
Synthesis and X-ray Crystal Structure of N’-Cyano-N,N’-dimethyl-4-nitrobenzohydrazide


1
Helmholtz-Zentrum Dresden-Rossendorf, Institut für Radiopharmazeutische Krebsforschung, Bautzner Landstraße 400, D-01328 Dresden, Germany
2
Anorganische Festkörperchemie, Institut für Chemie, Universität Rostock, Albert-Einstein-Straße 3a, D-18059 Rostock, Germany
*
Author to whom correspondence should be addressed.
Crystals 2017, 7(10), 290; https://doi.org/10.3390/cryst7100290
Submission received: 29 August 2017
/
Revised: 18 September 2017
/
Accepted: 19 September 2017
/
Published: 26 September 2017
(This article belongs to the Section Crystalline Materials)
Abstract
:Using a two-step procedure, N’-cyano-N,N’-dimethyl-4-nitrobenzohydrazide was synthesized. The structure was established using single crystal X-ray diffraction. It crystalized in the orthorhombic space group P212121 where a = 8.1974(6), b = 10.6696(7), and c = 12.9766(8) Å. The first reported crystal structure of an acyclic cyanohydrazide is discussed with a focus on the geometry of the hydrazide moiety, but intermolecular contacts in the crystal are also considered.
1. Introduction
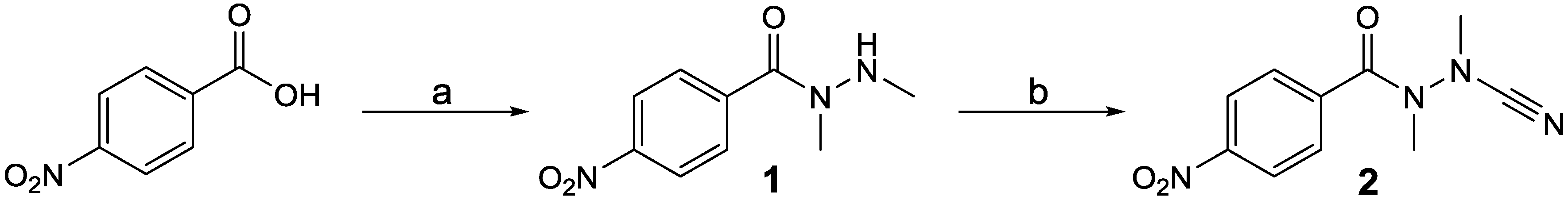
N’-Cyanohydrazides are only existent if both nitrogen atoms are at least methylated because unsubstituted or monoalkylated derivatives tend to cyclize [1,2]. Those analogues, which bear an amino acid-derived acyl moiety, so-called azapeptide nitriles, have been shown to be highly potent inhibitors of cysteine proteases [1,3,4,5]. Their high intrinsic reactivity towards thiols [6] renders this compound class potentially interesting for bioconjugation reactions to introduce reporter groups such as radiolabels. This seems to be especially valid for the site-specific modification of N-terminal cysteine residues, as has been demonstrated for other cyano-functionalized labeling reagents [7,8,9]. Despite their potential for enzyme inhibition and bioconjugate chemistry, crystal structures of N’-cyanohydrazides have not yet been reported, apart from one cyclic derivative in which the hydrazide moiety is integrated into a pyrazolidin-3-one ring system [10]. In addition, crystal structures of two cyclic cyanohydrazines have been published [11]. To enable labeling of cyanohydrazides with radiohalogens such as fluorine-18, their combination with para-substitued benzoyl moieties is obvious. During studies aiming at exploring the suitability of fluorinated cyanohydrazides for the labeling of peptides and proteins with fluorine-18, we prepared the title compound as a potential precursor for nucleophilic aromatic substitution with [18F]fluoride. Single crystals of this compound (2, Scheme 1) were grown and were found to be suitable for X-ray diffraction analysis. The results of this investigation, which reveals insight into the three-dimensional structure of linear N’-cyanohydrazides, will be communicated herein.
2. Results and Discussion
It has been demonstrated that the synthetic access to cyanohydrazides via conversion of N,N’-dialkylhydrazides with cyanogen bromide is highly feasible [1,5,12]. Therefore, N,N’-dimethyl-4-nitrobenzohydrazide (1) was prepared by C–N bond coupling between 4-nitrobenzoic acid and N,N’-dimethylhydrazine employing the mixed anhydride approach. This compound was prepared previously via an alternative approach involving hydrazone formation between the corresponding Nβ-unsubstituted hydrazide and formaldehyde followed by reduction with sodium cyanoborohydride [13]. Compound 1 was reacted with cyanogen bromide in the presence of sodium acetate as a base at room temperature using methanol as solvent. The final compound 2 was obtained in an overall yield of 33%. The synthetic sequence is shown in Scheme 1.
The crystal and instrumental parameters used in the unit cell determination together with the data collection and structure refinement parameters are summarized in Table 1.
