphology and Inner Microstructure of Various MicroPCMs
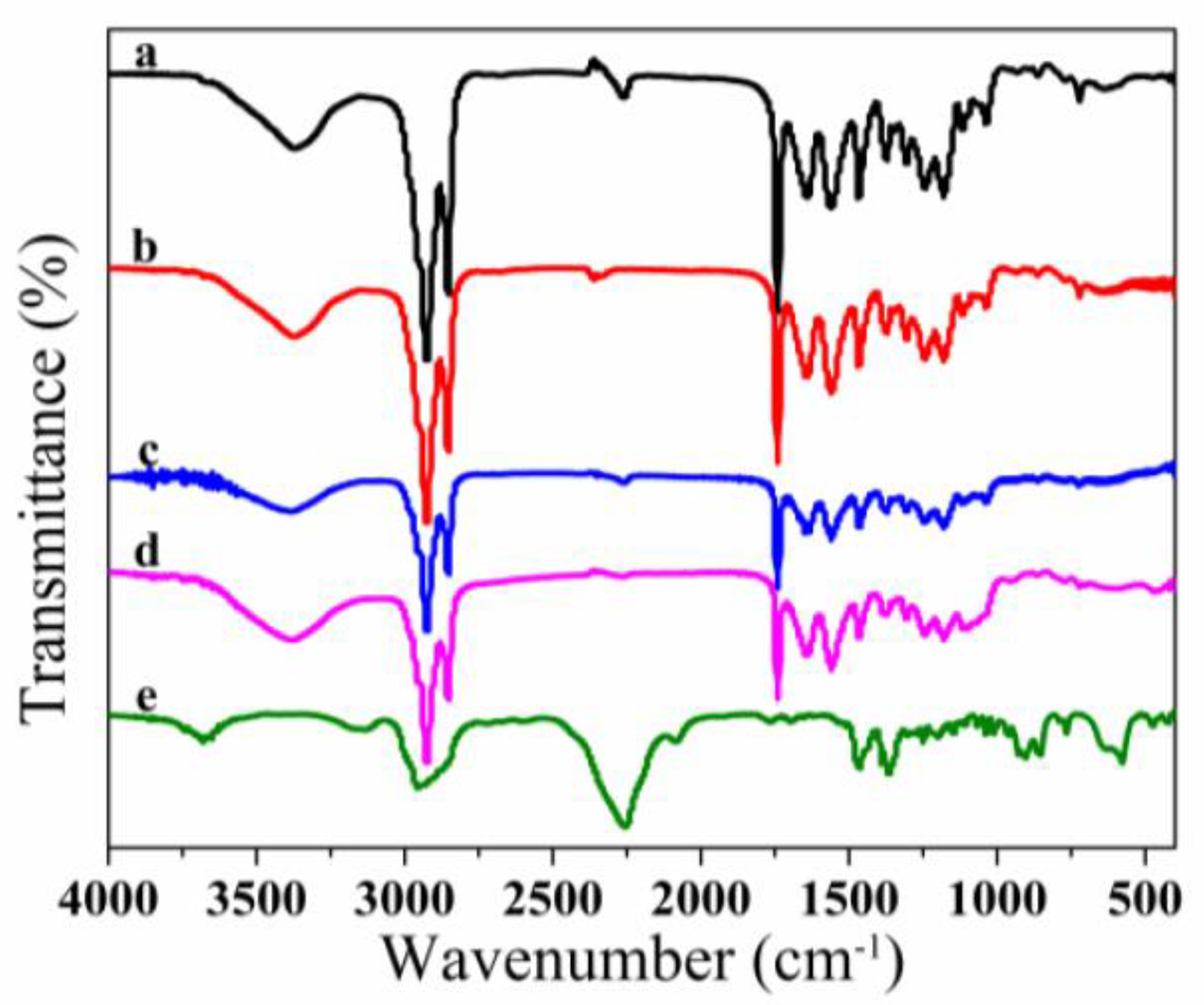
3.2. FT-IR Spectroscopy Analysis of Microcapsules
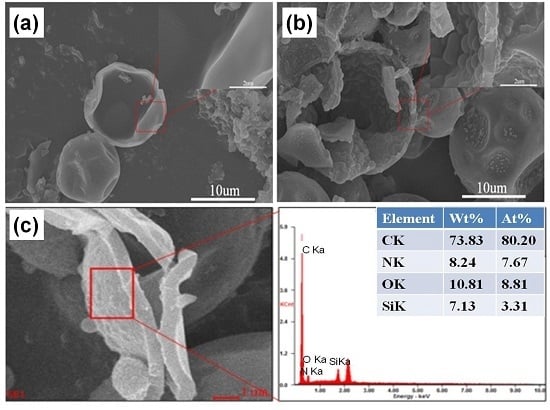
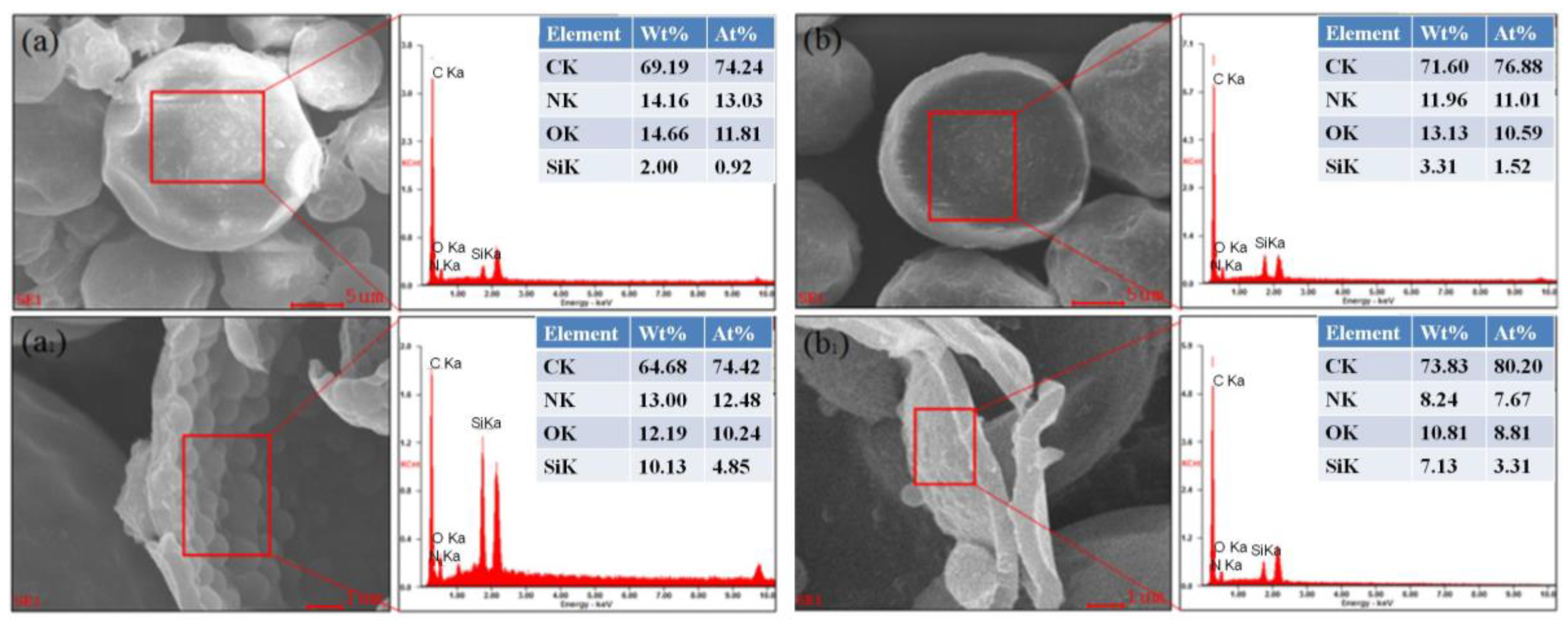
3.3. EDS Energy Spectra Analysis
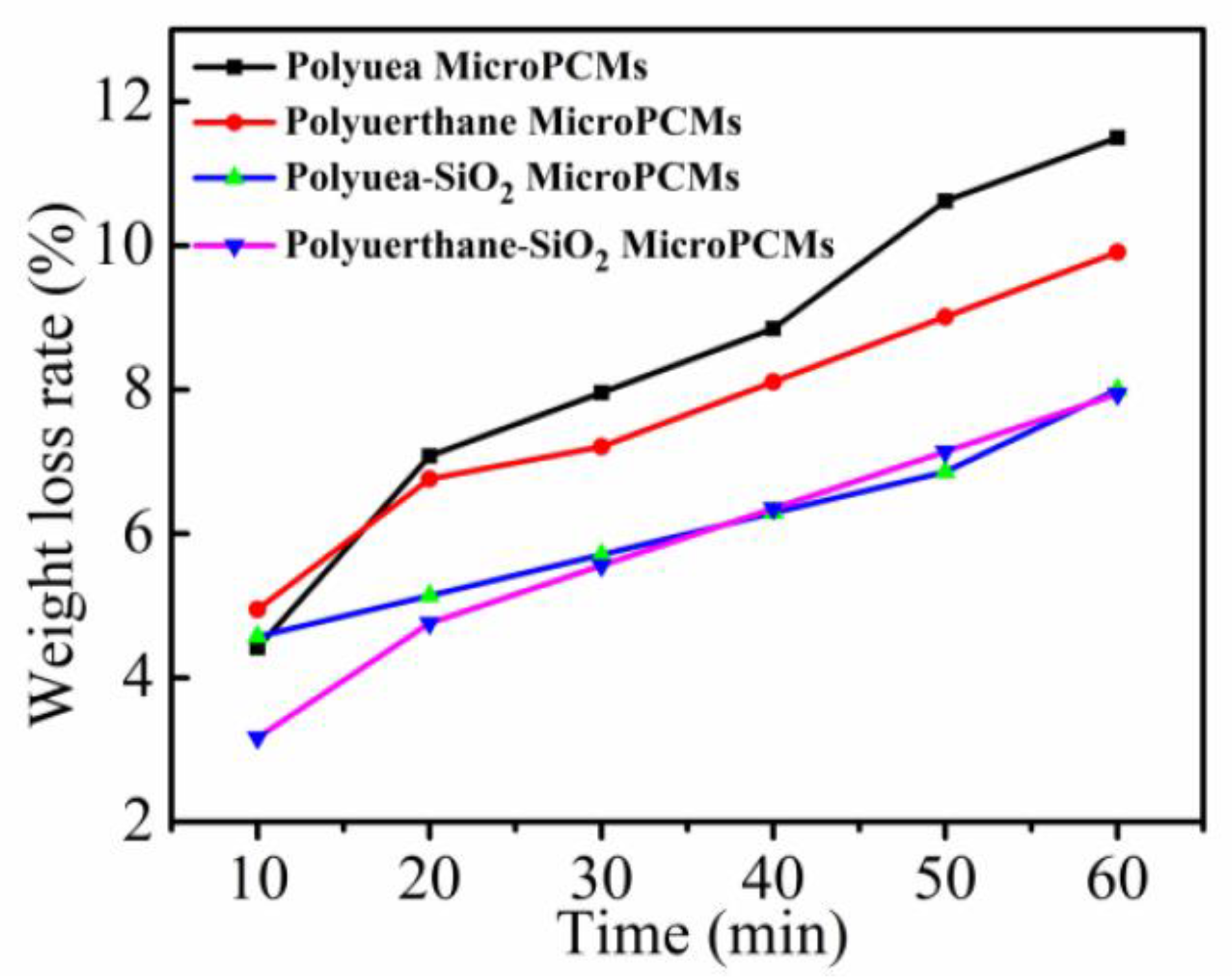
3.4. Compactness Analysis
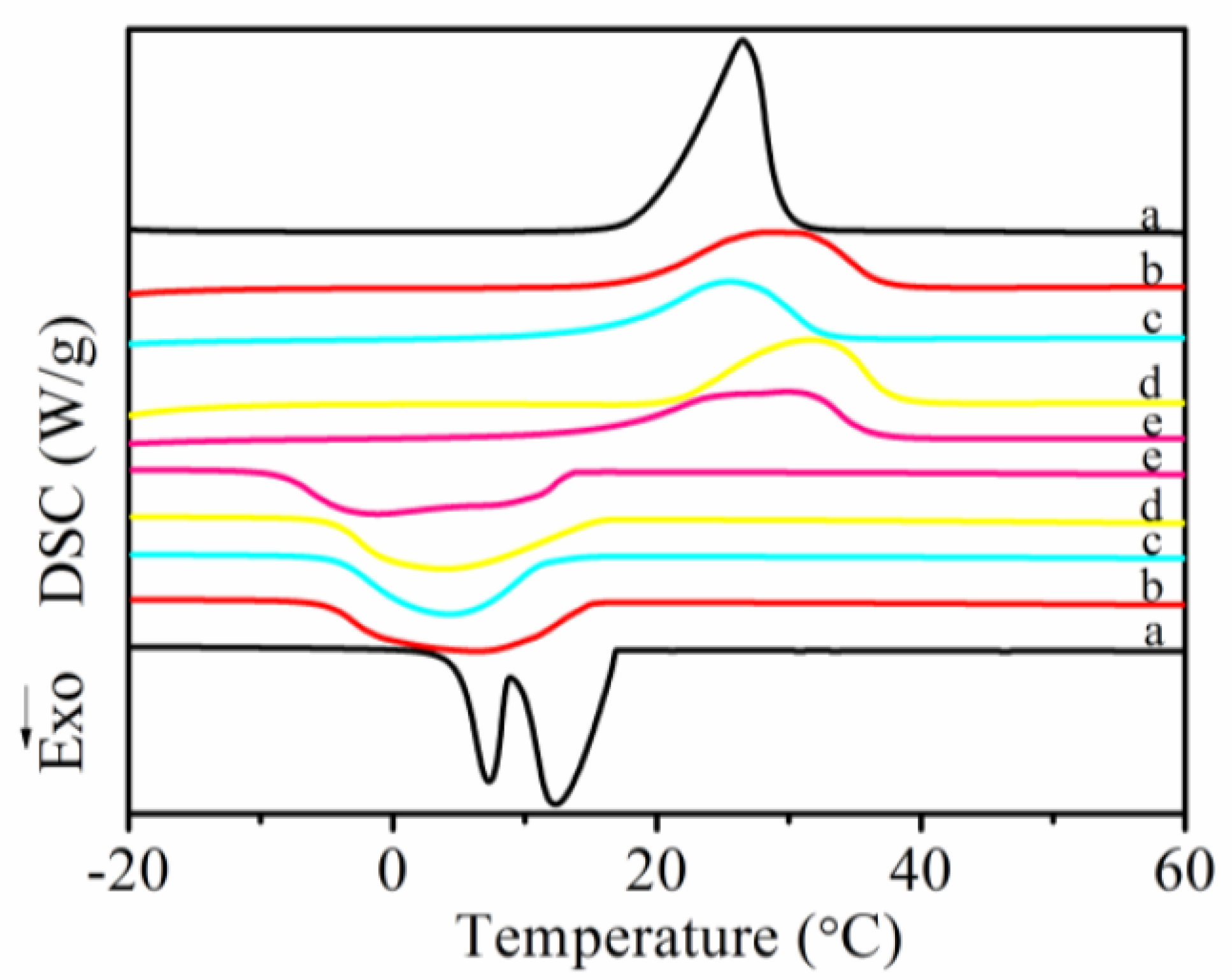
3.5. Melting and Crystallization Behaviors of MicroPCMs
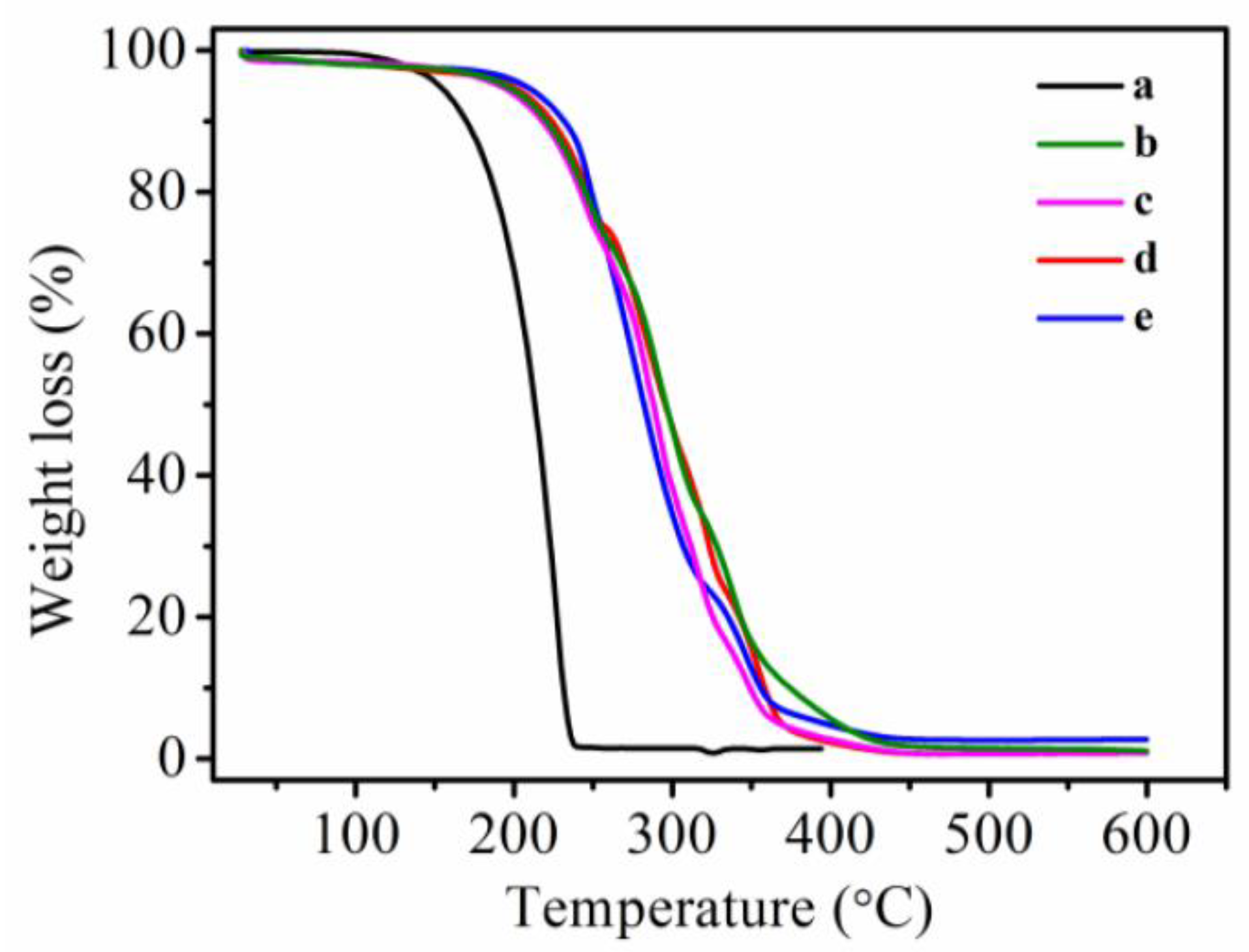
3.6. Thermal Stability of MicroPCMs
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Anghel, E.M.; Georgiev, A.; Petrescu, S.; Popov, R.; Constantinescu, M. Thermo-physical characterization of some paraffins used as phase change materials for thermal energy storage. J. Therm. Anal. Calorim. 2014, 117, 557–566. [Google Scholar] [CrossRef]
- Milian, Y.E.; Gutierrez, A.; Grageda, M.; Ushak, S. A review on encapsulation techniques for inorganic phase change materials and the influence on their thermophysical properties. Renew. Sustain. Energy Rev. 2017, 73, 983–999. [Google Scholar] [CrossRef]
- Chang, S.J.; Wi, S.; Jeong, S.G.; Kim, S. Analysis on phase transition range of the pure and mixed phase change materials (PCM) using a thermostatic chamber test and differentiation. J. Therm. Anal. Calorim. 2017, 131, 1999–2004. [Google Scholar] [CrossRef]
