
Strength and Character of R–X···π Interactions Involving Aromatic Amino Acid Sidechains in Protein-Ligand Complexes Derived from Crystal Structures in the Protein Data Bank


Abstract
1. Introduction
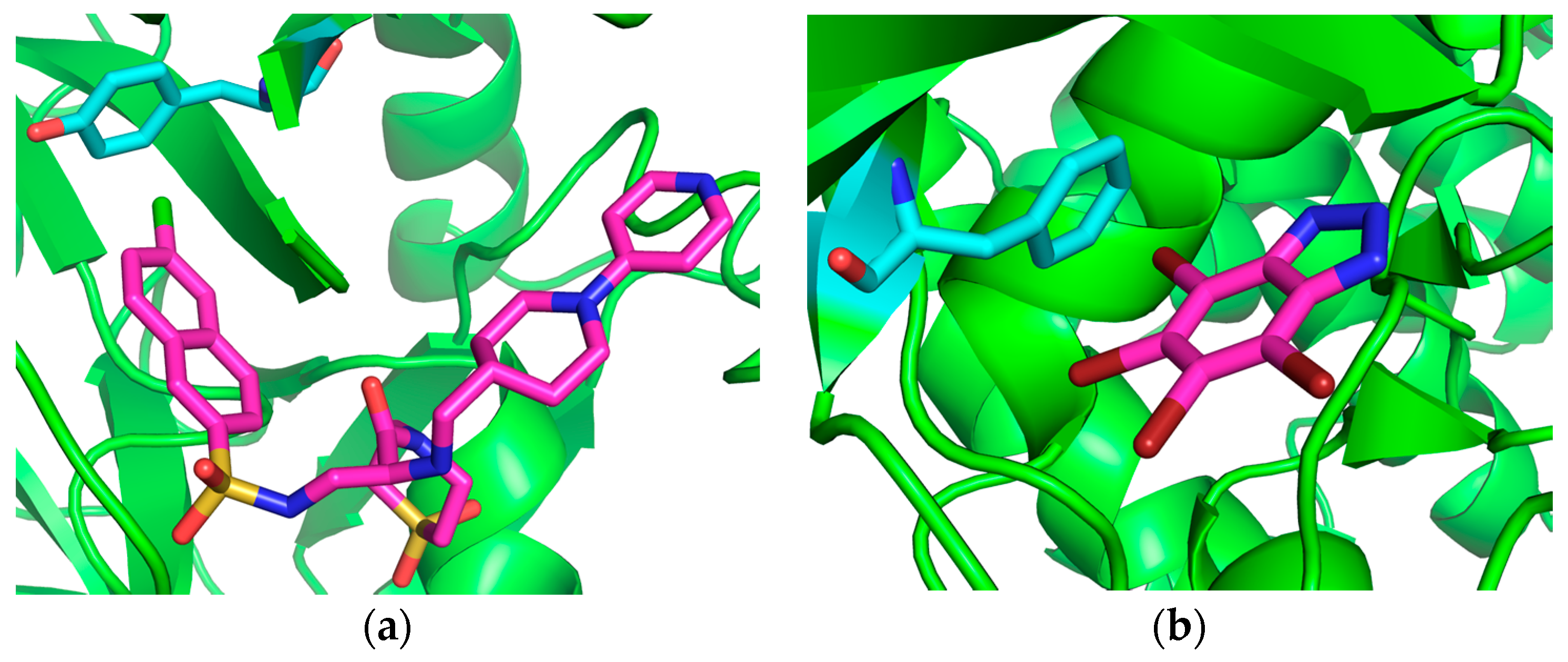
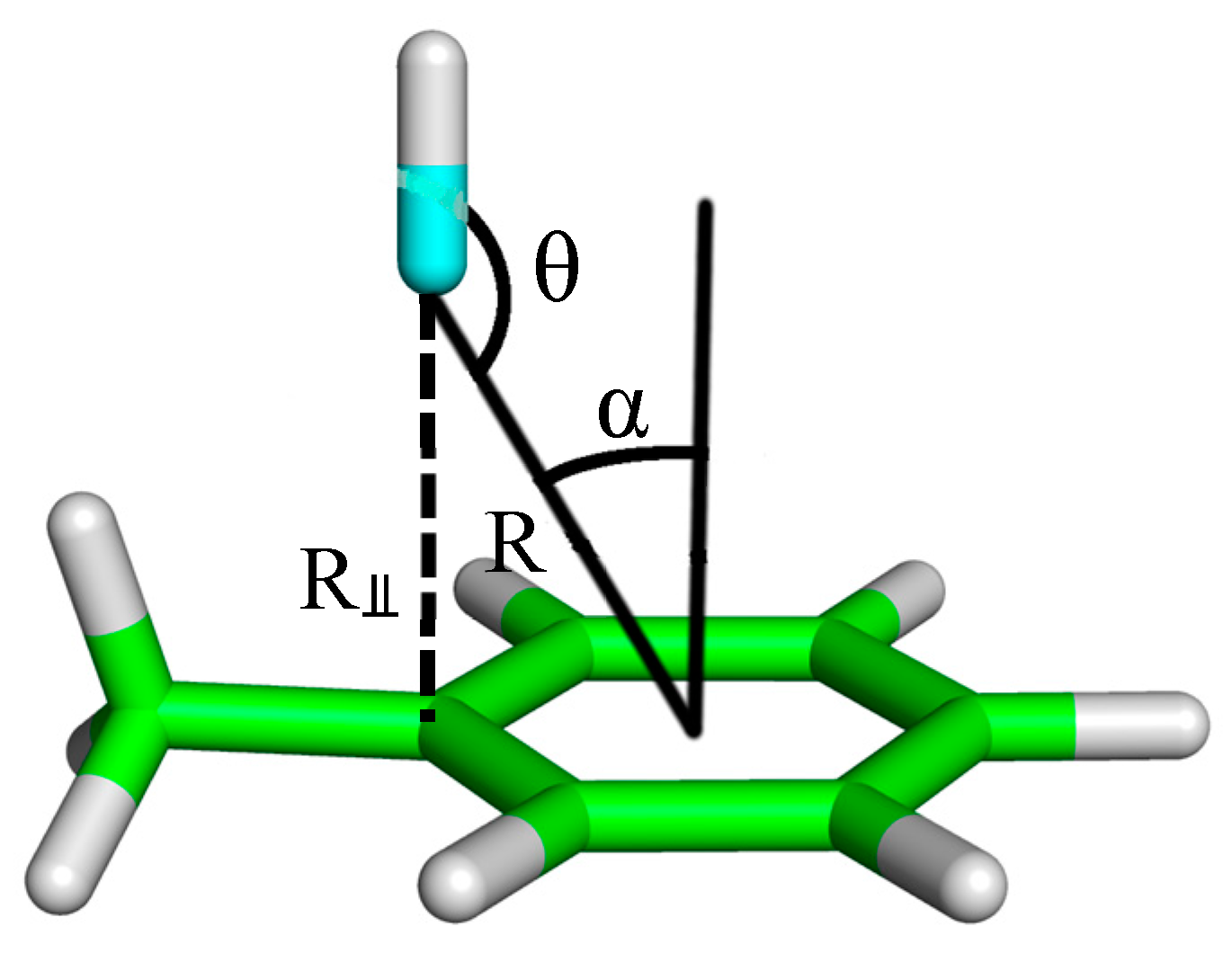
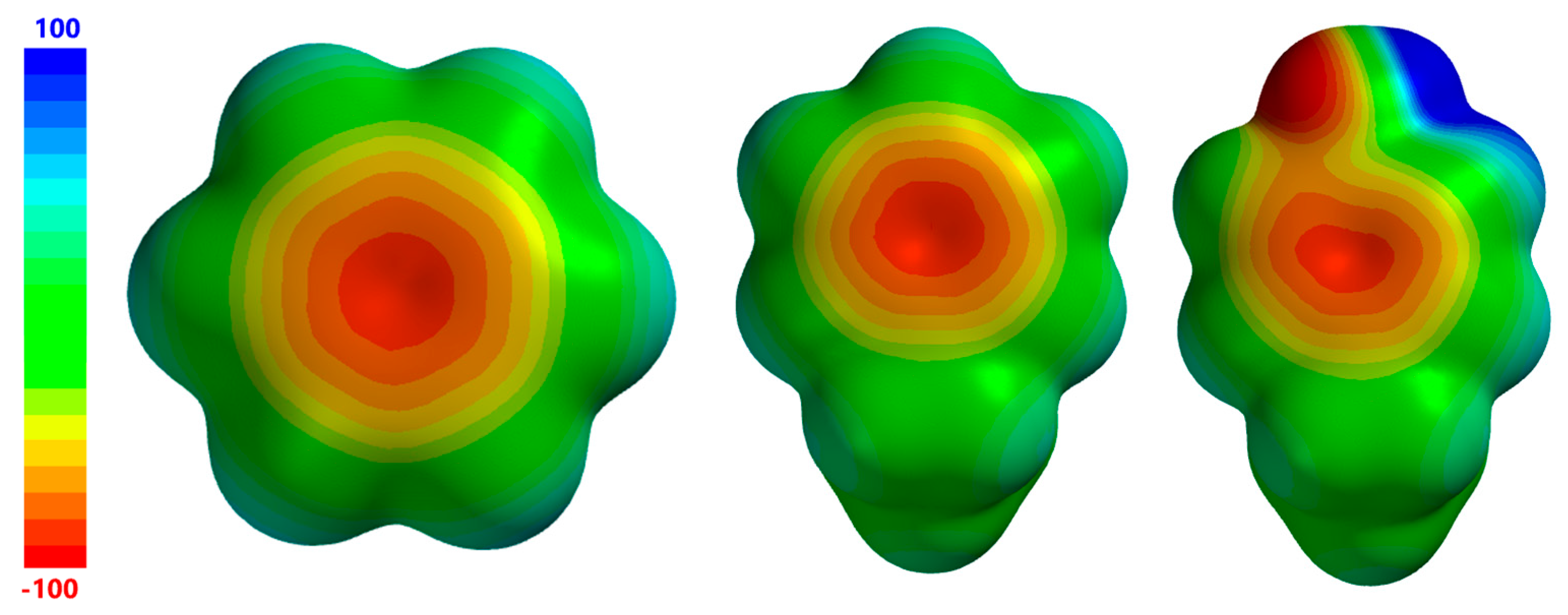
2. Results and Discussion
3. Conclusions
4. Computational Methods
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen Bonding in Supramolecular Chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef] [PubMed]
- Aackeröy, C.B.; Hurley, D.P.; Desper, J. Modulating Supramolecular Reactivity Using Covalent “Switches” on a Pyrazole Platform. Cryst. Growth Des. 2012, 12, 5806–5814. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Aackeröy, C.B.; Wijethunga, T.K.; Desper, J.; Đaković, M. Crystal Engineering with Iodoethynylmitrobenzenes: A Group of Highly Effective Halogen Bond Donors. Cryst. Growth Des. 2015, 15, 3853–3861. [Google Scholar] [CrossRef]
- Mukherjee, A.; Tothadi, S.; Desiraju, G.R. Halogen Bonds in Crystal Engineering: Like Hydrogen Bonds Yet Different. Acc. Chem. Res. 2014, 47, 2514–2524. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Hobza, P. Strength and Character of Halogen Bonds in Protein-Ligand Complexes. Cryst. Growth Des. 2011, 11, 4272–4278. [Google Scholar] [CrossRef]
- Parisini, E.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen Bonding in Halocarbon-Protein Complexes: A Structural Survey. Chem. Soc. Rev. 2011, 40, 2267–2278. [Google Scholar] [CrossRef] [PubMed]
- Scholfield, M.R.; Van der Zanden, C.M.; Carter, M.; Ho, P.S. Halogen Bonding (X-Bonding): A Biological Perspective. Protein Sci. 2013, 22, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, Y.; Zhu, W. Nonbonding Interactions of Organic Halogens in Biological Systems: Implications for Drug Discovery and Biomolecular Design. Phys. Chem. Chem. Phys. 2010, 12, 4543–4551. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.S. Halogen Bonding in Medicinal Chemistry: From Observation to Prediction. Future Sci. 2017, 9, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Lane, P.; Concha, M.C.; Ma, Y.; Murray, J.S. An Overview of Halogen Bonding. J. Mol. Model. 