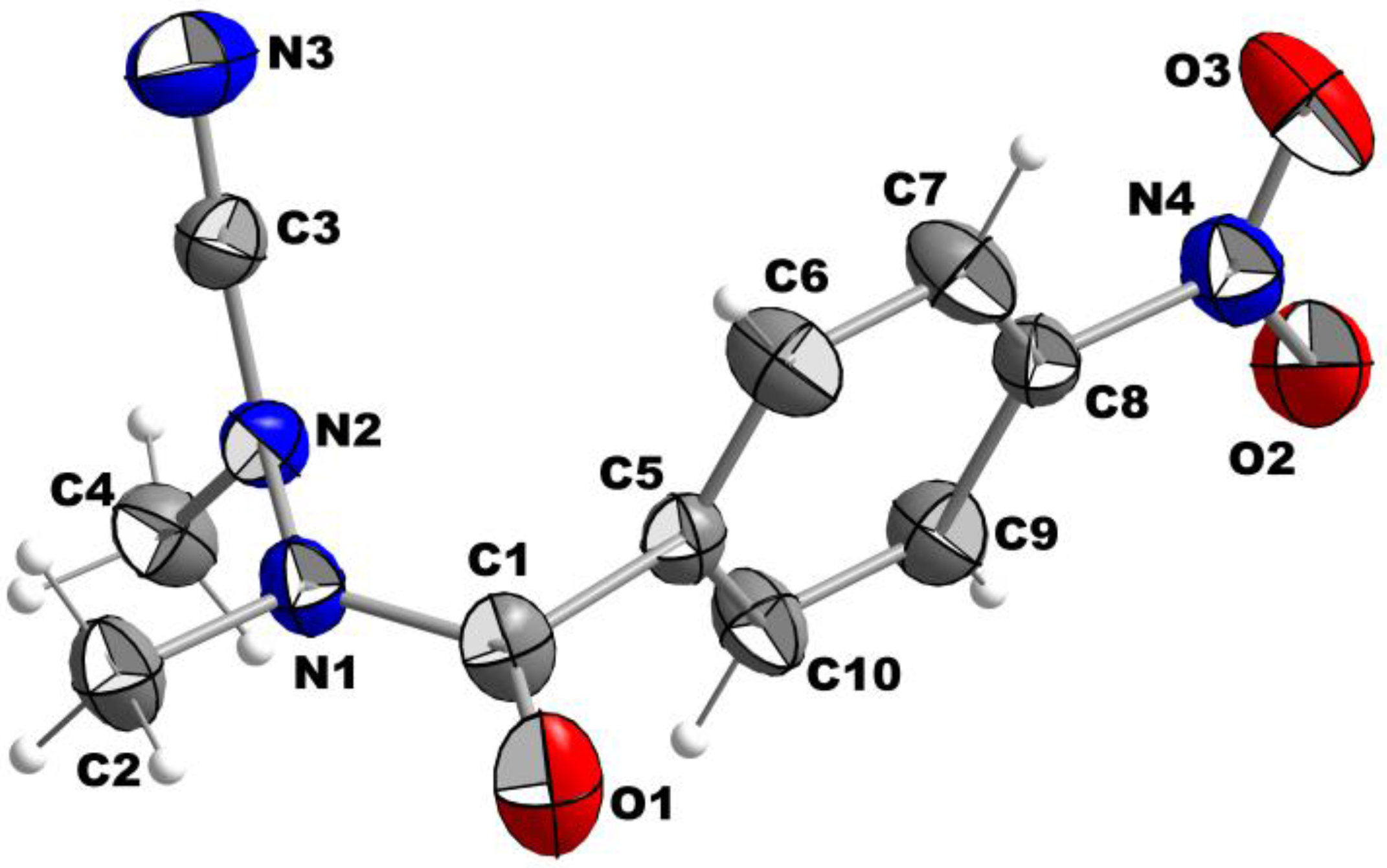
The molecular structure of Compound 2 with atom labeling scheme is shown in Figure 1 as an Ortep-style plot with 30% probability.
The nitro group is almost in plane with the phenyl ring (dihedral angle 7.4°). In contrast, the plane of the carbonyl group (defined by O1, C1, and C5) is almost perpendicularly oriented (87.9°) towards the plane of the ring system. This strong twist of the carbonyl axis against the aromatic plane is quite remarkable as it is significantly higher than that observed for all Nα-substituted 4-nitrobenzoylhydrazines included in the Cambridge structural database (CSD) (Table 2). The lengths of the bonds around atom C1 are less deviant to the values observed for related compounds. However, the double-bond character of the carbonyl group seems to be slightly stronger than in other 4-nitrobenzohydrazides, as the C1–O1 bond is with 1.214 Å shorter than the average value of 1.226 Å (Table 2). According to the length, the C1–C5 bond has largely single-bond character. The type of the C1–N1 bond is discussed below.
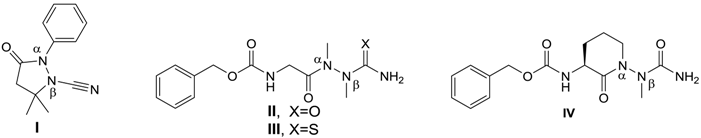
Considering the structural traits of the hydrazide moiety, the N–N distance (N1–N2) is comparable to that in the H2N–NH2 molecule (1.404(3) Å vs. 1.45 Å in hydrazine) and therefore in accordance with a single bond. The groups attached to the hydrazine moiety are twisted with an almost right angle (∢ (C1–N1–C2)/(C3–N2–C4) = 85.7°). Similar conformational preferences of the N–N bond were observed in the Nα,Nβ-dialkylated (thio)semicarbazides II–IV (Table 3) [14,15,16] and related azapeptides [17]. The methyl group of C2 is located almost in the plane of the carbonyl group (defined by C5, C1, and O1), as indicated by a dihedral angle of 177.2° (defined by the bonds C5–C1/N1–C2). In combination with the short C1–N1 bond (1.343(4) Å), a partial double bond character can be assumed for C1–N1. Accordingly, the out-of-plane angle for N1 is with 4.24° very small, which corresponds to a distance of 0.029 Å from the plane defined by C1, C2, and N2. Therefore, an almost integral sp2 hybridization state can be deduced for the N1 atom. The configuration of the C1–N1 bond is E, which is the preferred configuration of the amide bond in trisubstituted hydrazides [18]. It is worth noting that the geometry around the N2 atom is significantly pyramidalized, as indicated by the sum of the three bond angles defined by the carbon atoms surrounding N2, which is with 351.8° significantly less than 360°, and an out-of-plane angle of 19.4°. The corresponding nitrogen atom of Compound I, which is the only additional cyanohydrazide included in the CSD to date, appears to be even more pyramidalized. This is probably a result of the constraints imparted by the five-membered ring, as the out-of-plane angle is also larger for the corresponding Nα atom (Table 3). Thus, one can conclude that the hybridization of the N2 atom has a partial sp3 character and that its orbital overlap with the adjacent cyano carbon (C3) atom is weak. This conclusion is further supported by comparing the bond lengths N2–C3 and C3–N3, which are 1.323(5) and 1.140(5) Å, respectively. The cyano group is almost linear (N2–C3–N3: 176.5°). The slightly bended orientation of the two nitrogen atoms attached to the cyano carbon is in agreement with structural observations for other cyanohydrazines [10,11] and N,N-disubstituted cyanamides in general (see the crystal structures published in [19,20] for example). In contrast to the finding for Compound 2, the out-of-plane angles for the Nβ atoms in Nα,Nβ-dialkylated (thio)semicarbazides (II–IV, Table 3) are significantly smaller, which in turn indicates a more sp2-like hybridization state for these nitrogen atoms.
