New Ca2.90(Me2+)0.10(PO4)2 β-tricalcium Phosphates with Me2+ = Mn, Ni, Cu: Synthesis, Crystal-Chemistry, and Luminescence Properties


 , and
, and
Abstract
:
1. Introduction
2. Experimental
3. Results and Discussion
3.1. SEM-EDS Analysis
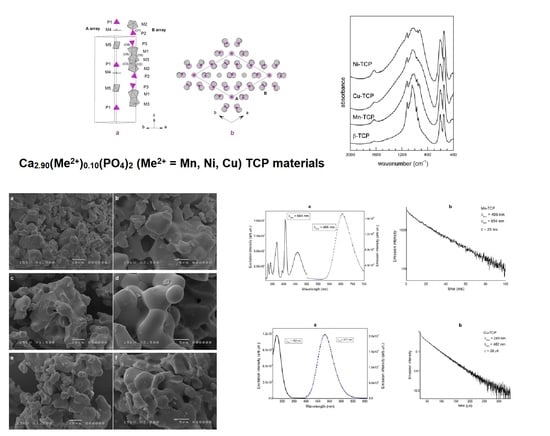
3.2. X-ray Structural Study
3.2.1. Structural Solution
Polyhedral coordination
3.2.2. Three-Dimensional Framework
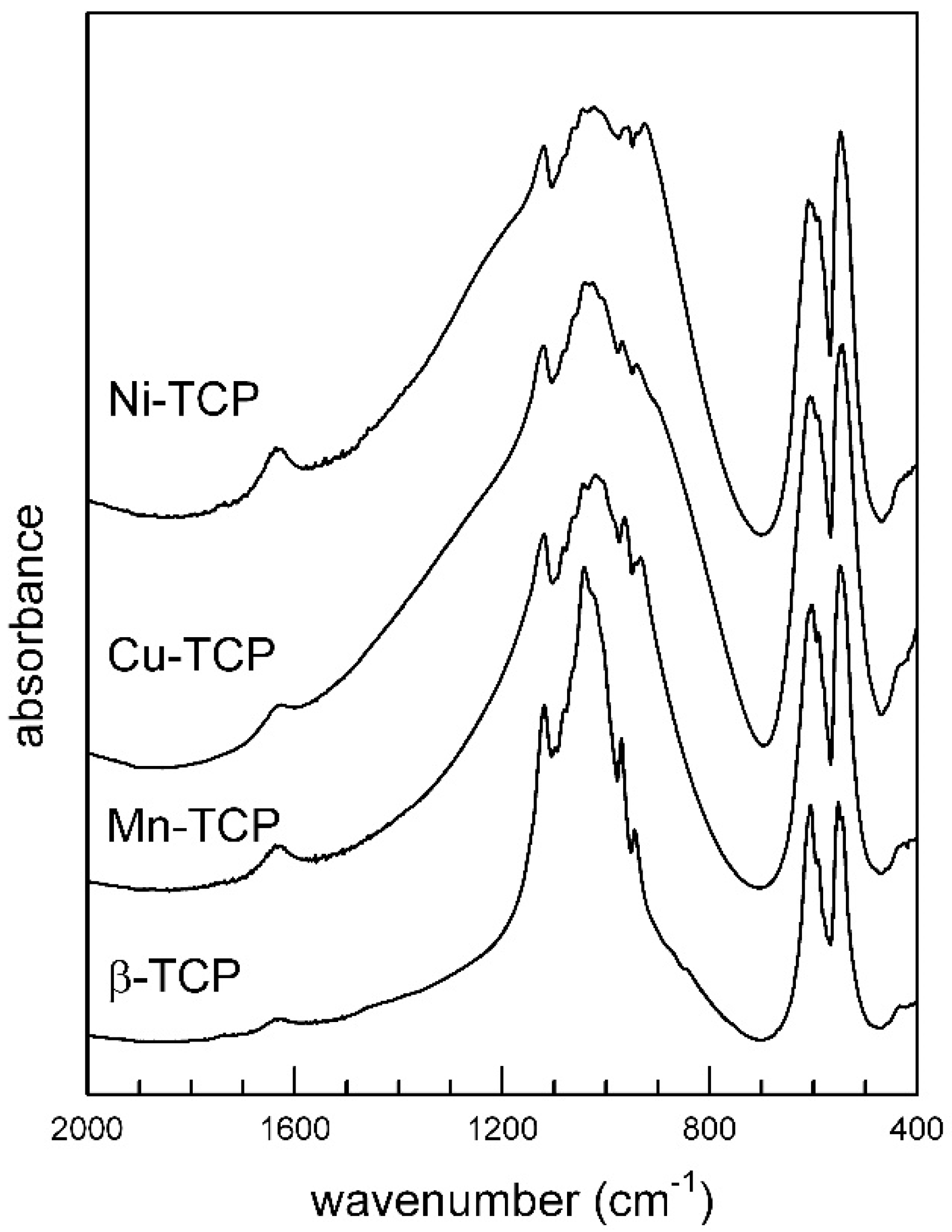
3.3. FTIR Spectroscopy
3.4. Luminescence Spectroscopy
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Dos Santos, M.L.; dos Santos Riccardi, C.; de Almeida Filho, E.; Guastaldi, A.C. Calcium phosphates of biological importance based coatings deposited on Ti-15Mo alloy modified by laser beam irradiation for dental and orthopedic applications. Ceram. Int. 2018, 44, 22432–22438. [Google Scholar] [CrossRef]
- Dorozkhin, S.V. Calcium Orthophosphates bioceramics. Ceram. Int. 2015, 41, 13913–13966. [Google Scholar] [CrossRef]
- Habraken, W.; Habibovic, P.; Epple, M.; Bohner, M. Calcium phosphates in biomedical applications: Materials for the future? Mater. Today 2016, 19, 69–87. [Google Scholar] [CrossRef]
- Hughes, J.M.; Rakovan, J.F. Structurally robust, chemically diverse: Apatite and apatite supergroup minerals. Elements 2015, 11, 165–170. [Google Scholar] [CrossRef]
- Renaudin, G.; Gomes, S.; Nedelec, J.-M. First-row transition metal doping in calcium phosphate bioceramics: A detailed crystallographic study. Materials 2017, 10, 92. [Google Scholar] [CrossRef]
- Agarwalla, A.; Puzzitiello, R.; Garcia, G.H.; Forsythe, B. Application of a Beta-Tricalcium Phosphate Graft to Minimize Bony Defect in Bone–Patella Tendon–Bone Anterior Cruciate Ligament Reconstruction. Arthrosc. Tech. 2018, 7, e725–e729. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Liu, X.; Gao, H.; Shi, X.; Zhao, N.; Wang, Y. Hydrothermal growth of whitlockite coating on β-tricalcium phosphate surfaces for enhancing bone repair potential. J. Mater. Sci. Technol. 2018, 34, 1054–1059. [Google Scholar] [CrossRef]
- Ogawa, K.; Miyaji, H.; Kato, A.; Kosen, Y.; Momose, T.; Yoshida, T.; Nishida, E.; Miyata, S.; Murakami, S.; Takita, H.; et al. Periodontal tissue engineering by nano beta-tricalcium phosphate scaffold and fibroblast growth factor-2 in one-wall infrabony defects of dogs. J. Periodont. Res. 2016, 51, 758–767. [Google Scholar] [CrossRef] [Green Version]