- Motahar, S.; Alemrajabi, A.A.; Khodabandeh, R. Experimental investigation on heat transfer characteristics during melting of a phase change material with dispersed TiO2 nanoparticles in a rectangular enclosure. Int. J. Heat Mass Transf. 2017, 109, 134–146. [Google Scholar]
- Geng, X.Y.; Li, W.; Wang, Y.; Lu, X.W.; Wang, J.P.; Wang, N.; Li, J.J.; Zhang, X.X. Reversible thermochromic microencapsulated phase change materials for thermal energy storage application in thermal protective clothing. Appl. Energy 2018, 217, 281–294. [Google Scholar] [CrossRef]
- Raghunanan, L.; Floros, M.C.; Narine, S.S. Thermal stability of renewable diesters as phase change materials. Thermochim. Acta 2016, 644, 61–68. [Google Scholar] [CrossRef]
- Guo, M.; Li, W.; Han, N.; Wang, J.; Su, J.; Li, J.; Zhang, X. Novel dual-component microencapsulated hydrophobic amine and microencapsulated isocyanate used for self-healing anti-corrosion coating. Polymers 2018, 10, 319. [Google Scholar] [CrossRef]
- Zhang, H.; Shi, Y.; Shentu, B.; Weng, Z. Synthesis and thermal performance of polyurea microcapsulated phase change materials by interfacial polymerization. Polym. Sci. Ser. B 2017, 59, 689–696. [Google Scholar] [CrossRef]
