3.1. X-ray Diffraction
After initial compression and gas-loading with helium to 0.96(5) GPa, the unit cell parameters of melamine were
a = 10.410(6) Å,
b = 7.465(2) Å,
c = 7.086(6) Å, and β = 113.89(4)° in the space group setting of P2
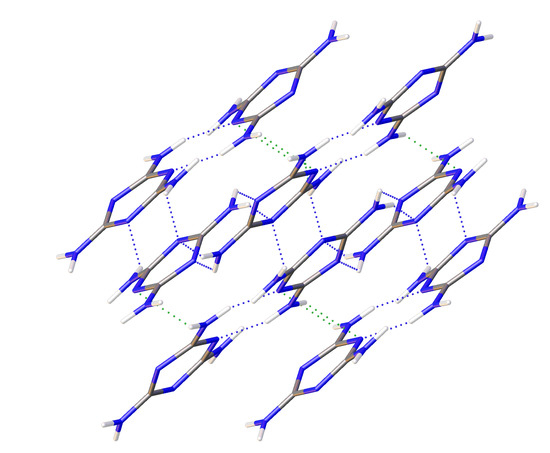
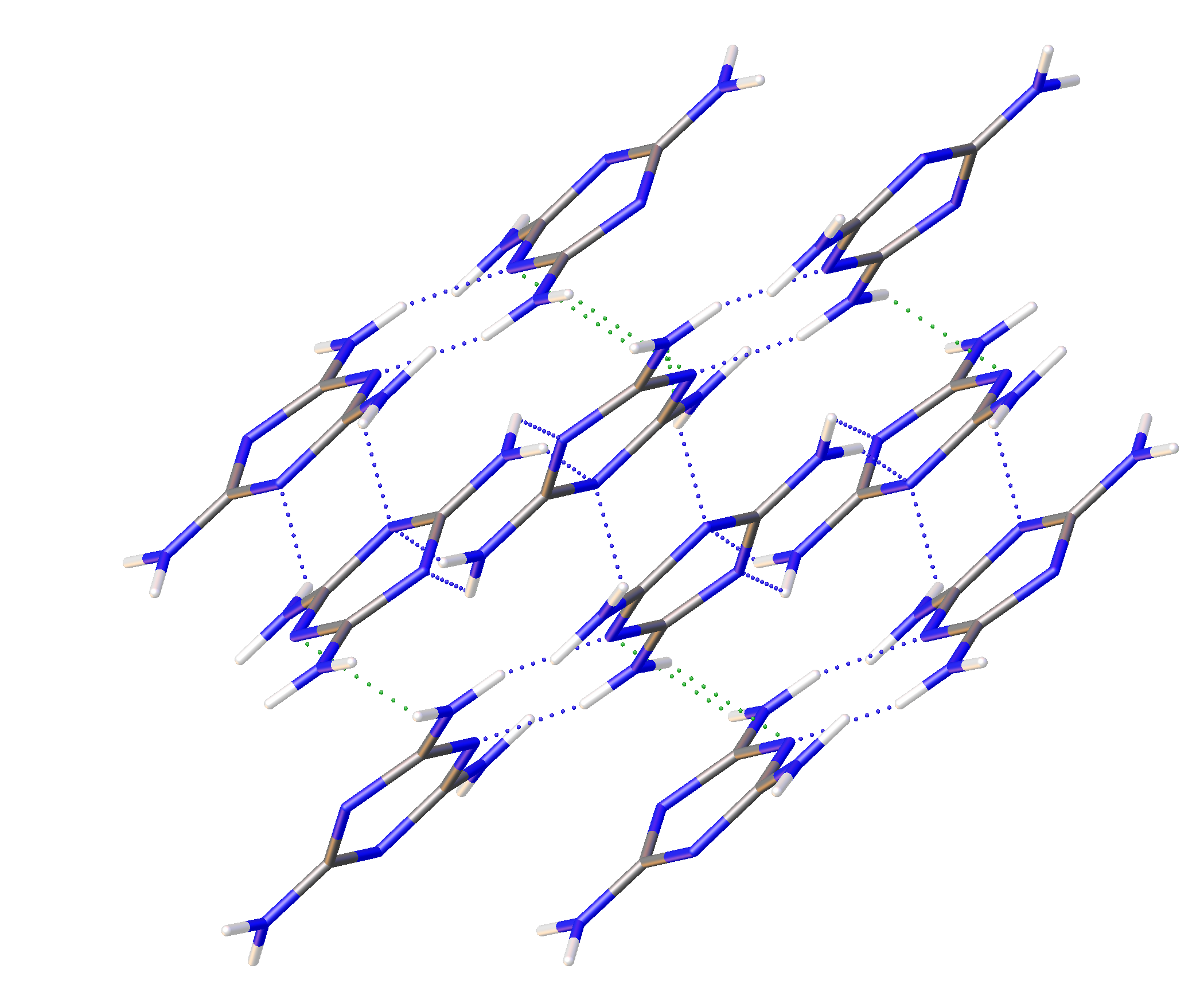
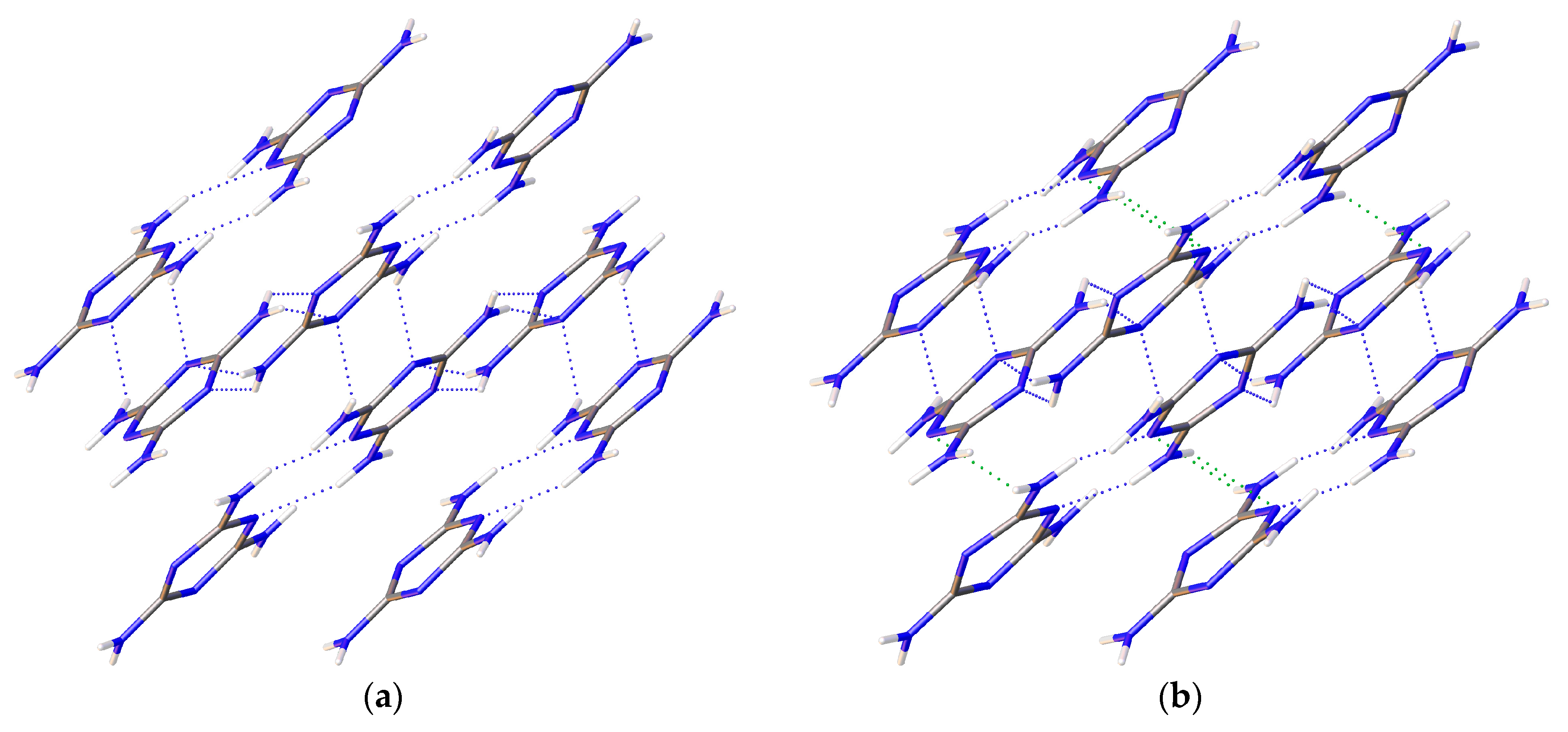
1/a. Multiple data sets, between 2010 and 2015, provide nineteen pressure points where the compressional effects on the structure of melamine was observed. All nineteen data points were used for equation of state fitting, whereas seventeen were of sufficient data quality to permit hydrogen bonding geometry analysis. The crystal structures obtained during the compression pathway show little deviation in the overall molecular geometry with pressure. At 0.96(5) GPa, each individual melamine unit is connected to its neighbors via eight N-H···N hydrogen bonds, as is the case at ambient pressure, forming layers of melamine molecules, approximately parallel to (010), that are hydrogen bonded within, and also linked to neighboring layers (
Figure 1a). Additionally, in neighboring layers, there are pairs of melamine molecules whose central rings lie within parallel planes and overlap. Neighboring pairs of molecules are offset by 33.8(9) degrees, forming the corrugated structural motif. With each pressure step up to 36.2(1) GPa, full structure refinement was conducted, initially based on the structure of melamine reported from ambient pressure neutron diffraction experiments [