- Bessière, A.; Lecointre, A.; Benhamou, R.A.; Suard, E.; Wallez, G.; Viana, B. How to induce red persistent luminescence in biocompatible Ca3(PO4)2. J. Mater. Chem. C 2013, 1, 1252–1259. [Google Scholar] [CrossRef]
- Bessière, A.; Benhamou, R.A.; Wallez, G.; Lecointre, A.; Viana, B. Site occupancy and mechanisms of thermally stimulated luminescence in Ca9Ln(PO4)7 (Ln = lanthanide). Acta Mater. 2012, 19, 6641–6649. [Google Scholar] [CrossRef]
- Piccinelli, F.; Trevisani, M.; Plaisier, J.R.; Bettinelli, M. Structural study of Yb3+, Eu3+ and Pr3+ doped Ca9Lu(PO4)7. J. Rare Earths 2015, 33, 977–982. [Google Scholar] [CrossRef]
- Hughes, J.M.; Jolliff, B.L.; Rakovan, J.F. The crystal chemistry of whitlockite and merrillite and the dehydrogenation of whitlockite to merrillite. Am. Mineral. 2008, 93, 1300–1305. [Google Scholar] [CrossRef]
- Xie, X.; Yang, H.; Gu, X.; Downs, R.T. Chemical composition and crystal structure of merrillite from the Suizhou meteorite. Am. Mineral. 2015, 100, 2753–2756. [Google Scholar] [CrossRef]
- El Khouri, A.; Elaatmani, M.; Della Ventura, G.; Sodo, A.; Rizzi, R.; Rossi, M.; Capitelli, F. Synthesis, structure refinement and vibrational spectroscopy of new rare-earth tricalcium phosphates Ca9RE(PO4)7 (RE = La, Pr, Nd, Eu, Gd, Dy, Tm, Yb). Ceram. Int. 2017, 43, 15645–15653. [Google Scholar] [CrossRef]
- Capitelli, F.; Rossi, M.; El Khouri, A.; Elaatmani, M.; Corriero, N.; Sodo, A.; Della Ventura, G. Synthesis, structural model and vibrational spectroscopy of lutetium tricalcium phosphate Ca9Lu(PO4)7. J. Rare Earths 2018, 36, 1162–1168. [Google Scholar] [CrossRef]
- Tahiti, A.A.; El Bali, B.; Lachkar, M.; Piniella, J.F.; Capitelli, F. Crystal structure of new lanthanide diphosphates KLnP2O7·2(H2O) (Ln = Gd, Tb, Yb). Z. Kristallogr. 2006, 221, 173–177. [Google Scholar] [CrossRef]
- Capitelli, F.; El Bali, B.; Essehli, R.; Lachkar, M.; Da Silva, I. New hybrid diphosphates Ln2(NH2(CH2)2NH2)2(HP2O7)2⋅4H2O [Ln = Eu, Tb, Er]: Synthesis, single crystal and powder X-ray crystal structure. Z. Kristallogr. 2006, 221, 788–794. [Google Scholar] [CrossRef]
- Matsunaga, K.; Kubota, T.; Toyoura, K.; Nakamura, A. First-principles calculations of divalent substitution of Ca(2+) in tricalcium phosphates. Acta Biomater. 2015, 23, 329–337. [Google Scholar] [CrossRef]
