
In Silico Prediction of Growth and Dissolution Rates for Organic Molecular Crystals: A Multiscale Approach


1
Chair for Process System Engineering, Technical University of Munich, 85354 Freising, Germany
2
Barry Callebaut Belgium N.V., 9280 Lebbeke-Wieze, Belgium
*
Author to whom correspondence should be addressed.
Crystals 2017, 7(10), 288; https://doi.org/10.3390/cryst7100288
Submission received: 27 June 2017
/
Revised: 14 September 2017
/
Accepted: 18 September 2017
/
Published: 25 September 2017
(This article belongs to the Special Issue Advances in Computer Simulation Studies on Crystal Growth)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Solution crystallization and dissolution are of fundamental importance to science and industry alike and are key processes in the production of many pharmaceutical products, special chemicals, and so forth. The ability to predict crystal growth and dissolution rates from theory and simulation alone would be of a great benefit to science and industry but is greatly hindered by the molecular nature of the phenomenon. To study crystal growth or dissolution one needs a multiscale simulation approach, in which molecular-level behavior is used to parametrize methods capable of simulating up to the microscale and beyond, where the theoretical results would be industrially relevant and easily comparable to experimental results. Here, we review the recent progress made by our group in the elaboration of such multiscale approach for the prediction of growth and dissolution rates for organic crystals on the basis of molecular structure only and highlight the challenges and future directions of methodic development.
1. Introduction
Crystal growth and dissolution processes are an area of vital interest for pharmaceuticals, agrochemicals, organic electronics and other technologies. Owing to their significance to many different fields, those processes have been studied for over a century [1,2,3]. However, the prediction of crystal growth and dissolution kinetics for novel organic compounds still presents a major challenge.
The theories of crystal growth and dissolution are extensively discussed in the literature [2,3,4,5]. Both processes proceed by analogous mechanisms [6,7] and involve two main steps: (1) surface reaction and integration/disintegration of the surface species and (2) mass transfer of this species from/toward the bulk solution across the diffusion layer that surrounds the crystal [8]. Thus, the actual process of crystal growth and dissolution occurs at the molecular level, which also concerns crystal packing. Crystal structure databases, which provide crystal packing information for a huge variety of molecular structures, are to a large extent based on experimental X-ray diffraction analysis. The major drawback of this method is that it requires a flawless crystal of very high quality. Thus, the compound has to be synthesized and crystallized before information about the crystal structure can be obtained. Knowledge of the crystal packing is essential for predicting growth and dissolution properties. Consequently, to predict growth and dissolution properties from nothing but the molecular structure, simulation techniques to predict the crystal structure must be available. However, not only the nanoscale aspects are significant for understanding the crystal growth and dissolution processes. There are many important phenomena associated with crystal growth that occur on a mesoscale comprising hundreds of nanometer to tens of microns and occurring over long time scales (microseconds and longer) [9], like, e.g., the evolution of crystal surface structures due to the formation of terraces, which range in size from 0.1 to 1 microns, or step bunches, which can be as large as 100 microns [9]. Even larger length scales and longer time scales are needed to incorporate concentration effects and calculate face displacement velocities. An in silico prediction of macroscopic growth and dissolution properties thus necessarily requires a multiscale modeling approach that integrates all of these aspects in one unified simulation framework.
The standard simulation technique for modeling molecular-level behavior to reveal how the molecular details influence the growth and dissolution kinetics is molecular dynamics (MD). In this method, Newton’s equations of motion are solved numerically for all atoms to track the time evolution of the systems and to derive the kinetic and thermodynamic properties of interest [10]. However, MD can, at most, only probe behavior on the nanometer and nanosecond scale. Thus, most investigations of growth and dissolution processes using MD are reported for relatively small and simple molecules like, e.g., urea [11,12,13] and glycine [14,15,16,17], or even for simple model systems, such as hard spheres and Lennard–Jones particles [18,19,20,21], whereas most organic molecules, especially those used as active pharmaceutical ingredients (APIs), form more complex crystal structures, and it is extremely challenging to capture their crystal growth using fully-atomistic simulations [22]. Replacing atomistic details with lower resolution, coarse-grained (CG) beads, in which groups of co-localized atoms are treated as a single interaction site, allows one to overcome the complexity of molecules and the long time scale associated with the crystallization [22,23]. However, the interpretation of time is problematic in CG models [24]. The time scale needs to be calibrated by directly comparing with experimental data or dynamics from atomic simulations for the system at hand [24]. Thus, mainly the usefulness of CG models to obtain relative characteristics, like, e.g., to estimate the role of additives on the crystal growth of different API molecules was demonstrated so far [22,23]. Enhanced-sampling methods such as umbrella sampling [25], metadynamics [26] and forward flux sampling [27] have emerged as useful tools for understanding the mechanisms involved in crystallization. A basic idea, common to these rare event sampling methods, is that a biased potential is added to the system either to drive it along a predefined reaction coordinate or to prevent it from repeating already explored trajectories [23]. However, the efficiency of these methods strongly depends on the accuracy of the choice of reaction coordinates and may be inefficient in the case of a large number of degrees of freedom of the molecule under consideration. Moreover, upon introduction of an extra term into the system Hamiltonian, the actual dynamics of the system is to some extent hampered [28]. Recently, Salvalaglio et al. [13] demonstrated that using well-tempered metadynamics, applied within MD, one can quantitatively estimate the ratio between growth rates and thus predict the crystal habits and their dependence on additive concentration and supersaturation. However, the authors stressed that the approach does not allow computing absolute growth rates. The Reuter group also established a method for a quick prediction of approximate dissolution rates at low undersaturation based on the combination of hyperdynamics and metadynamics approaches [29,30,31]. This method relies on the classic rotating spiral model of Burton, Cabrera and Frank (BCF) [32], which assumes that dissolution (growth) proceeds via rotating spirals of step edges at screw dislocations and that dissolution (incorporation) of molecular units takes place primarily at kink defects along these step edges.