30]. Selected data collection and refinement information from initial and final pressure steps for this structure are listed in
Table 1. The evolution of unit cell parameters with pressure is listed in
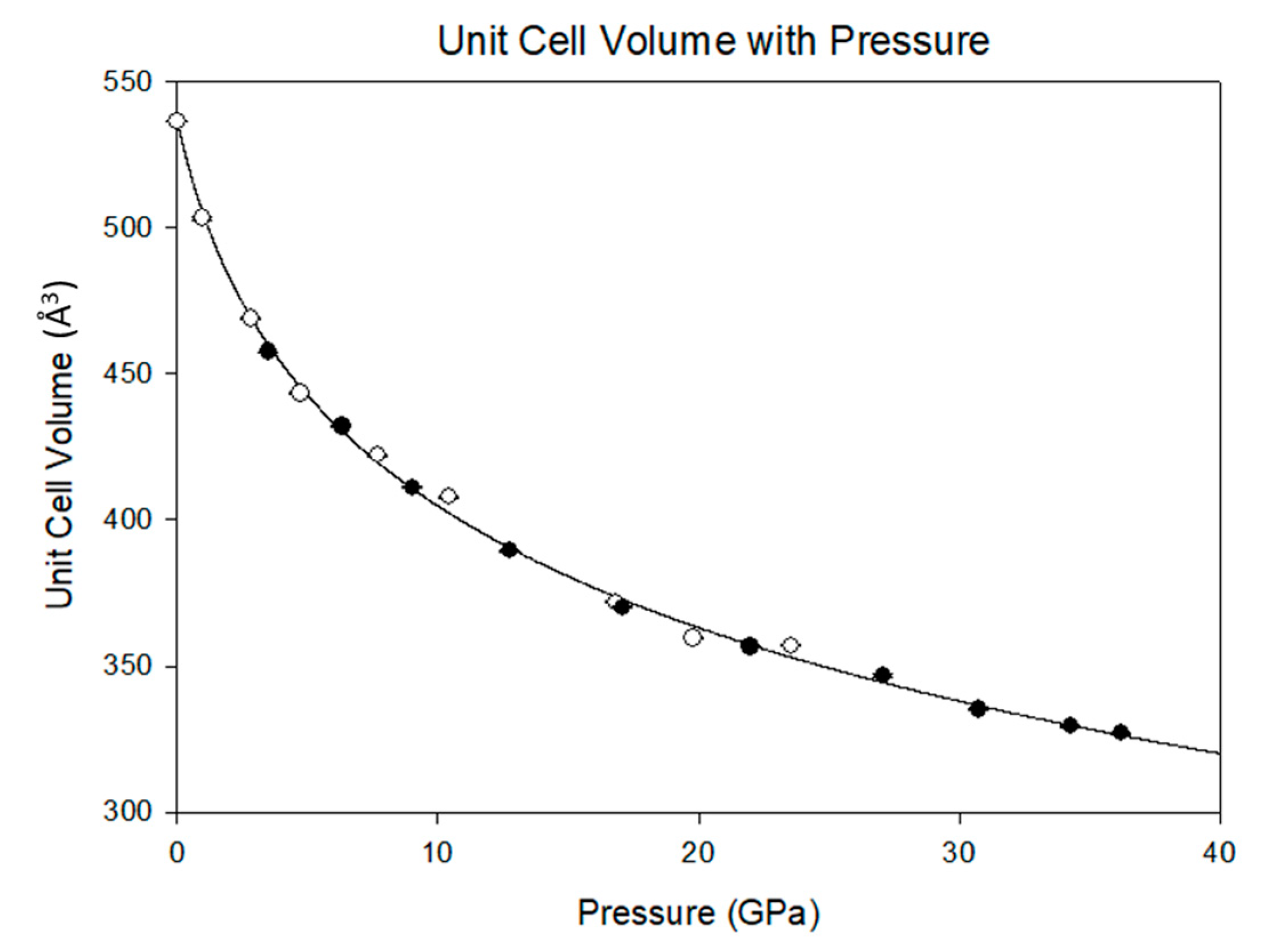
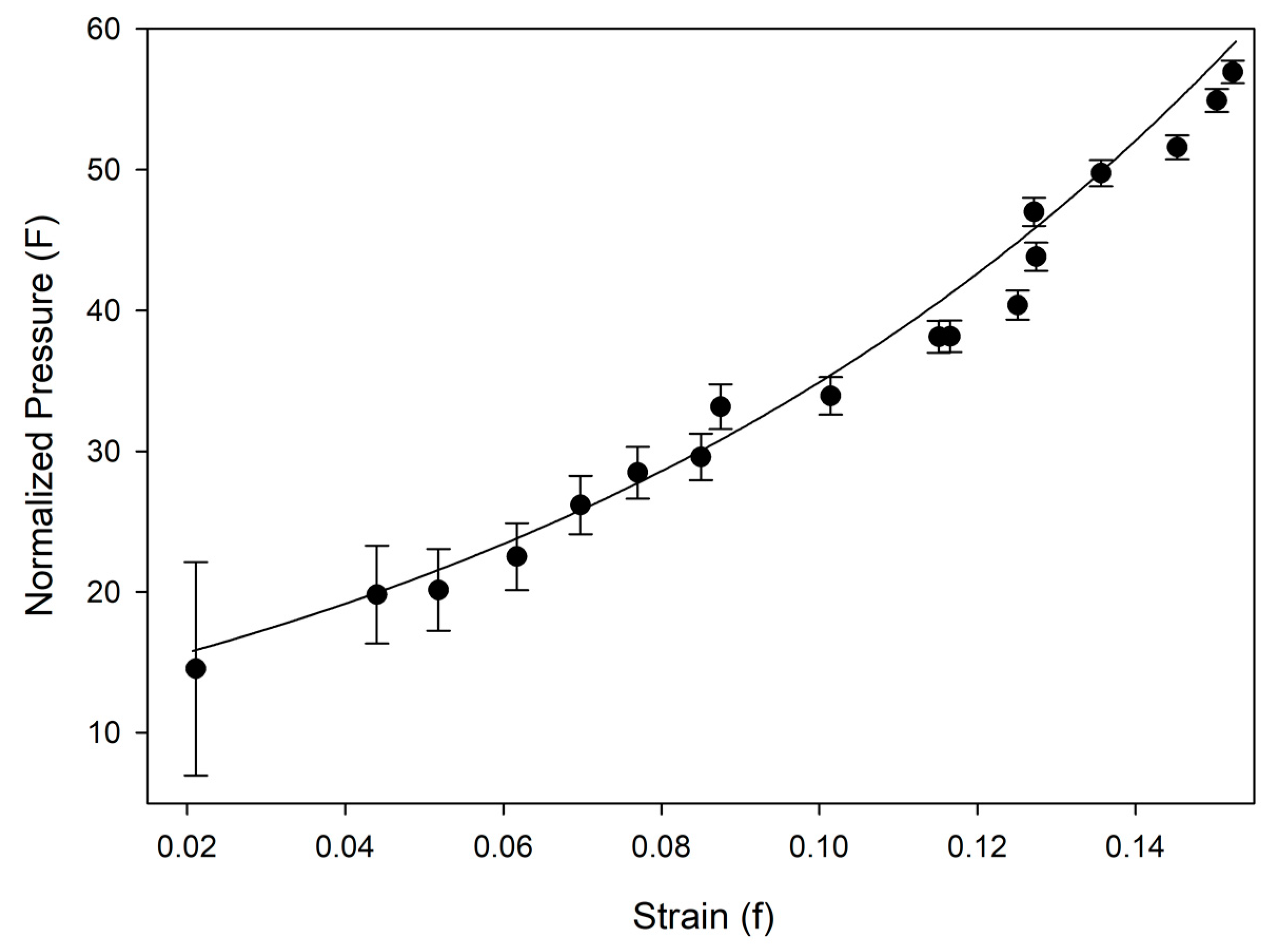
Table 2. Equation of state and linearized axial compressibilities are depicted in Figures 4 and 5, with the accompanying F-f plot depicted in
Figure A1. Additional information, including fractional atomic coordinates, symmetry operators, and selected bond lengths and angles are listed in
Appendix A Table A1,
Table A2,
Table A3 and
Table A4. On compression beyond 36.2(1) GPa in helium or 31.8(1) GPa in neon, a reversible phase transition to a twinned triclinic polymorph (where
a = 6.08(1) Å,
b = 7.267(2) Å,
c = 7.82(1) Å, and
α = 78.25(4)°, β = 80.1(2)°,
γ = 80.48(4)° at 38.9(1) GPa) was seen, with pronounced changes in the diffraction pattern, as shown in
Figure 2. This transition was reversible, however, further compression past approximately 45 GPa in helium resulted in irreversible amorphization, accompanied by loss of diffraction signal and easily identifiable change of the color and opacity of the crystal (
Figure 3).