2007, 13, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen Bonding and other σ-Hole Interactions: A Perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Riley, K.E.; Politzer, P.; Clark, T. Directional Weak Intermolecular Interactions: σ-Hole Bonding. Aust. J. Chem. 2010, 63, 1598–1607. [Google Scholar] [CrossRef]
- Riley, K.E.; Hobza, P. Investigations into the Nature of Halogen Bonding Including Symmetry Adapted Perturbation Theory Analyses. J. Chem. Theory Comput. 2008, 4, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Kolář, M.H.; Hobza, P. Computer Modeling of Halogen Bonding and other σ-Hole Interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, H.; Zhu, X.; Zhu, W.; Liu, H. How Does Halogen Bonding Behave in Solution? A Theoretical Study Using Implicit Solvation Model. J. Phys. Chem. A 2011, 115, 4467–4475. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.C.; Perutz, R.N.; Brammer, L.; Hunber, C. A Solvent-Resistant Halogen Bond. Chem. Sci. 2014, 5, 4179–4183. [Google Scholar] [CrossRef]
- Lu, Y.X.; Zou, J.W.; Wang, Y.H.; Yu, Q.S. Theoretical Investigations of the C–X/π Interactions between Benzene and Some Model Halocarbons. Chem. Phys. 2006, 334, 1–7. [Google Scholar] [CrossRef]
- Forni, A.; Pieraccini, S.; Rendine, S.; Gabas, F.; Sironi, M. Halogen-Bonding Interactions with π Systems: CCSD(T), MP2, and DFT Calculations. ChemPhysChem 2012, 13, 4224–4234. [Google Scholar] [CrossRef] [PubMed]
- Forni, A.; Pieraccini, S.; Rendine, S.; Sironi, M. Halogen Bonds with Benzene: An Assessment of DFT Functionals. J. Comput. Chem. 2014, 35, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Nagels, N.; Hauchecorne, D.; Herrebout, W.A. Exploring the C–X···π Halogen Bonding Motif: An Infrared and Raman Study of the Complexes of CF3X (X = Cl, Br, and I) with the Aromatic Model Compounds Benzene and Toluene. Molecules 2013, 18, 6829–6851. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, S.; Uchimaru, T.; Wakisaka, A.; Ono, T. Magnitude and Directionality of Halogen Bond of Benzene with C6F5X, C6H5X, and CF3X (X = I, Br, Cl, and F). J. Phys. Chem. A 2016, 120, 7020–7029. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Ford, C.L.; Demouchet, K. Comparison of Hydrogen Bonds, Halogen Bonds, C–H···π Interactions, and C–X···π Interactions using High-Level Ab Initio Methods. Chem. Phys. Lett. 2015, 621, 165–170. [Google Scholar] [CrossRef]
- Riley, K.E.; Vazquez, M.; Umemura, C.; Miller, C.; Tran, K.-A. Exploring the (Very Flat) Potential Energy Landscape of R–Br···π Interactions with Accurate CCSD(T) and SAPT Techniques. Chem. Eur. J. 2016, 22, 17690–17695. [Google Scholar] [CrossRef] [PubMed]
- Matter, H.; Nazaré, M.; Güssregen, S.; Will, D.S.; Schreuder, H.; Bauer, A.; Urmann, M.; Ritter, K.; Wagner, M.; Wehner, V. Evidence for C–Cl/C–Br···π Interactions as an Important Contribution to Protein-Ligand Binding Affinity. Ang. Chem. Int. Ed. 2009, 48, 2911–2916. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.N.; Inoue, Y.; Nakanishi, I.; Kitaura, K. Cl-π Interactions in Protein-Ligand Complexes. Protein Sci. 2008, 17, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Sirimulla, S.; Bailey, J.B.; Vegesna, R.; Narayan, M. Halogen Interactions in Protein-Ligand Complexes: Implications of Halogen Bonding for Rational Drug Design. J. Chem. Inf. Model. 2013, 53, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Sensitivity of Noncovalent Bonds to Intermolecular Separation: Hydrogen, Halogen, Chalcogen, and Pnicogen Bonds. CrystEngComm 2013, 15, 3119–3124. [Google Scholar] [CrossRef]
- Stone, A.J. Are Halogen Bonded Structures Electrostatically Driven. J. Am. Chem. Soc. 2013, 135, 7005–7009. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, U.; Scheiner, S. Sensitivity of Pnicogen, Chalcogen, Halogen, and H-Bonds to Angular Distortions. Chem. Phys. Lett. 2012, 532, 31–35. [Google Scholar] [CrossRef]
- Sedlák, R.; Kolář, M.H.; Hobza, P. Polar Flattening and the Strength of Halogen Bonding. J. Chem. Theory Comput. 2015, 11, 4727–4732. [Google Scholar] [CrossRef] [PubMed]