Kinetic Monte Carlo (kMC) is the method of choice in mesoscale modeling of dissolution or growth. In a kMC simulation, the growth or dissolution of a crystal is approximated by involving rare and independent state transition events, like, e.g., transition of molecule from solution to a kink or step site at the crystal surface. At each step in the simulation, the next event is determined on the basis of the probability proportional to the rate for that event. The time of the next event is determined by the overall rate for the microscopic surface processes and a suitably-defined random number. If a set of relevant states is defined and the transition rate constants are known, then the time evolution of the system can be modeled. Thus, the definition of a minimal set of distinct states and the estimation of corresponding transition rates are the main challenges for kMC simulations. Many kMC studies consider states on the basis of their nearest-neighbor coordination [33,34] or next-nearest-neighbor coordination [11,35]. Alternatively, the problem of state definition can be solved by identifying the most significant factors defining site reactivity with the help of electronic structure calculations and MD simulations for selected sites on the crystal surface [36]. Different approaches based on MD [11,20,33,34,35], accelerated MD [31,37], ab initio MD [38] or even DFT techniques [39] have been reported to determine rate constants, though the last three imply significant computational effort making them less attractive for systems with a high number of potential transitions, as well as for systems consisting of complex molecules with a high internal degree of freedom [40]. Significant steps toward the multiscale modeling of crystallization have been presented in studies conducted by Piana and co-workers [11,33,35], who first combined MD and kMC approaches to investigate the growth of a urea crystal from solution. Their simulations successfully predicted the different crystal morphologies of urea in solutions of methanol and water. However, as this only required relative rate constants, the model is still restricted in its applicability. Process time is an important factor in predicting crystallization processes, which demands that kinetic information including absolute rate constants is correctly accounted for. Further, in their studies, Piana et al. benefited from two special properties of urea: firstly, urea dissolution and growth has been shown to be fast in experiment and simulation, and secondly, urea is a small molecule, which has no facile torsional degrees of freedom [33]. For most substances, these simplifications are not necessarily true. Moreover, the concentration effects, relevant at the macroscopic scale, are not considered in their study, hindering the direct comparison with the experimental growth rates.
On the macroscopic scale, continuum methods are gainfully applied [41,42,43,44,45,46,47], handling physics expressed by continuum partial differential equations. These simulations are important for understanding the crystal growth and dissolution processes in their complexity accounting for advection and diffusion processes. They, however, do not shed light on how the molecular details influence the growth and dissolution kinetics. Molecular dynamics simulations are at most used only to predict physical parameters such as diffusivity or solubility, which are then employed to compute scale continuum transport models [48]. In most cases, the macroscopic simulation relied on experimental data; thus, the derivation of predictions for novel compounds still presents a major obstacle.
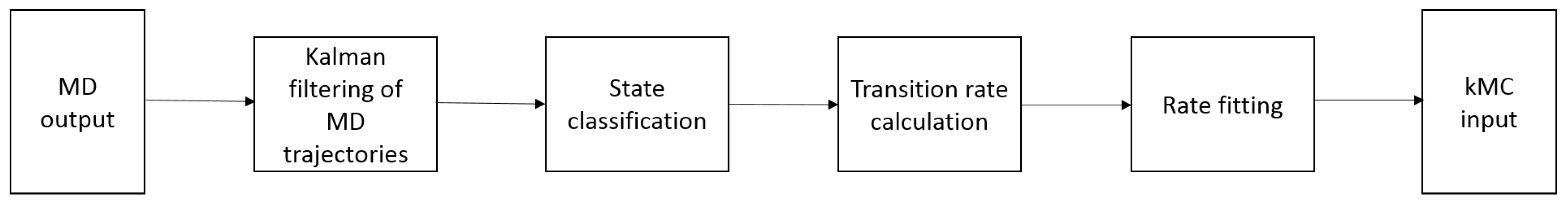
Thus, the protocol for in silico prediction of crystal growth/dissolution rates on the basis of molecular structure should comprise and join all of the corresponding steps from the prediction of molecular packing and crystal shape to continuum simulation of growth and dissolution processes. Figure 1 presents a multiscale protocol, elaborated based on our previously published studies and findings [40,49,50,51,52].