3.2. Equation of State and Bond Compressibility
Previously, Ma et al. [
15] documented the P-V equation of state of melamine, but only to approximately 15 GPa. Although that study describes a transformation to a triclinic structure below 2 GPa, there exist similarities between that study and the current results. In both cases, there is a precipitous drop in the unit cell volume below 5 GPa. However, in the present study, there is no evidence of a phase transformation. Rather, there is a smooth and continuous reduction in the unit cell volume that is well described by the Vinet equation of state [
25,
26].
where
,
V0 is the initial cell volume,
K0 is the isothermal bulk modulus, and
K′ is its derivative when the pressure is equal to zero. The output of this equation of state, with V
0 fixed to the ambiently determined value of 536.7(2) Å
3, is
K0 = 12.9(8) GPa, and
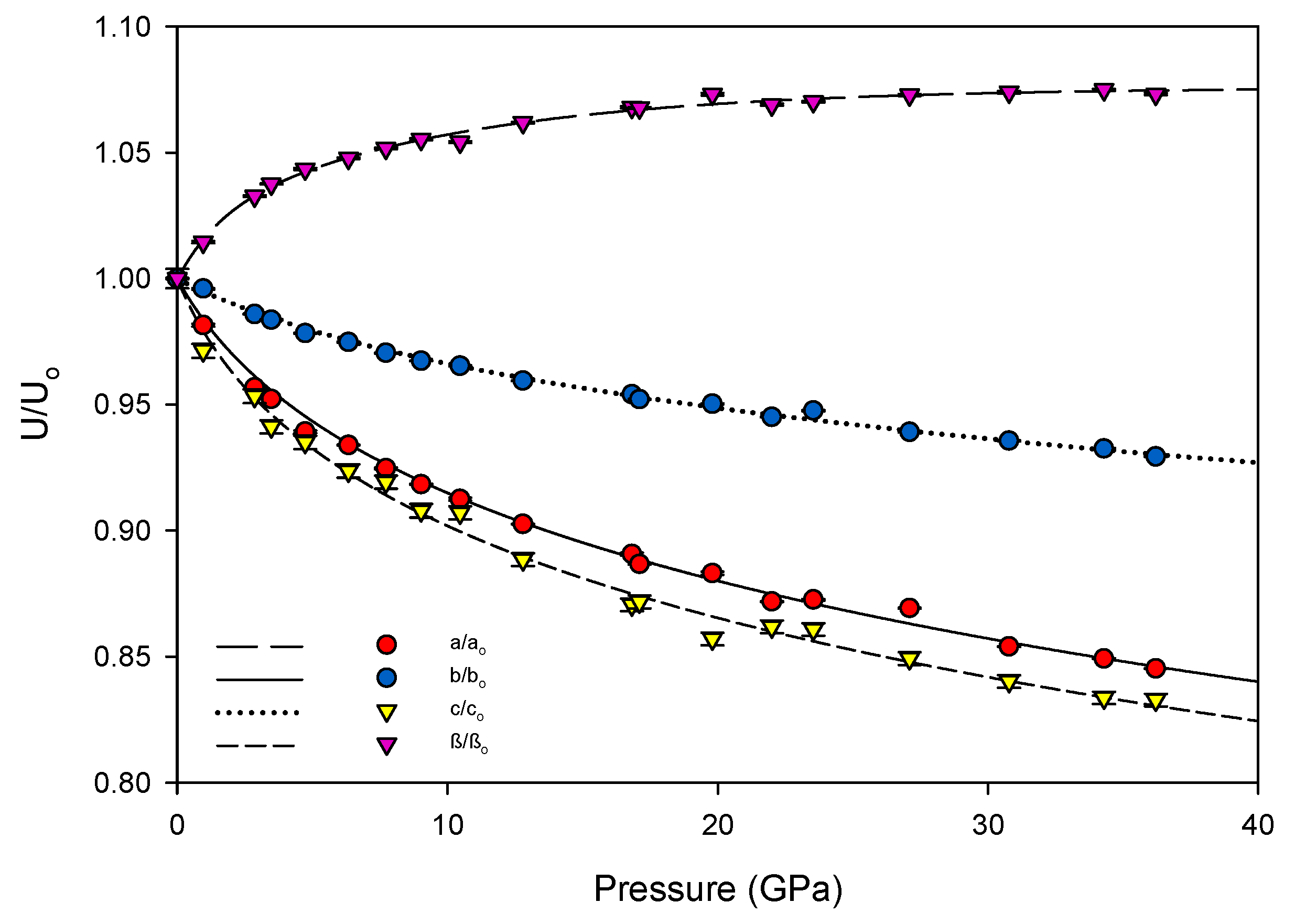
K′ = 7.4(3). The linear axial compressibilities (defined as β
l0 = 1/3
Kl0) [
31] were determined using a linearized version of the Vinet equation. The increase of the unit cell’s β angle was well-described by a three-parameter exponential rise-to-maximum function. The progression of each of these values with pressure can be seen in
Figure 4 and
Figure 5. By 36.2(1) GPa the unit cell volume has experienced a 40 percent collapse, driven primarily by the shortening of the
a and
c axes. As each axis shrinks the β angle opens, reflecting the shift of molecules with respect to one another, rising to a predicted maximum of 120.7(1) degrees at 40 GPa.
When comparing the bulk moduli other common six-membered ring molecules such as benzene (5.5 GPa) or aniline (5.44 GPa), as well as extended structures with ring motifs such as graphite (33.8 GPa), the compressibility of melamine is closer to the former [
32,
33,
34]. This can be readily explained by the type of dominating intermolecular interactions and arrangement of molecules with respect to each other, where graphite is held more rigidly in covalently-bonded planar sheets, while individual benzene or aniline molecules are not covalently bonded to their neighbors. Both polymorphs of aniline also form extended stacked layers held together by hydrogen bonding, where its singular amino group participates in N-H···N and N-H···π hydrogen bonds [
33,
35]. Individual aniline units do not experience significant structural or energetic modification in pressure, yet its hydrogen bonds decrease in length to the point of destabilization [
35]. Benzene does not share hydrogen atoms below a theorized point of metallization [
32,
36], and π-π interactions act as the driving intermolecular interaction with increasing pressure. For solid benzene-III, a recent theoretical study [
36] suggested that at 50 GPa, despite an almost two-fold reduction in unit cell volume, the intramolecular bond lengths stay basically unchanged. A similar situation appears to occur with melamine: while the intramolecular deformation is minimal, intermolecular hydrogen bonds accommodate pressure changes within the crystal without departing from the original space group and basic structure.
In comparison to energetic materials, the bulk modulus of melamine is analogous to or slightly less than β-HMX (12.4 GPa), α-RDX (13.9 GPa), and TATB (16.2 GPa) [
37]. Interestingly, these values were obtained using powder X-ray diffraction techniques in methanol-ethanol-water, argon, and hexane pressure media, respectively [
38,
39,
40]. The use of powders, as well as non-hydrostatic and non-inert pressure media likely introduces similar uncertainties and irregularities as those encountered with prior high-pressure experiments of melamine. Significant variations in compressibility for these compounds, including elastic constants and phase transformation behavior, have been shown to greatly depend on the hydrostatic character of the pressure media [
41,
42]. Hydrogen bonding has also been demonstrated to be the driving interaction in crystalline networks of RDX and TATB, where intermolecular hydrogen bonding networks could be disrupted by participating polar-solvent pressure media or inter-grain boundaries [
43]; when compressional hydrostaticity is ensured, highly energetic molecules like TATB have been shown to remain crystalline past 100 GPa [
44].