- Fredi, G.; Dorigato, A.; Fambri, L.; Pegoretti, A. Wax confinement with carbon nanotubes for phase changing epoxy blends. Polymers 2017, 9, 405. [Google Scholar] [CrossRef]
- Fredi, G.; Dorigato, A.; Fambri, L.; Pegoretti, A. Multifunctional epoxy/carbon fiber laminates for thermal energy storage and release. Compos. Sci. Technol. 2018, 158, 101–111. [Google Scholar] [CrossRef]
- Li, M.; Liu, J.P.; Shi, J.B. Synthesis and properties of phase change microcapsule with SiO2-TiO2 hybrid shell. Sol. Energy 2018, 167, 158–164. [Google Scholar] [CrossRef]
- Lian, Q.; Li, Y.; Sayyed, A.A.S.; Cheng, J.; Zhang, J.Y. Facile strategy in designing epoxy/paraffin multiple phase change materials for thermal energy storage applications. ACS Sustain. Chem. Eng. 2018, 6, 3375–3384. [Google Scholar] [CrossRef]
- Esapour, M.; Hosseini, M.J.; Ranjbar, A.A.; Pahamli, Y.; Bahrampoury, R. Phase change in multi-tube heat exchangers. Renew. Energy 2016, 85, 1017–1025. [Google Scholar] [CrossRef]
- Michaelides, E.E. A new model for the lifetime of fossil fuel resources. Nat. Resour. Res. 2016, 26, 161–175. [Google Scholar] [CrossRef]
