
Synthesis, Characterization and DFT Calculations of 4,5,12- and 1,8,12-trichloro-9,10-dihydro-9,10-ethanoanthracene-12-carbonitriles


 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
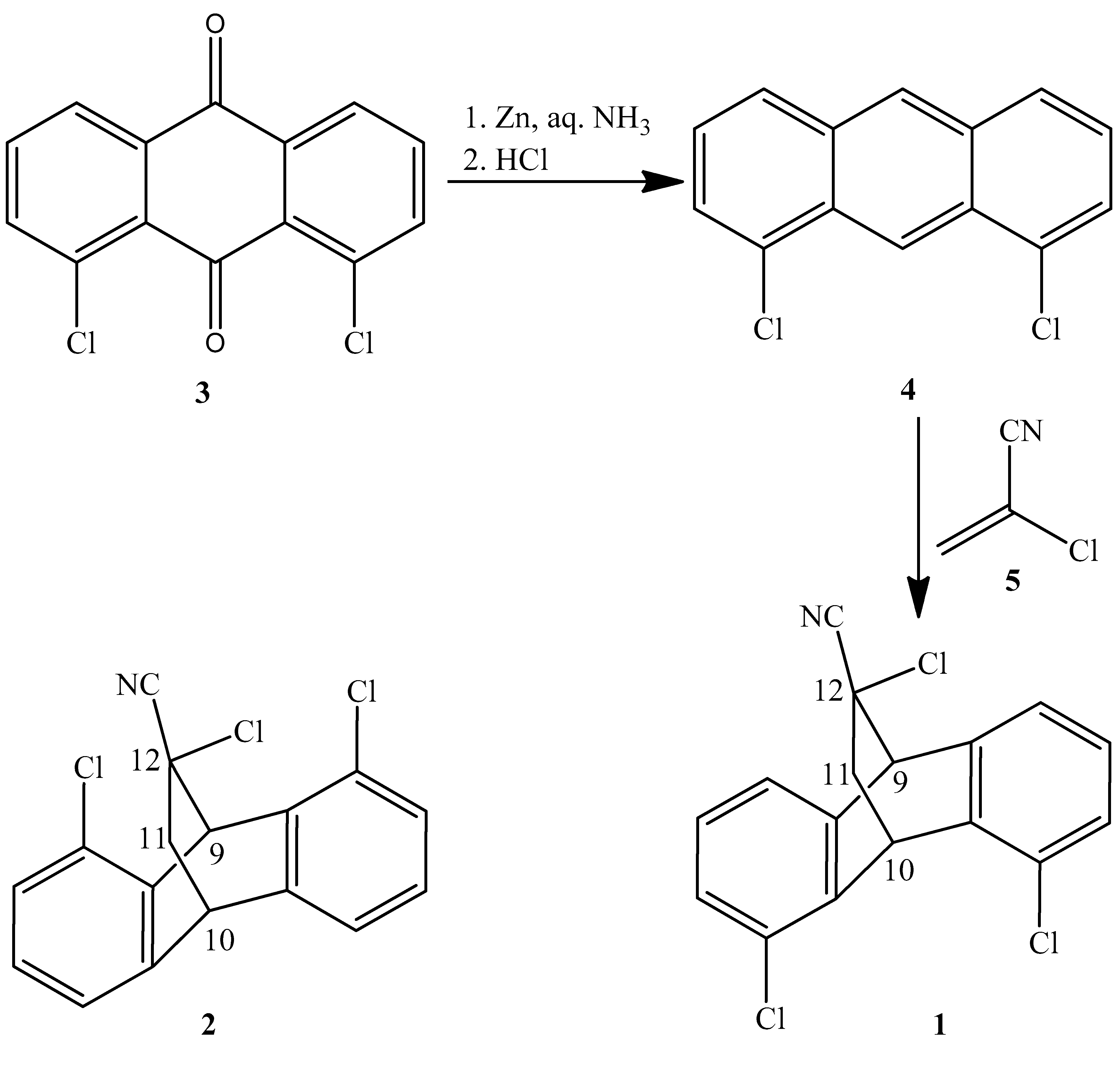
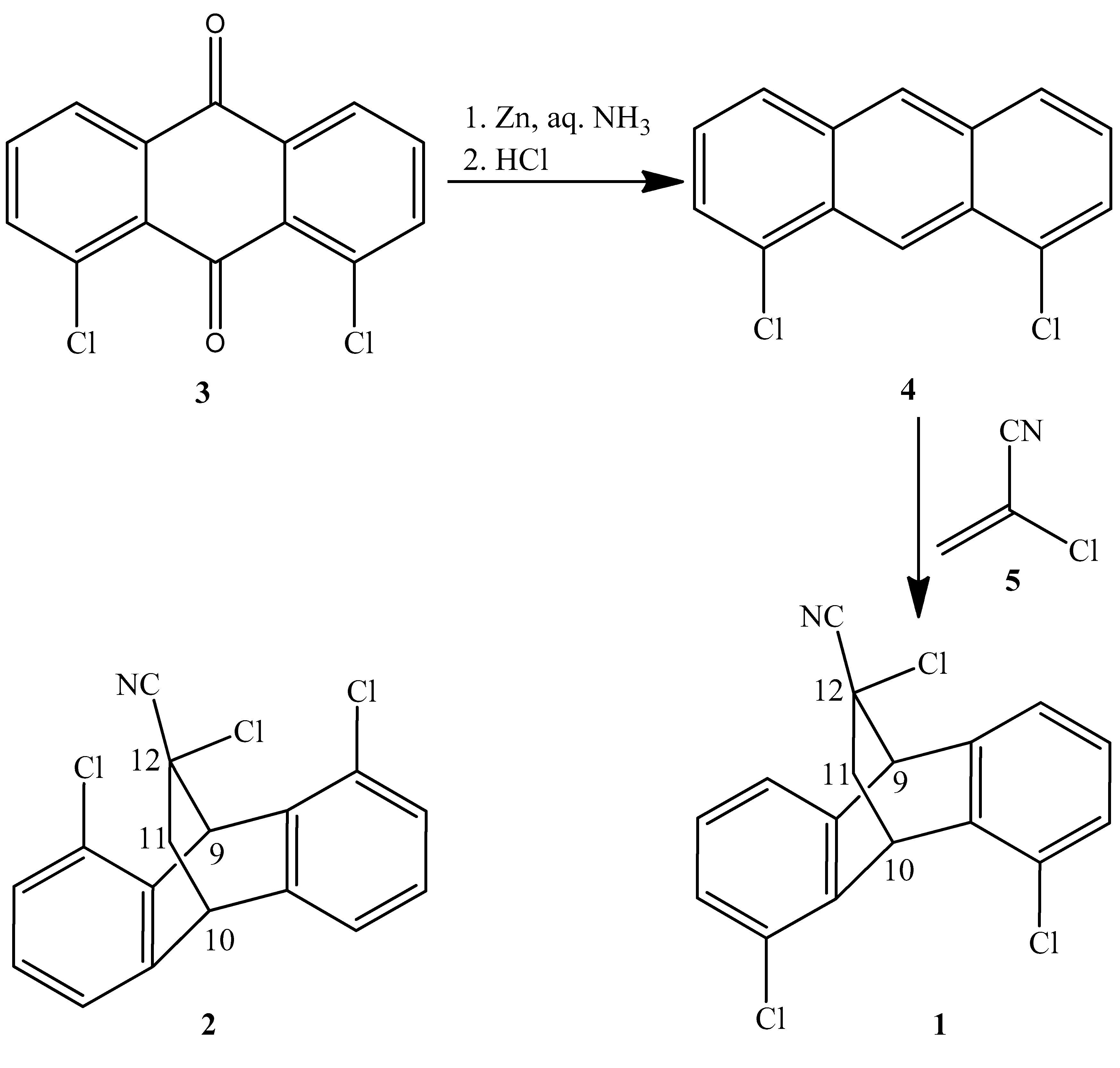
2.1. Synthesis
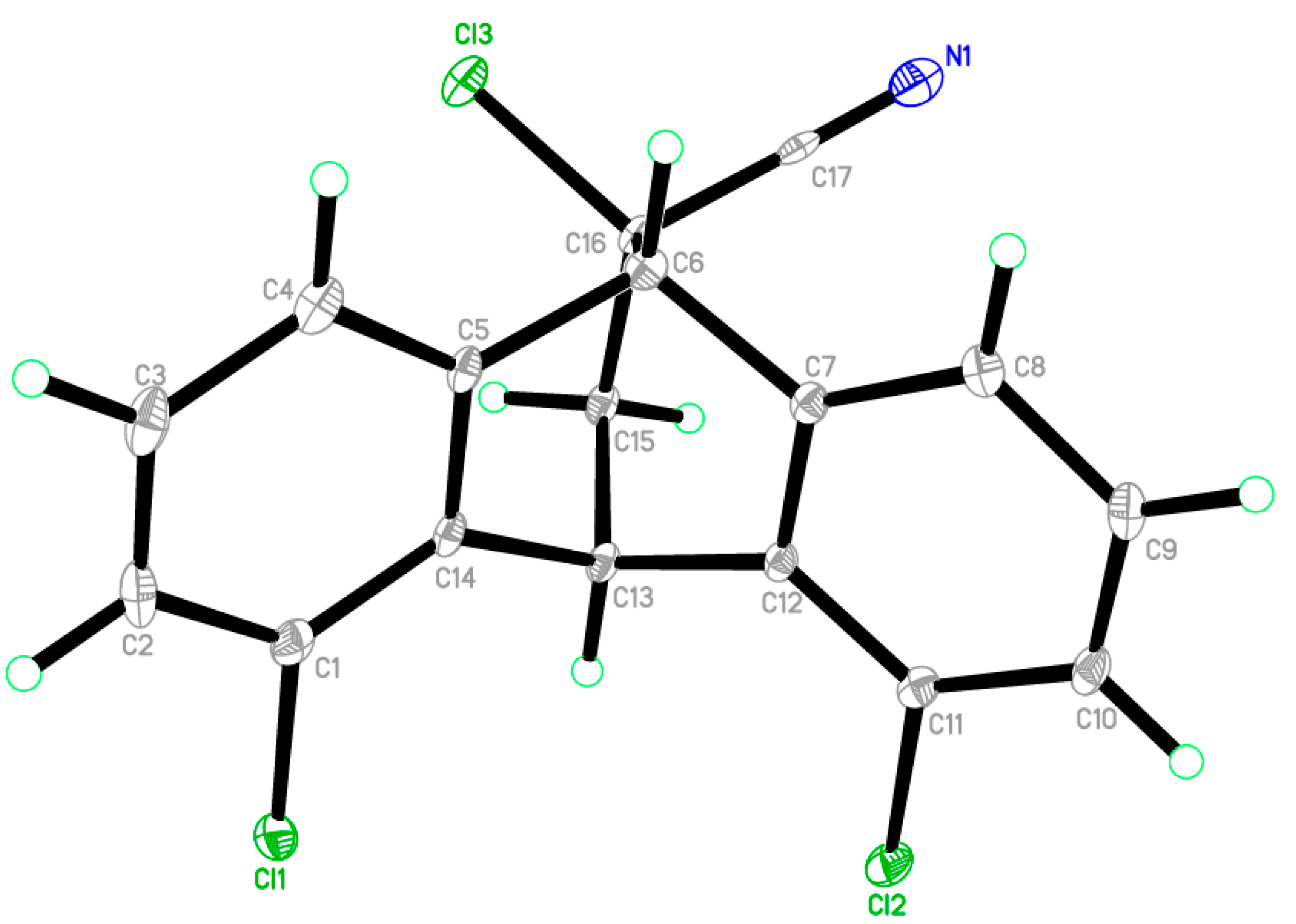
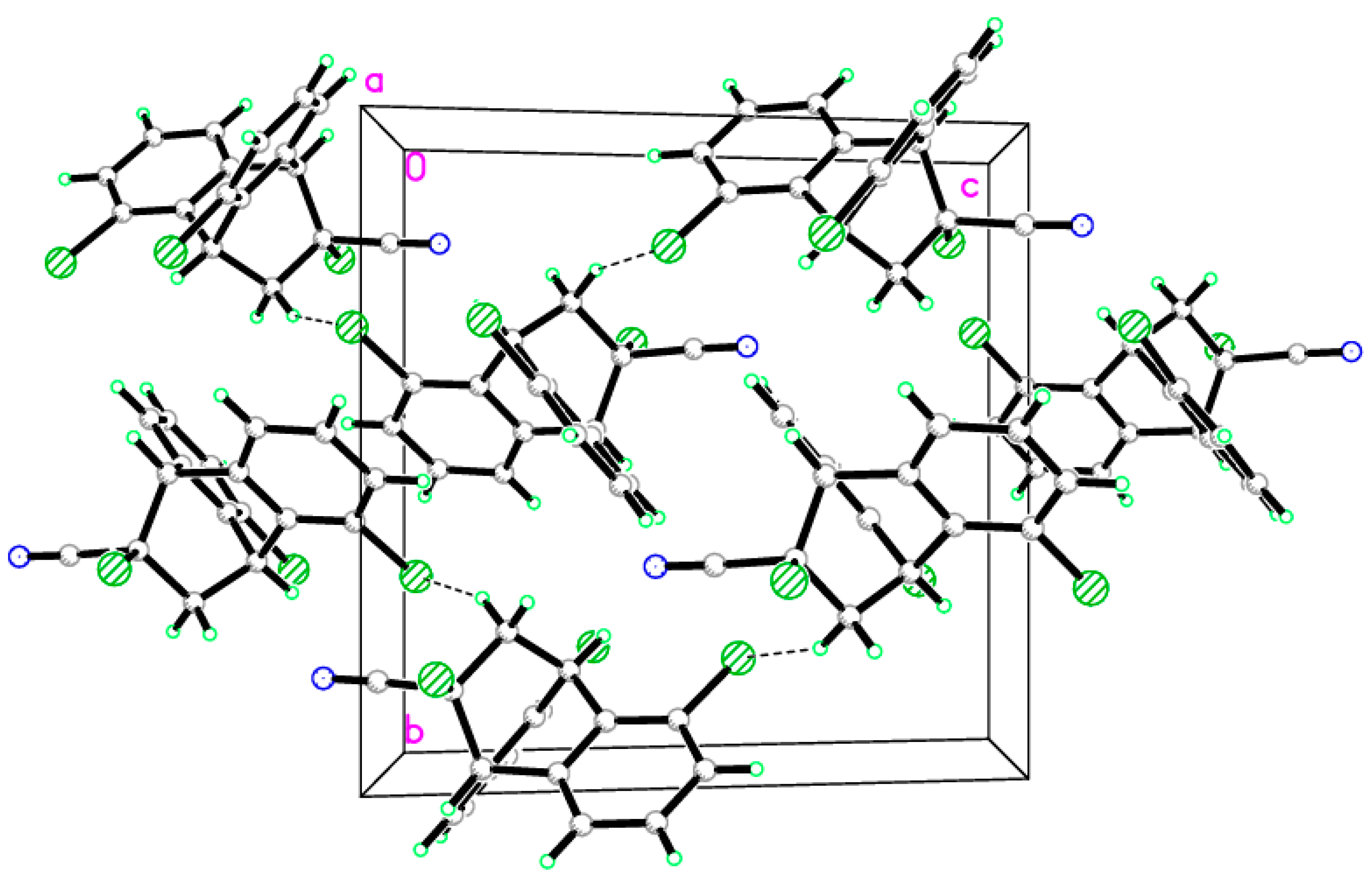
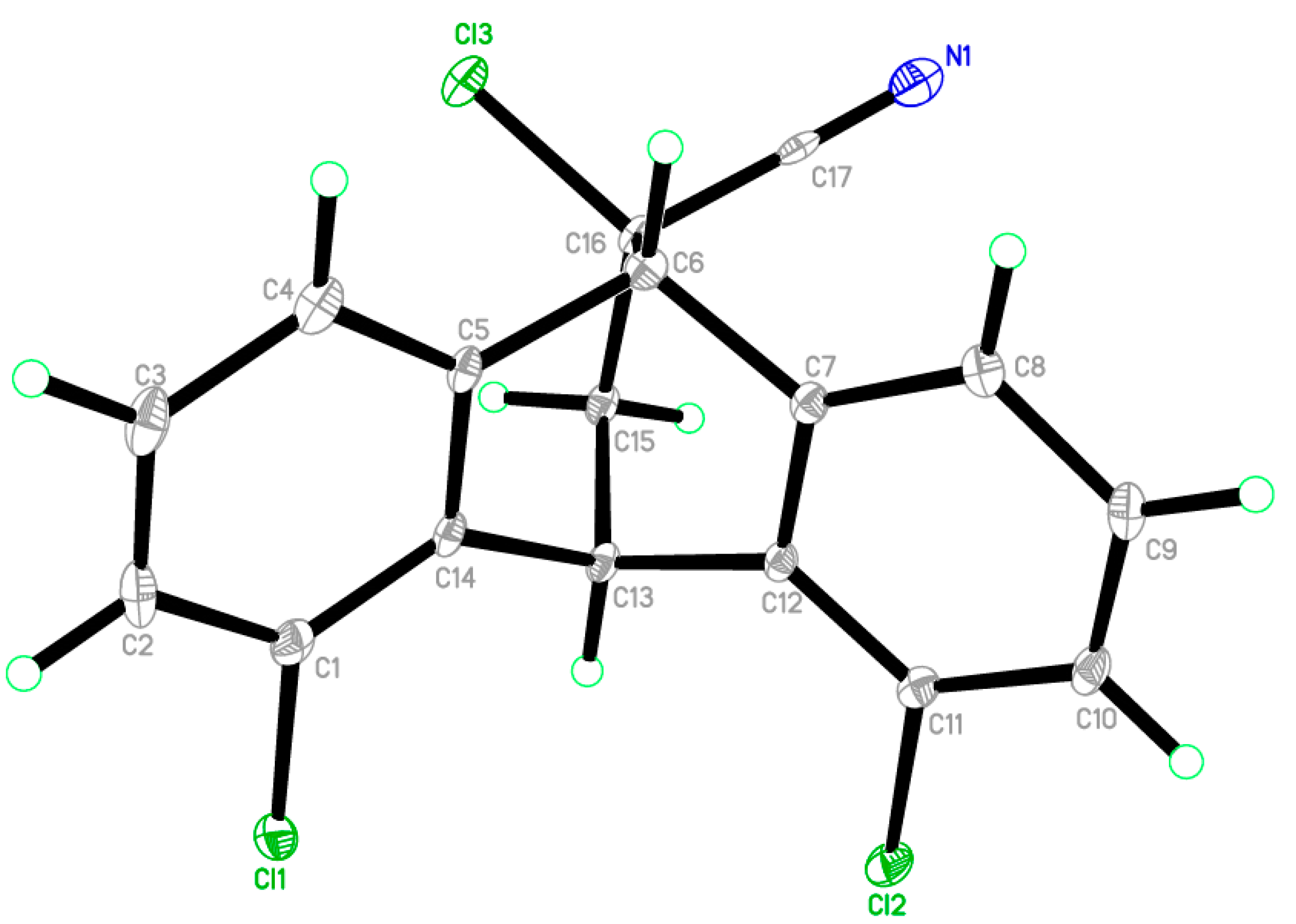
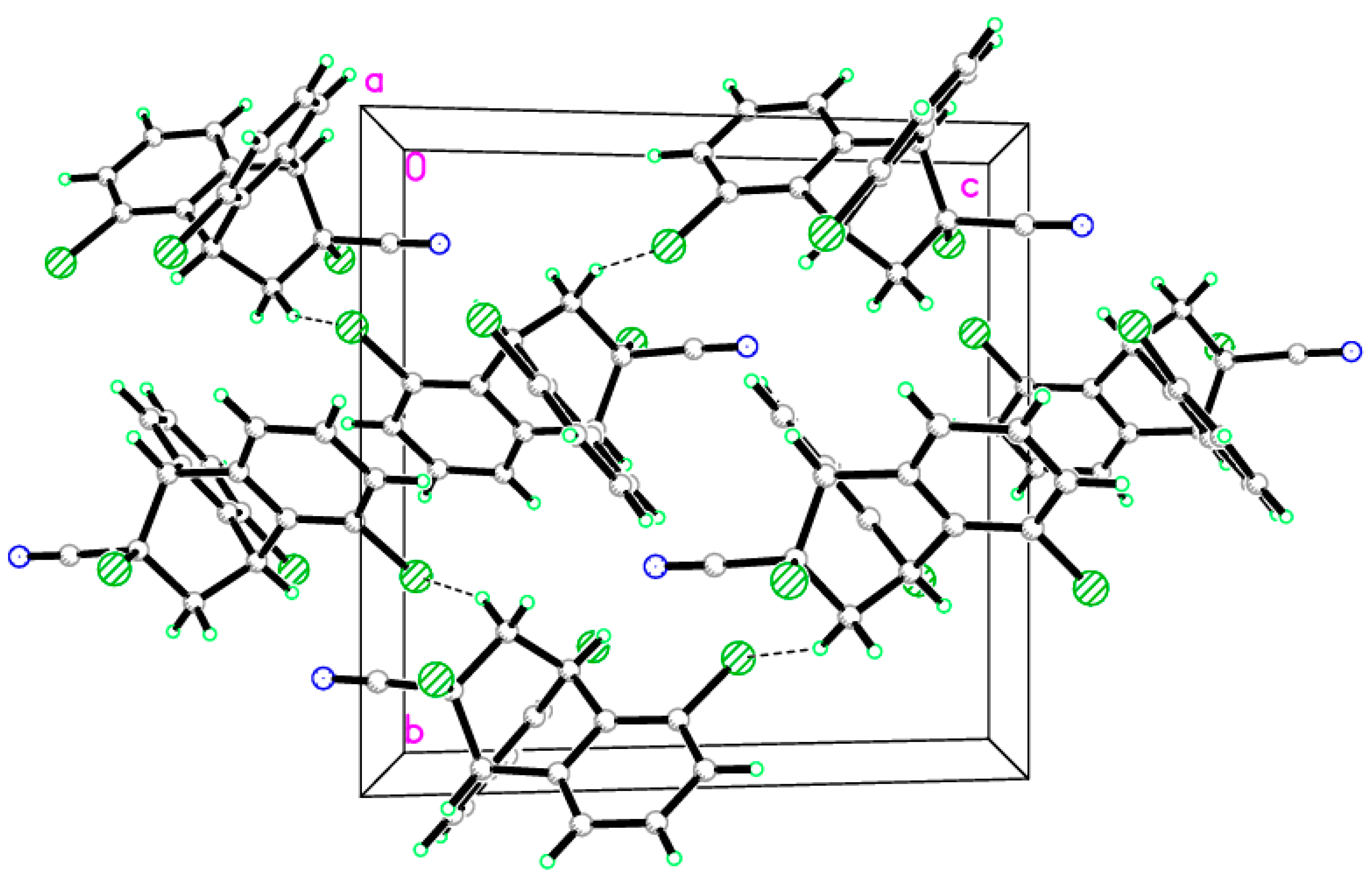
2.2. X-ray Single Crystal Structure Analysis of Isomer 1
2.3. Theoretical Calculations
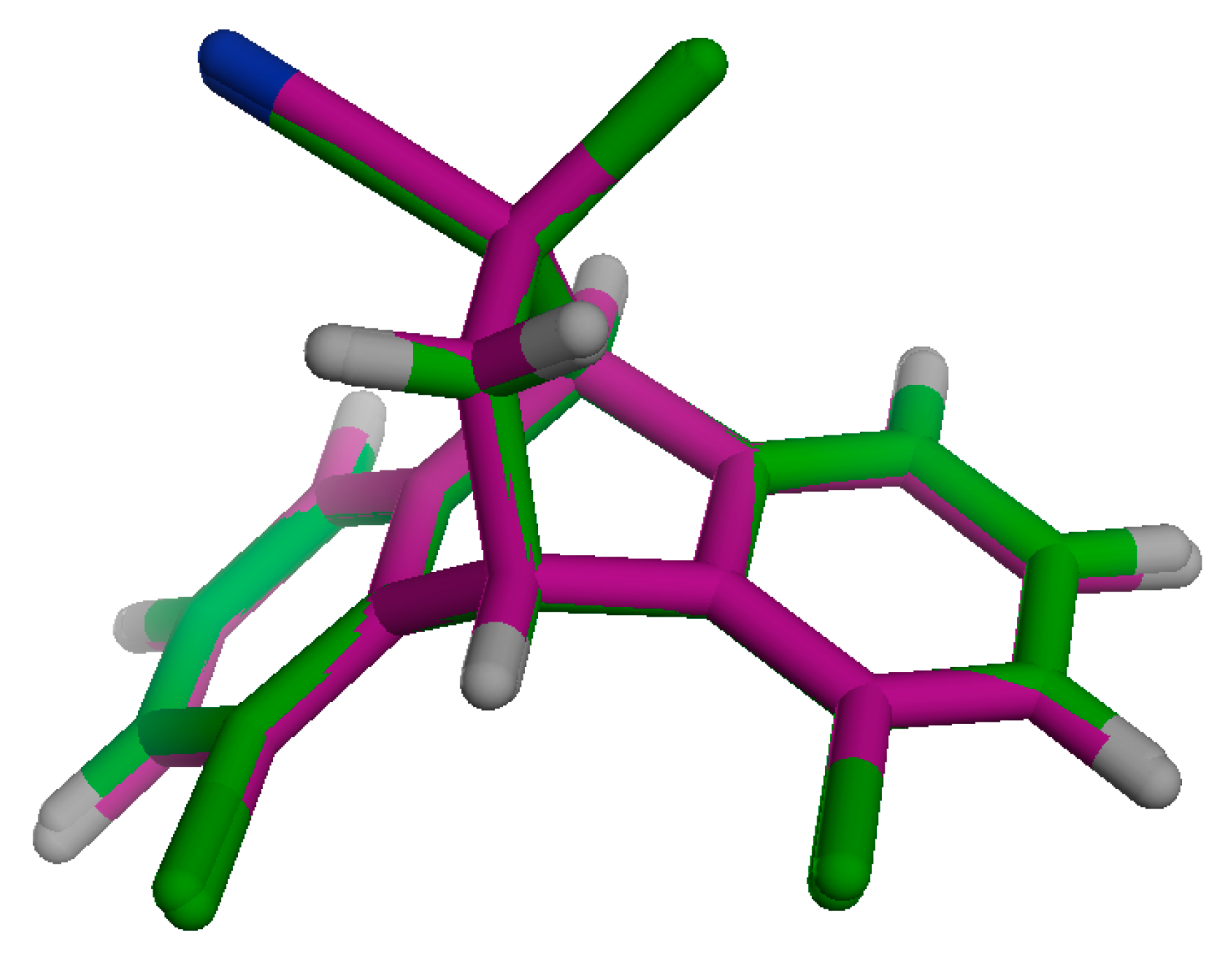
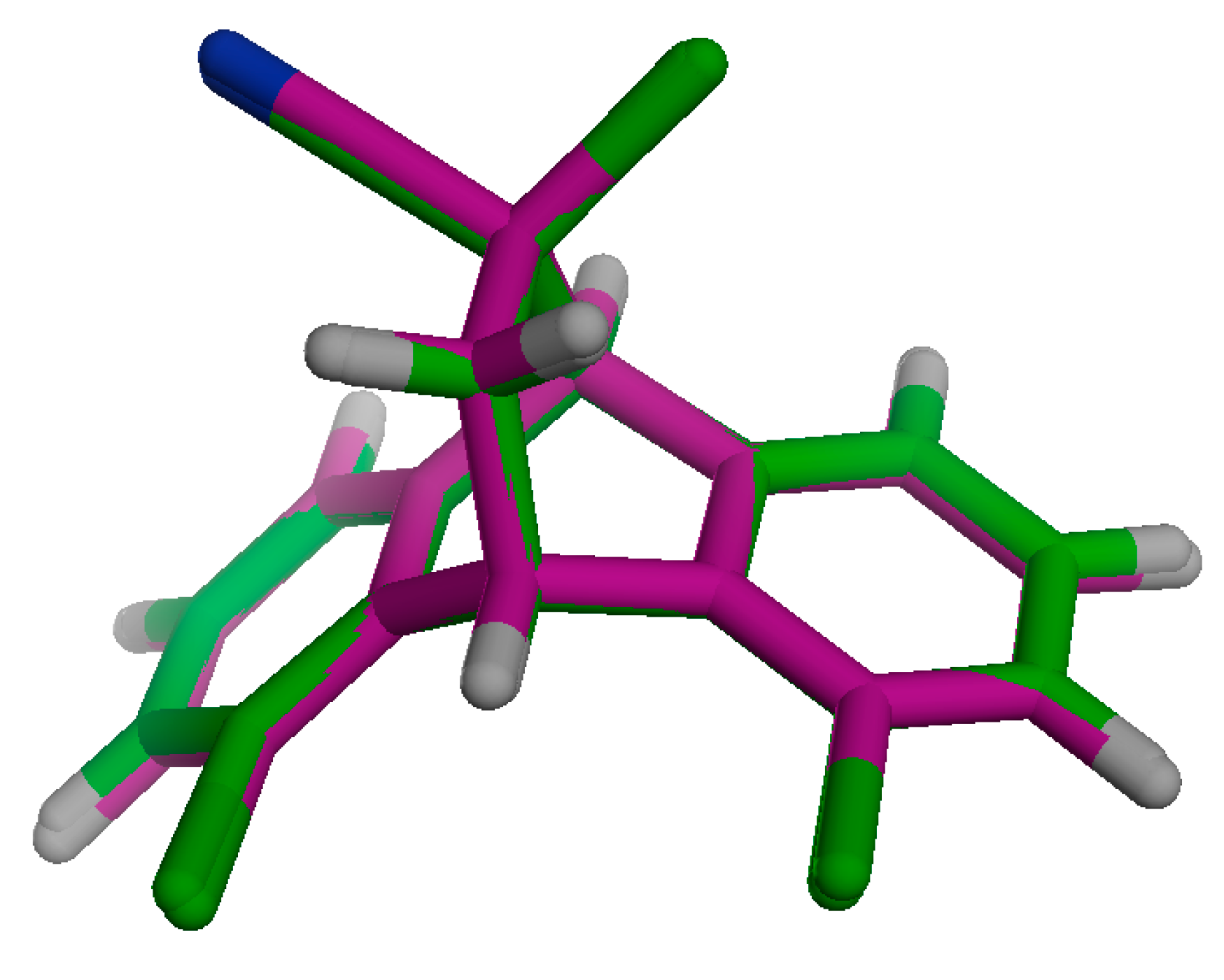
2.3.1. Energetic and Optimized Geometry
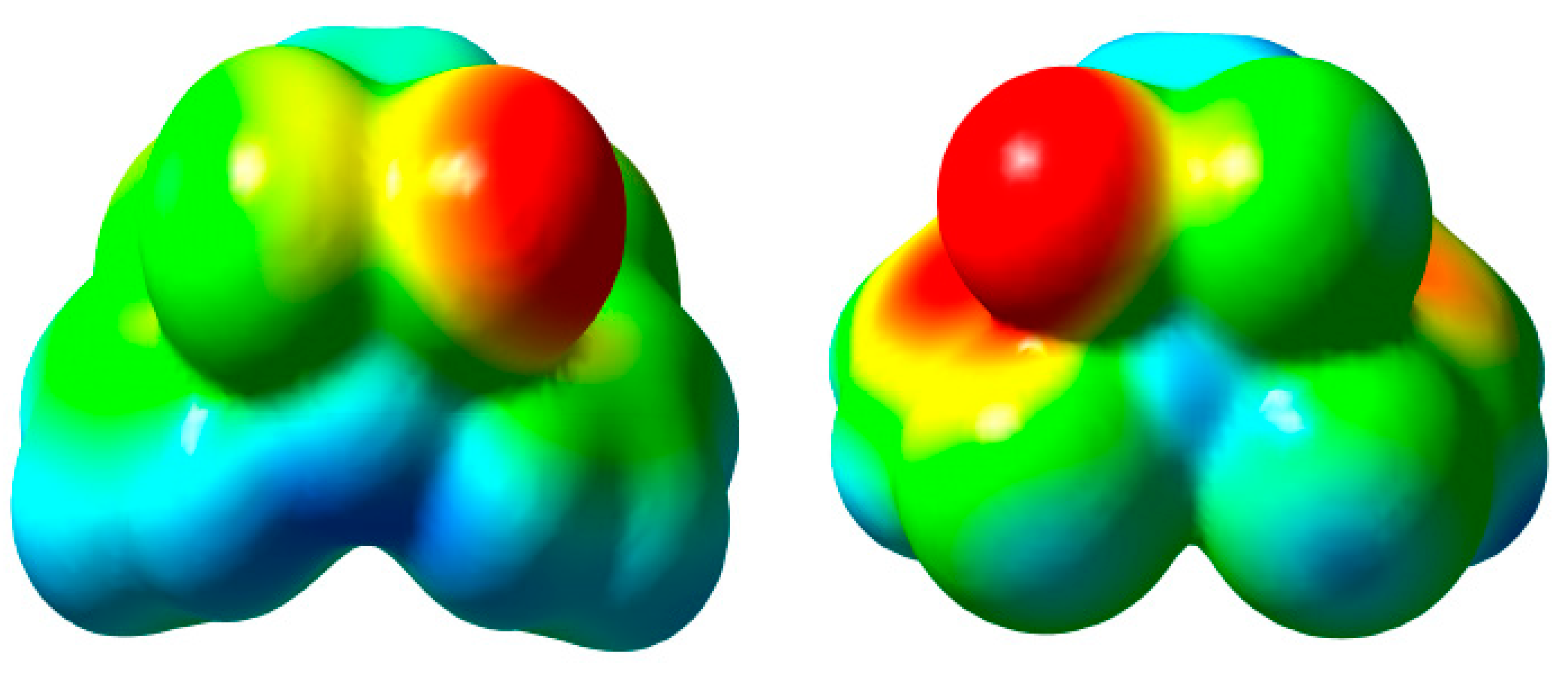
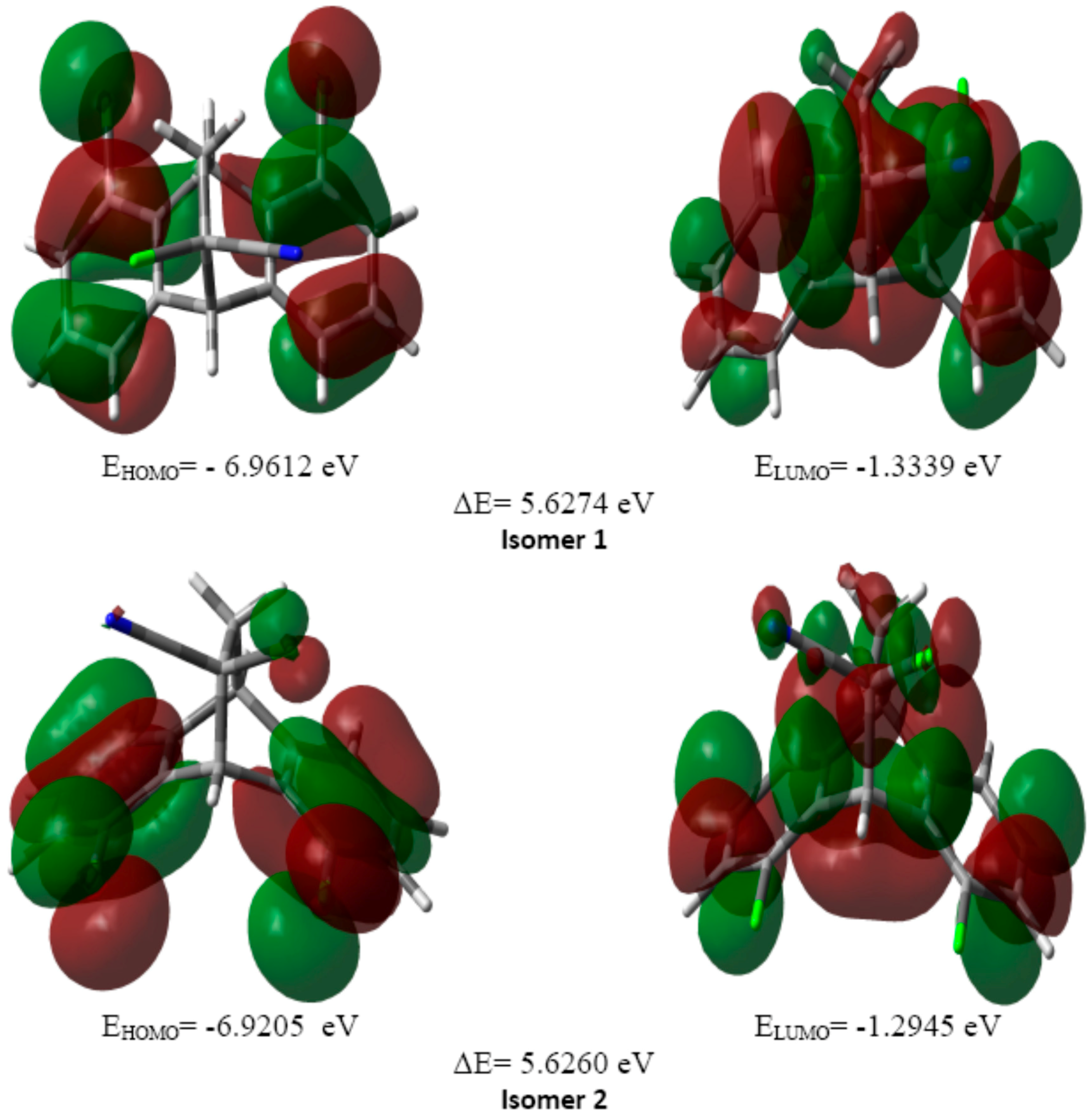
2.3.2. HOMO and LUMO Analysis
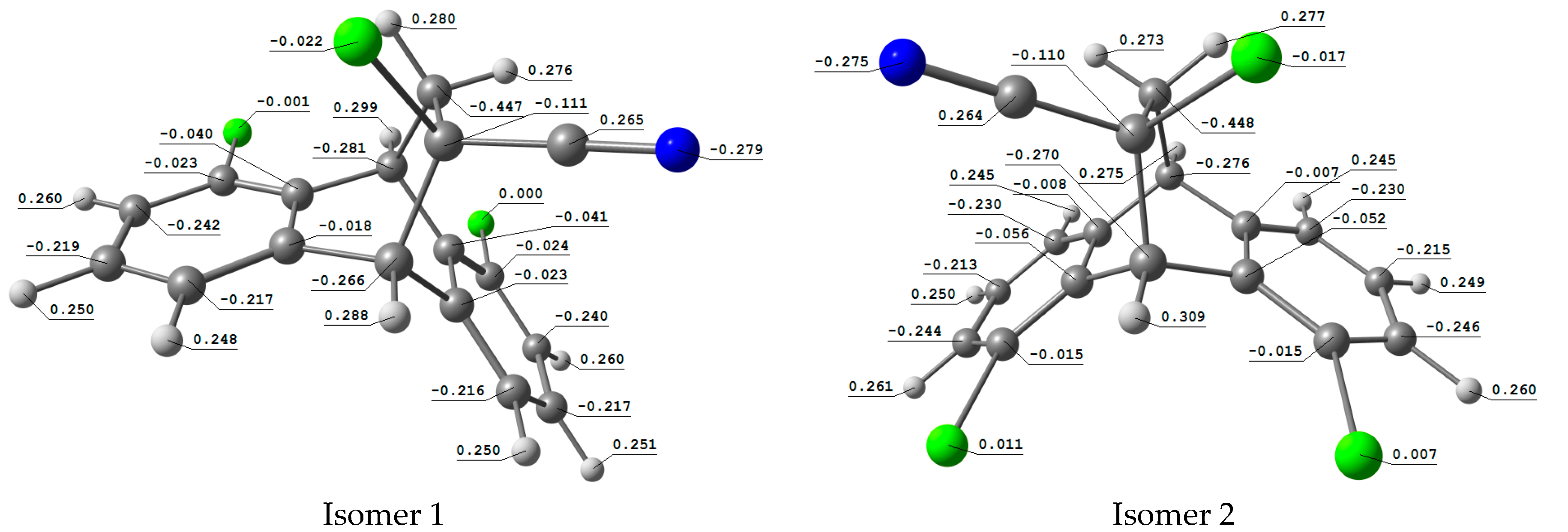
2.3.3. Atomic Charges
3. Materials and Methods
3.1. Synthesis
3.1.1. General Notes
3.1.2. Synthesis of 1,8-dichloroanthracene 4
3.1.3. Synthesis of 4,5,12-Trichloro-9,10-dihydro-9,10-ethanoanthracene-12-carbonitrile 1 and 1,8,12-trichloro-9,10-dihydro-9,10-ethanoanthracene-12-carbonitrile 2
3.2. Single-Crystal X-ray Diffraction of Isomer 1
3.3. Theoretical Calculations
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wadler, S.; Fuks, J.Z.; Wiernik, P.H. Phase I and II agents in cancer therapy: I. Anthracyclines and related compounds. J. Clin. Pharmacol. 1986, 26, 491–509. [Google Scholar] [PubMed]