For inorganic substances and minerals, a very common notion for understanding a compression mechanism is the Rigid Unit Mode model, in which it is assumed that each subunit (i.e., tetrahedra or octahedra) is very stiff compared to the framework in which it resides [
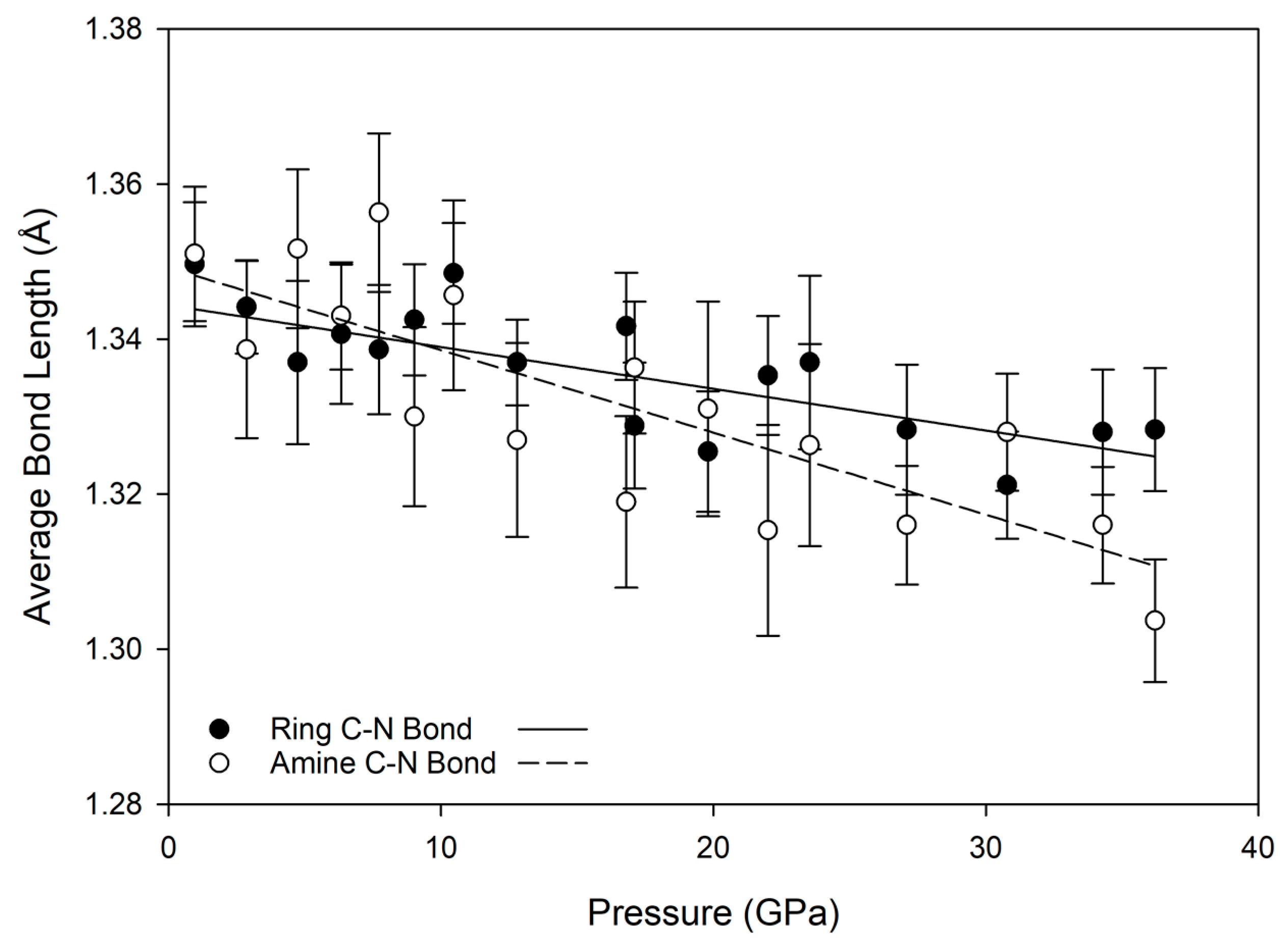
45]. As a result, rotation of whole units is preferable to alteration of bond lengths within a unit. Although it is an organic molecule, within this frame of reference melamine subunits (primarily the aromatic
s-triazine ring and amine nitrogen) can be considered as the inflexible subunit, and are relatively unchanged with pressure. In contrast, intermolecular hydrogen bonds greatly compress and shift position with pressure, and individual NH
2 units have some ability to rotate, in order to accommodate pressure changes and avoid repulsive H-H interactions. This is evident from the relative lack of change in carbon-nitrogen bond lengths with substantial increases in pressure, as shown in
Figure 6; for both ring and amine carbon-nitrogen bonds, the bond length decrease is less than about 0.05 Å, while the donor-acceptor lengths of hydrogen bonds decrease by nearly 0.5 Å over the same compression path.
The compressional behavior of melamine from the perspective of an individual molecular unit can also be visualized through Hirshfeld surfaces. This method of crystal analysis condenses properties such as interatomic angles and distances, crystal packing schemes, and intermolecular interactions into models that can be easily and qualitatively interpreted, yet are derived from quantitative analysis [
46,
47]. Hirshfeld surfaces are differentiated from other molecular surface representations such as electron density maps or van der Waals surfaces by accounting for both a molecule and its proximity to its nearest neighbors, making it well-suited for the analysis of molecular crystals. Two surfaces, the shape index and normalized contact distance (d
norm), are particularly useful for describing the packing of molecular crystals such as melamine.
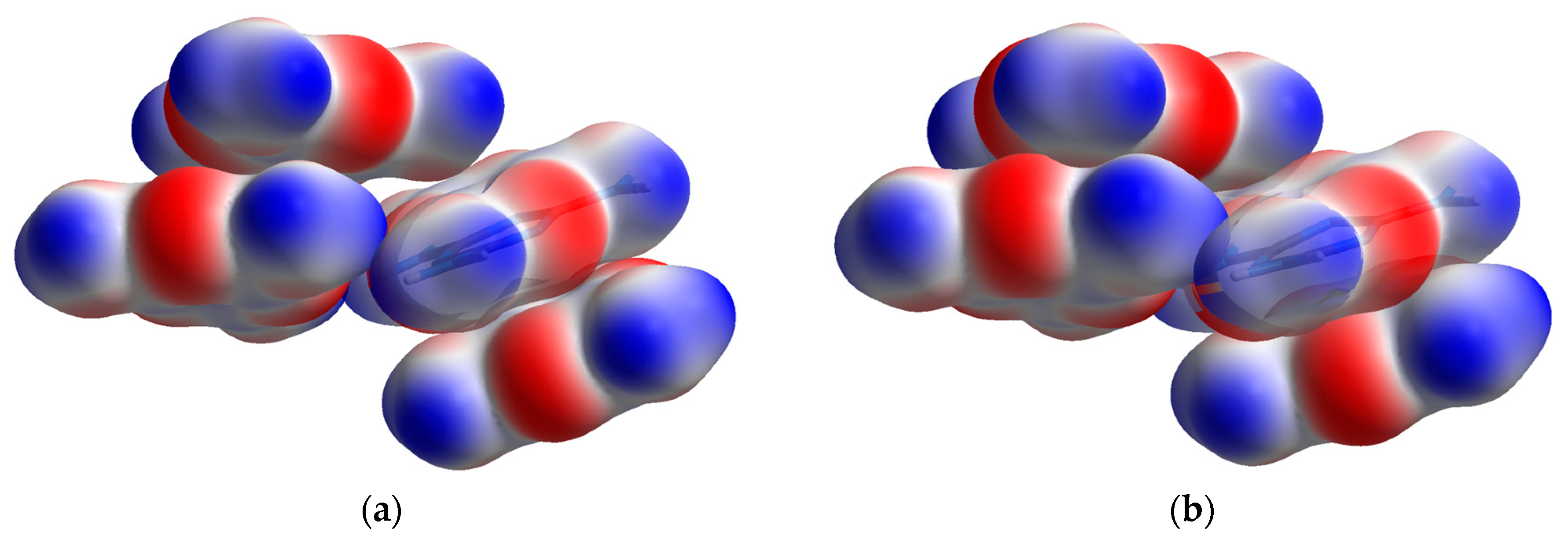
The shape index is a Hirshfeld surface that identifies concave or convex areas of a molecule’s surface based on charge density. Red areas indicate concave areas, whereas blue indicates convex. For the purpose of examining compressional behavior, any change in the intramolecular geometry is distinguishable by changes in color. For melamine, the relative lack of change between low and high pressure is apparent (
Figure 7), mirroring the small changes in covalent bond lengths and overall inflexibility of the aromatic component with pressure.
Hirshfeld surfaces of the normalized contact distances tell the other half of the story, and are shown in
Figure 8; this parameter describes the internal (d
i) and external (d
e) contact distances of the Hirshfeld surface to the nearest atomic nucleus, normalized by the van der Waals (vdW) radii of the atoms involved. The result is a surface where intermolecular contacts longer than the sum of the atoms’ vdW radii are displayed in blue, and contacts shorter than the vdW radii are displayed in red. At low pressures, this highlights the points of contact for the N-H···N hydrogen bonds, where the bond contracts the intermolecular distance. As pressure increases, contact points with distances shorter than the vdW radii appear on the previously non-interacting amine nitrogen and atoms of the central ring.
