Exploring the Long-Term Hydrolytic Behavior of Zwitterionic Polymethacrylates and Polymethacrylamides


Abstract
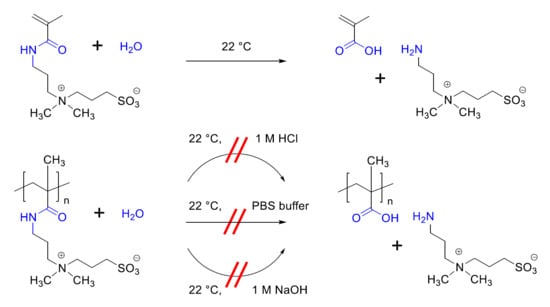
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthetic Methods and Procedures
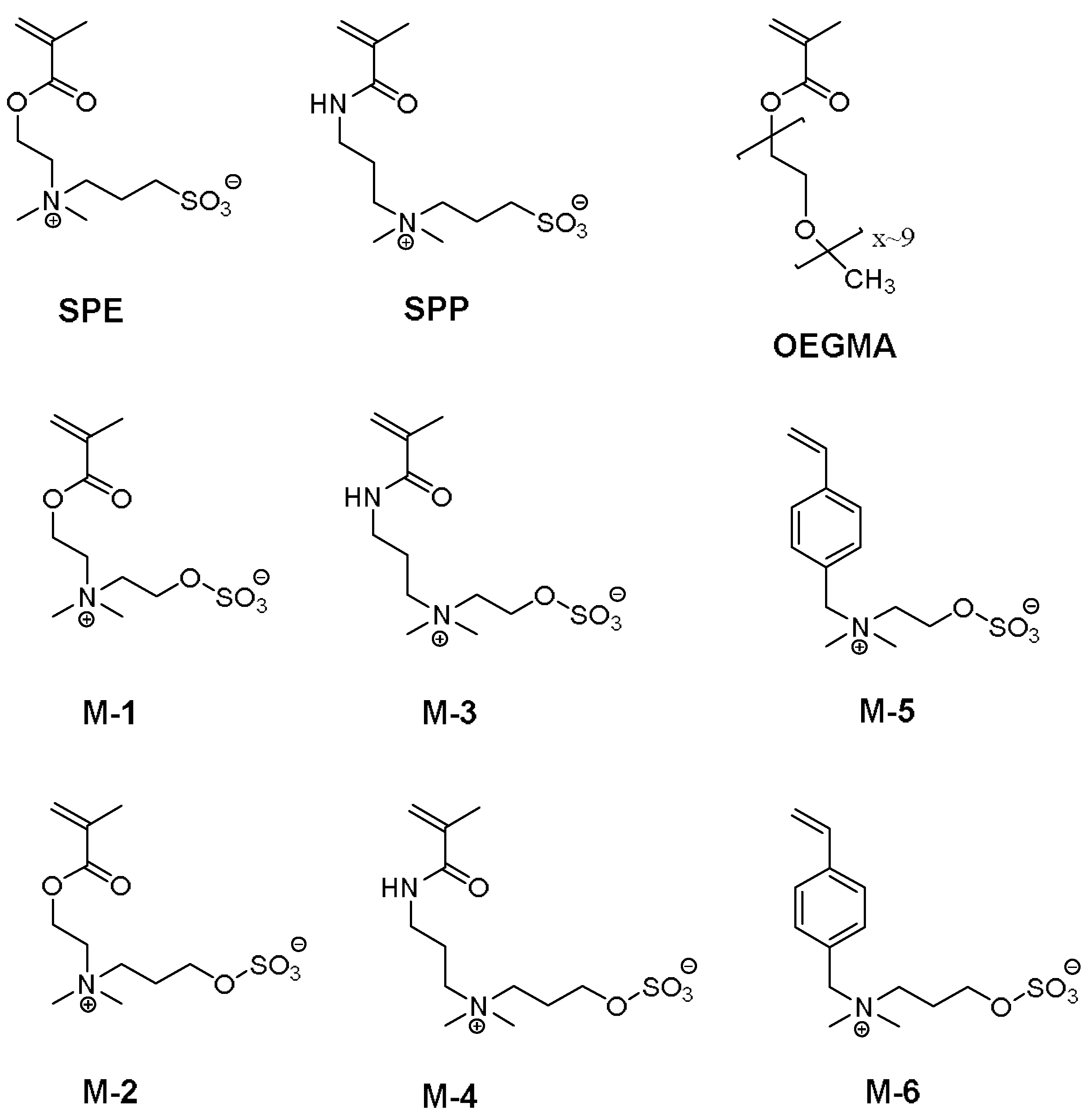
2.2.1. Synthesis of Sulfabetaine Monomers
2.2.2. Polymer Synthesis
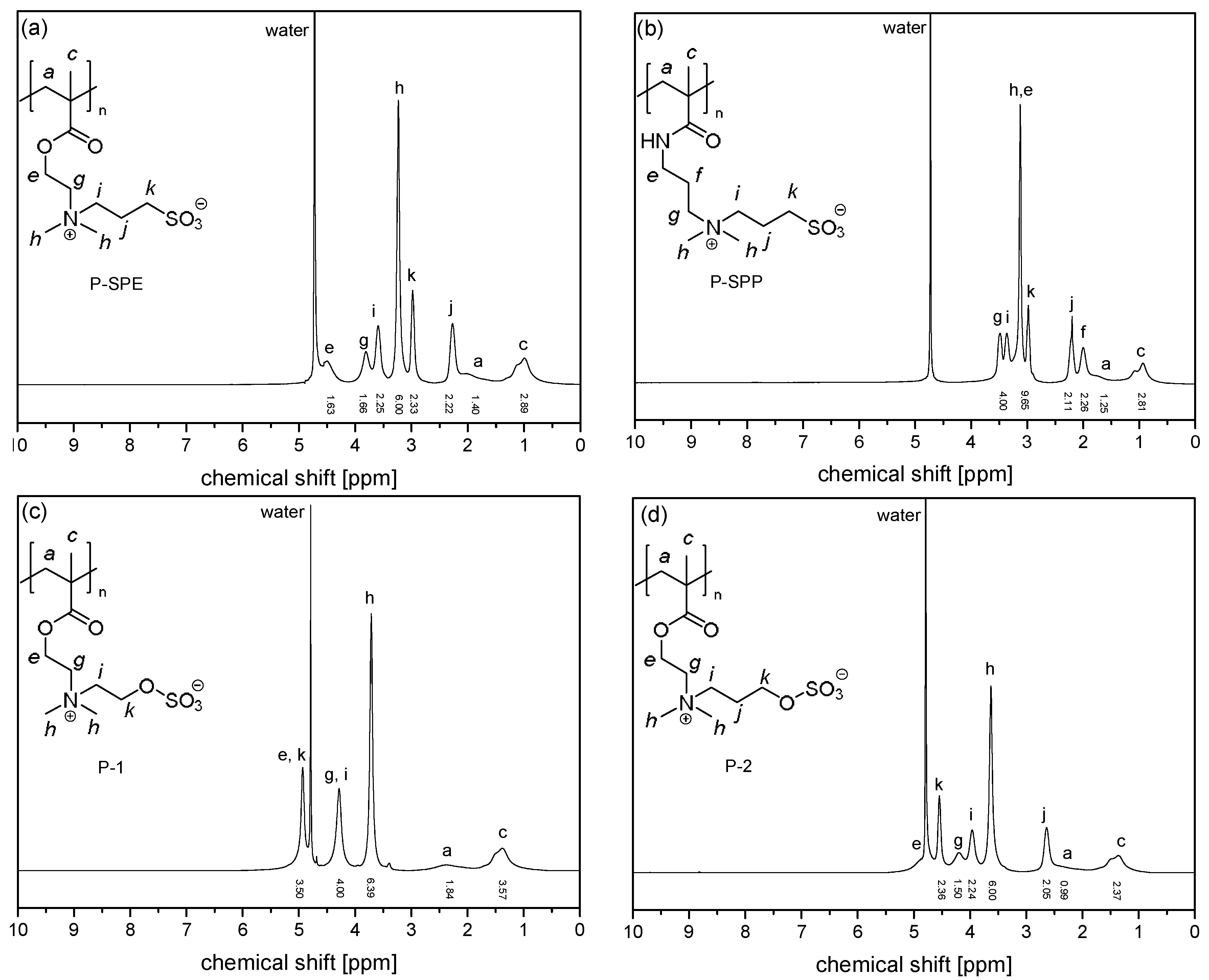
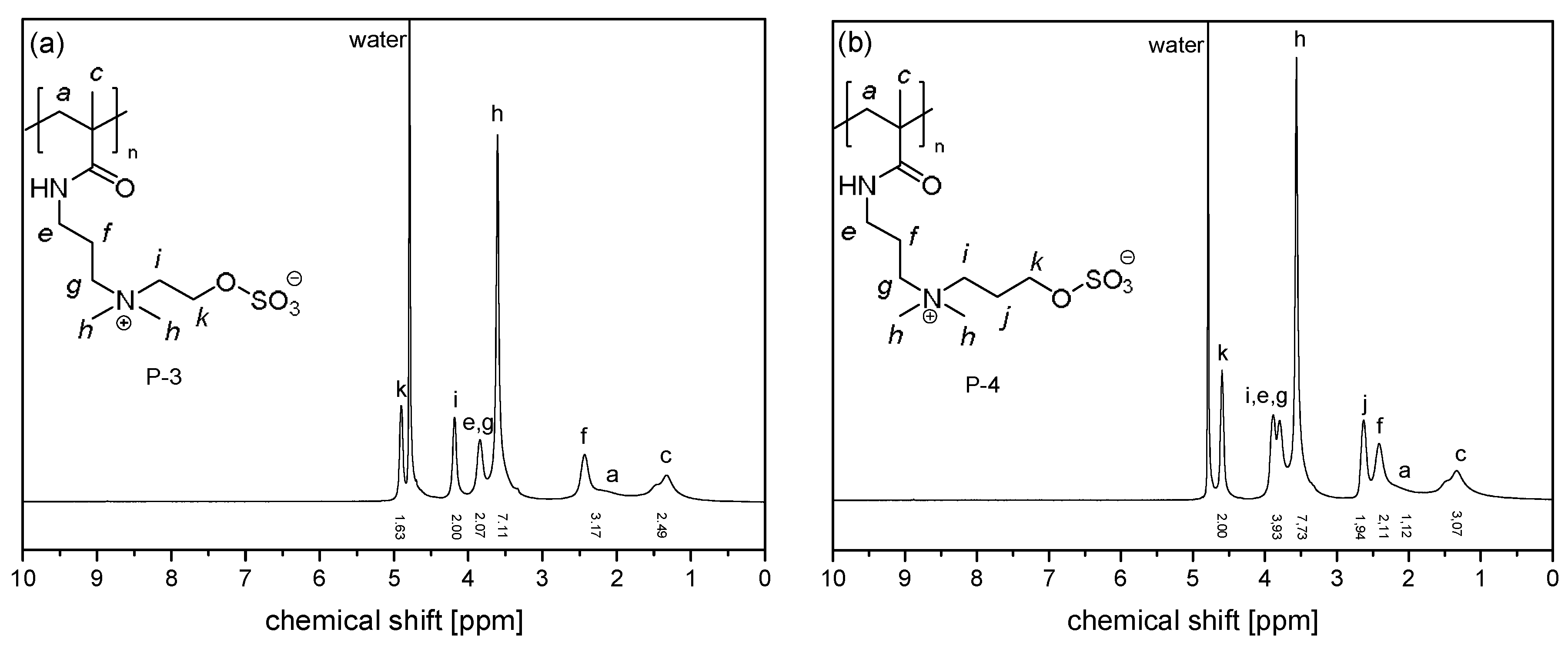
2.3. Instrumentation and Methods
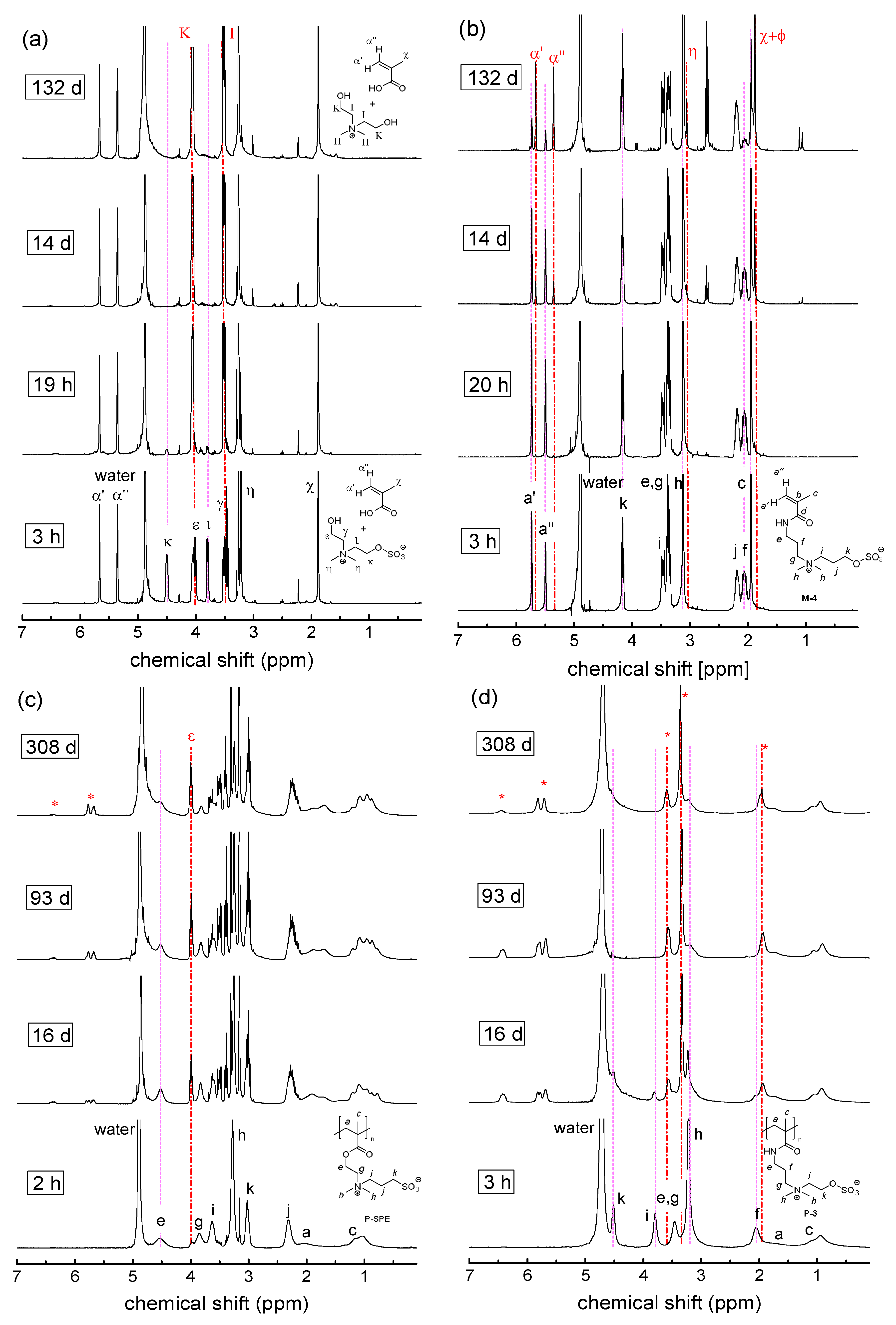
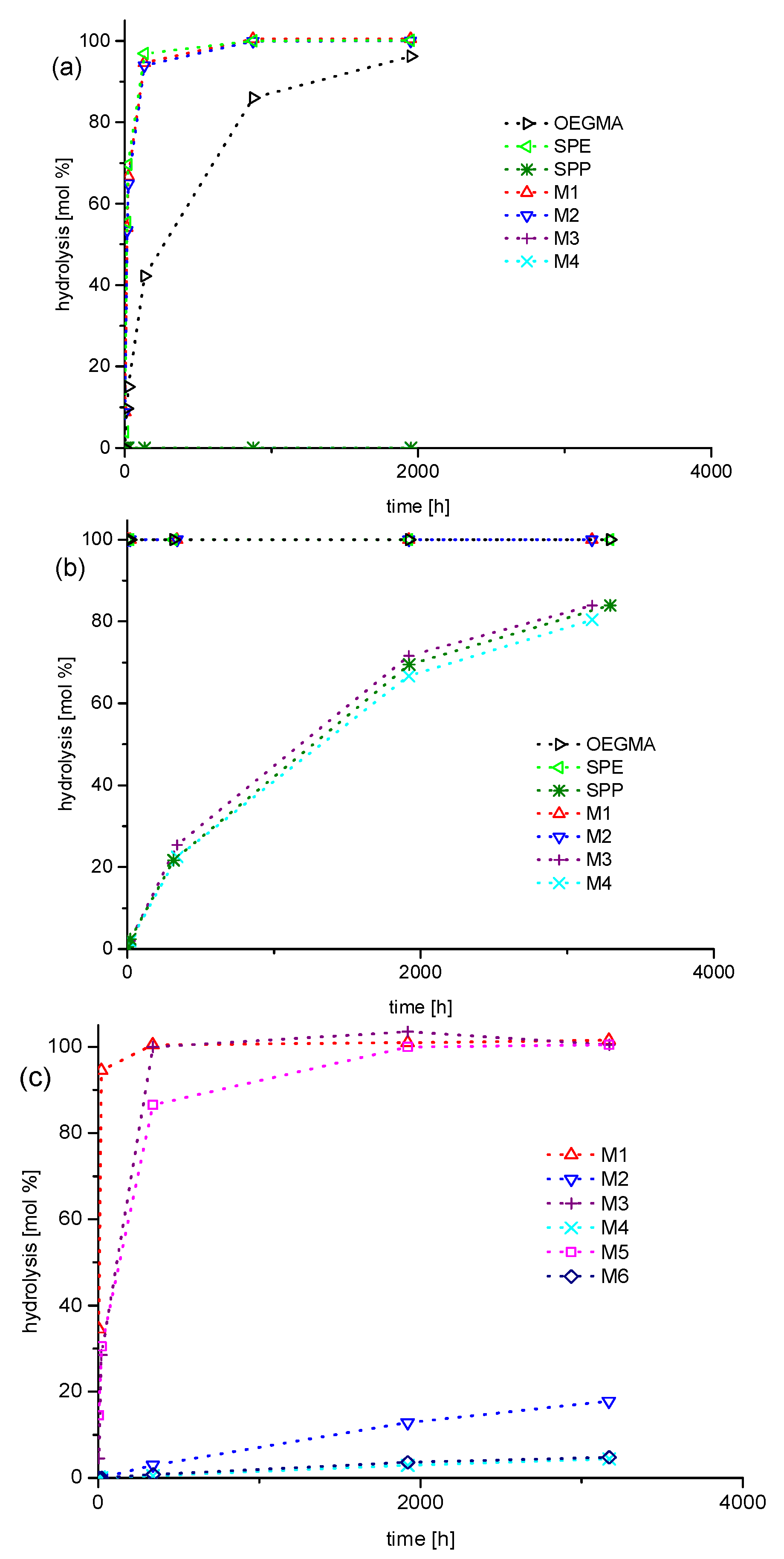
3. Results
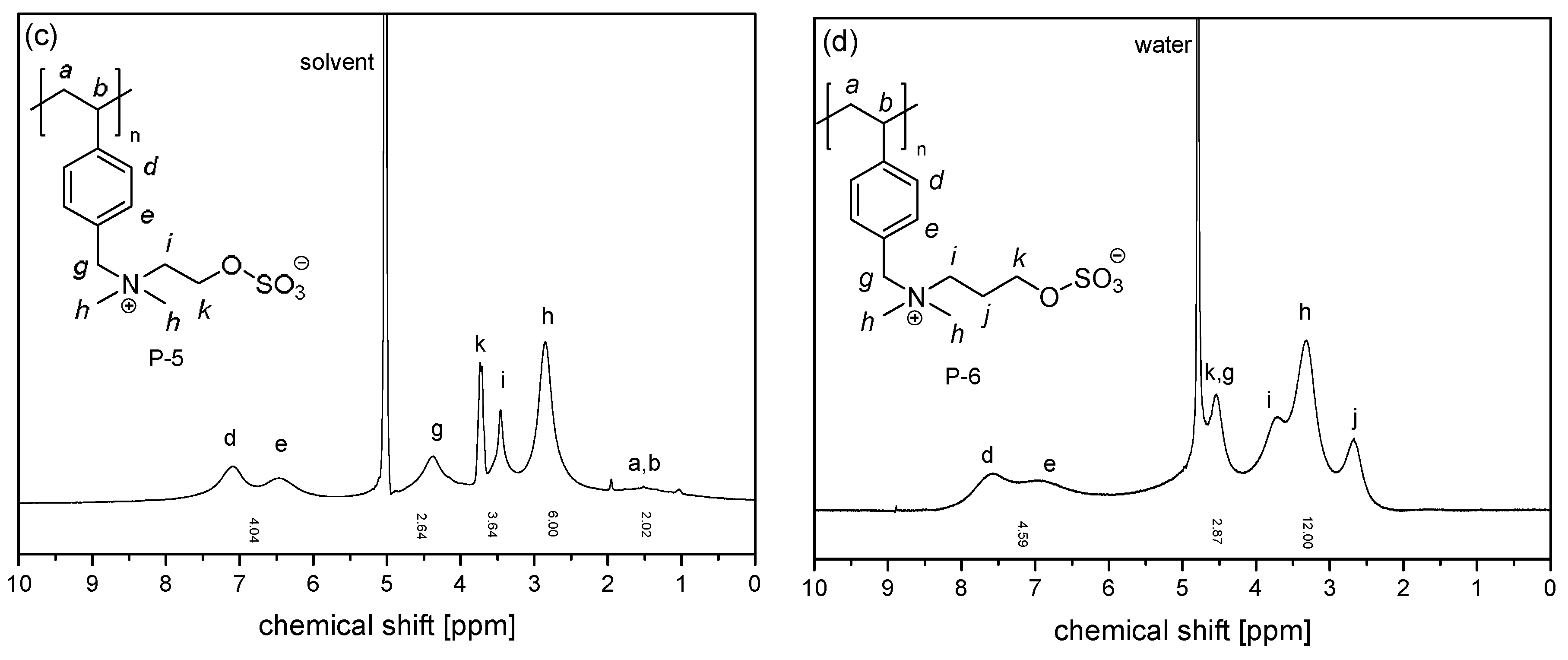
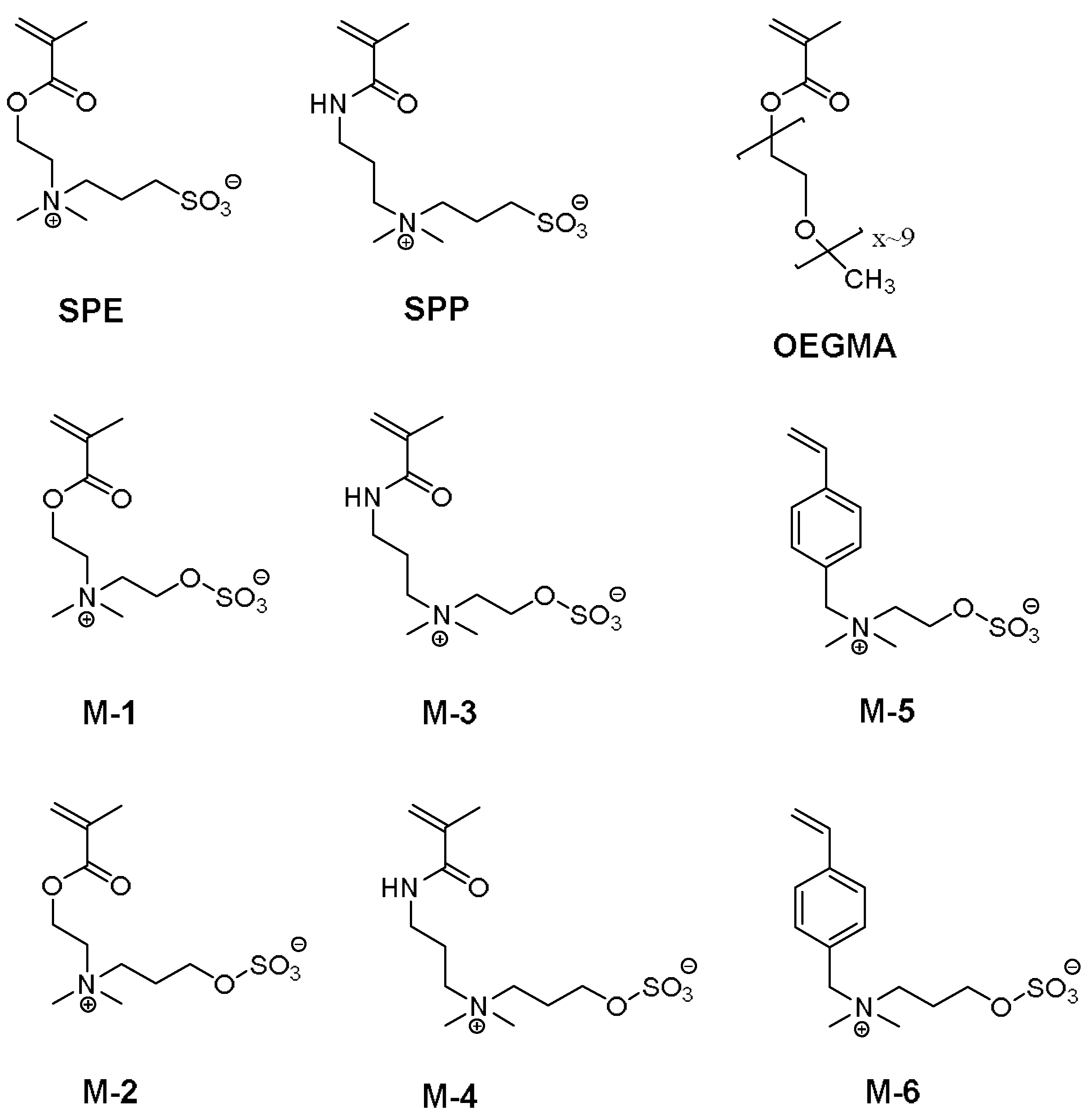
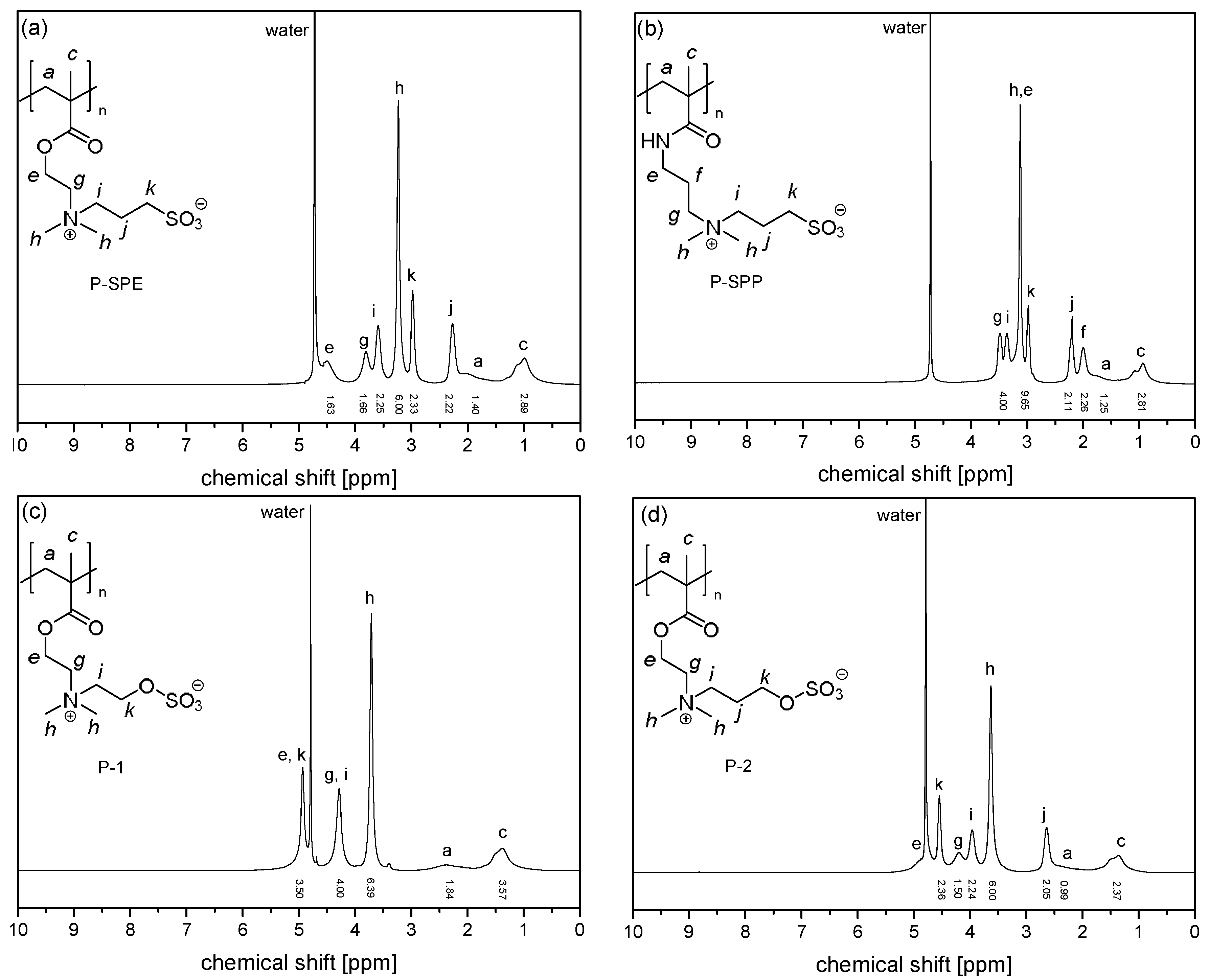
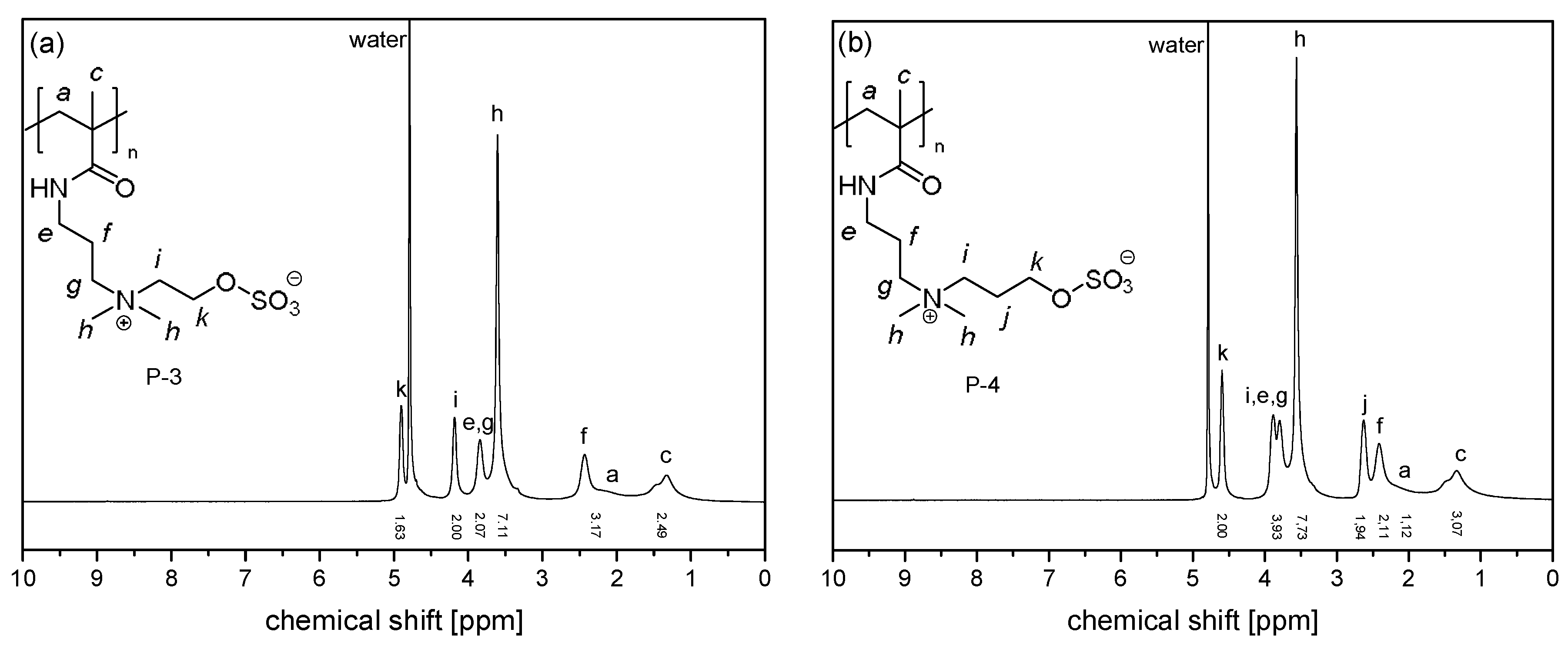
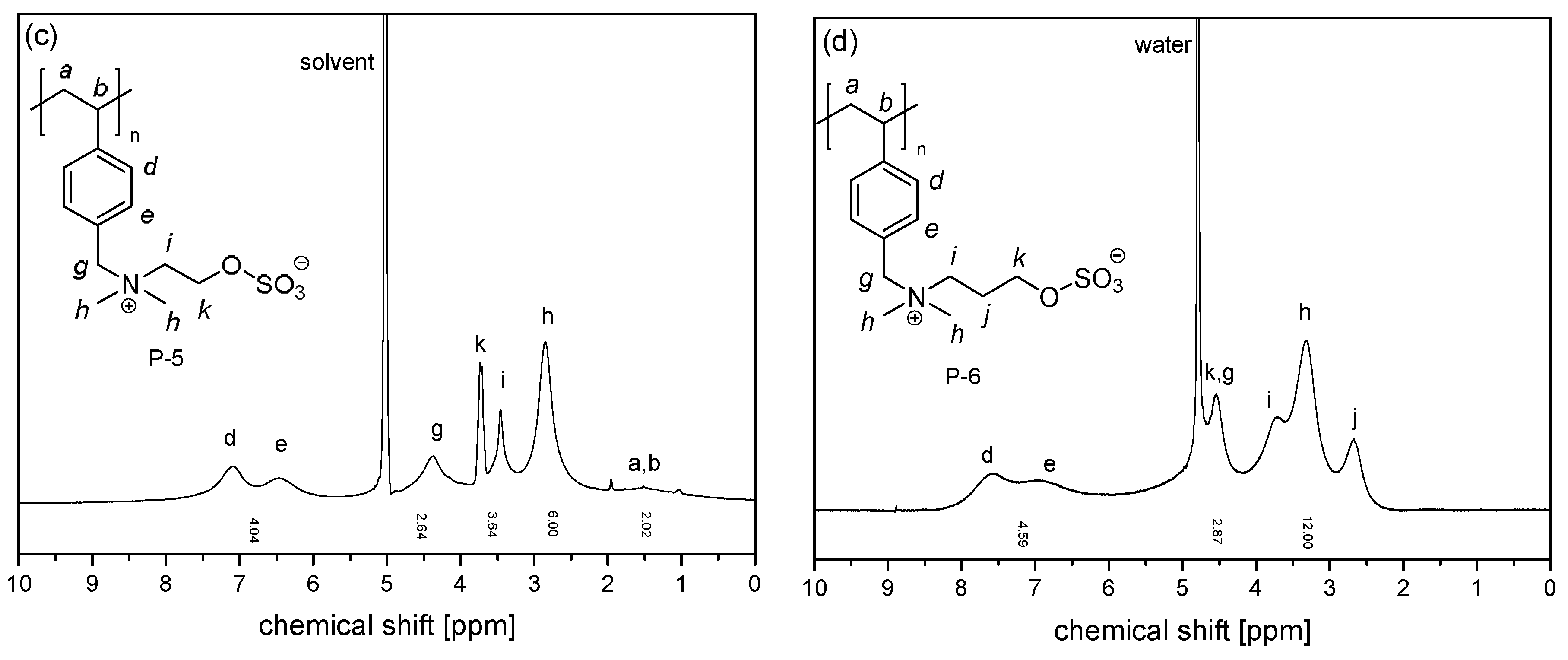
3.1. Synthesis of the Monomer and Polymers, and Their General Aqueous Solution Behavior
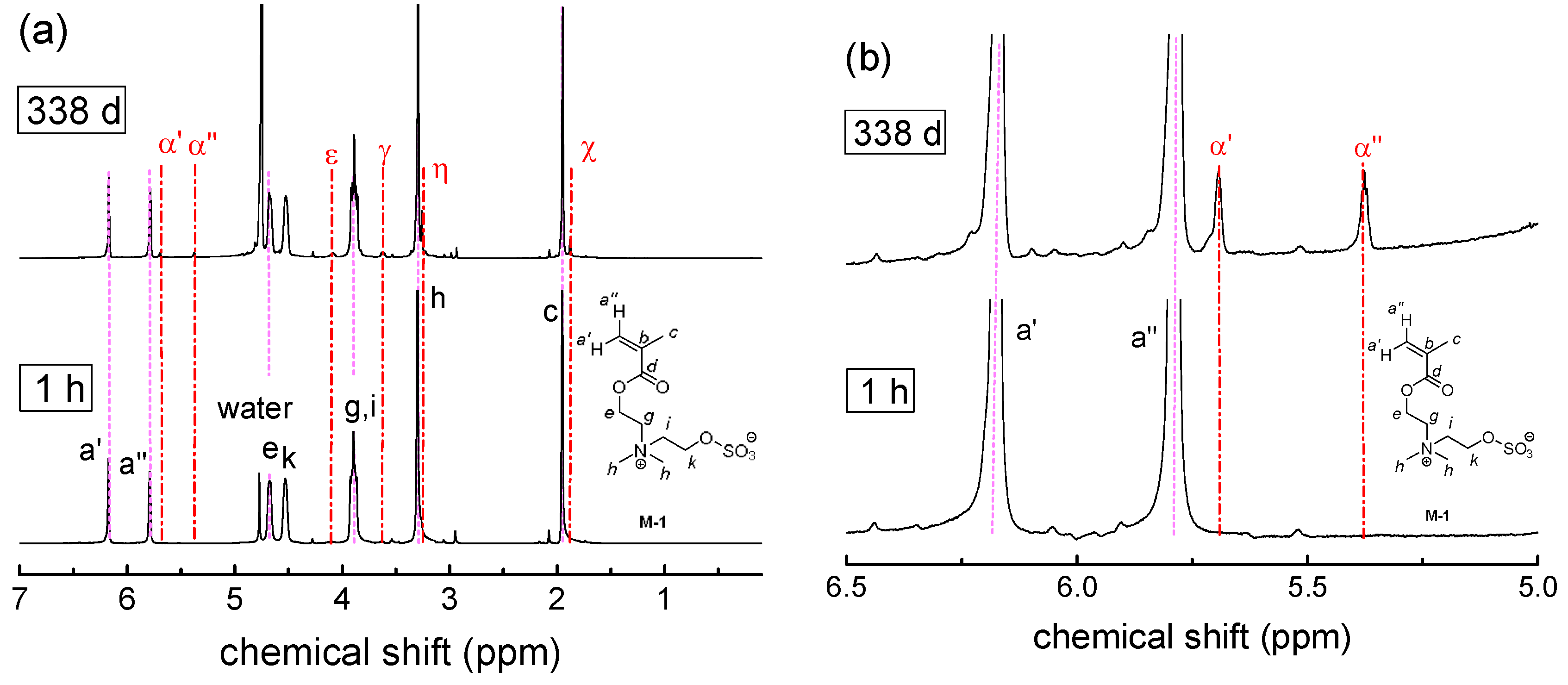
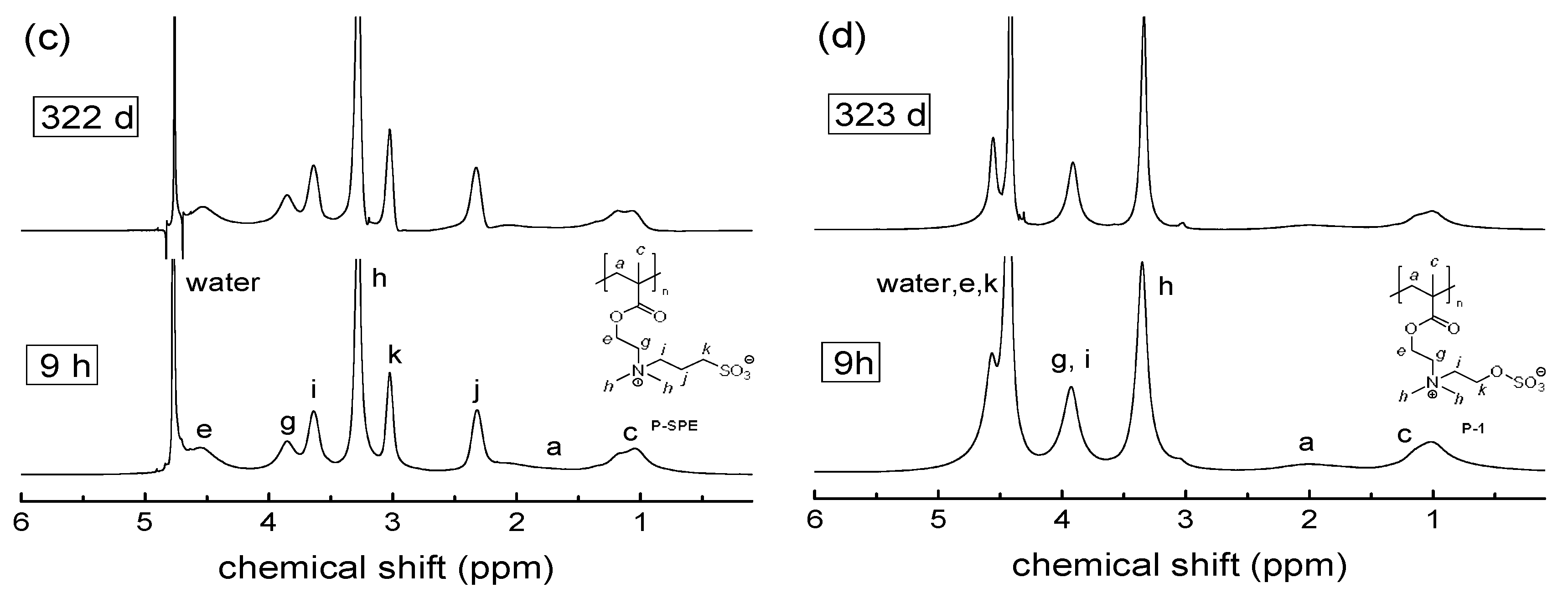
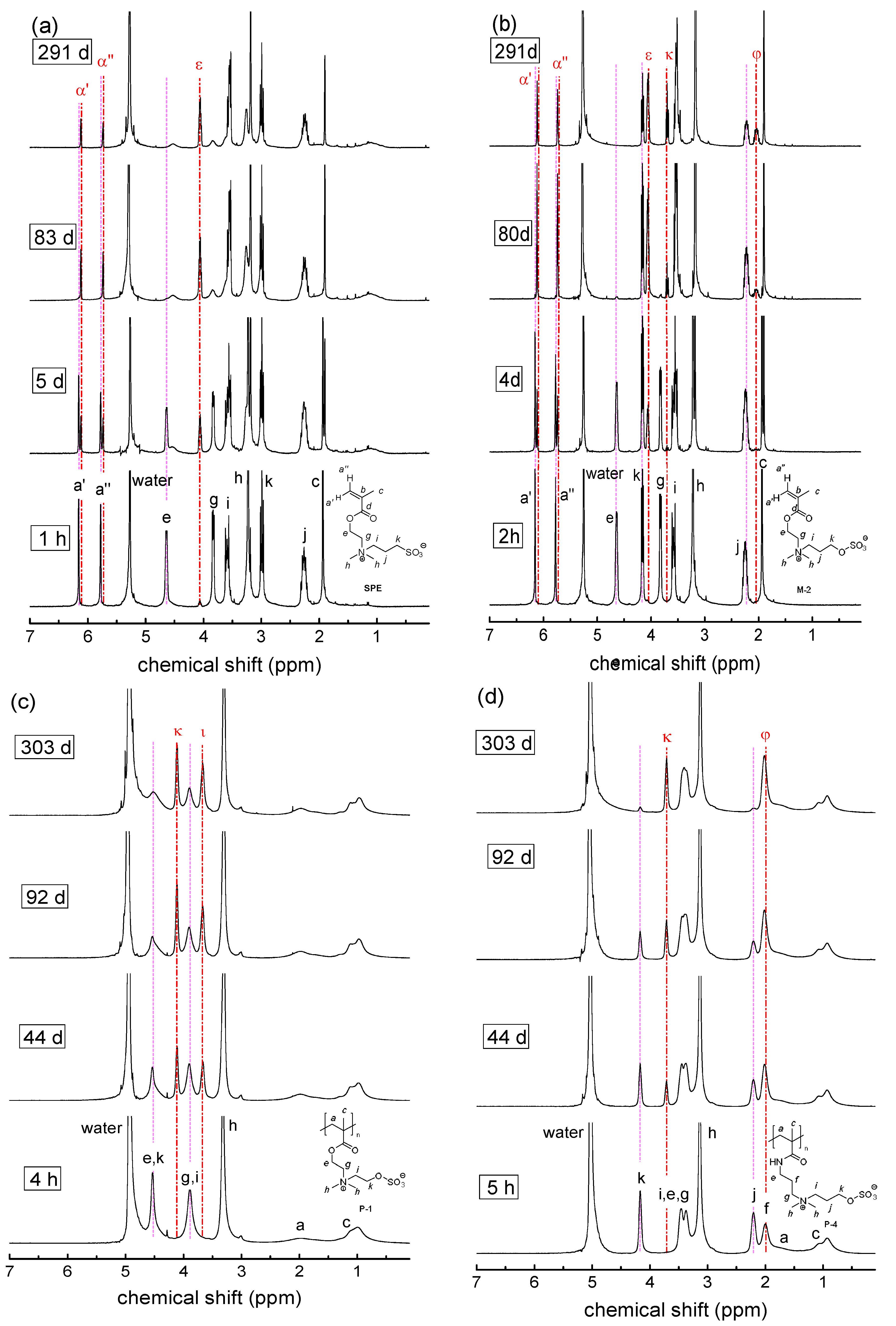
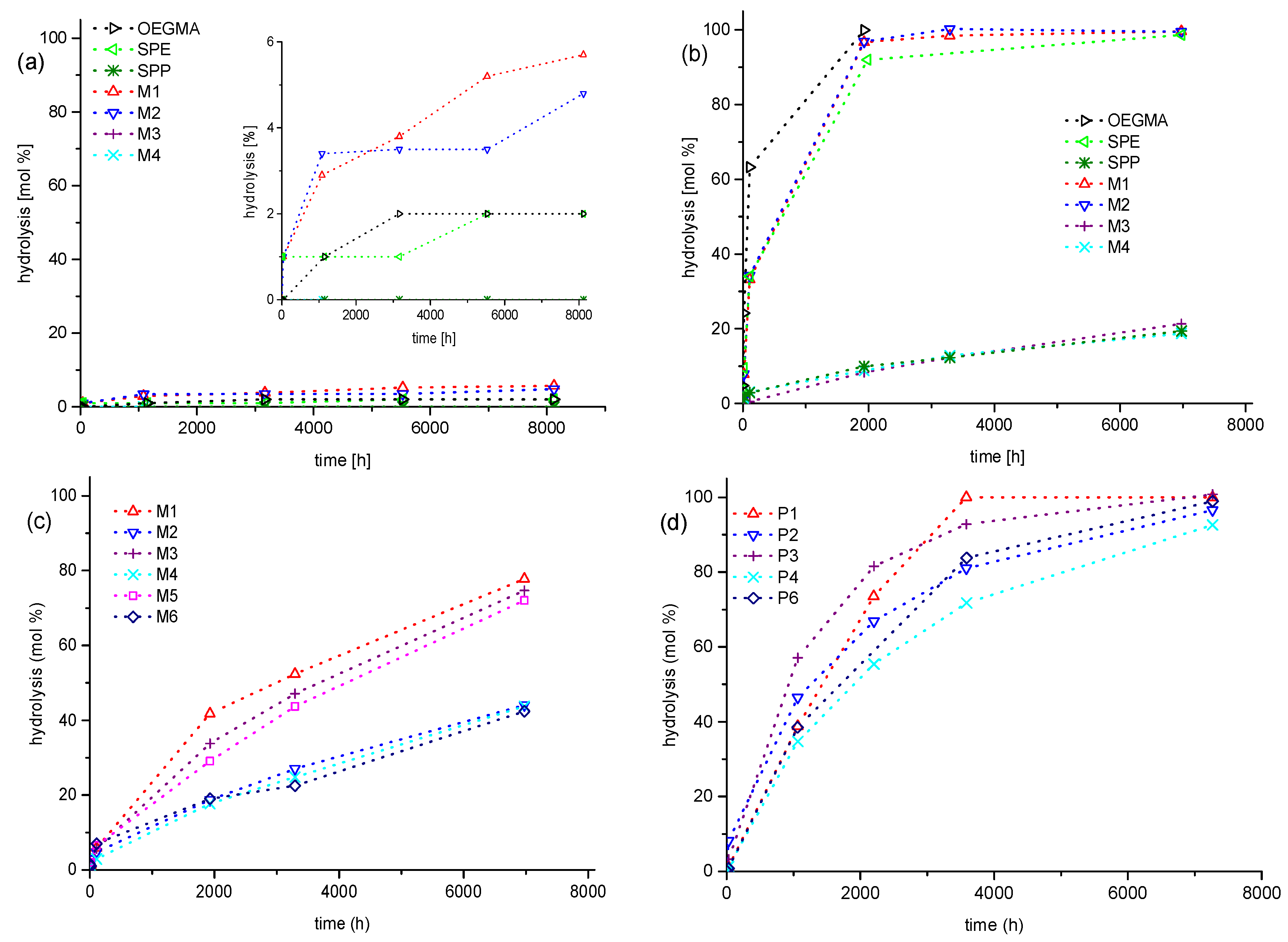
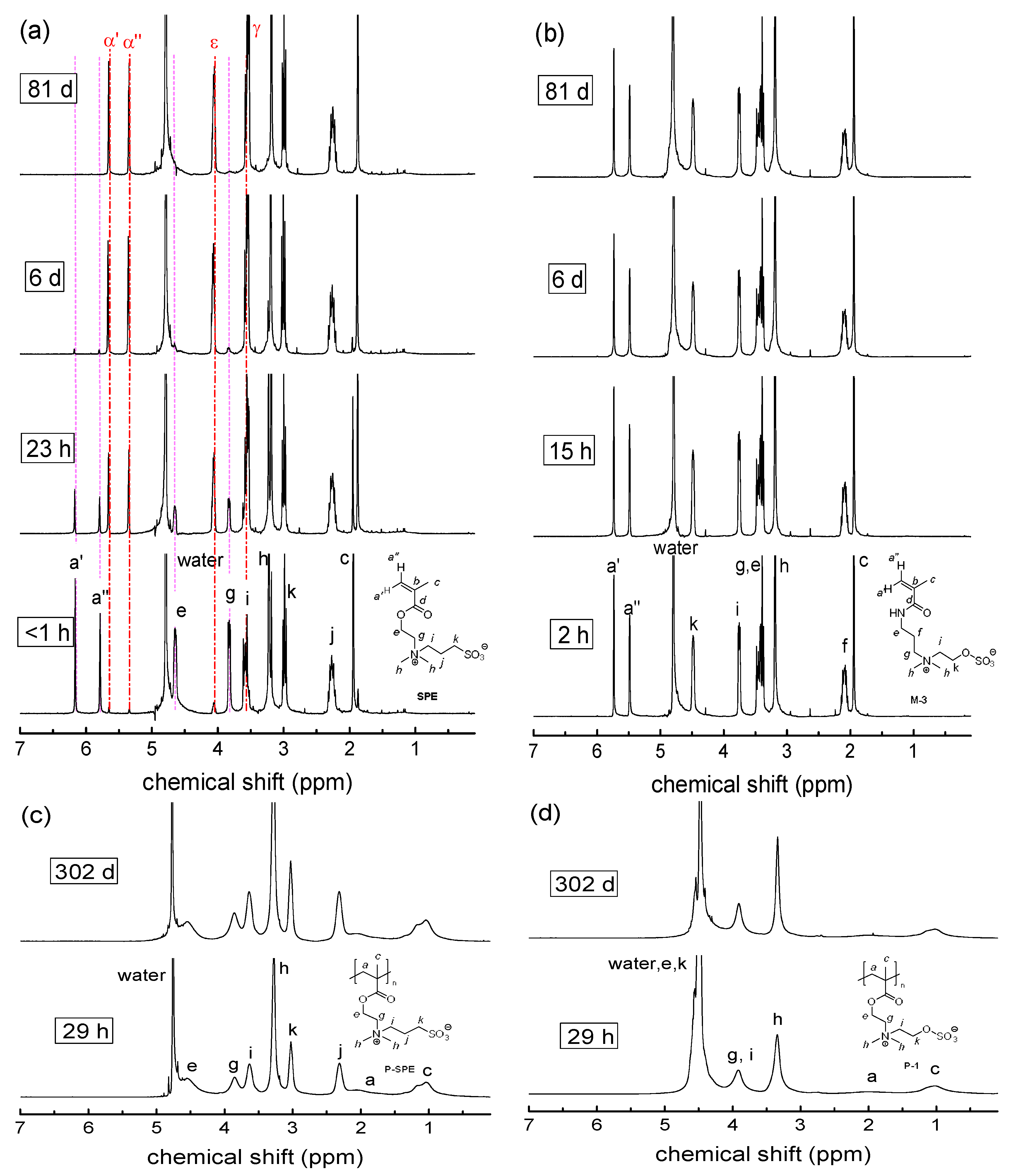
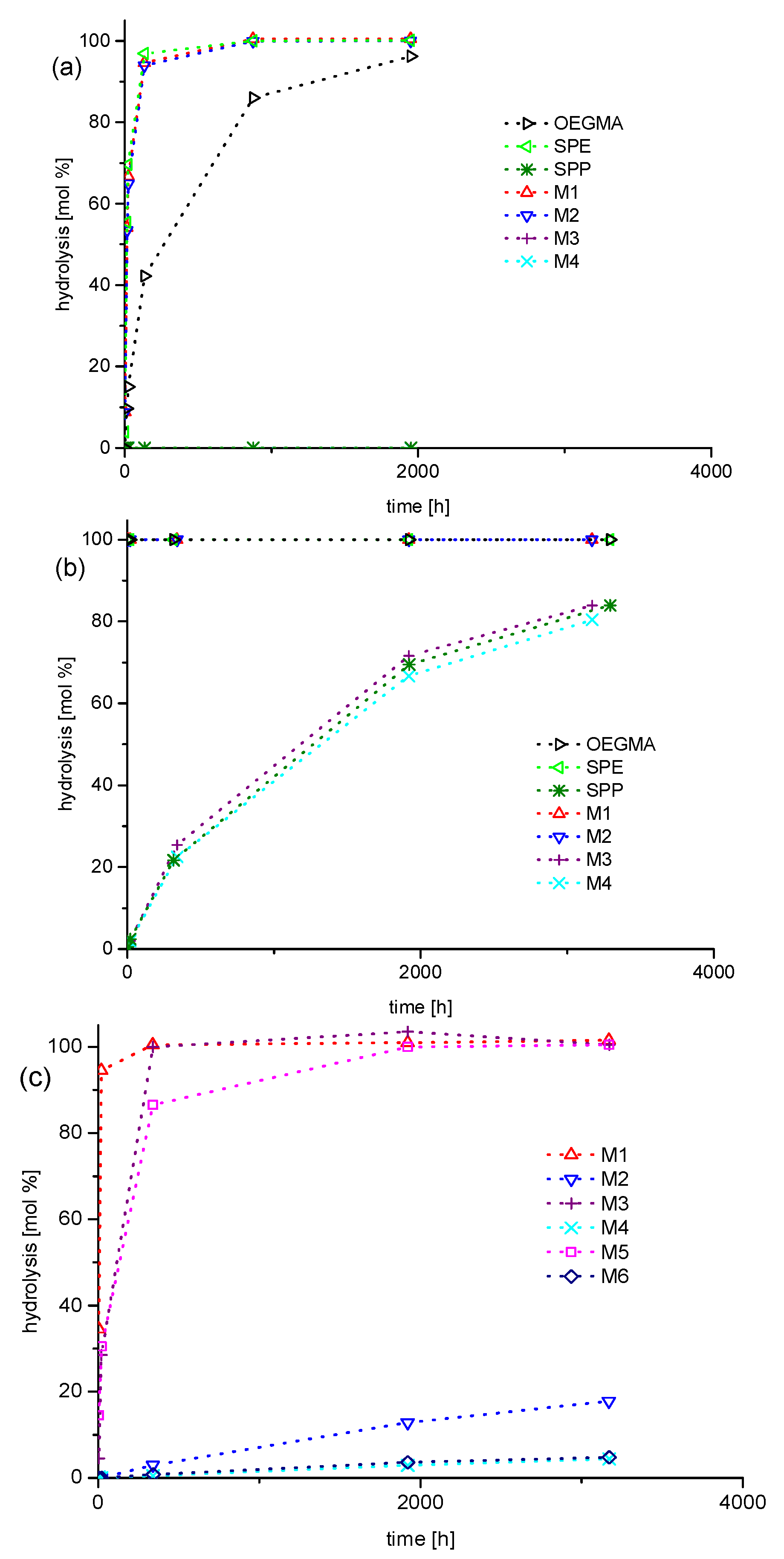
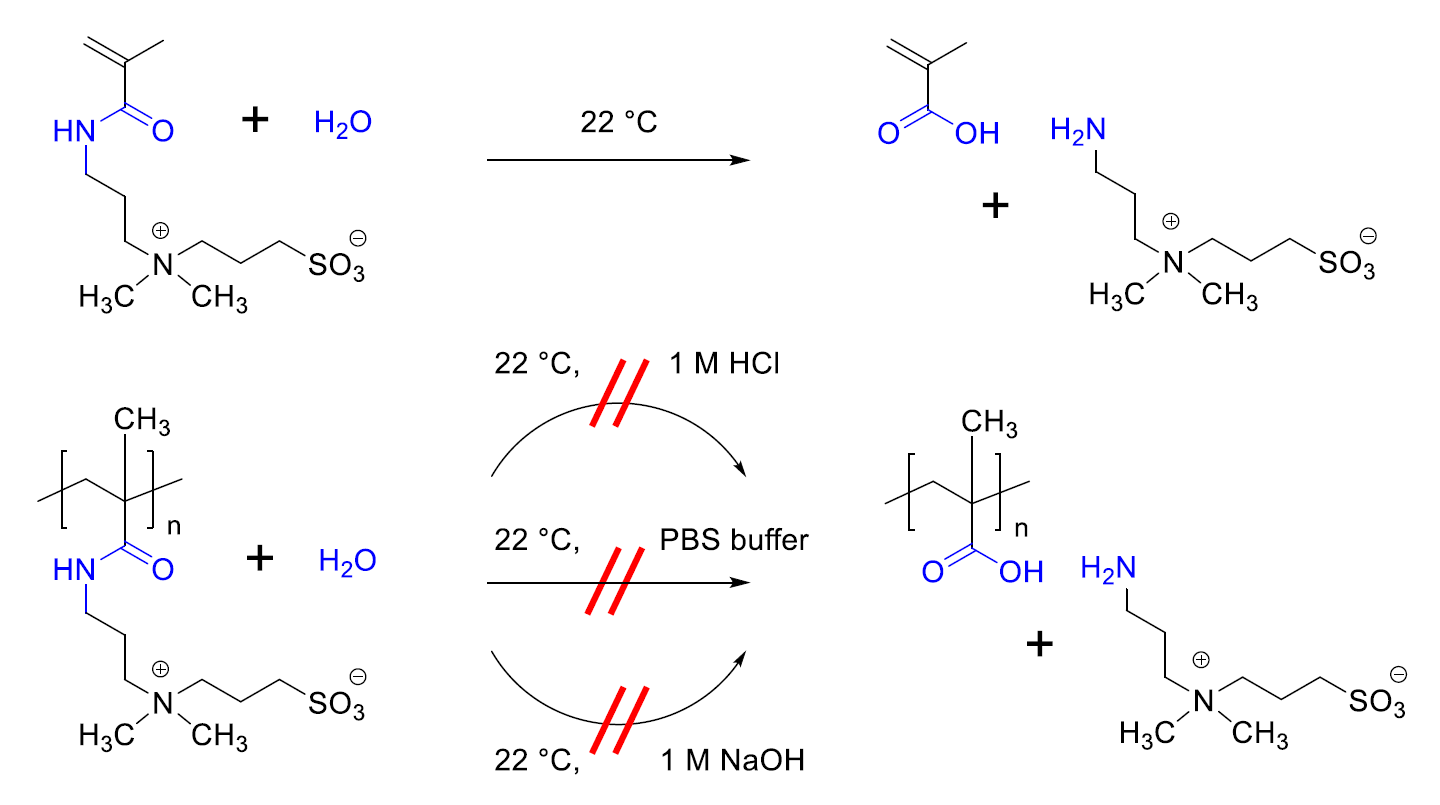
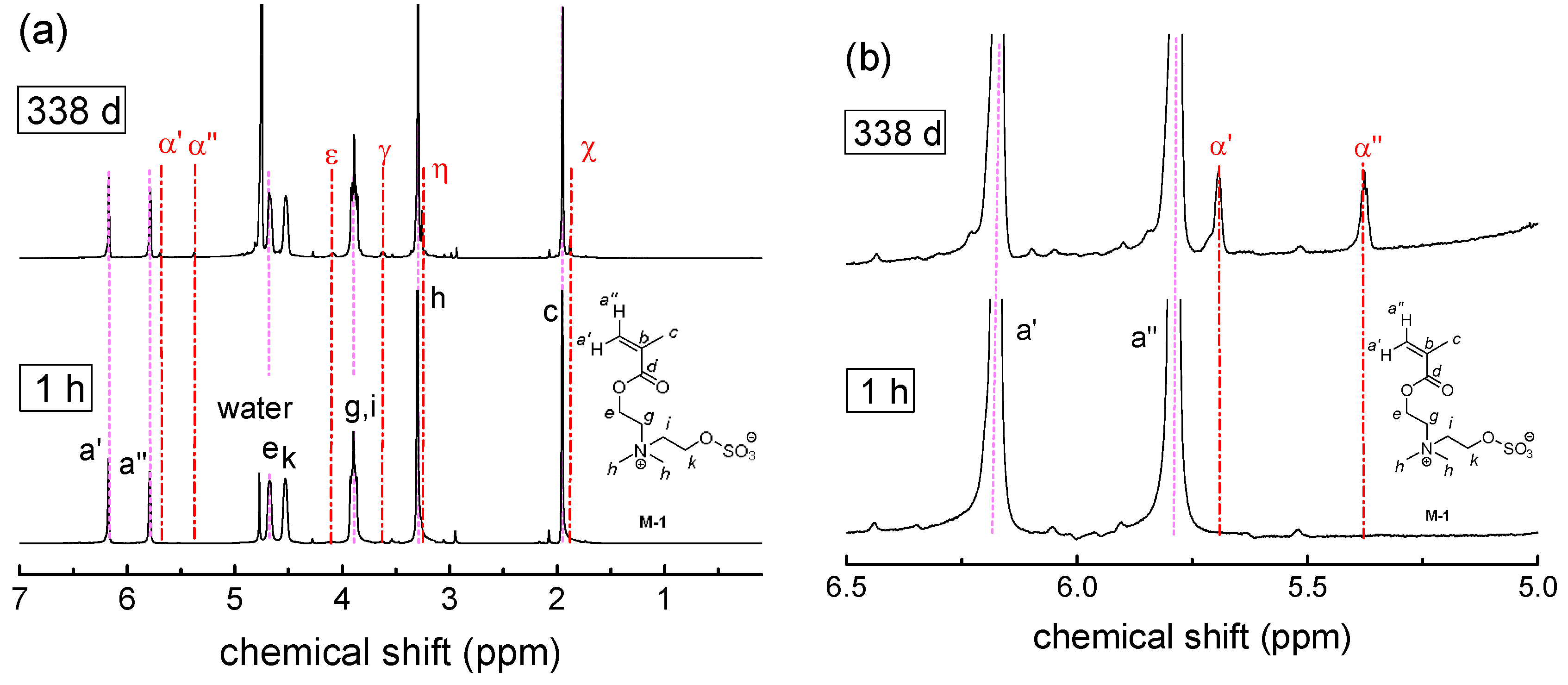
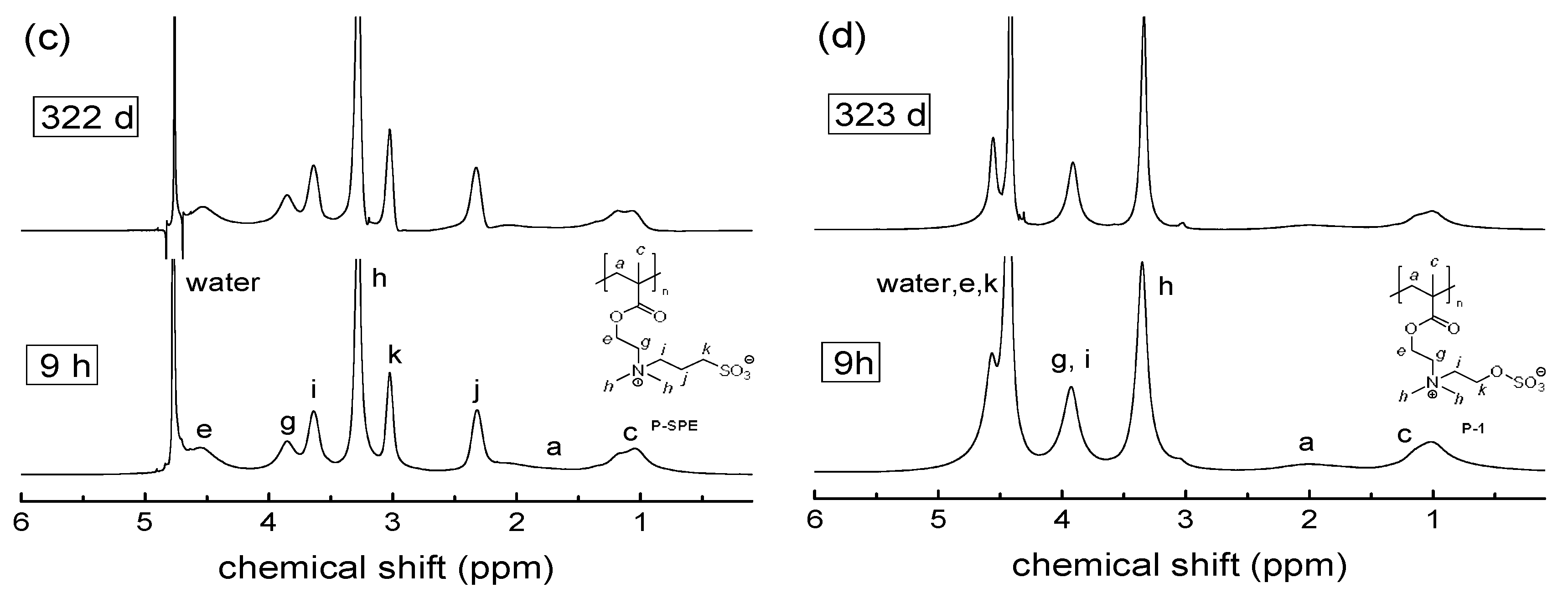
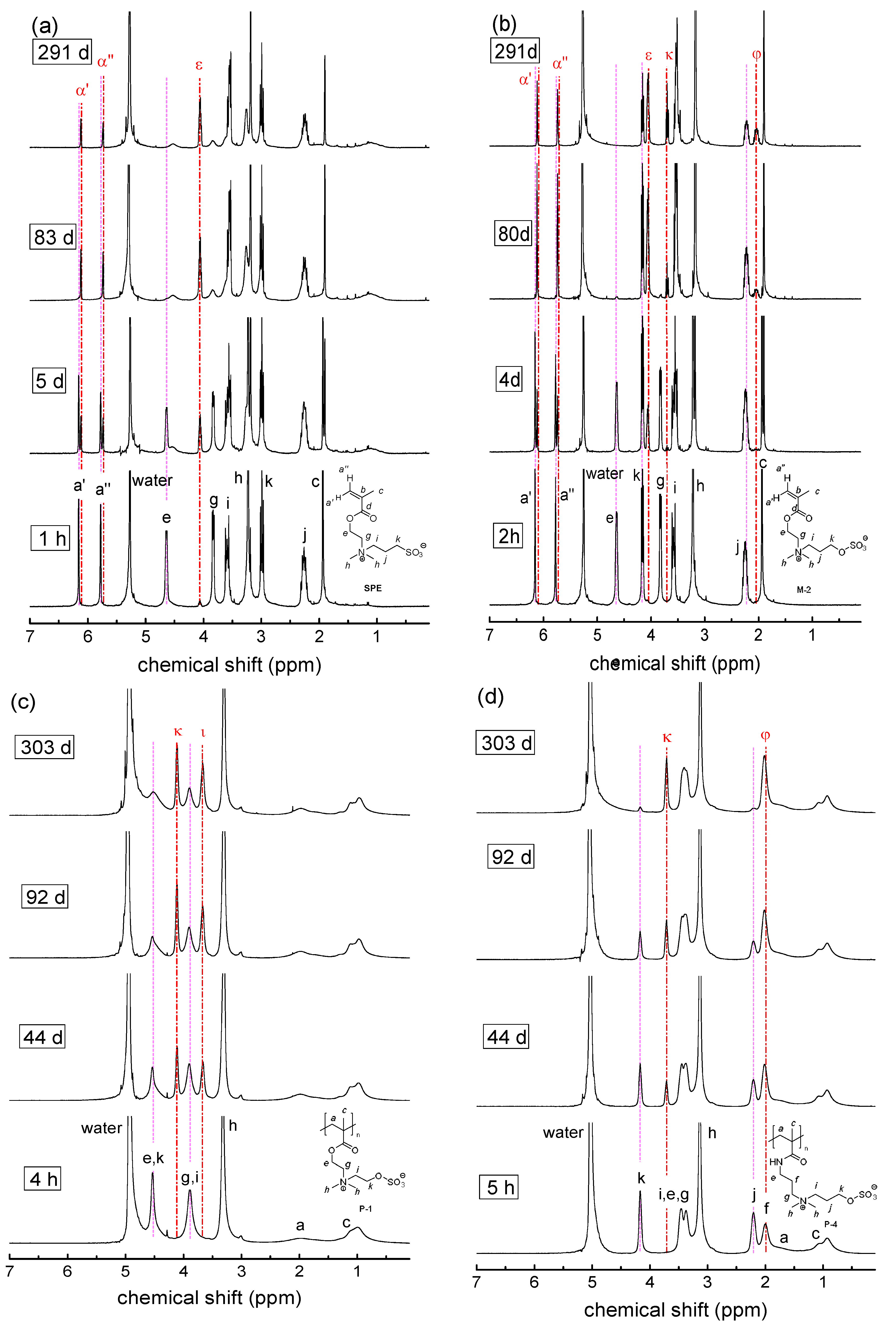
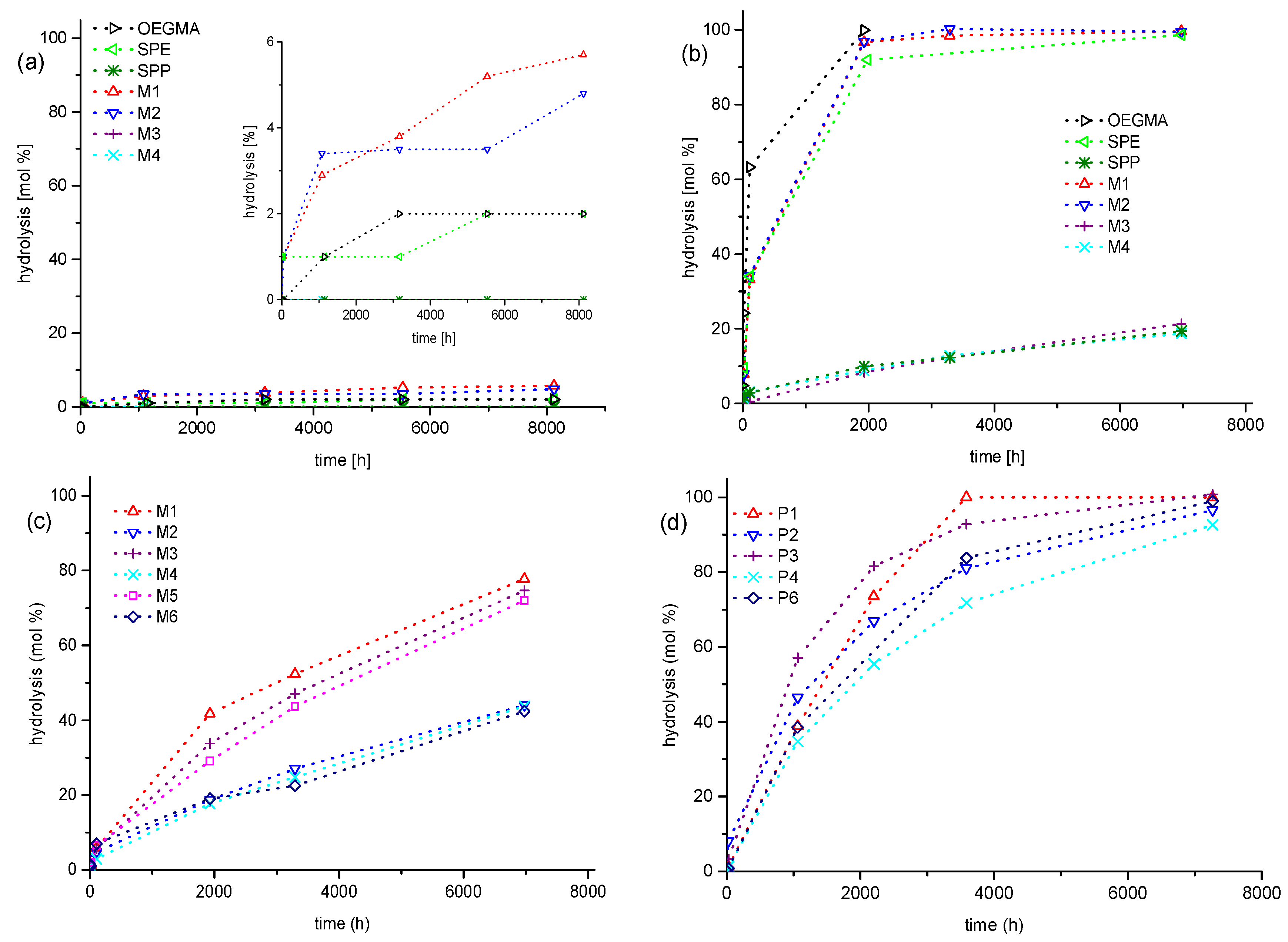
3.2. Investigation of Polymer Degradation in Aqueous Solution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lejars, M.; Margaillan, A.; Bressy, C. Fouling Release Coatings: A Nontoxic Alternative to Biocidal Antifouling Coatings. Chem. Rev. 2012, 112, 4347–4390. [Google Scholar] [CrossRef] [PubMed]
- Vaisocherová, H.; Brynda, E.; Homola, J. Functionalizable low-fouling coatings for label-free biosensing in complex biological media: Advances and applications. Anal. Bioanal. Chem. 2015, 407, 3927–3953. [Google Scholar] [CrossRef] [PubMed]
- Callow, J.A.; Callow, M.E. Trends in the development of environmentally friendly fouling-resistant marine coatings. Nat. Commun. 2011, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Schlenoff, J.B. Zwitteration: Coating Surfaces with Zwitterionic Functionality to Reduce Nonspecific Adsorption. Langmuir 2014, 30, 9625–9636. [Google Scholar] [CrossRef] [PubMed]
- Ostuni, E.; Chapman, R.G.; Holmlin, R.E.; Takayama, S.; Whitesides, G.M. A Survey of Structure-Property Relationships of Surfaces that Resist the Adsorption of Protein. Langmuir 2001, 17, 5605–5620. [Google Scholar] [CrossRef]