In this paper, we review all of the aspects relevant to establishing of such a protocol and highlight the challenges arising at each individual step. The first two steps, prediction of (1) crystal structure from molecular structure and (2) crystal shape, are necessary to provide the information for all further simulations and, thus, to initialize the multiscale protocol. These steps were completely omitted in our previous studies; however, in Section 2, we give a short survey of the actual state of knowledge and techniques in this field. The third step involves MD simulations to take into account molecular-level processes and to obtain process rate constants for kMC simulations, as was initially proposed by Piana et al. [11,33,35]. In comparison to Piana et al., we consider the three-dimensional crystal model in MD simulations, enabling dissolution to be seen on MD time scales even for highly hydrophobic and poorly water-soluble pharmaceutical ingredients. This, as well as a choice of force field for MD simulations and the necessity and ways to hold constant solution concentration in MD simulations are considered in Section 3 of this paper. Our approach to properly transfer the microscopic information from MD to kMC simulations and all procedures needed for that (state identification, detection of only rare, uncorrelated transition events in MD simulations, etc.) are described in Section 4. The fourth step of the multiscale protocol represents kMC simulations to calculate crystal face displacement velocities based on the MD rates and thus at the same constant solution concentration. This step is described in Section 5 of the paper. The fifth and last step involves continuum simulations to describe concentration-dependent crystal growth/dissolution on the macroscopic scale, where the results can be directly compared with the experimental ones. The scale continuum transport model, as well as our approach to transfer the mesoscopic information from kMC simulations to the macroscopic level are described in Section 6 of the paper. To demonstrate our approach, the dissolution rates for aspirin were predicted and validated by comparison with experimental assessment of aspirin dissolution using a Jamin-type interferometer [53], as described in Section 7 of the manuscript. Aspirin was chosen as a model substance as it is a well-studied compound, where the literature provides a broad array of information like crystal structure [54], polymorphs [55,56], morphology [57], critical nucleus size [58] needed to initialize the multiscale simulation protocol, as well as experimental data on crystal dissolution [59,60] for comparison with the simulation results and validation of the different steps of our protocol. Thus, all of the simulation results presented in the paper are for molecular aspirin crystals. However, our approach introduces the flexibility to handle different organic model compounds and not restricted to aspirin. A summary of our findings and some concluding remarks, as well as the information about future directions and challenges can be found in Section 8.
2. Crystal Structure and Shape Prediction
The prediction of crystal structure at the atomic level is one of the most fundamental challenges in condensed matter science [61]. Crystal structure prediction (CSP) approaches have been evolving rapidly in the past few decades and have now grown into an overwhelmingly vast, diversified and active field of research [62,63]. The ever increasing computer power allowed making many successful predictions of organic crystal structures [63]. The best starting point to keep track of the existing range of approaches is constituted by the accounts of the Blind Tests of CSP organized by the Cambridge Crystallographic Data Centre every few years [64]. These contests ask participants to predict the crystal structures of organic molecules starting from only the 2D molecular structure, which are then compared with the experimentally obtained, but not publicly released data to reveal the particularly effective techniques. Despite the excessive ramification of approaches developed to date, the whole CSP process can generally be subdivided into three distinct steps, each with their own methodologies and challenges: (1) conformational exploration of the molecule(s), (2) generation of candidate packing arrangements and (3) (re-)ranking of candidate structures using some form of fitness function [64]. Successful crystal engineering relies on the knowledge of molecular conformation, i.e., the overall shape of molecular building-blocks, as well as the relative arrangement of functional groups within a molecule that can participate in structure-directing interactions [65]. Molecular shape can be easily predicted for rigid molecules, but becomes more challenging as molecular flexibility is increased and the molecules of interest have a choice of conformers when self-assembling into a crystal [65]. Different conformers may lead to very different crystal packing arrangements, ultimately influencing the properties of the crystal [65]. Therefore, conformational flexibility can be seen as a principal obstacle to crystal engineering. Commercially available and industrially employed CSP approaches like the Polymorph Predictor within Accelrys’ Materials Studio rely on a rigid molecule approximation and only concentrate on the evaluation of different packing arrangements. This is clearly insufficient for flexible APIs, which in the crystal frequently adopt geometries that are significantly different from the stable gas-phase conformer. This is particularly relevant for the engineering of pharmaceutical materials, where solid form properties of active pharmaceutical ingredients may be manipulated, either by selection between polymorphs, or the design of multi-component crystals, such as salts or co-crystals. Thus, there is also still no general solution to the main CSP challenge, i.e., the existence of crystal polymorphism [62,63]. Each polymorph may differ from others in physicochemical properties, such as density, solubility, bioavailability, mechanical strength, dissolution rate, to name but a few [23]. Knowledge of different polymorphs is essential to the pharmaceutical industry for choosing the crystalline form with the most efficient therapeutic performance and to exclude the crystallization of any unwanted form.