The restricted electron transfer between N2 and C3 in Compound 2 might also explain why related azadipeptide nitriles are much more potent inhibitors of papain-like cysteine proteases compared to their carbon-based counterparts. While azadipeptide nitriles exhibit slower rates for the reaction with the active-site cysteine residue than their carba analogs, they display equilibrium inhibition constants that are smaller by about three orders of magnitude [1]. Nucleophilic attack of the nitrogen-bonded cyano group by the active-site thiol results in the formation of an enzyme-bound isothiosemicarbazide, in which the Nβ atom will probably adopt a more planar geometry. In turn, this will enhance resonance electron transfer to the adjacent cyano group-derived carbon atom. Therefore, the differences in the extent of the resonance electron transfer involving the Nβ lone pair between the enzyme-bound and free azadipeptide nitriles may account for their strong inhibitory potency.
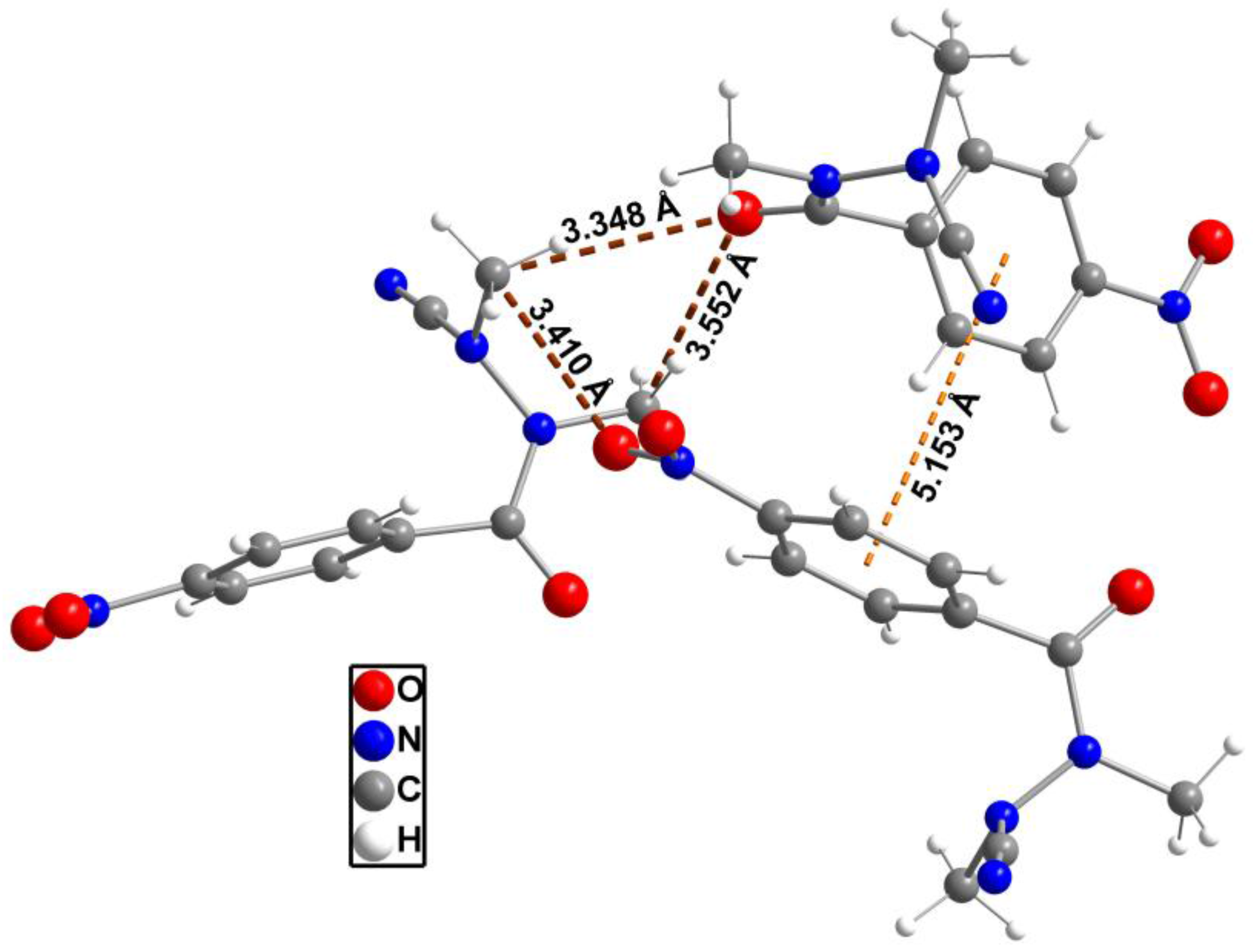
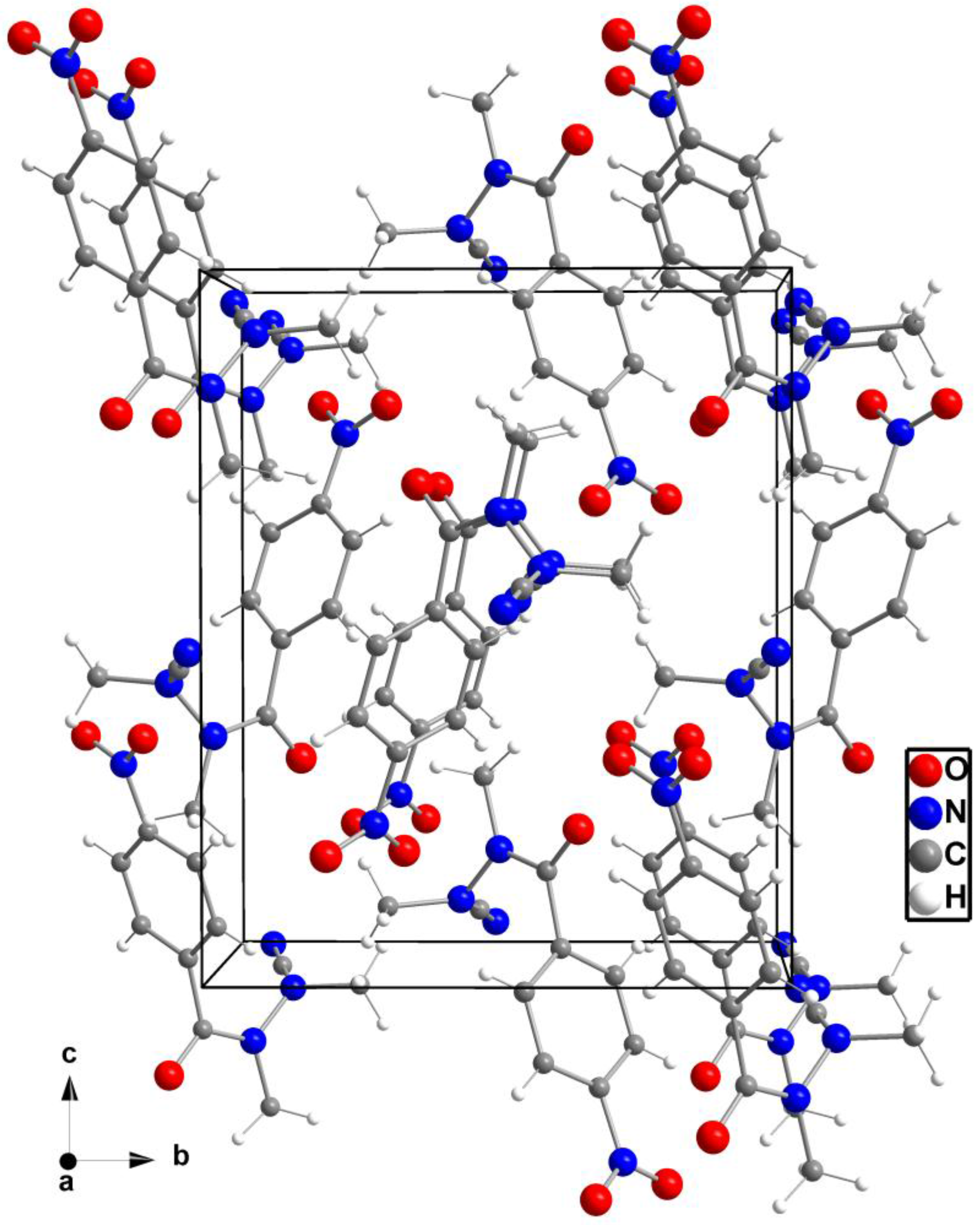
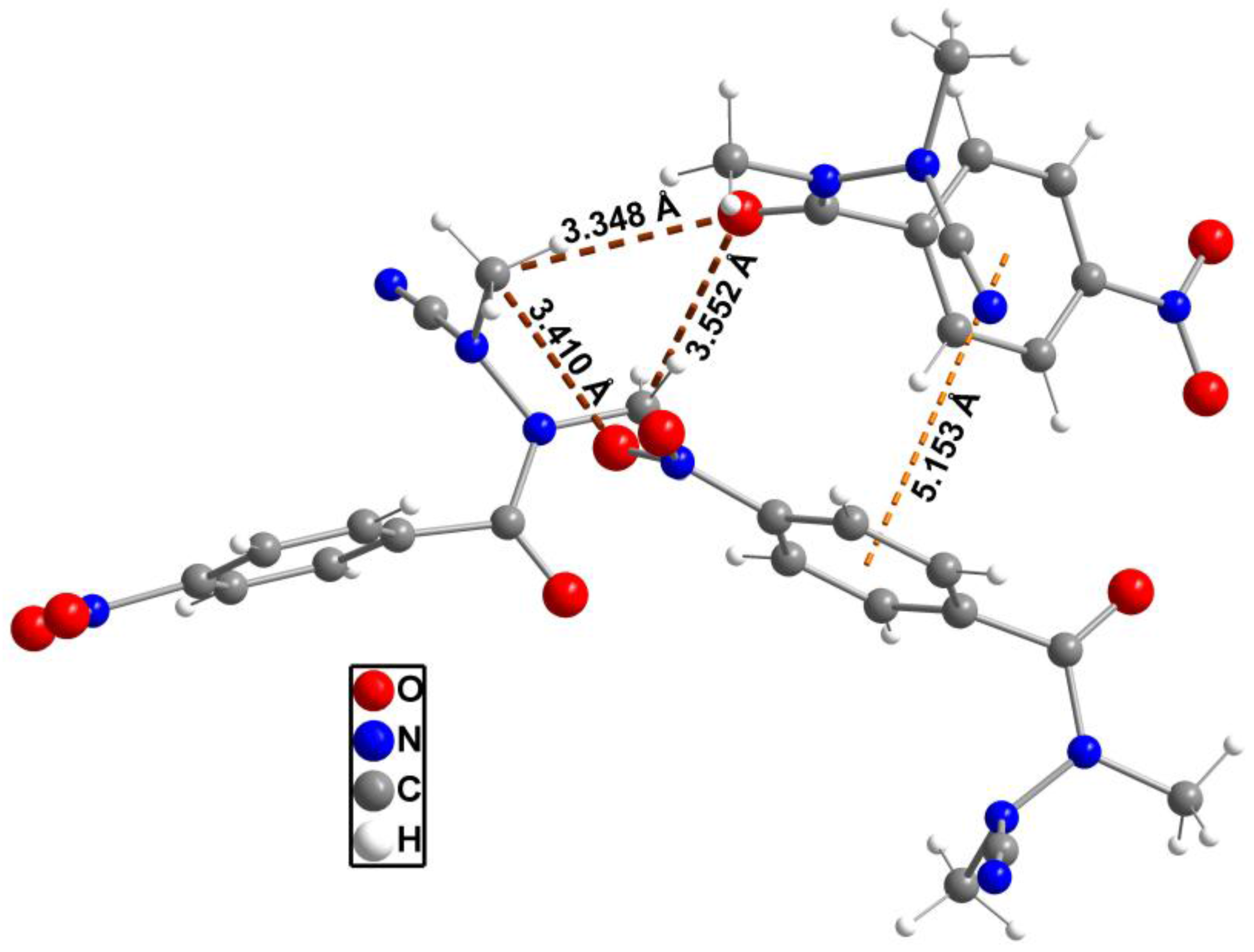
The packing of the molecules in the unit cell is shown in Figure 2. Compound 2 does not exhibit any polar hydrogen bonds, thus no classical hydrogen bonds exist. The shortest intermolecular distances are found between O1 and C4 (H4B, neighboring molecule) at 3.348(4) Å and O1 and C2 (H2B) at 3.552(4) Å (Figure 3). A similar contact can be observed between O2 and C4 (H4C, neighboring molecule) at a distance of 3.410(5) Å. Further intermolecular contacts are formed by π–π interactions of the edge-to-face type between the neighboring 1,4-disubstituted phenyl moieties as indicated by centroid-centroid distances of 5.1530(3) Å. The planes defined by the involved aromatic rings intersect each other with an angle of 87.0°, which is very close to the ideal angle of 90° [21].