- Glaser, R.; Murphy, R.F.; Sui, Y.; Barnesab, C.L.; Kim, H.S. Multifurcated Halogen Bonding involving Ph–Cl···H–CPhLN-R9 Interactions and its Relation to Idiotelamphiphile Layer Architecture. CrystEngComm 2006, 8, 372–376. [Google Scholar] [CrossRef]
- Riley, K.E.; Murray, J.S.; Fanfrlik, J.; Řezáč, J.; Solá, R.J.; Concha, M.C.; Ramos, F.M.; Politzer, P. Halogen Bond Tunability I: The Effects of Aromatic Fluorine Substitution on the Strengths of Halogen-Bonding Interactions Involving Chlorine, Bromine, and Iodone. J. Mol. Model. 2011, 17, 3309–3318. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Murray, J.S.; Fanfrlik, J.; Řezáč, J.; Solá, R.J.; Concha, M.C.; Ramos, F.M.; Politzer, P. Halogen Bond Tunability II: The Varying Roles of Electrostatics and Dispersion to Attraction in Halogen Bonds. J. Mol. Model. 2013, 19, 4651–4659. [Google Scholar] [CrossRef] [PubMed]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of Van Der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Hohenstein, E.G.; Sherrill, C.D. Wavefunction Methods for Noncovalent Interactions. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 304–326. [Google Scholar] [CrossRef]
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of Symmetry Adapted Perturbation Theory (SAPT) I. Efficiency and Performance for Interaction Energies. J. Chem. Phys. 2014, 140, 094106. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Hobza, P. Assessment of the MP2 Method, Along with Several Basis Sets, for the Computational of Interaction Energies of Biologically Relevant Hydrogen Bonded and Dispersion Bound Complexes. J. Phys. Chem. A 2007, 111, 8257–8263. [Google Scholar] [CrossRef] [PubMed]
- Turney, J.M.; Simmonett, A.C.; Parrish, R.M.; Hohenstein, E.G.; Evangelista, F.; Fermann, J.T.; Mintz, B.J.; Burns, L.A.; Wilke, J.J.; Abrams, M.L.; et al. PSI4: An Open-Source Ab Initio Electronic Structure Program. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 556. [Google Scholar] [CrossRef]