To see which surfaces are relevant to study growth and dissolution processes, not only molecular packing, but also crystal shape should be predicted in silico. They both are needed to initialize the multiscale growth and dissolution protocol. Crystals reveal a large variety of shapes, depending on their chemical composition and structure, as well as on the growth conditions, such as supersaturation, temperature, solvent and even impurities. For novel compounds, such external parameters will not be necessarily known, but one still needs to determine the facets that dominate the morphology from the known crystal structure for the next steps of the proposed multiscale protocol. Under conditions of extremely slow growth, the shape of a crystal is determined by thermodynamics: the crystal tends to grow to a shape of a polyhedron having minimum surface energy [66]. Such an equilibrium shape is thus obtained, according to the Gibbs thermodynamic principle, by minimizing the total surface free energy associated with the crystal-medium interface. The procedure is based on the Gibbs–Wulff theorem, also known as Wulff construction [67], and provides the equilibrium crystal shape from separately computed surface free energies of all low-index crystal facets. Critical for this shape are thereby not the absolute surface free energies, but only their relative ratios. Less stringent (and efficient) approaches may and have therefore been employed to their computation. One means to this end is to neglect (or crudely approximate) vibrational free energy contributions to the surface free energies and to use static surface energies obtained for optimized surface structures. This reduces the computational burden to such an extent that in fact first-principles electronic structure methods like DFT may directly be employed to produce these numbers. This approach is for instance commonly followed to determine nanoparticle shapes in heterogeneous catalysis [68,69,70]. It has also been used for growth applications from solutions, where also any solvent influences have been neglected [71,72]. This neglect extends over both the solvent influence on the surface vibrational properties and the electrostatic effects on the surface energies due to the solvent dielectric properties. However, these various levels of approximation are generally not deeply investigated, and further efforts should be made to conduct systematic analysis of the level of theory required to reliably predict the crystal shape. Moreover, one can notice that the Gibbs condition is not normally realized in practice as crystal growth in crystallizers is significantly removed from the equilibrium state [73]. Under most conditions, the shape of a crystal is determined by kinetics rather than thermodynamics [73]. Thus, it certainly might be a limitation for the whole protocol, if crystal structure and shape predictions completely fail. Other methods are also available to deduce morphology of a crystalline material from its internal crystal structure, like, e.g., the Bravais–Friedel–Donnay–Harker (BFDH) method [74,75,76], which uses the crystal lattice and symmetry to generate a list of possible growth faces and their relative growth rates, or the attachment energy model [77], which takes into account the energetics of crystal interactions in addition to the crystal geometry. These methods in general provide good predictions of vapor-grown crystals or crystals grown in systems in which the solvent does not interact strongly with the solute. However, in many instances, the simulated crystal shapes differ from the experimental ones because of the kinetic effects due to supersaturation, solvent and impurities dominating the crystal growth process [78]. The more is known about crystal growth conditions, the better the crystal shape can be predicted. For that, a number of sophisticated methods exists that account for the effects of such external factors [79,80,81,82,83]. However, for the next step of the protocol, e.g., MD simulations, it is essential that the dominant faces and edges are presented. Their actual size and relations to the surrounding surfaces are less important, as shown in the next section.
Currently, no simple general solution to predict crystal packing and crystal shape data for compounds with conformational flexibility exists; neither are we concerned with the elaboration of crystal structure and shape prediction methods. Instead, we relied on the experimental data for aspirin [57,84] in our work [40,52]. Aspirin was modeled in its protonated state using unit cell parameters of the polymorphic form I [84]. The aspirin nanocrystal for MD simulations was built according to the experimental morphology, typical for aspirin crystallized from ethanol-water solutions [57].
3. MD Simulations
MD simulations are widely used to study crystal growth and dissolution and are of the utmost importance in unraveling the microscopic details of these processes. However, simulations are presently affected by several shortcomings, which hinder a reliable comparison with experimental growth and dissolution rates and limit growth/dissolution studies to systems and conditions often far from those investigated experimentally [28]. These weaknesses can be classified into two main categories: (1) limitations related to the accuracy of the computational model used to represent the system and (2) shortcomings due to the finite-size effects. In this section, we describe our attempts to resolve these issues and to obtain reliable results in MD simulations: from the choice of the parameter set to describe intermolecular and intramolecular interactions and simulations of nanocrystal in its experimentally determined shape to avoiding finite-size effects during crystal growth and dissolution simulations.