3. Experimental Section
3.1. General
NMR spectra were recorded on a Varian Unity 400 MHz (Varian Inc., Palo Alto, CA, USA) or an Agilent DD2 400 MHz spectrometer (Agilent Technologies Inc., Santa Clara, CA, USA) equipped with ProbeOne probe. Chemical shifts of the 1H and 13C spectra were reported in parts per million (ppm) using tetramethylsilane (TMS) as internal standard. Assignments of signal to atoms are reported according to the atom numbering scheme of Compound 2 shown in Figure 1. Signal assignments for Compound 2 have been confirmed by heteronuclear single quantum coherence (HSQC) and heteronuclear multiple bond correlation (HMBC) spectra. Mass spectra were obtained on a Micromass Quattro/LC mass spectrometer (SpectraLab Scientific Inc., Markham, ON, Canada) by electrospray ionization. Elemental microanalysis was performed on a Euro EA 3000 Elemental Analyzer (Euro Vector Instruments & Software, Pavia, Italy). Melting points were determined on a Galen III Boetius apparatus (Cambridge Instruments, Cambridge, UK). Chromatographic separations and TLC detections were carried out with Merck Silica Gel 60 (63–200 μm) and Merck Silica Gel 60 F254 sheets, respectively. TLCs were developed by visualization under UV light (λ = 254 nm). Anhydrous THF was purchased from SigmaAldrich (Schnelldorf, Germany). All starting materials and reagents were commercially obtained and used without further purification.
Crystallographic data were collected with a Bruker–Nonius Apex-X8 CCD-diffractometer (Bruker, Madison, WI, USA) with Mo-Kα radiation (λ = 0.71073 Å) at 123 K. The structures were solved by direct methods using SHELXS-97 and refined against F2 on all data by full matrix least-squares refinements using the program suites from G.M. Sheldrick [22,23,24]. Data corrections including multi-scan absorption corrections were applied to the data sets using the Bruker AXS software [25]. All non-hydrogen atoms were refined anisotropically; all hydrogen atoms bonded to C atoms were placed on geometrically calculated positions and refined using riding models. CCDC 1567206 containing the supplementary crystallographic data of Compound 2 have been deposited with The Cambridge Crystallographic Data Centre. These data can be obtained free of charge via [26].
3.2. Synthesis
N,N’-Dimethyl-4-nitrobenzohydrazide (1). A solution of 4-nitrobenzoic acid (1.50 g, 8.98 mmol) in anhydrous THF (30 mL) was cooled to −30 °C and N-methylmorpholine (0.984 mL, 8.98 mmol) was added. In parallel, N,N’-dimethylhydrazine dihydrochloride (7.12 g, 44.9 mmol) was dissolved in water (6 mL) and 4 M NaOH (22.4 mL) was added under ice cooling. Isobutyl chloroformate (1.174 mL, 8.98 mmol) was added to the 4-nitrobenzoate solution under vigorous stirring. Immediately after the precipitation of N-methylmorpholine hydrochloride was completed, the aqueous solution of N,N’-dimethylhydrazine was added. The resulting reaction mixture was allowed to come to room temperature. After 3 h, THF was removed in vacuo and the remaining weakly alkaline aqueous solution was extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with water (10 mL), sat. NaHCO3 (2 × 10 mL), water (10 mL), brine (10 mL), dried over Na2SO4 and the solvent was completely evaporated. The obtained solid residue was subjected to purification by column chromatography on silica gel using CH2Cl2/methanol (100:1) as eluent to obtain 0.91 g (48%) of a pale-yellow solid. Mp 112–114 °C; 1H NMR (400 MHz, CDCl3, mixture of s-cis and s-trans rotamers), δ (ppm): 2.59, 2.70 (s, ∑ 3H, NHCH3), 3.11, 3.29 (s, ∑ 3H, NCH3), 7.66 (br s, 2H, H-6, H-10), 8.27–8.23 (br m, 2H, H-7, H-9); MS (ESI+) m/z: 210.25 ([M + H]+), 251.11 ([M + CH3CN + H]+), 419.25 ([2M + H]+).
N’-Cyano-N,N’-dimethyl-4-nitrobenzohydrazide (2). To a solution of Compound 1 (0.44 g, 2.11 mmol) in methanol (15 mL) were added sodium acetate (0.432 g, 5.27 mmol) and cyanogen bromide (0.335 g, 3.16 mmol) as solids. After stirring for 18 h at room temperature, the solvent was removed in vacuo. The remaining solid was suspended in 1 M HCl (10 mL) and extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with water (1 × 10 mL), sat. NaHCO3 (2 × 10 mL), water (1 × 10 mL), brine (10 mL), dried over Na2SO4 and the solvent was completely evaporated. The obtained solid residue was subjected to purification by column chromatography on silica gel using petroleum ether/ethyl acetate (1:1) as eluent to obtain 0.30 g (61%) of a pale-yellow solid. Mp 140–141 °C; 1H NMR (400 MHz, CDCl3), δ (ppm): 3.01 (s, 3H, H-4), 3.36 (s, 3H, H-2), 7.73 (d, 3J = 8.9 Hz, 2H, H-6, H-10), 8.33 (d, 3J = 8.9 Hz, 2H, H-7, H-9); 13C NMR (101 MHz, CDCl3), δ (ppm): 32.32 (br, C2), 40.80 (C4), 113.65 (C3), 123.95 (C7, C9), 128.33 (C6, C10), 139.72 (C5), 149.31 (C8), 169.76 (C1); MS (ESI+) m/z: 235.41 ([M + H]+), 273.32 ([M + K]+); elemental analysis C10H10N4O3, calcd. C: 51.28%, H: 4.30% N 23.92%, found C: 52.26%, H: 4.51% N 23.40%. Crystals that proved to be suitable for X-ray diffraction analysis were obtained by adding about 400–500 µL of cyclohexane to a solution of 5 mg of 2 in about 100 µL of chloroform. Chloroform was allowed to slowly evaporate, upon which crystals of 2 were slowly growing.