- Sunmonu, T.O.; Owolabi, O.D.; Oloyede, O.B. Anthracene-induced enzymatic changes as stress indicators in African catfish, Heterobranchus bidorsalis Geoffroy Saint Hilaire, 1809. Res. J. Environ. Sci. 2009, 3, 677–686. [Google Scholar]
- Lee, A.H.; Jang, Y.; Kim, G.H.; Kim, J.J.; Lee, S.S.; Ahn, B.J. Decolorizing an anthraquinone dye by phlebia brevispora: Intra-species characterization. Eng. Life Sci. 2017, 2, 125–131. [Google Scholar] [CrossRef]
- Varol, S.F.; Sayin, S.; Eymur, S.; Merdan, Z.; Ünal, D. Optical performance of efficient blue/near UV nitropyridine-conjugated anthracene (NAMA) based light emitting diode. Org. Electron. 2016, 31, 25–30. [Google Scholar] [CrossRef]
- Huang, H.-S.; Lee, K.-Y.; Shi, C.; Hsu, H. Synthesis and Pharmaceuticals of Novel 9-Substituted-1, 5-Dichloroanthracene Analogs. U.S. Patent 6,369,246 B2, 9 April 2001. [Google Scholar]
- Banerjee, A.K.; Giri, V.S.; Mukherjee, R.; Kapoor, K.K.; Desiraju, G.; Jaggi, M.; Singh, A.T.; Dutta, S.K.; Sairam, K.V. Hydroanthracene Based Compounds as Anticancer Agents. U.S. Patent 20,040,220,197 A1, 4 November 2004. [Google Scholar]
- Bair, K.W. Anthracene Derivatives. U.S. Patent 4,803,221 A, 7 February 1989. [Google Scholar]
- Rastinejad, F.; Foster, B.; Coffey, H.; Connell, R. Methods and Composition for Restoring Conformational Stability of a Protein of the p53 Family. U.S. Patent 20,020,048,271 A1, 23 May 2001. [Google Scholar]
- Karama, U.S.; Sultan, M.A.S.; Tahir, K.E.H.E. Antidepressant Compounds. U.S. Patent 9,125,866 B1, 8 September 2015. [Google Scholar]