- Holmlin, R.E.; Chen, X.; Chapman, R.G.; Takayama, S.; Whitesides, G.M. Zwitterionic SAMs that Resist Nonspecific Adsorption of Protein from Aqueous Buffer. Langmuir 2001, 17, 2841–2850. [Google Scholar] [CrossRef]
- Rosenhahn, A.; Schilp, S.; Kreuzer, H.J.; Grunze, M. The role of "inert" surface chemistry in marine biofouling prevention. Phys. Chem. Chem. Phys. 2010, 12, 4275–4286. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, L.; Zhao, C.; Zheng, J. Surface hydration: Principles and applications toward low-fouling/nonfouling biomaterials. Polymer 2010, 51, 5283–5293. [Google Scholar] [CrossRef]
- Wei, Q.; Becherer, T.; Angioletti-Uberti, S.; Dzubiella, J.; Wischke, C.; Neffe, A.T.; Lendlein, A.; Ballauff, M.; Haag, R. Protein Interactions with Polymer Coatings and Biomaterials. Angew. Chem. Int. Ed. 2014, 53, 8004–8031. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.; O’Brien-Simpson, N.M.; Connal, L.A. Antibiofouling polymer interfaces: Poly(ethylene glycol) and other promising candidates. Polym. Chem. 2015, 6, 198–212. [Google Scholar] [CrossRef]
- Leng, C.; Sun, S.; Zhang, K.; Jiang, S.; Chen, Z. Molecular level studies on interfacial hydration of zwitterionic and other antifouling polymers in situ. Acta Biomater. 2016, 40, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Higaki, Y.; Kobayashi, M.; Murakami, D.; Takahara, A. Anti-fouling behavior of polymer brush immobilized surfaces. Polym. J. Jpn. 2016, 48, 325–331. [Google Scholar] [CrossRef]
- Plegue, T.J.; Kovach, K.M.; Thompson, A.J.; Potkay, J.A. Stability of Polyethylene Glycol and Zwitterionic Surface Modifications in PDMS Microfluidic Flow Chambers. Langmuir 2018, 34, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.-N.; Manik, D.H.N.; Huang, C.-J.; Chau, L.-K. Effect of elimination on antifouling and pH-responsive properties of carboxybetaine materials. Chem. Commun. 2017, 53, 9143–9146. [Google Scholar] [CrossRef] [PubMed]
- Laycock, B.; Nikolić, M.; Colwell, J.M.; Gauthier, E.; Halley, P.; Bottle, S.; George, G. Lifetime prediction of biodegradable polymers. Prog. Polym. Sci. 2017, 71, 144–189. [Google Scholar] [CrossRef] [Green Version]