- Mayer, I.; Cohen, S.; Gdalya, S.; Burghaus, O.; Reinen, D. Crystal structure and EPR study of Mn-doped β-tricalcium phosphate. Mater. Res. Bull. 2008, 43, 447–452. [Google Scholar] [CrossRef]
- Belik, A.A.; Izumi, F.; Stefanovich, S.Yu.; Lazoryak, B.I.; Oikawa, K. Chemical and structural properties of a whitlockite-like phosphate, Ca9FeD(PO4)7. Chem. Mater. 2002, 14, 987–995. [Google Scholar] [CrossRef]
- Belik, A.A.; Morozov, V.A.; Khasanov, S.S.; Lazoryak, B.I. Crystal structures of new double calcium and cobalt phosphates. Mater. Res. Bull. 1998, 33, 987–995. [Google Scholar] [CrossRef]
- Lazoryak, B.I.; Khan, N.; Morozov, V.A.; Belik, A.A.; Khasanov, S.S. Preparation, structure determination, and redox characteristics of new calcium copper phosphates. J. Solid State Chem. 1999, 145, 345–355. [Google Scholar] [CrossRef]
- Zhu, G.; Ci, Z.; Shi, Y.; Que, M.; Wang, Q.; Wang, Y. Synthesis, crystal structure and luminescence characteristics of a novel red phosphor Ca19Mg2(PO4)14:Eu3+ for light emitting diodes and field emission displays. J. Mater. Chem. C 2013, 1, 5960–5969. [Google Scholar] [CrossRef]
- Bohner, M.; Tiainen, H.; Michel, P.; Döbelin, N. Design of an inorganic dual-paste apatite cement using cation exchange. J. Mater. Sci. Mater. Med. 2015, 26, 1–13. [Google Scholar] [CrossRef] [PubMed]
- El Bali, B.; Boukhari, A.; Holt, E.M.; Aride, J. Calcium nickel orthophosphate: Crystal structure of Ca8.5Ni9.5(PO4)12. Z. Kristallogr. 1995, 210, 838–842. [Google Scholar] [CrossRef]
- Lii, K.-H.; Shih, P.-F.; Chen, T.-M. AM3(P2O7)2 (A = alkaline-earth metals; M = Fe, Co, Ni): diphosphates containing infinite chains of edge-sharing MO6 octahedra. Inorg. Chem. 1993, 32, 4373–4377. [Google Scholar] [CrossRef]
- Funke, M.; Blum, M.; Glaum, R.; El Bali, B. Beiträge zum Koordinationsverhalten von Oxidionen in anorganischen Feststoffen. IV [1] Darstellung, Kristallstruktur und spektroskopische Charakterisierung von NiP4O11 und CaNiP2O7. Z. Anorg. Chem. 2004, 630, 1040–1047. [Google Scholar] [CrossRef]
- Capitelli, F.; Harcharras, M.; Assaaoudi, H.; Ennaciri, A.; Moliterni, A.G.G.; Bertolasi, V. Crystal structure of new hexahydrate dicobalt pyrophosphate Co2P2O7·6H2O: Comparison with Co2P2O7·2H2O, α-, β- and γ- Co2P2O7. Z. Kristallogr. 2003, 218, 345–350. [Google Scholar] [CrossRef]