- Cloonan, S.M.; Drozgowska, A.; Fayne, D.; Williams, D.C. The antidepressants maprotiline and fluoxetine have potent selective antiproliferative effects against Burkitt lymphoma independently of the norepinephrine and serotonin transporters. Leuk. Lymphoma 2010, 51, 523–539. [Google Scholar] [CrossRef] [PubMed]
- Szabó, D.; Szabó, G.; Ocsovszki, I.; Aszalos, A.; Molnár, J. Anti-psychotic drugs reverse multidrug resistance of tumor cell lines and human AML cells ex-vivo. Cancer Lett. 1999, 139, 115–119. [Google Scholar] [CrossRef]
- Bitonti, A.J.; Sjoerdsma, A.; McCann, P.P.; Kyle, D.E.; Oduola, A.; Rossan, R.N.; Milhous, W.K.; Davidson, D.E. Reversal of chloroquine resistance in malaria parasite Plasmodium falciparum by desipramine. Science 1988, 242, 1301–1303. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, S.M.; Williams, D.C. The antidepressants maprotiline and fluoxetine induce type II autophagic cell death in drug-resistant Burkitt’s lymphoma. Int. J. Cancer 2011, 128, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Stockdale, T.P.; Williams, C.M. Pharmaceuticals that contain polycyclic hydrocarbon scaffolds. Chem. Soc. Rev. 2015, 44, 7737–7763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brocksom, T.J.; Nakamura, J.; Ferreira, M.L.; Brocksom, U. The Diels-Alder reaction: An update. J. Braz. Chem. Soc. 2001, 12, 597–622. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Snyder, S.A.; Montagnon, T.; Vassilikogiannakis, G. The Diels–Alder reaction in total synthesis. Angew. Chem. Int. Ed. 2002, 41, 1668–1698. [Google Scholar] [CrossRef]