3.3. Hydrogen Bonding Behavior with Pressure
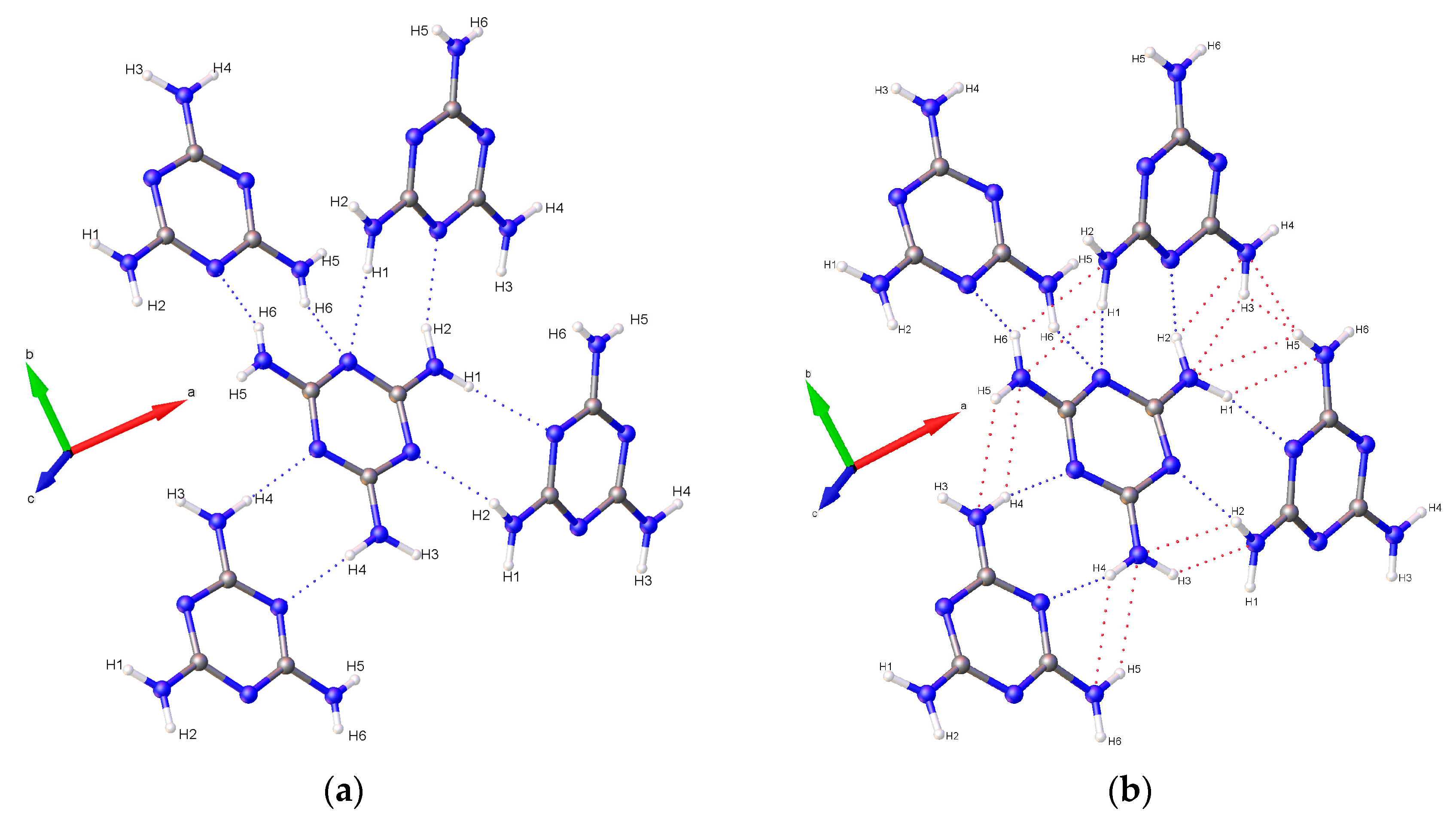
At ambient and low-pressure conditions, individual melamine molecules are linked with neighbors through pairs of complementary hydrogen bonds connecting amino hydrogens exclusively to ring-based nitrogen atoms. In previous ambient-pressure X-ray and neutron diffraction studies, it was observed that of the six symmetry independent hydrogen atoms in NH
2 groups, only four strongly participate in hydrogen bonding [
30]. The two remaining hydrogen atoms are subject to hindrances that prevent strong hydrogen bonding interactions, and are denoted in this study as H3 and H5. In the case of H5, the hydrogen atom is in close contact with another H5 on a neighboring molecule, and repulsive interaction occurs as distance decreases. For H3, steric hindrance prevents it from being sufficiently close to a ring nitrogen acceptor atom, allowing only weak interaction with a NH
2 group on a neighboring molecule. Each ring nitrogen atom also acts as a hydrogen bond acceptor for a total of four bonds per ring, with one nitrogen (denoted here as N4) acting as acceptor for two bonds (
Figure 1a).
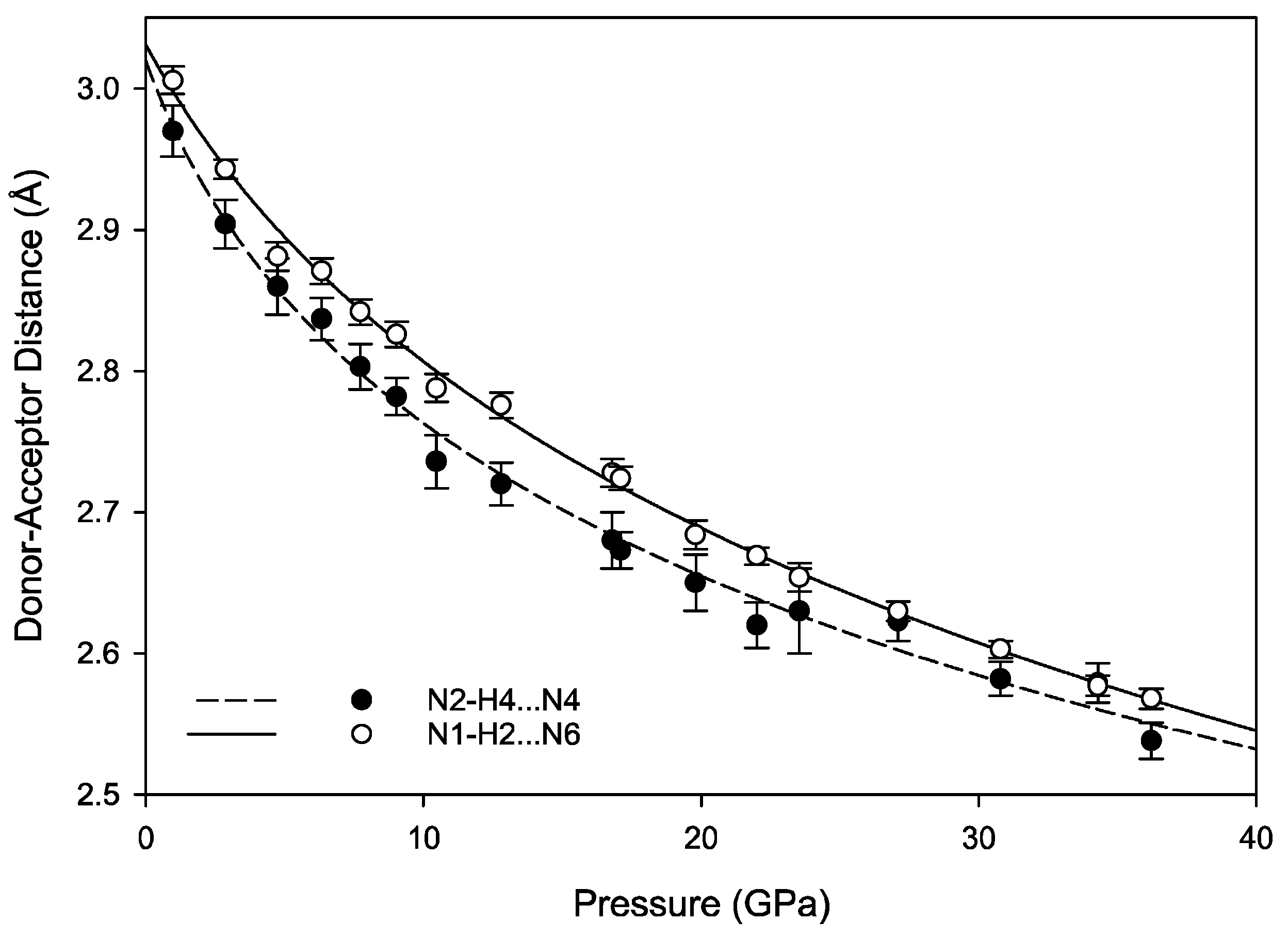
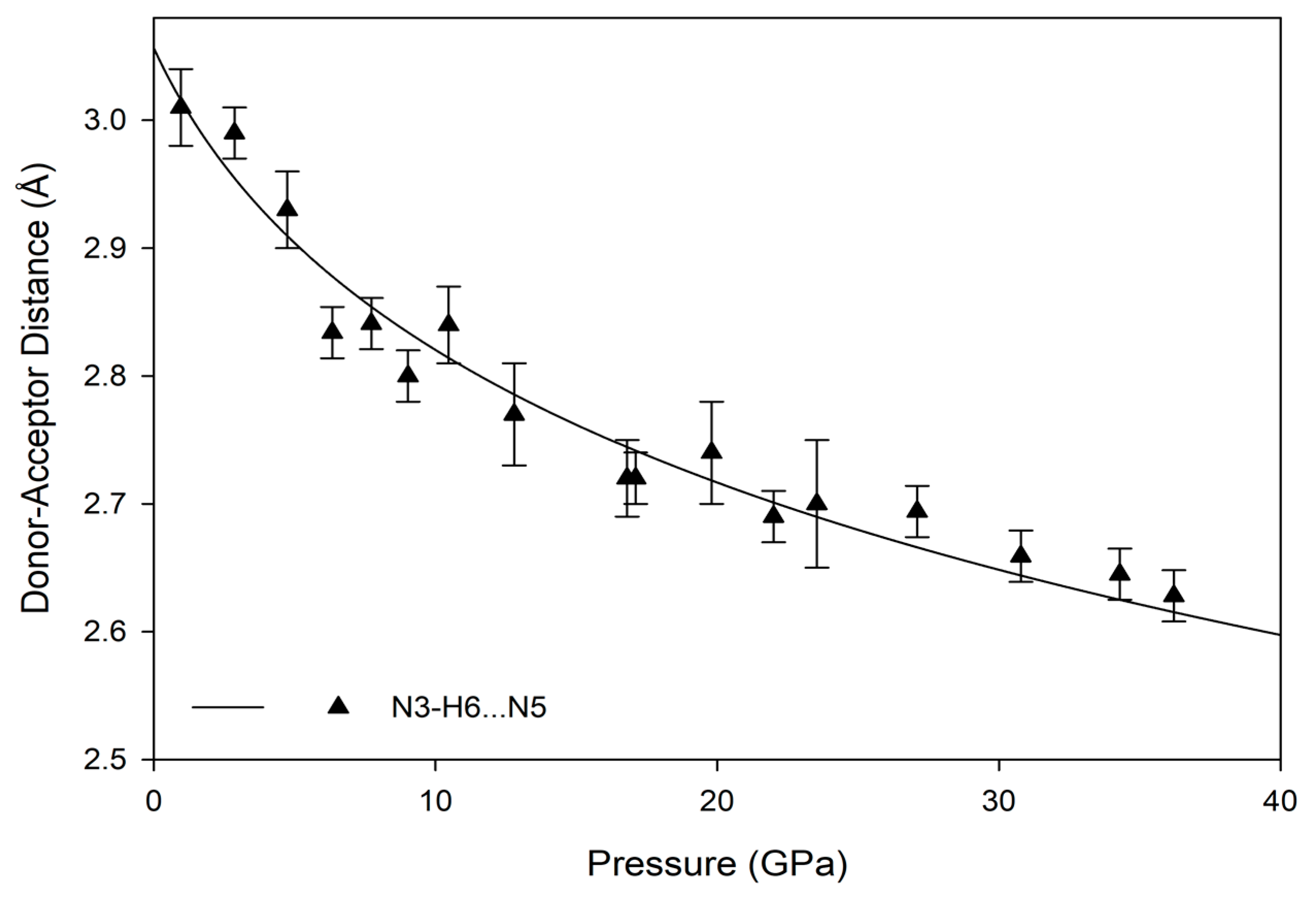
The hydrogen bonds in melamine can also be distinguished by whether they link molecules within a given corrugated plane or between them; for instance, the decrease in donor-acceptor distance for intra-plane molecules decreases smoothly as a function of pressure akin to the behavior of the unit cell parameters, while the inter-plane behavior is less consistent (
Figure 9 and
Figure 10). This is likely caused by the larger influence of the intra-plane bonding, as there are more hydrogen bonds within a layer, as well as small amounts of rotation and torsion to accommodate the increased intra-layer bonding and repulsive interactions between close-contact hydrogens. Ultimately, the intermolecular hydrogen bonds are capable of significant shortening with response to pressure, without any significant changes in the pattern of the original hydrogen bonds. Compression through 36.2(1) GPa decreases donor-acceptor distances substantially, which increases the covalent character of a bond and increases its strength [
48].
Notably, none of the original hydrogen bonds present at ambient pressures are broken below the phase transition pressure; instead, new hydrogen bonds between amino hydrogens and ring nitrogens form by 9.0(1) GPa and persist until at least 36.2(1) GPa (