4. Conclusions
In this article we reported on the synthesis and crystal structure of cyanohydrazide 2, a member of a crystallographically underexplored compound class. The results confirm that the hybridization states of the hydrazide N atoms are different, despite substituents that both exert-M effects are attached to each nitrogen atom.
Acknowledgments
We wish to thank Karin Landrock for carrying out elemental microanalysis. Partial funding to R.L. by the Fonds der Chemischen Industrie/Germany is gratefully acknowledged.
Author Contributions
R.L. conceived and designed the experiments; R.P., M.K. and R.L. performed the experiments; M.K. and R.L. analyzed the data; R.L. and M.K. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Löser, R.; Frizler, M.; Schilling, K.; Gütschow, M. Azadipeptide nitriles: Highly potent and proteolytically stable inhibitors of papain-like cysteine proteases. Angew. Chem. Int. Ed. 2008, 47, 4331–4334. [Google Scholar] [CrossRef] [PubMed]
- Löser, R.; Nieger, M.; Gütschow, M. Synthesis and crystal structure of benzyl [(1S)-1-(5-amino-1,3,4-oxadiazol-2-yl)-2-phenylethyl]carbamate. Crystals 2012, 2, 1201–1209. [Google Scholar] [CrossRef]
- Loh, Y.; Shi, H.; Hu, M.; Yao, S.Q. “Click” synthesis of small molecule-peptide conjugates for organelle-specific delivery and inhibition of lysosomal cysteine proteases. Chem. Commun. 2010, 46, 8407–8409. [Google Scholar] [CrossRef] [PubMed]
- Frizler, M.; Lohr, F.; Furtmann, N.; Kläs, J.; Gütschow, M. Structural optimization of azadipeptide nitriles strongly increases association rates and allows the development of selective cathepsin inhibitors. J. Med. Chem. 2011, 54, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.F.; Li, H.W.; Fang, X.; Wu, Y.; Wang, L.; Zou, S. Highly selective azadipeptide nitrile inhibitors for cathepsin K: Design, synthesis and activity assays. Org. Biomol. Chem. 2013, 11, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Löser, R.; Bergmann, R.; Frizler, M.; Mosch, B.; Dombrowski, L.; Kuchar, M.; Steinbach, J.; Gütschow, M.; Pietzsch, J. Synthesis and radiopharmacological characterisation of a fluorine-18-labelled azadipeptide nitrile as a potential PET tracer for in vivo imaging of cysteine cathepsins. ChemMedChem 2013, 8, 1330–1344. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Xiao, F.; Zhan, K.; Kim, Y.P.; Xie, H.; Xia, Z.; Rao, J. A biocompatible condensation reaction for the labeling of terminal cysteine residues on proteins. Angew. Chem. Int. Ed. 2009, 48, 9658–9662. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Jeon, J.; Gambhir, S.S.; Rao, J.; Chin, F.T. 18F-Cyanobenzolthiol ([18F]CBT): A novel 18F-prosthetic group for labeling peptide or protein. J. Label. Compd. Radiopharm. 2011, 54, S503. [Google Scholar] [CrossRef]
- Inkster, J.A.; Colin, D.J.; Seimbille, Y. A novel 2-cyanobenzothiazole-based 18F prosthetic group for conjugation to 1,2-aminothiol-bearing targeting vectors. Org. Biomol. Chem. 2015, 13, 3667–3676. [Google Scholar] [CrossRef] [PubMed]
- Bird, C.W.; Cobb, J.; Nyburg, S.C.; Parkins, A.W. Some stereochemical aspects of the Pellizzari rearrangement. Tetrahedron 1995, 51, 13161–13166. [Google Scholar] [CrossRef]
- Bilewicz, E.; Malecka, M.; Grabowski, S.J.; Mloston, G. 2′-(4-Chlorophenyl)-2,3,4,5,6,7-hexahydro-4′,7′-methanospiro[9H-fluorene-9,3′-1H-indazole]-1′-carbo-nitrile and methyl 4′-chloro-2′-(4-chlorophenyl)-1′-cyanospiro[9H-fluorene-9,3′-pyrazolidine]-4′-carboxylate. Acta Cryst. 2007, C63, o739–o742. [Google Scholar] [CrossRef]