- Diels, O.; Alder, K. Synthesen in der hydroaromatischen reihe. Justus Liebigs Ann. Chem. 1928, 460, 98–122. [Google Scholar] [CrossRef]
- Sultan, M.A.; Karama, U. Substituent effects on regioselectivity of the Diels-Alder reactions: Reactions of 10-allyl-1,8-dichloroanthracene with 2-chloroacrylonitrile, 1-cyanovinyl acetate and phenyl vinyl sulfone. J. Chem. 2016, 5. [Google Scholar] [CrossRef]
- Phutdhawong, W.; Buddhasukh, D. Facile microwave-assisted synthesis of 9, 10-dihydro-9, 10-ethanoanthracene-11-carboxylic acid methyl ester. Molecules 2005, 10, 1409–1412. [Google Scholar] [CrossRef] [PubMed]
- Atherton, J.; Jones, S. Diels-Alder reactions of anthracene, 9-substituted anthracenes and 9, 10-disubstituted anthracenes. Tetrahedron 2003, 59, 9039–9057. [Google Scholar] [CrossRef]
- Singh, M.D.; Ningombam, A. Diels-Alder reaction of 9-anthracenemethanol and dimethylacetylene-dicarboxy-late; potential route for the synthesis of regiospecific products of 9-substituted anthracene with unsymmetrical. Indian J. Chem. 2010, 49, 77–83. [Google Scholar]
- Singh, M.D.; Ningombam, A. High stereoselectivity in the Diels-Alder reaction of substituted anthracenes: Reactions of 1-succinimidoanthracene and 1-phthalimidoanthracene with maleic anhydride. Indian J. Chem. 2010, 49, 789–794. [Google Scholar] [CrossRef]
- Khan, R.; Singh, T.P.; Singh, M.D. Highly regioselective Diels-Alder reaction of 9-substituted anthracenes with citraconic anhydride. Synlett 2014, 25, 696–700. [Google Scholar] [CrossRef]