- Guégain, E.; Michel, J.-P.; Boissenot, T.; Nicolas, J. Tunable Degradation of Copolymers Prepared by Nitroxide-Mediated Radical Ring-Opening Polymerization and Point-by-Point Comparison with Traditional Polyesters. Macromolecules 2018, 51, 724–736. [Google Scholar] [CrossRef]
- Xie, Q.; Xie, Q.; Pan, J.; Ma, C.; Zhang, G. Biodegradable Polymer with Hydrolysis-Induced Zwitterions for Antibiofouling. ACS Appl. Mater. Interfaces 2018, 10, 11213–11220. [Google Scholar] [CrossRef] [PubMed]
- Favresse, P.; Laschewsky, A. New poly(carbobetaine)s made from zwitterionic diallylammonium monomers. Macromol. Chem. Phys. 1999, 200, 887–895. [Google Scholar] [CrossRef]
- Vasantha, V.A.; Jana, S.; Parthiban, A.; Vancso, J.G. Water swelling, brine soluble imidazole based zwitterionic polymers-synthesis and study of reversible UCST behaviour and gel-sol transitions. Chem. Commun. 2014, 50, 46–48. [Google Scholar] [CrossRef] [PubMed]
- Laschewsky, A. Structures And Synthesis Of Zwitterionic Polymers. Polymers 2014, 6, 1544–1601. [Google Scholar] [CrossRef]
- Letteri, R.A.; Santa Chalarca, C.F.; Bai, Y.; Hayward, R.C.; Emrick, T. Forming Sticky Droplets from Slippery Polymer Zwitterions. Adv. Mater. 2017, 29, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Longenecker, R.; Mu, T.; Hanna, M.; Burke, N.A.D.; Stöver, H.D.H. Thermally Responsive 2-Hydroxyethyl Methacrylate Polymers: Soluble–Insoluble and Soluble–Insoluble–Soluble Transitions. Macromolecules 2011, 44, 8962–8971. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Baggerman, J.; Paulusse, J.M.J.; van Rijn, C.J.M.; Zuilhof, H. Stable Protein-Repellent Zwitterionic Polymer Brushes Grafted from Silicon Nitride. Langmuir 2011, 27, 2587–2594. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.; Falentin-Daudre, C.; Jerome, C.; Detrembleur, C. Mussel-inspired protein-repelling ambivalent block copolymers: Controlled synthesis and characterization. Polym. Chem. 2015, 6, 2919–2933. [Google Scholar] [CrossRef]
- Ren, P.-F.; Fang, Y.; Wan, L.-S.; Ye, X.-Y.; Xu, Z.-K. Surface modification of polypropylene microfiltration membrane by grafting poly(sulfobetaine methacrylate) and poly(ethylene glycol): Oxidative stability and antifouling capability. J. Membr. Sci. 2015, 492, 249–256. [Google Scholar] [CrossRef]
- Vatankhah-Varnoosfaderani, M.; Ina, M.; Adelnia, H.; Li, Q.; Zhushma, A.P.; Hall, L.J.; Sheiko, S.S. Well-Defined Zwitterionic Microgels: Synthesis and Application as Acid-Resistant Microreactors. Macromolecules 2016, 49, 7204–7210. [Google Scholar] [CrossRef]