3.1. Choosing the Force Field
To successfully simulate crystal growth and dissolution, i.e., to obtain the results corresponding to the experimental ones quantitatively or at least qualitatively, the molecular interactions in both crystal and solution environments have to be accurately described. Thus, the choice of force field with a suitable combination of a mathematical formula and associated parameters that are used to describe molecular energy plays a pivotal role. In the literature, a broad variety of force field parameter sets can be found. The difficulty in choosing a suitable force field mainly arises from the strict focus of most common force fields on particular properties. Moreover, some of these are designed to be compound specific; thus, their applicability to novel compounds is limited. For screening purposes those force fields are favored for which software packages and online resources are provided to facilitate the generation of force field parameter files for common simulation packages. Thus, the most general approach to find the parameters for novel compounds like, e.g., small-molecule drugs is to use common biomolecular force fields designed for protein interactions and ligand-docking simulations. Here, small-molecule drugs of varying configuration and containing various functional groups are included in the force fields parameter sets. Examples for such force fields are, besides others, the CHARMM [85] (Chemistry at HARvard Molecular Mechanics) force field, the AMBER [86] (Assisted Model Building and Energy Refinement) force field or, in an extended form, the Generalized AMBER Force Field [87] (GAFF), and the OPLS [88] (Optimized Potential for Liquid Simulations) force field. These force fields are designed to adequately describe protein interactions, where the screening of multiple small-molecule compounds is an important field of interest. For that, force field generating tools have been developed, e.g., the SWISS PARAM server [89] for the CHARMM-compatible force field based on the Merck Molecular Force Field (MMFF) [90], the ACPYPE (AnteChamber PYthon Parser interfacE) [91] wrapper script around the ANTECHAMBER software [92] for the GAFF or the PRODRG server [93] for the GROMOS [94] (GROningen MOlecular Simulation package) force field. The latter, however, has been shown to produce sub-optimal results [95], especially when it comes to calculating the charges. Additionally, force fields can be obtained directly from software packages in commercial software, such as Maestro from the Schrödinger software package creating OPLS force field parameters.
Choosing an appropriate force field remains a challenge and is crucial to the results obtained. For example, studying the crystal growth of glycine from an aqueous solution, Banerjee and Briesen [14] monitored dissolution, although growth was expected for the applied supersaturated conditions. Cheong and Boon [16] compared different force fields and charges for the simulation of glycine crystal growth and found the heat of solution to be an important criterion when choosing the force field for crystal growth simulation. Heat of solution is the change in the enthalpy when 1 mol of a substance is dissolved in a solvent, thus an important quantity for crystallization, as it incorporates both the crystal and the solution phase and quantifies the tendency for crystallization and dissolution. A positive value indicates that the crystal dissolution is an endothermic process. The crystalline state is then favored over the dissolved state, and one can also expect that crystal growth would be obtainable with a suitable supersaturation.