- Wise, K.E.; Wheeler, R.A. Donor-acceptor-assisted diels-alder reaction of anthracene and tetracyanoethylene. J. Phys. Chem. A 1999, 103, 8279–8287. [Google Scholar] [CrossRef]
- Karama, U.S.; Sultan, M.A.S.; Tahir, K.E.H.E.; Almansour, A.I. Halogenated Tetracyclic Compounds. U.S. Patent 9,498,460 B1, 22 November 2016. [Google Scholar]
- Karama, U.; Sultan, M.A.; Almansour, A.I.; El-Taher, K.E. Synthesis of chlorinated tetracyclic compounds and testing for their potential antidepressant effect in mice. Molecules 2016, 21, 61. [Google Scholar] [CrossRef] [PubMed]
- Sultan, M.A.; Karama, U.; Almansour, A.I.; Al-saeedi, A.; Ghabbour, H.A. Crystal structure of 9-allyl-4, 5-dichloro-12-cyano-9, 10-dihydro-9, 10-ethanoanthracen-12-yl acetate, C22H17Cl2NO2. Z. Krist.-New Cryst. Struct. 2016, 231, 801–803. [Google Scholar]
- Verma, S.M.; Singh, M.D. Structural elucidation with nuclear magnetic resonance spectroscopy. Diels-alder adducts of 1-aminoanthracene and maleic anhydride: Restricted rotation about the aryl C (1)-N bond and intrinsic asymmetry about the imide (Nsp2-Csp3) system. J. Org. Chem. 1977, 42, 3736–3740. [Google Scholar]
- Kaplan, F.; Conroy, H. Electronic effects on the stereochemistry of the Diels-Alder reaction1. J. Org. Chem. 1963, 28, 1593–1596. [Google Scholar] [CrossRef]
- House, H.O.; Koepsell, D.; Jaeger, W. Derivatives of 1, 8-diphenylanthracene. J. Org. Chem. 1973, 38, 1167–1173. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXTL. Version 6.12 for Windows NT; Bruker AXS Inc.: Madison, WI, USA, 2008. [Google Scholar]