- Yang, P.Y.; Wang, M.; Li, L.; Wu, H.; He, C.Y.; Yao, S.Q. Design, synthesis and biological evaluation of potent azadipeptide nitrile inhibitors and activity-based probes as promising anti-Trypanosoma brucei agents. Chem. Eur. J. 2012, 18, 6528–6541. [Google Scholar] [CrossRef] [PubMed]
- McNab, H.; Hulme, A.; Benstead, D.; Wight, P. An efficient synthesis of substituted hydrazides. Synlett 2005, 1571–1574. [Google Scholar] [CrossRef]
- Ottersbach, P.A.; Schnakenburg, G.; Gütschow, M. Induction of chirality: Experimental evidence of atropisomerism in azapeptides. Chem. Commun. 2012, 48, 5772–5774. [Google Scholar] [CrossRef] [PubMed]
- Ottersbach, P.A.; Schnakenburg, G.; Gütschow, M. Atropisomerism in azadipeptides: Evaluation of N1-methylation and thioamide introduction. Tetrahedron Lett. 2015, 56, 4889–4891. [Google Scholar] [CrossRef]
- Ottersbach, P.A.; Schmitz, J.; Schnakenburg, G.; Gütschow, M. An access to aza-Freidinger lactams and E-locked analogs. Org. Lett. 2013, 15, 448–451. [Google Scholar] [CrossRef] [PubMed]
- Chingle, R.; Lubell, W.D. Azopeptides: Synthesis and pericyclic chemistry. Org. Lett. 2015, 17, 5400–5403. [Google Scholar] [CrossRef] [PubMed]
- Licandro, E.; Perdicchia, D. N-Acylhydrazines: Future perspectives offered by new syntheses and chemistry. Eur. J. Org. Chem. 2004, 665–675. [Google Scholar] [CrossRef]
- Panaka, S.; Trivedi, R.; Sony, T.; Prabhakar, S.; Raju Chowhan, L. Silver(I) catalyzed intramolecular cyclization of N-(2-(alk-1-yn-1-yl))-1H-tetrazoles leading to the formation of N-cyano-2-substituted indoles under ambient conditions. Org. Chem. Front. 2017, 4, 1574–1579. [Google Scholar] [CrossRef]
- Paciaroni, N.G.; Ratnayake, R.; Matthews, J.H.; Norwood, V.M., IV; Arnold, A.C.; Dang, L.H.; Luesch, H.; Huigens, R.W., III. A tryptoline ring-distortion strategy leads to complex and diverse biologically active molecules from the indole alkaloid yohimbine. Chem. Eur. J. 2017, 23, 4327–4335. [Google Scholar] [CrossRef] [PubMed]
- Meyer, E.A.; Castellano, R.K.; Diederich, F. Interactions with aromatic rings in chemical and biological recognition. Angew. Chem. Int. Ed. 2003, 42, 1210–1250. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Scheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXL 2014/1; University of Göttingen: Göttingen, Germany, 2014. [Google Scholar]
- Bruker AXS Inc. APEX-II (ver. 2008.1-0), SAINT (ver. 7.51A) and SADABS (ver. 2007/4); Bruker AXS Inc.: Madison, WI, USA, 2008. [Google Scholar]
- The Cambridge Crystallographic Data Centre. Available online: http://www.ccdc.cam.ac.uk/conts/retrieving.html (accessed on 20 September 2017).
Scheme 1.
Synthesis of N’-cyano-N,N’-dimethyl-4-nitrobenzohydrazide (2). Reagents and conditions: (a) i: N-Methylmorpholine, THF, −30 °C; ii: (CH3NH)2 × 2 HCl, H2O, 4N NaOH, −30° C to room temperature; (b) BrCN, NaCH3COO, methanol, room temperature.
Scheme 1.
Synthesis of N’-cyano-N,N’-dimethyl-4-nitrobenzohydrazide (2). Reagents and conditions: (a) i: N-Methylmorpholine, THF, −30 °C; ii: (CH3NH)2 × 2 HCl, H2O, 4N NaOH, −30° C to room temperature; (b) BrCN, NaCH3COO, methanol, room temperature.
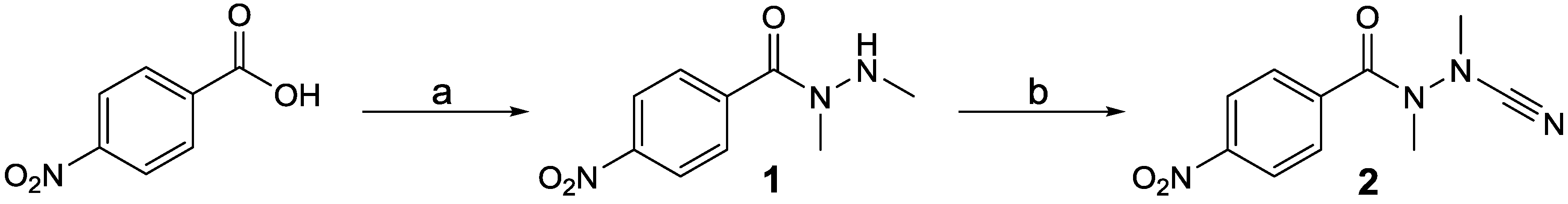
Figure 1.
The molecular structure of Compound 2 as observed in the crystal state.
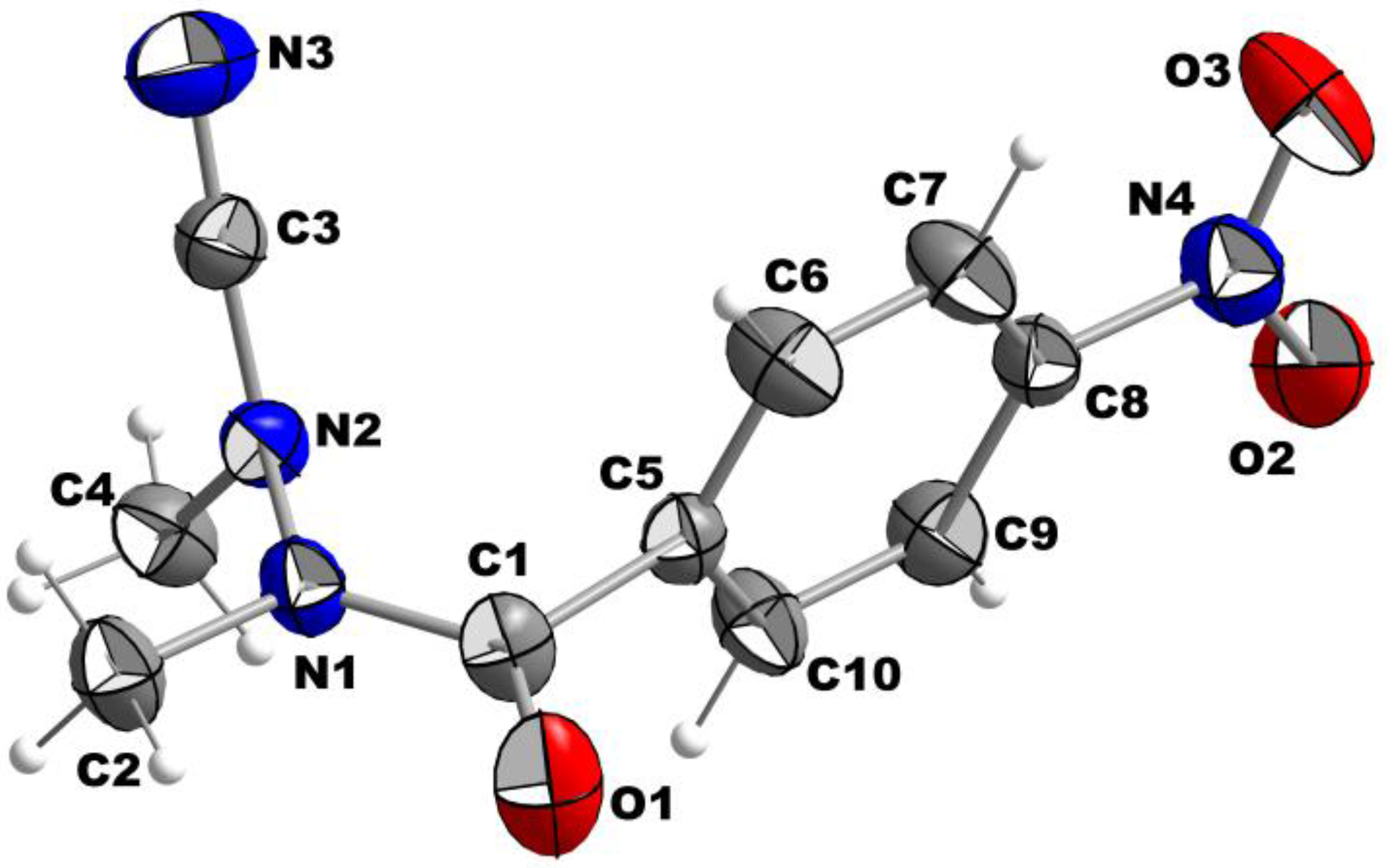
Figure 2.