- Khan, Z.; Khan, Z.; Ghafoor, A. A review of performance enhancement of PCM based latent heat storage system within the context of materials, thermal stability and compatibility. Energy Convers. Manag. 2016, 115, 132–158. [Google Scholar] [CrossRef]
- Abdollahzadeh, M.; Esmaeilpour, M. Enhancement of phase change material (PCM) based latent heat storage system with nano fluid and wavy surface. Int. J. Heat Mass Transf. 2015, 80, 376–385. [Google Scholar] [CrossRef]
- Pasupathy, A.; Velraj, R.; Seeniraj, R.V. Phase change material-based building architecture for thermal management in residential and commercial establishments. Renew. Sustain. Energy Rev. 2008, 12, 39–64. [Google Scholar] [CrossRef]
- Goeke, J.; Henne, A. Time-temperature charge function of a high dynamic thermal heat storage with phase change material. Energy Power Eng. 2015, 7, 41–54. [Google Scholar] [CrossRef]
- Luo, J.; Zhao, L.; Yang, Y.; Song, G.; Liu, Y.; Chen, L.; Tang, G. Emulsifying ability and cross-linking of silk fibroin microcapsules containing phase change materials. Sol. Energy Mater. Sol. Cells 2016, 147, 144–149. [Google Scholar] [CrossRef]
- Sun, N.; Xiao, Z. Paraffin wax-based phase change microencapsulation embedded with silicon nitride nanoparticles for thermal energy storage. J. Mater. Sci. 2016, 51, 8550–8561. [Google Scholar] [CrossRef]