- Capitelli, F.; Chita, G.; Ghiara, M.R.; Rossi, M. Crystal chemical investigation of Fe3(PO4)2∙8H2O vivianite minerals. Z. Kristallogr. 2012, 227, 92–101. [Google Scholar] [CrossRef]
- Rossi, M.; Ghiara, M.R.; Chita, G.; Capitelli, F. Crystal-chemical and structural characterization of fluorapatites in ejecta from Somma-Vesuvius volcanic complex. Am. Mineral. 2011, 96, 1828–1837. [Google Scholar] [CrossRef]
- Dorozkhin, S.V. Calcium Orthophosphates: Occurrence, properties and major applications. Bioceram. Dev. Appl. 2014, 4. [Google Scholar] [CrossRef]
- Belik, A.A.; Yanov, O.V.; Lazoryak, B.I. Synthesis and crystal structure of Ca9Cu1.5(PO4)7 and reinvestigation of Ca9.5Cu(PO4)7. Mater. Res. Bull. 2001, 36, 1863–1871. [Google Scholar] [CrossRef]
- Rietveld, H.M. A profile refinement method for nuclear and magnetic structures. J. Appl. Cryst. 1969, 2, 65–71. [Google Scholar] [CrossRef]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R.; Corriero, N.; Falcicchio, A. EXPO2013: A kit of tools for phasing crystal structures from powder data. J. Appl. Cryst. 2013, 46, 1231–1235. [Google Scholar] [CrossRef]
- Horiba Jobin, Y. DAS6 Fluorescence Decay Analysis Software User Guide: Version 2744.F.; Edison: New Jersey, NJ, USA, 2008; pp. 29–31. [Google Scholar]
- Yashima, M.; Sakai, A.; Kamiyama, T.; Hoshikawa, A. Crystal structure analysis of β-tricalcium phosphate Ca3(PO4)2 by neutron powder diffraction. J. Solid State Chem. 2003, 175, 272–277. [Google Scholar] [CrossRef]
- Sakae, T.; Yamamoto, H.; Mishima, H.; Matsumoto, T.; Kozawa, Y. Morphology and chemical composition of dental calculi mainly composed of whitlockite. Scanning Microsc. 1989, 3, 855–860. [Google Scholar]
- Fadeeva, I.V.; Gafurov, M.R.; Filippov, Y.Y.; Davydova, G.A.; Savintseva, I.V.; Fomin, A.S.; Petrakova, N.V.; Antonova, O.S.; Akhmetov, L.I.; Gabbasov, B.F.; et al. Copper-substituted tricalcium phosphates. Dokl. Chem. 2016, 471, 384–387. [Google Scholar] [CrossRef]
- Altomare, A.; Campi, G.; Cuocci, C.; Eriksson, L.; Giacovazzo, C.; Moliterni, A.; Rizzi, R.; Werner, P.-E. Advances in powder diffraction pattern indexing: N-TREOR09. J. Appl. Cryst. 2009, 42, 768–775. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Inorganic Crystal Structure Database (ICSD); Version 2018–2; Fachinformationszentrum: Karlsruhe, Germany, 2018.