- Kazantsev, O.A.; Shirshin, K.V.; Sivokhin, A.P.; Tel’nov, S.V.; Zhiganov, I.V.; Kuznetsov, A.E.; Mironycheva, Y.L. Hydrolysis of 2-Hydroxyethyl Methacrylate in Concentrated Aqueous Solutions. Russ. J. Appl. Chem. 2003, 76, 1296–1298. [Google Scholar] [CrossRef]
- Nishiyama, N.; Suzuki, K.; Yoshida, H.; Teshima, H.; Nemoto, K. Hydrolytic stability of methacrylamide in acidic aqueous solution. Biomaterials 2004, 25, 965–969. [Google Scholar] [CrossRef]
- Cao, B.; Li, L.; Tang, Q.; Cheng, G. The impact of structure on elasticity, switchability, stability and functionality of an all-in-one carboxybetaine elastomer. Biomaterials 2013, 34, 7592–7600. [Google Scholar] [CrossRef] [PubMed]
- Kazantsev, O.A.; Orekhov, D.V.; Sivokhin, A.P.; Kamorin, D.M.; Savinova, M.V. Concentration effects in the base-catalyzed hydrolysis of oligo(ethylene glycol)- and amine-containing methacrylic monomers. Des. Monomers Polym. 2017, 20, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, D.V.; Kazantsev, O.A.; Sivokhin, A.P.; Savinova, M.V. Features of the acid-catalyzed hydrolysis of mono- and poly(ethylene glycol) methacrylates. Eur. Polym. J. 2018, 100, 18–24. [Google Scholar] [CrossRef]
- van de Wetering, P.; Zuidam, N.J.; van Steenbergen, M.J.; van der Houwen, O.A.G.J.; Underberg, W.J.M.; Hennink, W.E. A Mechanistic Study of the Hydrolytic Stability of Poly(2-(dimethylamino)ethyl methacrylate). Macromolecules 1998, 31, 8063–8068. [Google Scholar] [CrossRef]
- Zhao, W.; Fonsny, P.; FitzGerald, P.; Warr, G.G.; Perrier, S. Unexpected behavior of polydimethylsiloxane/poly(2-(dimethylamino)ethyl acrylate) (charged) amphiphilic block copolymers in aqueous solution. Polym. Chem. 2013, 4, 2140–2150. [Google Scholar] [CrossRef]
- Cotanda, P.; Wright, D.B.; Tyler, M.; O’Reilly, R.K. A comparative study of the stimuli-responsive properties of DMAEA and DMAEMA containing polymers. J. Polym. Sci. Part A 2013, 51, 3333–3338. [Google Scholar] [CrossRef]
- Zhu, Y.; Noy, J.-M.; Lowe, A.B.; Roth, P.J. The synthesis and aqueous solution properties of sulfobutylbetaine (co)polymers: Comparison of synthetic routes and tuneable upper critical solution temperatures. Polym. Chem. 2015, 6, 5705–5718. [Google Scholar] [CrossRef]