- Riley, K.E.; Tran, K.A.; Lane, P.; Murray, J.S.; Politzer, P. Comparative Analysis of Electrostatic Potential Maxima and Minima on Molecular Surfaces, As Determined by Three Methods and a Variety of Basis Sets. J. Comput. Sci. 2016, 17, 273–284. [Google Scholar] [CrossRef]
- Shao, Y.; Molnar, L.F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S.T.; Gilbert, A.T.B.; Slipchenko, L.V.; Levchenko, S.V.; O’Neill, D.P.; et al. Advances in Methods and Algorithms in a Modern Quantum Chemistry Program Package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Complex Geometry VS,max (kcal/mol) | SAPT Interaction Energy Terms (kcal/mol) (% attraction) | Model Complex | R–Cl···π Donor ESP |
|---|---|---|---|
| 1T4V |  |  | |
| R = 3.78 Å | E(elec): −1.05 (22.6) | ||
| α = 13.4° | E(disp): −3.35 (71.9) | ||
| θ = 137.1° | E(ind): −0.26 (5.5) | ||
| R⫫ = 3.68 Å | E(exch): 2.01 | ||
| VS,max = 32 | ∆E(int): −2.66 | ||
| 1IQE |  |  | |
| R = 3.47 Å | E(elec): −1.73 (27.3) | ||
| α = 14.7° | E(disp): −4.15 (65.3) | ||
| θ = 169.1° | E(ind): −0.47 (7.4) | ||
| R⫫ = 3.36 Å | E(exch): 3.80 | ||
| VS,max = 53 | ∆E(int): −2.56 | ||
| 1NFY |  |  | |
| R = 3.70 Å | E(elec): −1.19 (24.2) | ||
| α = 21.9° | E(disp): −3.36 (68.5) | ||
| θ = 177.0° | E(ind): −0.36 (7.3) | ||
| R⫫ = 3.43 Å | E(exch): 2.57 | ||
| VS,max = 53 | ∆E(int): −2.34 | ||
| 1NFU |  |  | |
| R = 3.57 Å | E(elec): −1.78 (27.9) | ||
| α = 22.4° | E(disp): −4.09 (63.9) | ||
| θ = 173.9° | E(ind): −0.52 (8.2) | ||
| R⫫ = 3.30 Å | E(exch): 4.09 | ||
| VS,max = 54 | ∆E(int): −2.30 | ||
| 2BQW |  |  | |
| R = 3.50 Å | E(elec): −1.86 (27.9) | ||
| α = 19.9° | E(disp): −4.29 (64.3) | ||
| θ = 174.6° | E(ind): −0.52 (7.8) | ||
| R⫫ = 3.29 Å | E(exch): 4.47 | ||
| VS,max = 25 | ∆E(int): −2.20 | ||
| 2BMG |  |  | |
| R = 3.71 Å | E(elec): −1.06 (21.9) | ||
| α = 22.4° | E(disp): −3.41 (70.4) | ||
| θ = 167.5° | E(ind): −0.37 (7.7) | ||
| R⫫ = 3.43 Å | E(exch): 2.75 | ||
| VS,max = 37 | ∆E(int): −2.10 |
| Complex Geometry VS,max (kcal/mol) | SAPT Interaction Energy Terms (kcal/mol) (% attraction) | Model Complex | R–Br···π Donor ESP |
|---|---|---|---|
| 1P5E |  |  | |
| R = 3.68 Å | E(elec): −4.35 (35.6) | ||
| α = 30.3° | E(disp): −6.57 (53.8) | ||