In addition to the heat of solution, we considered several other criteria to make a proper choice in force field selection for crystal growth and dissolution simulations [49]. Structural, thermodynamic and interface-specific parameters were evaluated for several API ingredients (aspirin, ibuprofen, paracetamol). Apart from the importance of the heat of solution for the evaluation of the solid-to-solution phase transitions, it was concluded that the interaction energies might give valuable information about the choice of the force field whenever a dominant interaction within the crystal was present, such as hydrogen bond pairs between carboxyl groups in the case of aspirin and ibuprofen. Moreover, the lattice parameters of the unit cell after its relaxation with the corresponding force field (further referred to as a relaxed unit cell) have been shown to provide reliable information, which is important for the stability of the crystal structure. The distance threshold and the orientation tolerance parameters, characterizing maximal deviations from the reference crystal molecular positions and orientations, have also proven to be appropriate indicators of a correct representation of the crystal-water interfaces by the force fields [49]. The consideration of all of these aspects led us to the choice of the Merck molecular force field for the simulation of aspirin crystal dissolution in our further works [40,50].
3.2. Superiority of Three-Dimensional over Two-Dimensional Dissolution Simulations
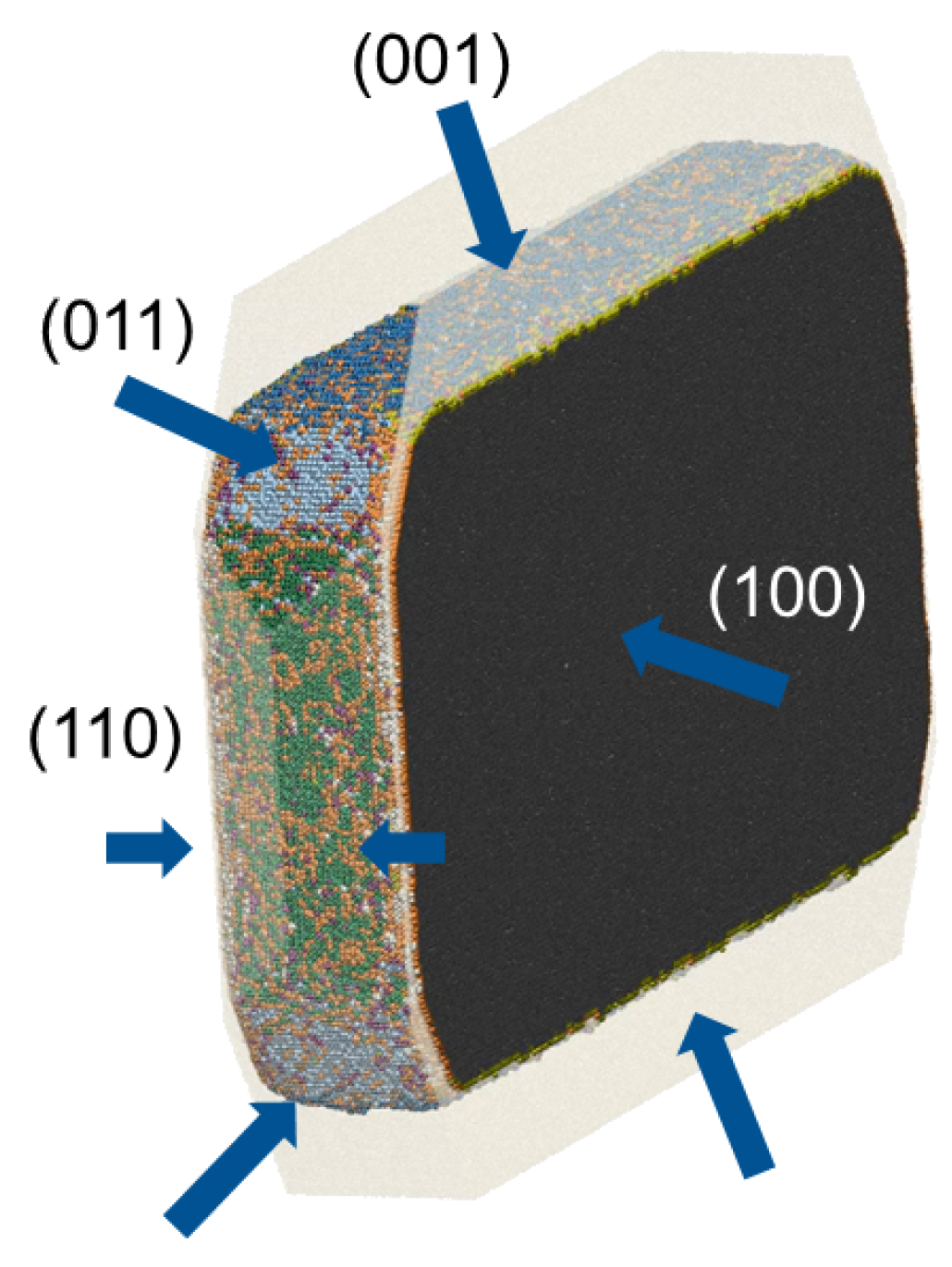
Most investigations of crystallization processes using MD consider the distinct faces of the crystal in contact with the solvent [11,12,14,15,16,33,35,96,97,98]. However, time scales accessible in regular MD simulation are often not sufficient to resolve dissolution from the perfectly flat interfaces. Recently published experimental and MD simulation data on the dissolution of paracetamol highlighted the significance of the so-called “corner and edge effect” [99], indicating that in particular corners and edges between facets serve as the initial sites for dissolution and may be accessed by MD. The crystal representation used in the paracetamol study [99], however, was in no way related to the true experimental morphology, and due to its limited size, the employed crystal was not stable over the whole simulation time. This prevents a direct comparison to experiment or use as a predictive-quality protocol. We overcame these limitations with MD simulations of an entire aspirin nanocrystal in its experimentally determined shape revealing the (100), (001), (011) and (110) faces, cf. Figure 2a [40,50]. It was cut from a pre-equilibrated supercell (310 K and ambient pressure of 1 bar) using the VMD [100] program. The size of the nanocrystal was around 10 × 18 × 14 nm [40] and thus exceeded the size of the critical diameter for stable nuclei [58], enabling one to properly sample dissolution while the crystal bulk remained in the same configuration as obtained from the relaxed unit cell. The system consisted of a total number of 9407 aspirin molecules surrounded with 177589 TIP3P water molecules [40]. The dissolution was simulated over 280 ns using version 4.6 of the GROMACS package and took about ten days on 1024 cores of SuperMUC System of Leibniz Supercomputing Centre [40]. Pronounced differences were observed in the face-specific dissolution behavior. A dissolution mechanism via receding edges was found for the (001) plane, which is in good agreement with experimental results [40,50]. However, while the proposed dissolution mechanism for the (100) plane is terrace sinking on a rough surface, no pronounced dissolution of the perfectly flat face was seen. The most obvious reason why there is almost no dissolution of the (100) aspirin crystal face in the MD simulations is that simulation times were too short. While this is necessarily true, another reason for the stability of the (100) plane might be the strong deviation of the initial surface structure used in the simulations from experimentally observed ones. Danesh et al. [60] have shown that the (100) faces are very rough, which is in good agreement with etching patterns obtained by Wen et al. [101] for pure water as an etching medium. The roughness of the (100) crystal face is expected to be of major importance for crystal dissolution. Unfortunately, the feasible simulation cell size does not allow for the construction of proper, rough surfaces for MD simulations.