- Whitfield, R.; Anastasaki, A.; Truong, N.P.; Wilson, P.; Kempe, K.; Burns, J.A.; Davis, T.P.; Haddleton, D.M. Well-Defined PDMAEA Stars via Cu(0)-Mediated Reversible Deactivation Radical Polymerization. Macromolecules 2016, 49, 8914–8924. [Google Scholar] [CrossRef] [Green Version]
- Rolph, M.S.; Pitto-Barry, A.; O’Reilly, R.K. The hydrolytic behavior of N,N′-(dimethylamino) ethyl acrylate-functionalized polymeric stars. Polym. Chem. 2017, 8, 5060–5070. [Google Scholar] [CrossRef]
- Mary, P.; Bendejacq, D.D.; Labeau, M.-P.; Dupuis, P. Reconciling Low- and High-Salt Solution Behavior of Sulfobetaine Polyzwitterions. J. Phys. Chem. B 2007, 111, 7767–7777. [Google Scholar] [CrossRef] [PubMed]
- Nizardo, N.; Schanzenbach, D.; Schönemann, E.; Laschewsky, A. Exploring Poly(ethylene glycol)-Polyzwitterion Diblock Copolymers as Biocompatible Smart Macrosurfactants Featuring UCST-Phase Behavior in Normal Saline Solution. Polymers 2018, 10, 325. [Google Scholar] [CrossRef]
- Xue, W.; Huglin, M.B.; Russell, A.T. Unusual behaviour of crosslinked and linear forms of a zwitterionic polymer in aqueous alkali. Macromol. Rapid Commun. 1999, 20, 239–243. [Google Scholar] [CrossRef]
- Marino, M.G.; Kreuer, K.D. Alkaline Stability of Quaternary Ammonium Cations for Alkaline Fuel Cell Membranes and Ionic Liquids. ChemSusChem 2015, 8, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, R.; Yokota, N.; Otsuji, K.; Miyatake, K. Structurally Well-Defined Anion Conductive Aromatic Copolymers: Effect of the Side-Chain Length. Macromolecules 2018, 51, 3394–3404. [Google Scholar] [CrossRef]
- Asonova, T.A.; Zezin, A.B.; Razvodovskii, Y.F. Synthesis and properties of poly-β-[N,N-dimethyl-N-(β-methacryloxyethyl)]-propiobetain. Polym. Sci. USSR 1974, 16, 896–906. [Google Scholar] [CrossRef]
- Aldred, N.; Li, G.; Gao, Y.; Clare, A.S.; Jiang, S. Modulation of barnacle (Balanus amphitrite Darwin) cyprid settlement behavior by sulfobetaine and carboxybetaine methacrylate polymer coatings. Biofouling 2010, 26, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Couturier, J.-P.; Sütterlin, M.; Laschewsky, A.; Hettrich, C.; Wischerhoff, E. Responsive Inverse Opal Hydrogels for the Sensing of Macromolecules. Angew. Chem. Int. Ed. 2015, 54, 6641–6644. [Google Scholar] [CrossRef] [PubMed]