- Bruker. SMART, Version 5.625 for Windows NT; Bruker AXS Inc.: Madison, WI, USA, 2000. [Google Scholar]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Montgomery, J., Jr.; Vreven, T.; Kudin, K.; Burant, J. Gaussian 03, Revision c. 02; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. Gauss View, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Data | |
|---|---|
| Chemical formula | C17H10Cl3N |
| Formula weight | 334.61 |
| Crystal system, space group | Monoclinic, P21/n |
| Temperature (K) | 100 |
| a, b, c (Å) | 9.2332 (5), 12.4617 (6), 12.8375 (7) |
| β () | 105.678 (2) |
| V (Å3) | 1422.15 (13) |
| Z | 4 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.63 |
| Crystal size (mm) | 0.55 × 0.38 × 0.12 |
| Data collection | |
| Diffractometer | Bruker APEX-II D8 VENTURE diffractometer |
| Absorption correction | Multi-scan, SADABS Bruker 2014 |
| Tmin, Tmax | 0.723, 0.926 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 16403, 3250, 2462 |
| Rint | 0.089 |
| Refinement | |
| R[F2 > 2σ(F2)], wR(F2), S | 0.049, 0.124, 1.04 |
| No. of reflections | 3250 |
| No. of parameters | 190 |
| No. of restraints | 0 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinements |
| Δρmax, Δρmin (e Å−3) | 0.49, −0.61 |
| Gas | CHCl3 | |||
|---|---|---|---|---|
| Isomer 1 | Isomer 2 | Isomer 1 | Isomer 2 | |
| E (a.u.) | −2089.1773 | −2089.1753 | −2089.1885 | −2089.1871 |
| ZPVE (a.u.) | 0.222732 | 0.222619 | 0.221657 | 0.221574 |
| Ecorr (a.u.) | −2088.95453 | −2088.95268 | −2088.96683 | −2088.96549 |