- Wei, H.; Zhao, Z.; Wei, C.; Yu, G.; Liu, Z.; Zhang, B.; Bian, J.; Bian, Z.; Huang, C. Antiphotobleaching: A type of structurally rigid chromophore ready for constructing highly luminescent and highly photostable europium complexes. Adv. Funct. Mater. 2016, 26, 2085–2096. [Google Scholar] [CrossRef]
- Tillet, G.; Boutevin, B.; Ameduri, B. Chemical reactions of polymer crosslinking and post-crosslinking at room and medium temperature. Prog. Polym. Sci. 2011, 36, 191–217. [Google Scholar] [CrossRef]
- Zhu, Y.; Qin, Y.; Wei, C.; Liang, S.; Luo, X.; Wang, J.; Zhang, L. Nanoencapsulated phase change materials with polymer-SiO2 hybrid shell materials: Compositions, morphologies, and properties. Energy Convers. Manag. 2018, 164, 83–92. [Google Scholar] [CrossRef]
- Lee, S.M.; Park, J.Y.; Park, S.B. Comparative study of the storage stability between a melamine-urea-formaldehyde and a urea-formaldehyde resin. For. Prod. J. 2012, 62, 146–149. [Google Scholar] [CrossRef]
- Despres, A.; Pizzi, A. Colloidal aggregation of aminoplasticpolycondensation resins: Urea-formaldehyde versus melamine-formaldehyde and melamine-urea-formaldehyde resins. J. Appl. Polym. Sci. 2006, 100, 1406–1412. [Google Scholar] [CrossRef]