Packing of the molecules in the unit cell in a view along a.

Figure 3.
Network of intermolecular contacts in the crystal as represented by three neighboring molecules.
Figure 3.
Network of intermolecular contacts in the crystal as represented by three neighboring molecules.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Crystal data and structure refinement parameters of Compound 2.
| Formula | C10H10N4O3 |
| Formula weight (g·mol−1) | 234.22 |
| Temperature (K) | 296 |
| Wavelength (Å) | 0.71073 |
| Crystal system | orthorhombic |
| Space group | P212121 |
| Unit cell dimensions | |
| a (Å) | 8.1974(6) |
| b (Å) | 10.6696(7) |
| c (Å) | 12.9766(8) |
| Volume (Å3) | 1135.0(1) |
| Z | 4 |
| Density (calcd.) (g·cm–3) | 1.371 |
| Absorpt. coeff. (mm–1) | 0.10 |
| F(000) | 488 |
| Crystal size (mm3) | 0.31 × 0.06 × 0.06 |
| Refinement method | Full matrix least-squares on F2 |
| Data/restraints/param. | 2719/0/155 |
| Measured reflections | 18,484 |
| 2 θmax (°) | 28.0 |
| Rint | 0.069 |
| GoF on F2 | 0.99 |
| R1 [I > 2σ(I)] | 0.046 |
| wR2 (all data) | 0.131 |
Table 2.
Solid-state structural parameters for the amide moieties of Compound 2 and related Nα-substituted 4-nitrobenzoylhydrazines *.
Table 2.
Solid-state structural parameters for the amide moieties of Compound 2 and related Nα-substituted 4-nitrobenzoylhydrazines *.
| Compound 2 | Other Nα-substituted 4-nitrobenzoylhydrazines | ||||
|---|---|---|---|---|---|
| Range | Mean ± SEM | Median | n | ||
| Carbonyl-phenyl twist (°) | 87.9 | 23.74–66.42 | 44.37 ± 2.70 | 43.99 | 13 |
| Distance C1–O1 (Å) | 1.213(4) | 1.203–1.243 | 1.226 ± 0.004 | 1.231 | |
| Distance C1–C5 (Å) | 1.491(5) | 1.476–1.516 | 1.499 ± 0.003 | 1.498 | |
| Distance C1–N1 (Å) | 1.343(4) | 1.324–1.418 | 1.358 ± 0.009 | 1.358 | |
* Data are taken from the CCDC entries 1110329, 1453241, 1465377, 909014, 909013, 1442944, 1442945, 814973, 834529, and 1012886.
Table 3.
Selected structural data of Compound 2 and comparison with cyclic cyanohydrazide I and related azapeptidic N,N’-dialkylated (thio)semicarbazides II–IV.
Table 3.
Selected structural data of Compound 2 and comparison with cyclic cyanohydrazide I and related azapeptidic N,N’-dialkylated (thio)semicarbazides II–IV.
| Compound | Nα–Nβ Distance | Torsion of the N–N Bond 1 | Out-of-Plane Angle | CCDC Number | Ref. | |
|---|---|---|---|---|---|---|
| Nα | Nβ | |||||
| 2 | 1.404 Å | 85.7° | 2.40° | 19.4° | 1567206 | this work |
| I | 1.425 Å | 52.1° | 10.82° | 28.91° | 1315768 | [10] |
| II | 1.394 Å | 82.8° | 7.60° | 10.73° | 837002 | [14] |
| III | 1.396 Å | 85.6° | 2.33° | 9.56° | 1401837 | [15] |
| IV | 1.400 Å | 85.9° | 0° | 14.26° | 896854 | [16] |
 | ||||||
1 Defined as angle between the planes defined by each nitrogen atom and the adjacent carbon atoms.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Löser, R.; Pitzschler, R.; Köckerling, M. Synthesis and X-ray Crystal Structure of N’-Cyano-N,N’-dimethyl-4-nitrobenzohydrazide. Crystals 2017, 7, 290. https://doi.org/10.3390/cryst7100290
AMA Style
Löser R, Pitzschler R, Köckerling M. Synthesis and X-ray Crystal Structure of N’-Cyano-N,N’-dimethyl-4-nitrobenzohydrazide. Crystals. 2017; 7(10):290. https://doi.org/10.3390/cryst7100290
Chicago/Turabian StyleLöser, Reik, Riccardo Pitzschler, and Martin Köckerling. 2017. "Synthesis and X-ray Crystal Structure of N’-Cyano-N,N’-dimethyl-4-nitrobenzohydrazide" Crystals 7, no. 10: 290. https://doi.org/10.3390/cryst7100290
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.