- Vasantha, V.A.; Jana, S.; Parthiban, A.; Vancso, J.G. Halophilic polysulfabetaines - synthesis and study of gelation and thermoresponsive behavior. RSC Adv. 2014, 4, 22596–22600. [Google Scholar] [CrossRef]
- Arjunan Vasantha, V.; Junhui, C.; Ying, T.B.; Parthiban, A. Salt-Responsive Polysulfabetaines from Acrylate and Acrylamide Precursors: Robust Stabilization of Metal Nanoparticles in Hyposalinity and Hypersalinity. Langmuir 2015, 31, 11124–11134. [Google Scholar] [CrossRef] [PubMed]
- Vasantha, V.A.; Zainul Rahim, S.Z.; Jayaraman, S.; Junyuan, G.H.; Puniredd, S.R.; Ramakrishna, S.; Teo, S.L.-M.; Parthiban, A. Antibacterial, electrospun nanofibers of novel poly(sulfobetaine) and poly(sulfabetaine)s. J. Mater. Chem. B 2016, 4, 2731–2738. [Google Scholar] [CrossRef]
- Vasantha, V.A.; Jana, S.; Lee, S.S.-C.; Lim, C.-S.; Teo, S.L.-M.; Parthiban, A.; Vancso, J.G. Dual hydrophilic and salt responsive schizophrenic block copolymers-synthesis and study of self-assembly behavior. Polym. Chem. 2015, 6, 599–606. [Google Scholar] [CrossRef]
- Wen, J.; Weinhart, M.; Lai, B.; Kizhakkedathu, J.; Brooks, D.E. Reversible hemostatic properties of sulfabetaine/quaternary ammonium modified hyperbranched polyglycerol. Biomaterials 2016, 86, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Monroy Soto, V.M.; Galin, J.C. Poly(sulphopropylbetaines): 2. Dilute solution properties. Polymer 1984, 25, 254–262. [Google Scholar] [CrossRef]
- Köberle, P.; Laschewsky, A.; Lomax, T.D. Interactions of a zwitterionic polysoap and its cationic analog with inorganic salts. Makromol. Chem. Rapid Commun. 1991, 12, 427–433. [Google Scholar] [CrossRef]
- Delgado, J.D.; Schlenoff, J.B. Static and Dynamic Solution Behavior of a Polyzwitterion Using a Hofmeister Salt Series. Macromolecules 2017, 50, 4454–4464. [Google Scholar] [CrossRef]
- Hildebrand, V.; Laschewsky, A.; Päch, M.; Müller-Buschbaum, P.; Papadakis, C.M. Effect of the Zwitterion Structure on the Thermo-responsive Behaviour of Poly(Sulfobetaine Methacrylate)s. Polym. Chem. 2017, 8, 310–322. [Google Scholar] [CrossRef]
- Lowe, A.B.; McCormick, C.L. Synthesis and solution properties of zwitterionic polymers. Chem. Rev. 2002, 102, 4177–4189. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, V.; Laschewsky, A.; Wischerhoff, E. Modulating the solubility of zwitterionic poly((3-methacrylamidopropyl)ammonioalkane sulfonate)s in water and aqueous salt solutions via the spacer group separating the cationic and the anionic moieties. Polym. Chem. 2016, 7, 731–740. [Google Scholar] [CrossRef] [Green Version]