Figure 11). As pressure increases further, amino groups are pushed into close contact with one another, and interactions between oppositely aligned N-H atoms occur as each hydrogen is brought closer to an opposing nitrogen’s lone pair of electrons. These interactions, although primarily electrostatic in character, are stabilized by their anti-parallel orientation to one another, with pressure overriding unfavorably shallow D-H-A angles, steric crowding, and repulsive interactions. At 36.2(1) GPa, sufficiently short bond distances indicate bi- and tri-furcation of these bonds, as shown in
Figure 1b, by the previously un-bonded H3 and H5 atoms.
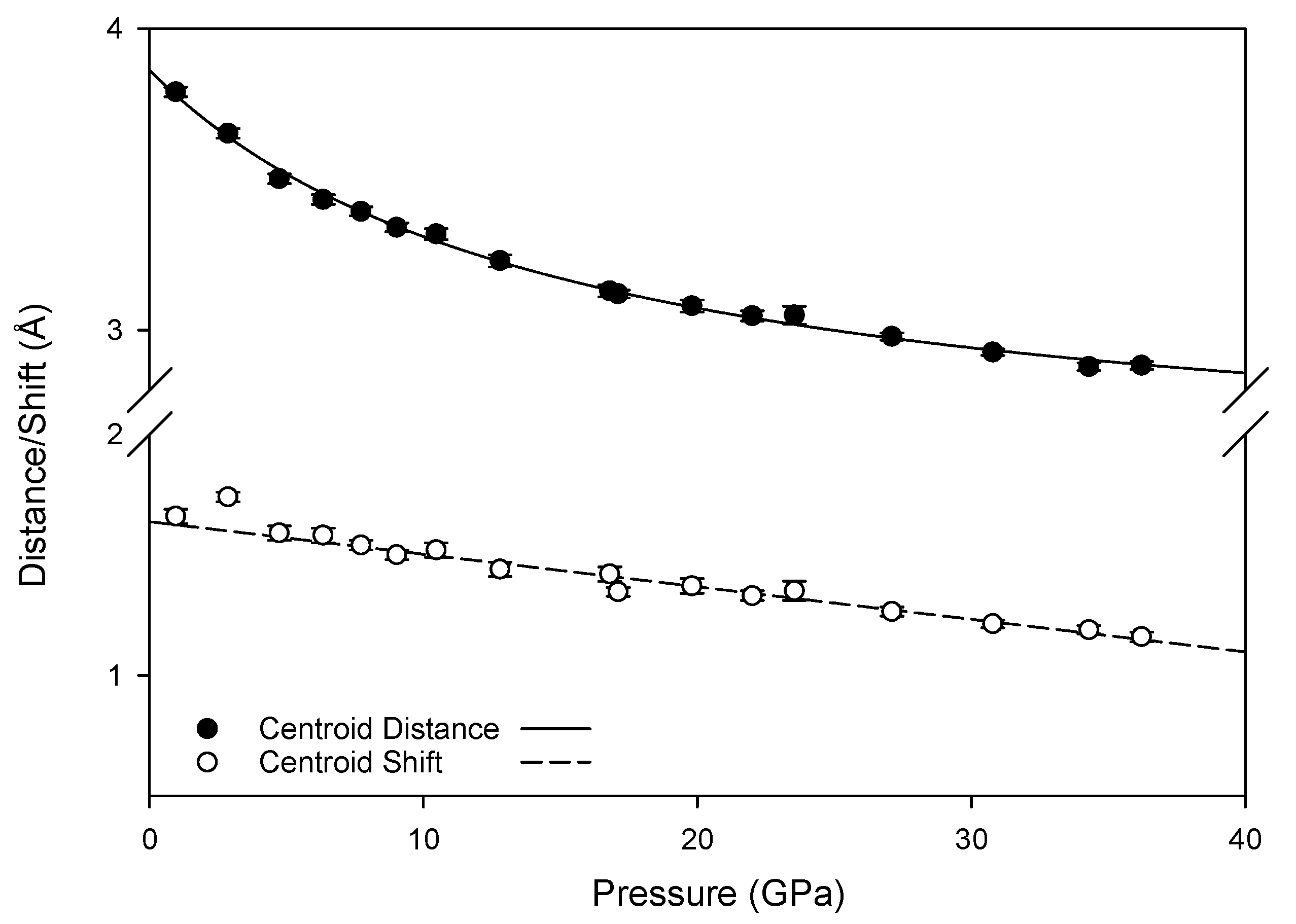
The new pressure-induced hydrogen bonds are not sufficiently strong to create a stable high-pressure configuration, as the transformation between monoclinic and triclinic phases is reversible. This is However, the new pressure-induced hydrogen bonds with ring nitrogen atoms may foster changes in the intermolecular interactions of π electrons in the high pressure phase; at ambient conditions, inter-layer hydrogen bonds link molecules where the centroid planes of the rings are parallel to one another, resulting in a skewed parallel-displaced arrangement where an electron-rich ring nitrogen is roughly aligned with a moderately electron-deficient ring center. At high pressures, the new hydrogen bonds also link molecules whose planes are at an offset to one another, introducing an interaction between these rings not experienced at lower pressures. Furthermore, compression reduces the distance and shift between the ring centroids (depicted in
Figure A2), as well as reducing the angle between offset pairs of molecules (
Figure 12), increasing the likelihood of extended interactions between multiple pairs of molecules. Although the term “π-π stacking” does not correctly describe the contact between neighboring melamine molecules [
49,
50], the distances and angles between ring centroids are within limits for attractive electrostatic π-σ interactions at both ambient and high pressures [
49,
51,
52]. However, the N-H···N hydrogen bonds ultimately direct the supramolecular changes in the melamine crystal; this provides exceptional stability when compared to un-substituted
s-triazine, which does not have the ability to act as a hydrogen bond donor [
49,
53,
54].