| ΔE (kcal/mol) | −1.16290 | −0.84192 | ||
| H (a.u.) | −2088.9369 | −2088.935 | −2088.9492 | −2088.9478 |
| S (cal/mol K) | 131.624 | 131.532 | 131.826 | 131.812 |
| G (a.u.) | −2088.9994 | −2088.9975 | −2089.0118 | −2089.0104 |
| ΔG (kcal/mol) | −1.194778 | −0.8571779 | ||
| Parameter | Calc. | X-ray | Parameter | Calc. | X-ray |
|---|---|---|---|---|---|
| R(1-5) | 1.761 | 1.746 | A(6-5-26) | 120.7 | 120.8 |
| R(2-22) | 1.760 | 1.743 | A(5-6-8) | 119.7 | 119.4 |
| R(3-30) | 1.841 | 1.811 | A(5-26-12) | 118.7 | 118.4 |
| R(5-6) | 1.398 | 1.395 | A(5-26-24) | 128.1 | 127.8 |
| R(5-26) | 1.392 | 1.383 | A(6-8-10) | 120.6 | 120.8 |
| R(6-8) | 1.394 | 1.389 | A(8-10-12) | 119.0 | 118.6 |
| R(8-10) | 1.398 | 1.388 | A(10-12-13) | 125.8 | 125.4 |
| R(10-12) | 1.390 | 1.383 | A(10-12-26) | 121.3 | 121.9 |
| R(12-13) | 1.517 | 1.517 | A(13-12-26) | 112.9 | 112.7 |
| R(12-26) | 1.405 | 1.396 | A(12-13-15) | 108.2 | 108.6 |
| R(13-15) | 1.521 | 1.519 | A(12-13-30) | 107.4 | 106.6 |
| R(13-30) | 1.578 | 1.566 | A(12-26-24) | 113.2 | 113.8 |
| R(15-16) | 1.390 | 1.382 | A(15-13-30) | 105.6 | 104.6 |
| R(15-23) | 1.406 | 1.398 | A(13-15-16) | 125.8 | 125.5 |
| R(16-18) | 1.398 | 1.394 | A(13-15-23) | 112.9 | 113.1 |
| R(20-22) | 1.398 | 1.398 | A(13-30-27) | 108.9 | 110.1 |
| R(22-23) | 1.392 | 1.386 | A(13-30-31) | 110.3 | 109.2 |
| R(23-24) | 1.520 | 1.514 | A(16-15-23) | 121.4 | 121.4 |
| R(24-26) | 1.519 | 1.519 | A(15-16-18) | 119.0 | 118.9 |
| R(24-27) | 1.561 | 1.550 | A(15-23-22) | 118.7 | 118.7 |