- Calle, M.; Lligadas, G.; Ronda, J.C.; Galia, M.; Cadiz, V. Non-isocyanate route to biobased polyurethanes and polyureas via AB-type self-polycondensation. Eur. Polym. J. 2016, 84, 837–848. [Google Scholar] [CrossRef]
- Zhang, F.; Jiang, X.; Zhu, X.; Chen, Z.; Kong, X.Z. Preparation of uniform and porous polyurea microspheres of large size through interfacial polymerization of toluene diisocyanate in water solution of ethylene diamine. Chem. Eng. J. 2016, 303, 48–55. [Google Scholar] [CrossRef]
- Tatiya, P.D.; Hedaoo, R.K.; Mahulikar, P.P.; Gite, V.V. Novel polyurea microcapsules using dendritic functional monomer: Synthesis, characterization, and its use in self-healing and anticorrosive polyurethane coatings. Ind. Eng. Chem. Res. 2013, 52, 1562–1570. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Tmoa (°C) | Tmpb (°C) | Tmec (°C) | ΔHm (J·g−1) | Tcod (°C) | Tcpe (°C) | Tcef (°C) | ΔHc (J·g−1) | ΔHi g (J·g−1) | PCM content h (wt%) |
|---|---|---|---|---|---|---|---|---|---|---|
| a | 19.6 | 26.5 | 29.3 | 203.9 | 16.9 | 12.3 | 8.8 | 202.7 | 203.3 | — |
| b | 18.7 | 28.5 | 36.9 | 123.4 | 15.0 | 6.2 | −5.0 | 128.6 | 126.0 | 62.0 |
| c | 16.9 | 25.5 | 32.4 | 110.5 | 11.5 | 4.2 | −4.1 | 113.7 | 112.1 | 55.1 |