Thus, one can notice that the principal limitation to the range of applicability of the given multiscale approach is determined by the rate of the molecular crystallization and dissolution steps, as well as size limitations in MD. In order to calculate a reliable rate for a transition event, this event should occur at least a few times during the MD simulation [35]. A too small number of events of a certain type would lead to zero or almost zero rates. As the MD rates are input rates for the kMC algorithm, they have a strong influence on the end results primarily affecting a given slow growing/dissolving surface, as well as the crystal habit, determined by the relative growth/dissolution rates. The problem is that the typical times currently accessible to a single MD simulation are s, which might prevent the observation of some of the slowest steps. This also prevents the observation of the formation of new faces and edges, which are not present in the experimental morphology, but could be expected to evolve during the kMC simulations, like, e.g., the development of a new (010) face due to easy detachment of molecules with a low number of neighbors from the tip formed by the (110) faces. However, as the basic assumption of the given multiscale approach is that individual molecular steps are independent of each other, it is possible to combine the results of multiple independent MD simulations to calculate the transition rates. Thus, to obtain all rates for kMC simulations, three different starting configurations were considered for aspirin nanocrystal, cf. Figure 2: (a) crystal in its experimental shape, (b) crystal obtained by cutting the tip formed by the (110) faces and, thus, with the (010) face exposed and (c) block structure revealing only the (100), (010) and (001) faces to obtain rates for (010)/(100) and (010)/(001) edges [40]. Thereby, we also were able to sample dissolution events for the slowest (100) face. We assumed that it is the orientation of the hydrogen bonds that plays an important role for stabilization of the (100) face. As the hydrogen bonds are directed perpendicular to the (100) plane, the molecules have strong interactions with the underlying layer of aspirin molecules. Thus, to increase the sampling of dissolution events, alternative terminations were applied to this slowly dissolving face in the starting configuration (b), cf. Figure 2b: from the two opposite (100) faces, one face was constructed such that the surface layer had hydrogen-bonding partners, whereas on the opposite side, the surface layer was cut such that no hydrogen-bonding partners were present. Additionally, another approach was applied to improve the sampling of dissolution events for the (100) faces in this structure: voids were introduced to reduce the number of nearest neighbors for the molecules on the surface, thus reducing their stability in the crystal lattice, cf. Figure 2b. While the second approach led to no significant differences in the dissolution properties on the (100) face, cutting the crystal so that the hydrogen-bonding partners were removed allowed us to register much more transition events [40].
3.3. Constant Chemical Potential
With the presently available computational resources, classical MD calculations can typically study systems of size up to – atoms. However, such size limitations are particularly dramatic in the simulation of phase transformations, such as crystal growth from solution or dissolution of crystals [13,102,103,104,105]. While for a macroscopic system, the solutions’ chemical potential does not change in the time scale accessible by MD simulation, the standard MD simulations cannot guarantee this. During the growth or dissolution, the concentration of the solute changes, resulting in a change of chemical potential eventually affecting the process itself. In MD, this should be handled by finite-size corrections. Recently, some works appeared, which discuss finite-size effects and methods to expand the information from MD simulations of phase transformations towards the limit of a macroscopic system [13,102,103,104,105]. However, most of them consider only nucleation processes, where the correction term is introduced based on the classical nucleation theory and modified liquid drop model for describing nucleation in a finite closed model [102], sharing many approximations and shortcomings. Thus, the direct application of this method to the crystal growth and dissolution problem is not trivial. Grand-canonical simulations could in principle eliminate finite-size effects by imposing a constant chemical potential in the solution phase. This is achieved by means of trial insertion and deletion of molecules. However, low acceptance rates render such approaches computationally infeasible [106]. Furthermore, insertions of particles near the growing crystal or insertion of fractional particles may lead to unphysical effects, which may ultimately obscure the true growth mechanisms and rates [106]. An approach to allow for simulations under constant chemical potential was proposed by Perego et al. [105]. Their CMD method allows to maintain a region containing the growing/dissolving crystal and its immediate surrounding at constant solution concentration, while the remainder of the simulation box acts as a molecular reservoir. This is achieved by implementing an external force, which regulates the passage of molecules from and to the control region. Consequently the time scale accessible for this method is limited by the amount of solute molecules in the molecular reservoir. The method was successfully applied to study planar crystal-solution interfaces. However, significant changes are needed to extend the method to other geometries [105]. Moreover, the parametrization of the CMD method is not trivial and needs a preliminary tuning and testing to provide an effective decoupling between the reservoir region and the growing crystal [105].