| R(27-30) | 1.566 | 1.553 | A(15-23-24) | 113.2 | 113.4 |
| R(30-31) | 1.465 | 1.511 | A(16-18-20) | 120.6 | 120.9 |
| A(1-5-6) | 118.7 | 118.8 | A(18-20-22) | 119.7 | 119.3 |
| A(1-5-26) | 120.6 | 120.4 | A(20-22-23) | 120.7 | 120.8 |
| A(2-22-20) | 118.7 | 118.5 | A(22-23-24) | 128.2 | 127.9 |
| A(2-22-23) | 120.6 | 120.8 | A(23-24-26) | 107.4 | 107.7 |
| A(3-30-13) | 109.3 | 105.0 | A(23-24-27) | 106.9 | 106.9 |
| A(3-30-27) | 110.7 | 110.2 | A(26-24-27) | 106.9 | 106.1 |
| A(3-30-31) | 105.9 | 105.0 | A(24-27-30) | 109.4 | 108.9 |
| A(4-31-30) | 179.2 | 177.4 | A(27-30-31) | 111.8 | 112.5 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sultan, M.A.; Karama, U.; Almansour, A.I.; Soliman, S.M.; Ghabbour, H.A.; Mabkhot, Y.N. Synthesis, Characterization and DFT Calculations of 4,5,12- and 1,8,12-trichloro-9,10-dihydro-9,10-ethanoanthracene-12-carbonitriles. Crystals 2017, 7, 259. https://doi.org/10.3390/cryst7090259
Sultan MA, Karama U, Almansour AI, Soliman SM, Ghabbour HA, Mabkhot YN. Synthesis, Characterization and DFT Calculations of 4,5,12- and 1,8,12-trichloro-9,10-dihydro-9,10-ethanoanthracene-12-carbonitriles. Crystals. 2017; 7(9):259. https://doi.org/10.3390/cryst7090259
Chicago/Turabian StyleSultan, Mujeeb A., Usama Karama, Abdulrahman I. Almansour, Saied M. Soliman, Hazem A. Ghabbour, and Yahia N. Mabkhot. 2017. "Synthesis, Characterization and DFT Calculations of 4,5,12- and 1,8,12-trichloro-9,10-dihydro-9,10-ethanoanthracene-12-carbonitriles" Crystals 7, no. 9: 259. https://doi.org/10.3390/cryst7090259