3.4. Raman Spectroscopy
In general, Raman spectroscopy is an excellent tool for detecting pressure-induced structural phase transitions in solids, which usually manifest themselves as discontinuous changes in vibration mode behavior. For instance, symmetry lowering related to displacive phase transitions typically results in splitting of Raman peaks. The case of molecular crystals, however, is often more complicated than simple inorganic solids. The starting crystal symmetry is often lower, and the number of Raman modes can be very significant. At the same time, there are more types of competing interatomic and intermolecular interactions (e.g., hydrogen bonds, van der Waals forces, electrostatic interactions, charge transfer), which affect the vibration force constants. At high pressure the balance between these various interactions changes and may cause discontinuities in the Raman mode behavior unrelated to first-order structural phase transitions. An example of such was found in benzene, for which Raman experiments [
55] described the existence of phase transitions between the II-III and III-IV phases at about 4 and 11 GPa. However, later studies combining both IR spectroscopy and powder X-ray diffraction cast doubt on tose proposed transitions, as the observed discontinuities and changes in vibrational modes did not correspond to symmetry-altering first-order structural changes [
32].
The ambient pressure Raman spectrum of melamine was first quantitatively interpreted in terms of mode assignment by Schneider and Schrader [
56]. The assignment of the collective ring vibration modes can be made by analogy to the unsubstituted parent-molecule of
s-triazine [
57]. There are also several recent Raman studies of solid salts of melamine [
58,
59] that are useful in interpretation of individual vibration modes, such as those from hydrogen bonds. In the spectral range covered by our experiments, the Raman spectrum of melamine can be divided into three regions: the 250–1200 relative cm
−1 is the collective ring vibration mode region, from 1400–1900 cm
−1 is the C-NH
2 vibration mode range, and the 3000–3700 cm
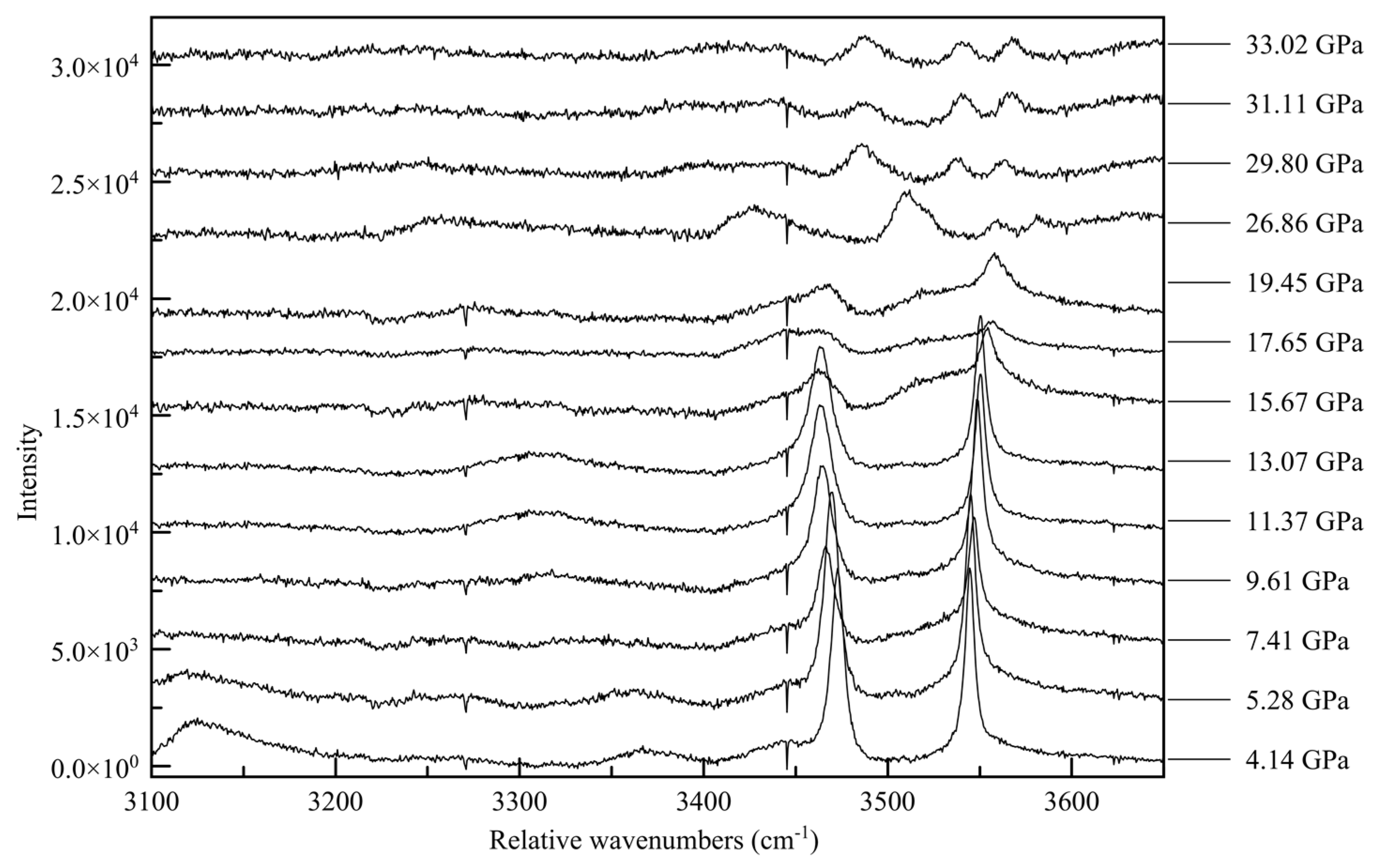
−1 range is the N-H vibration mode range. This last region proves to be the most informative; at ambient pressure there are four distinct Raman peaks, at 3128, 3333, 3420, and 3471 relative cm
−1 [
16]. The two peaks at lower wavenumbers are very broad and quite asymmetric, whereas the peaks at higher wavenumbers are sharp and symmetric, as seen in
Figure 13.
The non-uniformity of the hydrogen bonding interactions can also be seen in the Raman spectra. The sharp Raman peaks of 3420 and 3471 cm−1 at close to ambient pressure conditions can be associated with the non-hydrogen bonded N-H vibrations, such as from H3 and H5. The broad features, reminiscent of the O-H peak shapes in other hydrogen-bonded crystals, e.g., solid H2O, correspond to the two groups of hydrogen-bonded NH2. With increasing pressure, as intermolecular distances are reduced and hydrogens are forced into closer vicinity of nitrogen atoms, these peaks broaden and fade. As these previously non-interacting atoms are forced into hydrogen bonding interactions, the signal for each is muddied until the point of transition.