- Henning, P.A.; Adolfsson, E.; Grins, J. The Chalcogenide Phosphate Apatites Ca10(PO4)6S, Sr10(PO4)6S, Ba10(PO4)6S and Ca10(PO4)6Se. Z. Kristallogr. 2000, 215, 226–230. [Google Scholar] [CrossRef]
- Mayer, I.; Gdalya, S.; Burghaus, O.; Reinen, D. A spectroscopic and structural study of M(3d)2+-doped β-tricalcium phosphate—The binding properties of Ni2+ and Cu2+ in the pseudo-octahedral Ca(5)O6 host-sites. Z. Anorg. Chem. 2009, 635, 2039–2045. [Google Scholar] [CrossRef]
- Altomare, A.; Corriero, N.; Cuocci, C.; Falcicchio, A.; Moliterni, A.; Rizzi, R. OChemDb: The free on-line Open Chemistry Database portal for searching and analysing crystal structure information. J. Appl. Cryst. 2018, 51, 1229–1236. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Yin, X.; Stott, M.J.; Rubio, A. α and β-tricalcium phosphate: A density functional study. Phys. Rev. B 2003, 68, 205205. [Google Scholar] [CrossRef]
- Zatovsky, I.V.; Strutynska, N.Y.; Baumer, V.N.; Shishkin, O.V.; Slobodyanik, N.S. The whitlockite-related phosphate Ca9Cr(PO4)7. Acta Crystallogr. E 2007, 63, i180–i181. [Google Scholar] [CrossRef]
- Lazoryak, B.I.; Morozov, V.A.; Belik, A.A.; Khasanov, S.S.; Shekhtman, V.S. Crystal structures and characterization of Ca9Fe(PO4)7and Ca9FeH0.9(PO4)7. J. Solid State Chem. 1996, 122, 15–21. [Google Scholar] [CrossRef]
- De Aza, P.N.; Guitián, F.; Santos, C.; De Aza, S.; Cuscó, R.; Artús, L. Vibrational Properties of Calcium Phosphate Compounds. 2. Comparison between Hydroxyapatite and β-Tricalcium Phosphate. Chem. Mater. 1997, 9, 916–922. [Google Scholar] [CrossRef]
- Jillavenkatesa, A.; Condrate, R.A., Sr. The Infrared and Raman Spectra of α- and β-Tricalcium Phosphate (Ca3(PO4)2). Spectrosc. Lett. 1998, 31, 1619–1634. [Google Scholar] [CrossRef]
- Ross, S.D. Phosphates and Other Oxy-Anions of Group V. In Mineralogical Society Monograph 4: The Infrared Spectra of Minerals; Farmer, V.C., Ed.; Mineralogical Society of Great Britain and Ireland: London, UK, 1974. [Google Scholar]
- Meejoo, S.; Maneeprakorn, W.; Winotai, P. Phase and thermal stability of nanocrystalline hydroxyapatite prepared via microwave heating. Thermochim. Acta 2006, 447, 115–120. [Google Scholar] [CrossRef]
- Strens, R.G.J. The Common Chain, Ribbon, and Ring Silicates. In Mineralogical Society Monograph 4: The Infrared Spectra of Minerals; Farmer, V.C., Ed.; Mineralogical Society of Great Britain and Ireland: London, UK, 1974. [Google Scholar]
- Aguilar, G.M.; Osendi, M.I. Fluorescence of Mn2+ in CaCO3. J. Lumin. 1982, 27, 365–375. [Google Scholar] [CrossRef]
- Flores, J.M.C.; Caldino, G.U.; Hernández, A.J.; Camarillo, G.E.; Cabrera, B.E.; Del Castillo, H.; Speghini, A.; Bettinelli, M.; Murrieta, S.H. Study of Mn2+ luminescence in Zn(PO3)2 glasses. Phys. Status Solidi C 2007, 4, 922–925. [Google Scholar] [CrossRef]