| θ = 174.8° | E(ind): −1.28 (10.5) | ||
| R⫫ = 3.18 Å | E(exch): 8.60 | ||
| VS,max = 118 | ∆E(int): −3.60 | ||
| 1ZOE |  |  | |
| R = 3.93 Å | E(elec): −2.18 (30.5) | ||
| α = 28.7° | E(disp): −4.36 (61.1) | ||
| θ = 163.0° | E(ind): −0.60 (8.4) | ||
| R⫫ = 3.45 Å | E(exch): 4.04 | ||
| VS,max = 86 | ∆E(int): −3.10 | ||
| 3JTK |  |  | |
| R = 3.82 Å | E(elec): −1.57 (23.4) | ||
| α = 20.3° | E(disp): −4.71 (70.3) | ||
| θ = 127.5° | E(ind): −0.42 (6.3) | ||
| R⫫ = 3.58 Å | E(exch): 4.21 | ||
| VS,max = 61 | ∆E(int): −2.49 | ||
| 2AF6 |  |  | |
| R = 3.80 Å | E(elec): −1.70 (28.7) | ||
| α = 23.1° | E(disp): −3.70 (62.4) | ||
| θ = 164.7° | E(ind): −0.53 (8.9) | ||
| R⫫ = 3.50 Å | E(exch): 3.80 | ||
| VS,max = 57 | ∆E(int): −2.12 | ||
| 1O27 |  |  | |
| R = 3.98 Å | E(elec): −0.45 (13.7) | ||
| α = 16.3° | E(disp): −2.61 (78.4) | ||
| θ = 162.4° | E(ind): −0.27 (8.0) | ||
| R⫫ = 3.82 Å | E(exch): 1.32 | ||
| VS,max = 71 | ∆E(int): −2.01 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riley, K.E.; Tran, K.-A. Strength and Character of R–X···π Interactions Involving Aromatic Amino Acid Sidechains in Protein-Ligand Complexes Derived from Crystal Structures in the Protein Data Bank. Crystals 2017, 7, 273. https://doi.org/10.3390/cryst7090273
Riley KE, Tran K-A. Strength and Character of R–X···π Interactions Involving Aromatic Amino Acid Sidechains in Protein-Ligand Complexes Derived from Crystal Structures in the Protein Data Bank. Crystals. 2017; 7(9):273. https://doi.org/10.3390/cryst7090273
Chicago/Turabian StyleRiley, Kevin E., and Khanh-An Tran. 2017. "Strength and Character of R–X···π Interactions Involving Aromatic Amino Acid Sidechains in Protein-Ligand Complexes Derived from Crystal Structures in the Protein Data Bank" Crystals 7, no. 9: 273. https://doi.org/10.3390/cryst7090273
APA StyleRiley, K. E., & Tran, K.-A. (2017). Strength and Character of R–X···π Interactions Involving Aromatic Amino Acid Sidechains in Protein-Ligand Complexes Derived from Crystal Structures in the Protein Data Bank. Crystals, 7(9), 273. https://doi.org/10.3390/cryst7090273