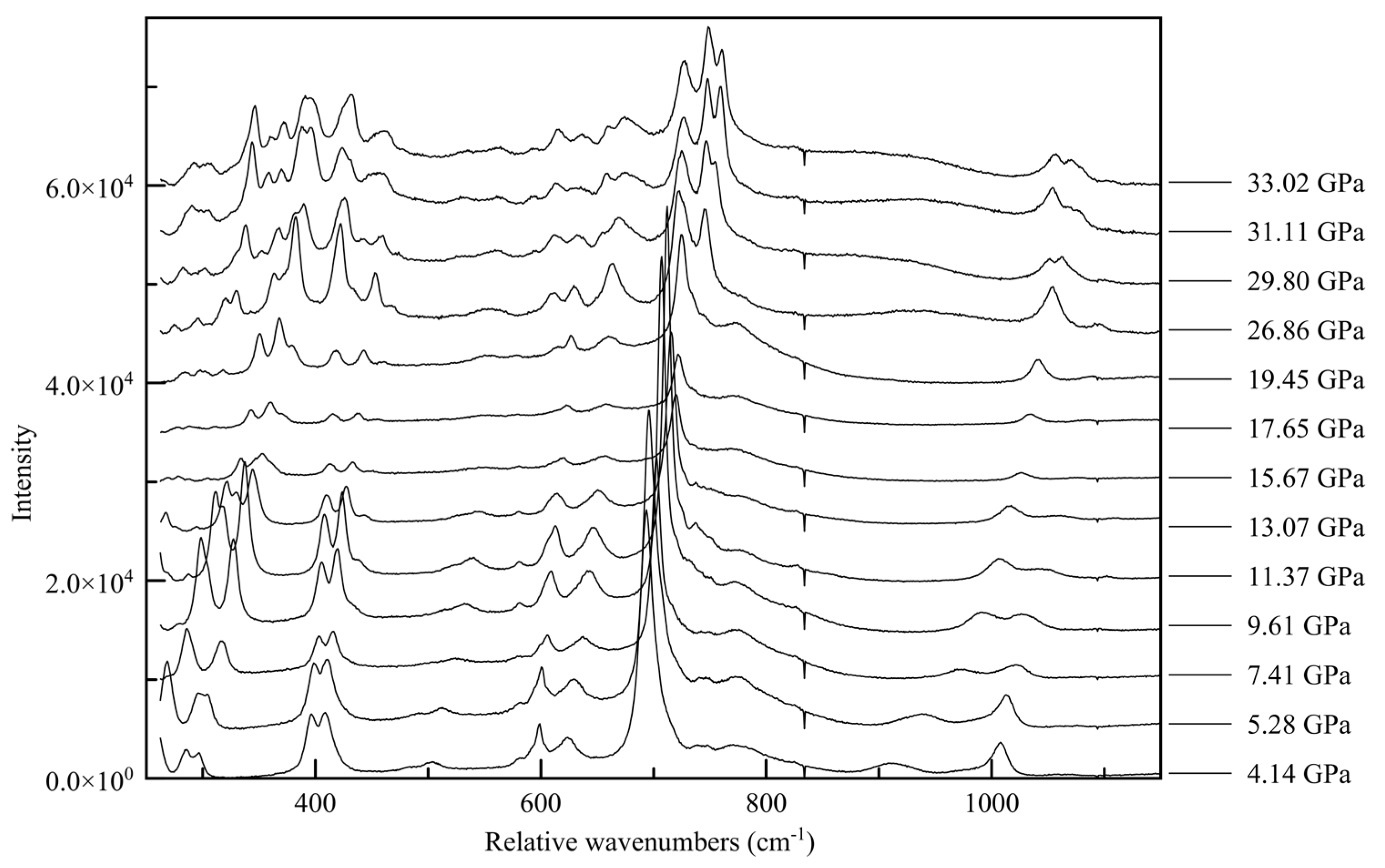
The changes in the Raman spectra accompanying the monoclinic-to-triclinic phase transition, observed in diffraction data at approximately 30 GPa in Ne and 38 GPa in He, are quite pronounced in all three spectral ranges, with the appearance of new spectral features often occurring slightly before the observed transition pressure. In the ring breathing mode range the high wave number component of the 750 cm
−1 peak splits into a doublet, as does the 1100 cm
−1 peak, shown in
Figure 14. This is indicative of a change in the interaction between inter-layer molecules, potentially between newly crystallographically and energetically inequivalent ring systems after the phase transition [
60,
61,
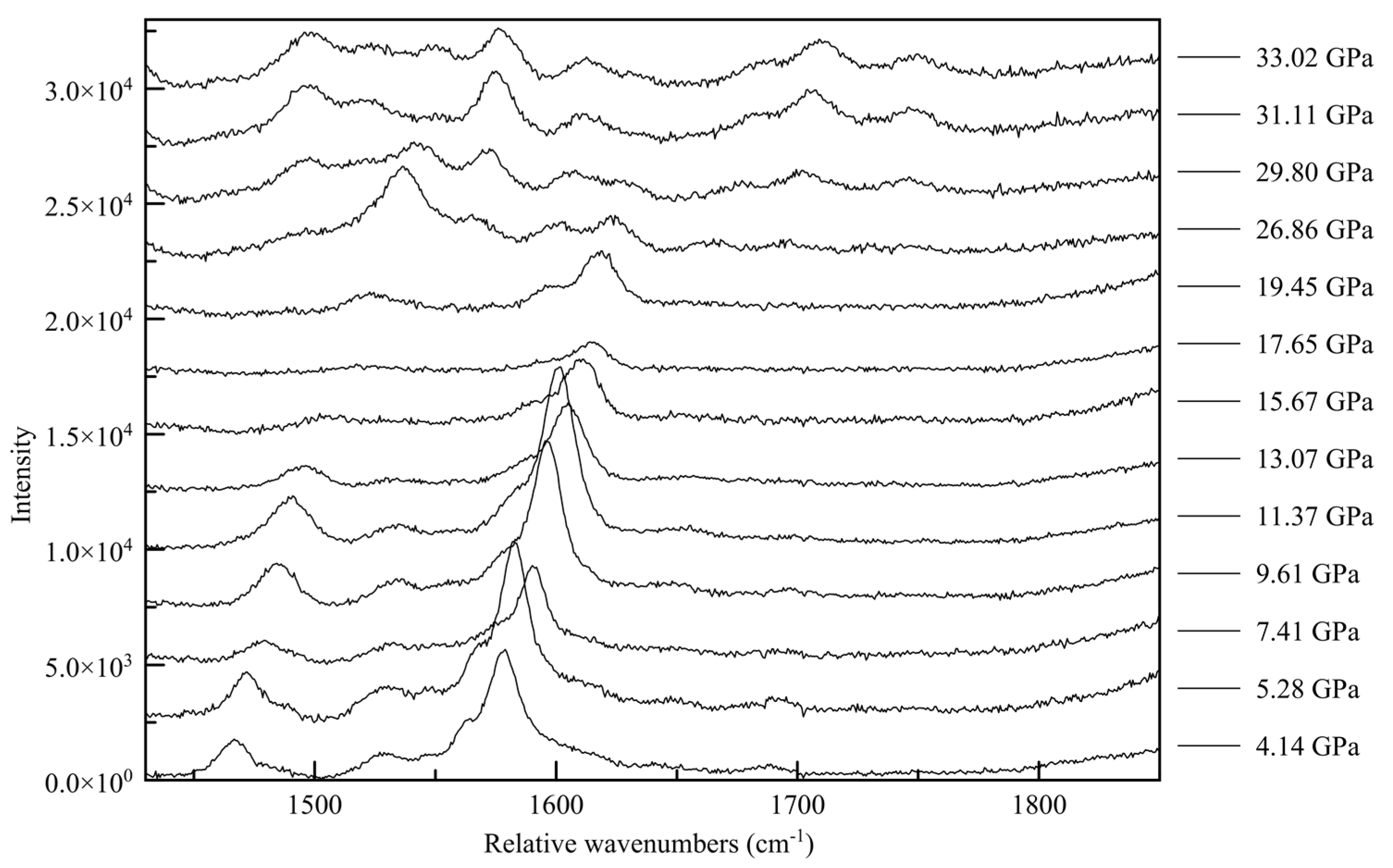
62]. In the C-N vibration range (
Figure 15) a whole new family of peaks appear before the point of transition between 1500 and 1600 relative cm
−1, with new peaks forming at approximately 1700 and 17900 rel cm
−1 above approximately 30 GPa. This corroborates a change in the electronic state beginning at 26.86(5) GPa, culminating in a change in symmetry. Similarly, in the N-H range two new high wavenumber peaks appear between 3500 and 3600 cm
−1 at approximately 30 GPa, and strengthen with increasing pressure.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
