- Wanmaker, W.L.; Bakker, C. Luminescence of Copper-Activated Calcium and Strontium Orthophosphates. J. Electrochem. Soc. 1959, 106, 1027–1032. [Google Scholar] [CrossRef]
- Verwey, J.W.M.; Coronado, J.M.; Blasse, G. The luminescence of Cu(I) in strontium tetraborate. J. Solid State Chem. 1991, 92, 531–536. [Google Scholar] [CrossRef]
- Boutinaud, P.; Duloisy, E.; Pedrini, C.; Moine, B.; Parent, C.; Le Flem, G. Fluorescence properties of Cu+ ion in phosphate glasses of the BaLiPO4-P2O5 system. J. Solid State Chem. 1991, 94, 236–243. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Formula | Ca2.90Mn0.10(PO4)2 | Ca2.90Ni0.10(PO4)2 | Ca2.90Cu0.10(PO4)2 |
|---|---|---|---|
| Formula weight | 363.35 | 363.79 | 364.45 |
| Color | colorless | colorless | colorless |
| Temperature (K) | 293 | 293 | 293 |
| Wavelength (Å) | 1.54056 | 1.54056 | 1.54056 |
| 2θ range; step (deg) | 8–120°; (0.02) | 8–120°; (0.02) | 8–120°; (0.02) |
| System, space group | Rhombohedral, R3c | Rhombohedral, R3c | Rhombohedral, R3c |
| a = b (Å) | 10.41012(19) | 10.39447(10) | 10.40764(8) |
| c (Å) | 37.2984(14) | 37.2901(8) | 37.3158(6) |
| V (Å3) | 3500.52(16) | 3489.22(9) | 3500.48(7) |
| Z; Densitycalc. (Mg·m−3) | 21; 3.10 | 21; 3.12 | 21; 3.11 |
| Refinement method | FMLQ | FMLQ | FMLQ |
| Profile function | Pearson VII | Pearson VII | Pearson VII |
| Bragg refl., parameters | 587; 81 | 587; 79 | 587; 79 |
| Rp; Rwp; Rexp (%) | 2.93; 4.43; 3.97 | 2.56; 4.04; 3.92 | 1.85; 2.82; 4.71 |
| RF; RBragg (%) | 10.34; 16.76 | 8.09; 11.27 | 5.72; 7.90 |
| Mn-TCP | Ni-TCP | Cu-TCP | |
|---|---|---|---|
| CaO | 51.8(8) | 51.7(5) | 51.1(1) |
| MeO | 2.5(1) | 2.7(3) | 2.9(2) |
| P2O5 | 45.7(8) | 45.6(7) | 45.9(3) |
| Tot. | 99.9 | 99.9 | 99.9 |
| Ca | 2.87 | 2.87 | 2.85 |
| Me | 0.11 | 0.11 | 0.12 |
| P | 2.01 | 2.01 | 2.03 |
| Site | x | y | z | Uiso | Occ | |
|---|---|---|---|---|---|---|
| Ca1 | 18b | 0.1435(5) | 0.2761(5) | 0.3408 | 0.0013(2) | 1 |
| Ca2 | 18b | 0.2837(4) | 0.1296(6) | 0.4739(1) | 0.0035(2) | 1 |
| Ca3 | 18b | 0.4813(4) | 0.5450(4) | 0.4007(1) | 0.0018(2) | 1 |
| Ca4 | 6a | 2/3 | 1/3 | 0.4253(3) | 0.0090(2) | 1/2 |
| Ca5 | 6a | 0 | 0 | 0.4082(2) | 0.0006(2) | 0.69(2) |
| Ni5 | 6a | 0 | 0 | 0.4082(2) | 0.0006(2) | 0.31(2) |
| P1 | 6a | 2/3 | 1/3 | 0.5065(2) | 0.0038(4) | 1 |
| P2 | 18b | −0.1945(7) | −0.3539(5) | 0.3755(2) | 0.0023(4) | 1 |
| P3 | 18b | 0.1939(6) | 0.3447(6) | 0.4404(2) | 0.0000(4) | 1 |
| O1 | 18b | 0.4009(12) | 0.1493(12) | 0.4165(4) | 0.0200(4) | 1 |
| O2 | 18b | −0.1007(16) | −0.4270(18) | 0.3607(4) | 0.0087(4) | 1 |
| O3 | 18b | 0.2886(16) | 0.2757(17) | 0.5205(4) | 0.0165(4) | 1 |
| O4 | 18b | −0.0894(19) | −0.1789(12) | 0.3702(4) | 0.0060(4) | 1 |