- Shao, Q.; Jiang, S. Influence of Charged Groups on the Properties of Zwitterionic Moieties: A Molecular Simulation Study. J. Phys. Chem. B 2014, 118, 7630–7637. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, P.G.; Lindman, B.; Laughlin, R.G. The upper consolute boundary in zwitterionic surfactant-water systems. J. Phys. Chem. 1984, 88, 6357–6362. [Google Scholar] [CrossRef]
- Wielema, T.A.; Engberts, J.B.F.N. Zwitterionic polymers—I. Synthesis of a novel series of poly(vinylsulphobetaines). Effect of structure of polymer on solubility in water. Eur. Polym. J. 1987, 23, 947–950. [Google Scholar] [CrossRef]
- Mertoglu, M.; Garnier, S.; Laschewsky, A.; Skrabania, K.; Storsberg, J. Stimuli responsive amphiphilic block copolymers for aqueous media synthesised via reversible addition fragmentation chain transfer polymerisation (RAFT). Polymer 2005, 46, 7726–7740. [Google Scholar] [CrossRef]
- Isobe, Y.; Yamada, K.; Nakano, T.; Okamoto, Y. Stereospecific Free-Radical Polymerization of Methacrylates Using Fluoroalcohols as Solvents. Macromolecules 1999, 32, 5979–5981. [Google Scholar] [CrossRef]
- Neradovic, D.; van Steenbergen, M.J.; Vansteelant, L.; Meijer, Y.J.; van Nostrum, C.F.; Hennink, W.E. Degradation Mechanism and Kinetics of Thermosensitive Polyacrylamides Containing Lactic Acid Side Chains. Macromolecules 2003, 36, 7491–7498. [Google Scholar] [CrossRef]
- Babazadeh, M. Synthesis, characterization, and in vitro drug-release properties of 2-hydroxyethyl methacrylate copolymers. J. Appl. Polym. Sci. 2007, 104, 2403–2409. [Google Scholar] [CrossRef]
- Alfurhood, J.A.; Sun, H.; Kabb, C.P.; Tucker, B.S.; Matthews, J.H.; Luesch, H.; Sumerlin, B.S. Poly(N-(2-hydroxypropyl)methacrylamide)-valproic acid conjugates as block copolymer nanocarriers. Polym. Chem. 2017, 8, 4983–4987. [Google Scholar] [CrossRef] [PubMed]
- Hauptmann, S.; Bier, H.; Faust, H. Beiträge zur Chemie des 2-Chloräthyltrimethylammoniumchlorids. J. Prakt. Chem. 1969, 311, 705–711. [Google Scholar] [CrossRef]
- Heyna, J. Reactive Dyes Containing Vinylsulfonyl Groups. Angew. Chem. Int. Ed. 1963, 2, 20–23. [Google Scholar] [CrossRef]
- Kratzer, D.; Barner, L.; Friedmann, C.; Bräse, S.; Lahann, J. A Synthetic Route to Sulfobetaine Methacrylates with Varying Charge Distance. Eur. J. Org. Chem. 2014, 36, 8064–8071. [Google Scholar] [CrossRef]