| d | 21.9 | 31.6 | 37.5 | 120.1 | 15.3 | 3.7 | −4.4 | 121.0 | 120.5 | 59.3 |
| e | 16.5 | 29.8 | 35.8 | 122.1 | 13.3 | −1.2 | −8.3 | 126.1 | 124.1 | 61.0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, Q.; Zhu, Z.; Li, W.; Guo, M.; Wang, Y.; Wang, J.; Zhang, X. Fabrication and Performance of Composite Microencapsulated Phase Change Materials with Palmitic Acid Ethyl Ester as Core. Polymers 2018, 10, 726. https://doi.org/10.3390/polym10070726
Yin Q, Zhu Z, Li W, Guo M, Wang Y, Wang J, Zhang X. Fabrication and Performance of Composite Microencapsulated Phase Change Materials with Palmitic Acid Ethyl Ester as Core. Polymers. 2018; 10(7):726. https://doi.org/10.3390/polym10070726
Chicago/Turabian StyleYin, Qing, Zhenguo Zhu, Wei Li, Maolian Guo, Yu Wang, Jianping Wang, and Xingxiang Zhang. 2018. "Fabrication and Performance of Composite Microencapsulated Phase Change Materials with Palmitic Acid Ethyl Ester as Core" Polymers 10, no. 7: 726. https://doi.org/10.3390/polym10070726
APA StyleYin, Q., Zhu, Z., Li, W., Guo, M., Wang, Y., Wang, J., & Zhang, X. (2018). Fabrication and Performance of Composite Microencapsulated Phase Change Materials with Palmitic Acid Ethyl Ester as Core. Polymers, 10(7), 726. https://doi.org/10.3390/polym10070726