| O5 | 18b | 0.3512(16) | 0.3964(14) | 0.4535(4) | 0.0074(4) | 1 |
| O6 | 18b | 0.1127(17) | 0.4287(18) | 0.4595(4) | 0.0252(4) | 1 |
| O7 | 18b | 0.0976(16) | 0.1752(13) | 0.4499(4) | 0.0117(4) | 1 |
| O8 | 18b | 0.1912(13) | 0.3686(9) | 0.3985(3) | 0.0094(4) | 1 |
| O9 | 18b | 0.5146(11) | 0.1948(10) | 0.4945(5) | 0.0101(4) | 1 |
| O10 | 6a | 2/3 | 1/3 | 0.5490(5) | 0.0087(4) | 1 |
| Mn-TCP | Ni-TCP | Cu-TCP | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Bond | Distance | BVP | BVS | Distance | BVP | BVS | Distance | BVP | BVS |
| M1-O8 | 2.331(12) | 0.37 | 2.309(10) | 0.4 | 2.383(9) | 0.33 | |||
| M1-O9 | 2.434(19) | 0.28 | 2.455(9) | 0.27 | 2.477(13) | 0.25 | |||
| M1-O4 | 2.48(2) | 0.25 | 2.478(14) | 0.25 | 2.489(17) | 0.24 | |||
| M1-O4 | 2.386(19) | 0.32 | 2.35 | 2.411(18) | 0.3 | 2.33 | 2.481(12) | 0.25 | 2.06 |
| M1-O5 | 2.420(17) | 0.29 | 2.435(17) | 0.28 | 2.457(12) | 0.27 | |||
| M1-O3 | 2.430(15) | 0.29 | 2.45(2) | 0.27 | 2.479(11) | 0.25 | |||
| M1-O6 | 2.255(16) | 0.46 | 2.249(18) | 0.47 | 2.313(11) | 0.39 | |||
| <M1-O> | 2.39 | 2.4 | 2.44 | ||||||
| M2-O7 | 2.39(2) | 0.32 | 2.385(19) | 0.32 | 2.343(9) | 0.36 | |||
| M2-O7 | 2.47(2) | 0.26 | 2.430(12) | 0.28 | 2.434(16) | 0.28 | |||
| M2-O9 | 2.287(19) | 0.42 | 2.276(13) | 0.43 | 2.355(11) | 0.35 | |||
| M2-O5 | 2.661(16) | 0.15 | 2.34 | 2.612(16) | 0.18 | 2.40 | 2.672(15) | 0.15 | 2.18 |
| M2-O3 | 2.322(15) | 0.38 | 2.290(17) | 0.42 | 2.349(14) | 0.36 | |||
| M2-O2 | 2.311(17) | 0.39 | 2.348(16) | 0.36 | 2.441(13) | 0.28 | |||
| M2-O1 | 2.435(18) | 0.28 | 2.422(14) | 0.29 | 2.418(12) | 0.29 | |||
| M2-O6 | 2.706(14) | 0.14 | 2.76(2) | 0.12 | 2.803(16) | 0.1 | |||
| <M2-O> | 2.45 | 2.44 | 2.48 | ||||||
| M3-O8 | 2.505(11) | 0.23 | 2.503(9) | 0.24 | 2.503(11) | 0.24 | |||
| M3-O8 | 2.643(17) | 0.16 | 2.634(12) | 0.17 | 2.614(15) | 0.17 | |||
| M3-O10 | 2.542(7) | 0.21 | 2.526(7) | 0.22 | 2.529(5) | 0.22 | |||
| M3-O5 | 2.411(19) | 0.3 | 1.83 | 2.451(15) | 0.27 | 1.86 | 2.414(14) | 0.3 | 1.85 |
| M3-O3 | 2.36(2) | 0.35 | 2.349(15) | 0.36 | 2.371(15) | 0.34 | |||
| M3-O2 | 2.526(16) | 0.22 | 2.535(18) | 0.22 | 2.561(11) | 0.2 | |||
| M3-O1 | 2.618(17) | 0.17 | 2.609(14) | 0.18 | 2.659(13) | 0.15 | |||
| M3-O6 | 2.594(18) | 0.18 | 2.534(15) | 0.22 | 2.514(12) | 0.23 | |||
| <M3-O> | 2.53 | 2.52 | 2.52 | ||||||
| M4-O1 | 2.431(14) | 0.29 | 2.472(12) | 0.26 | 2.524(11) | 0.22 | |||
| M4-O1 | 2.431(14) | 0.29 | 0.86 | 2.472(12) | 0.26 | 0.76 | 2.524(11) | 0.22 | 0.67 |
| M4-O1 | 2.431(14) | 0.29 | 2.472(12) | 0.26 | 2.524(11) | 0.22 | |||
| <M4-O> | 2.43 | 2.47 | 2.52 | ||||||
| M5-O7 | 2.194(19) | 0.47 | 2.217(17) | 0.42 | 2.296(13) | 0.36 | |||
| M5-O7 | 2.194(19) | 0.47 | 2.217(17) | 0.42 | 2.296(13) | 0.36 | |||
| M5-O7 | 2.194(19) | 0.47 | 2.217(17) | 0.42 | 2.296(13) | 0.36 | |||