- Hashmi, S.; Vatankhah-Varnoosfaderani, M.; GhavamiNejad, A.; Obiweluozor, F.O.; Du, B.; Stadler, F.J. Self-associations and temperature dependence of aqueous solutions of zwitterionically modified N-isopropylacrylamide copolymers. Rheol. Acta 2015, 54, 501–516. [Google Scholar] [CrossRef]
- Zehm, D.; Laschewsky, A.; Gradzielski, M.; Prévost, S.; Liang, H.; Rabe, J.P.; Schweins, R.; Gummel, J. Amphiphilic Dual Brush Block Copolymers as “Giant Surfactants” and Their Aqueous Self-Assembly. Langmuir 2010, 26, 3145–3155. [Google Scholar] [CrossRef] [PubMed]
- Abid, S.K.; Hamid, S.M.; Sherrington, D.C. Micellization and surface activity of long-chain monoquaternary and diquaternary ammonium salts. J. Colloid Interface Sci. 1987, 120, 245–255. [Google Scholar] [CrossRef]
- Cochin, D.; Laschewsky, A.; Nallet, F. Emulsion Polymerization of Styrene Using Conventional, Polymerizable, and Polymeric Surfactants. A Comparative Study. Macromolecules 1997, 30, 2278–2287. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Mnapp (kg·mol−1) a | Dispersity Ð a | CP in water (°C) | CP in normal saline (°C) b | CP in PBS (°C) c | CP in brine (°C) d |
|---|---|---|---|---|---|---|
| P-OEGMA | 130 e | 3.8 (e) | 85 f | 80 f | 79 f | j |
| P-SPE | 210 | 2.2 | 55 g | <0 | <0 | <0 |
| P-SPP | 180 | 2.2 | 21 g | <0 | <0 | <0 |
| P-1 | 400 | 1.8 | h | h | h | <0 |
| P-2 | 420 | 1.6 | h | h | h | <0 |
| P-3 | j | j | h | h | h | <0 |
| P-4 | 220 | 2.3 | h | h | h | <0 |
| P-5 | j | j | h | h | h | j |
| P-6 | j | j | h | h | h | <0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schönemann, E.; Laschewsky, A.; Rosenhahn, A. Exploring the Long-Term Hydrolytic Behavior of Zwitterionic Polymethacrylates and Polymethacrylamides. Polymers 2018, 10, 639. https://doi.org/10.3390/polym10060639
Schönemann E, Laschewsky A, Rosenhahn A. Exploring the Long-Term Hydrolytic Behavior of Zwitterionic Polymethacrylates and Polymethacrylamides. Polymers. 2018; 10(6):639. https://doi.org/10.3390/polym10060639
Chicago/Turabian StyleSchönemann, Eric, André Laschewsky, and Axel Rosenhahn. 2018. "Exploring the Long-Term Hydrolytic Behavior of Zwitterionic Polymethacrylates and Polymethacrylamides" Polymers 10, no. 6: 639. https://doi.org/10.3390/polym10060639