| M5-O4 | 2.18(2) | 0.48 | 2.85 | 2.144(16) | 0.52 | 2.82 | 2.165(14) | 0.45 | 2.43 |
| M5-O4 | 2.18(2) | 0.48 | 2.144(16) | 0.52 | 2.165(14) | 0.45 | |||
| M5-O4 | 2.18(2) | 0.48 | 2.144(16) | 0.52 | 2.165(14) | 0.45 | |||
| <M5-O> | 2.19 | 2.18 | 2.23 | ||||||
| P1-O10 | 1.52(2) | 1.28 | 1.58(2) | 1.09 | 1.562(15) | 1.16 | |||
| P1-O9 | 1.589(15) | 1.08 | 1.580(11) | 1.1 | 1.509(12) | 1.34 | |||
| P1-O9 | 1.589(15) | 1.08 | 4.51 | 1.580(11) | 1.11 | 4.41 | 1.509(12) | 1.34 | 5.17 |
| P1-O9 | 1.589(15) | 1.08 | 1.580(11) | 1.1 | 1.509(12) | 1.34 | |||
| <P1-O> | 1.57 | 1.58 | 1.52 | ||||||
| P2-O4 | 1.606(16) | 1.03 | 1.598(11) | 1.05 | 1.536(10) | 1.25 | |||
| P2-O3 | 1.61(2) | 1.03 | 1.637(18) | 0.95 | 1.571(14) | 1.13 | |||
| P2-O2 | 1.663(18) | 0.88 | 3.86 | 1.60(2) | 1.04 | 4.02 | 1.514(18) | 1.32 | 4.84 |
| P2-O1 | 1.644(18) | 0.92 | 1.625(14) | 0.98 | 1.571(13) | 1.14 | |||
| <P2-O> | 1.63 | 1.62 | 1.62 | ||||||
| P3-O7 | 1.562(16) | 1.16 | 1.571(13) | 1.13 | 1.544(16) | 1.21 | |||
| P3-O8 | 1.562(14) | 1.17 | 1.583(12) | 1.1 | 1.521(10) | 1.29 | |||
| P3-O5 | 1.56(2) | 1.16 | 4.53 | 1.524(17) | 1.29 | 4.23 | 1.481(12) | 1.44 | 5.05 |
| P3-O6 | 1.60(2) | 1.05 | 1.65(2) | 0.92 | 1.581(12) | 1.1 | |||
| <P3-O> | 1.57 | 1.58 | 1.53 | ||||||
| Mn-TCP | Ni-TCP | Cu-TCP | β-TCP | |
|---|---|---|---|---|
| 1121 | 1119 | 1121 | 1118 | ν3 (P-O) |
| 1096 | 1096 | 1098 | 1098 | |
| 1081 | 1081 | 1081 | 1080 | |
| 1063 | 1062 | 1062 | 1064 | |
| 1043 | 1044 | 1041 | 1042 | |
| 1021 | 1023 | 1024 | 1022 | |
| 1005 | 1005 | 1005 | 1007 | |
| 965 945 933 | 966 957 940 924 | 969 957 945 933 | 969 943 | ν1 (P-O) |
| 610 603 589 579 554 546 | 609 602 590 580 547 | 609 604 590 580 546 | 610 606 591 579 552 544 | ν4 (P-O) O-P-O |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altomare, A.; Rizzi, R.; Rossi, M.; El Khouri, A.; Elaatmani, M.; Paterlini, V.; Della Ventura, G.; Capitelli, F. New Ca2.90(Me2+)0.10(PO4)2 β-tricalcium Phosphates with Me2+ = Mn, Ni, Cu: Synthesis, Crystal-Chemistry, and Luminescence Properties. Crystals 2019, 9, 288. https://doi.org/10.3390/cryst9060288
Altomare A, Rizzi R, Rossi M, El Khouri A, Elaatmani M, Paterlini V, Della Ventura G, Capitelli F. New Ca2.90(Me2+)0.10(PO4)2 β-tricalcium Phosphates with Me2+ = Mn, Ni, Cu: Synthesis, Crystal-Chemistry, and Luminescence Properties. Crystals. 2019; 9(6):288. https://doi.org/10.3390/cryst9060288
Chicago/Turabian StyleAltomare, Angela, Rosanna Rizzi, Manuela Rossi, Asmaa El Khouri, Mohammed Elaatmani, Veronica Paterlini, Giancarlo Della Ventura, and Francesco Capitelli. 2019. "New Ca2.90(Me2+)0.10(PO4)2 β-tricalcium Phosphates with Me2+ = Mn, Ni, Cu: Synthesis, Crystal-Chemistry, and Luminescence Properties" Crystals 9, no. 6: 288. https://doi.org/10.3390/cryst9060288