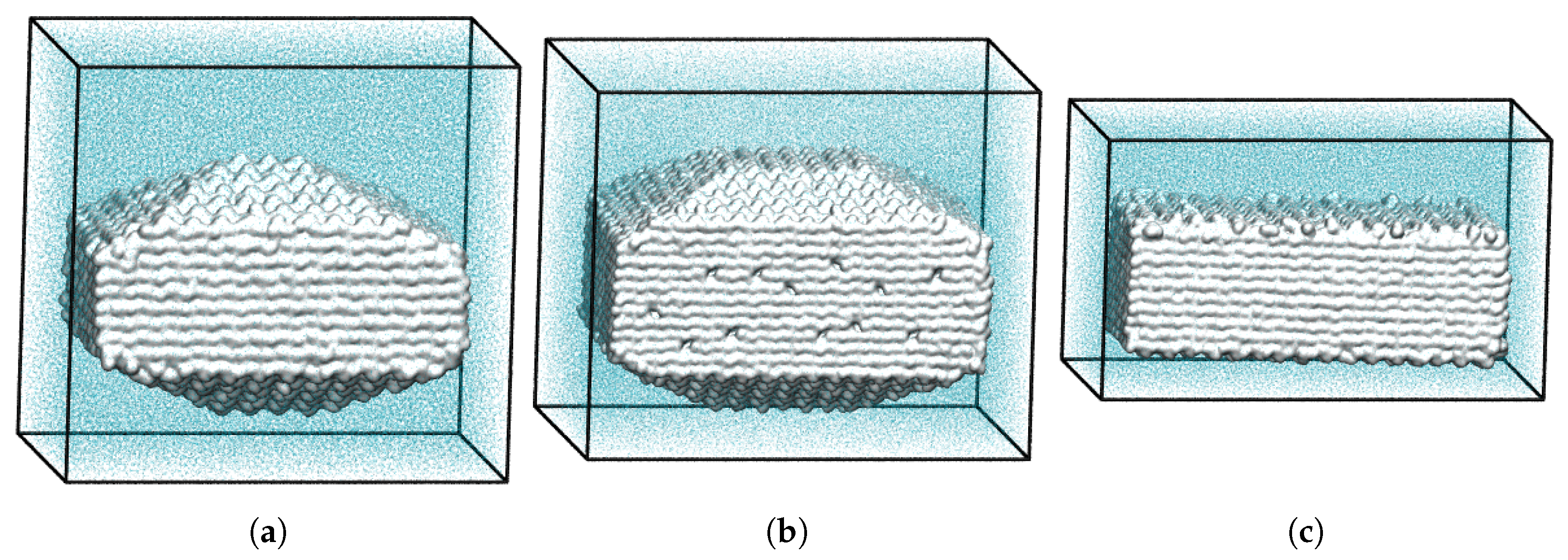
A simple approach to simulate crystal dissolution at constant undersaturation of the surrounding medium at comparably low computational cost was devised and demonstrated by our group [50]. In our MD simulations of aspirin nanocrystal dissolution [40,50], we proposed to use virtual atoms (sticky dummy atoms), which have a strong interaction potential with dissolved aspirin molecules, whereas interactions with water are excluded. To ensure that the virtual atoms do not interfere with the crystal bulk and thus have no impact on the crystal dissolution behavior, a plane of 100 sticky dummy atoms is introduced into the water slab at a distance of at least two cutoffs from the crystal (see Figure 3a). This still allows for free diffusion of aspirin molecules in the solvent. As was shown in our work [50], almost exactly half of the molecules, which have been in the liquid once, have returned to the crystal, where they could adsorb or reintegrate into the crystal lattice, which agrees nicely with the random walk expected for free diffusion. The other half reach within the cutoff radius of sticky dummy atoms and get trapped immediately and irreversibly, cf. Figure 3b, where the final configuration after 280 ns of simulation is shown. Thus, the number of aspirin molecules diffusing freely in solution is kept at a low value over the whole simulation time, and continuous dissolution of the crystal at almost zero concentration conditions can be monitored.
4. Linking Nanoscale and Microscale
The principal objective of MD simulations in the multiscale strategy by Piana et al. [33] is to obtain state transition rates for kMC simulations. The main task for a quantification of these rate constants from the obtained MD trajectories is thereby the unambiguous definition and identification of distinct molecular states. To characterize the crystal growth and dissolution process, order parameters need to be established that discriminate between “crystal-like” and “solution-like” states. For aspirin dissolution, structural order parameters in the form of the orientation and number of neighbors proved to be sufficient [51]. Crystal-like molecules are thereby defined as those having their orientations within a certain threshold value from the orientations of the molecules from the relaxed unit cell used to construct the crystal (called reference orientations) and at least one neighbor among the crystal molecules, as described in detail in our papers [40,51]. However, the identification of an appropriate set of order parameters can be far from trivial in many other cases [107], and a significant number of approaches to differentiate one state from the other can be found in the literature [12,20,33,96,97,98,108,109,110,111,112,113,114,115,116,117].
Crystalline molecules can then be further subdivided in different substates according to their local coordination environment, e.g., on the basis of the number of neighboring crystalline molecules. The state transition rate constants for the kMC simulations can be calculated from MD simulations by just counting the corresponding transition events, as well as the possibilities for these transitions to occur [40]:
where is the number of transitions from state A to state B and is the number of molecules in state A calculated at each time interval , while is the number of time intervals during the whole simulation.
The central idea behind a kMC simulation is a coarse-graining of the time evolution to discrete rare events and focusing on the corresponding Markovian state-to-state dynamics [118]. Thus, only rare, uncorrelated events are of interest. The analysis of processes occurring at the solid/solution interface during crystal growth and dissolution simulations requires an effective way to detect only rare, uncorrelated transitions from one state to another. Because of the oscillatory behavior of molecules, this is not a trivial problem. The oscillatory behavior of molecules (especially of surface molecules interacting with the solution) leads to spurious recrossings at the boundary between liquid and solid state, which severely complicate the reliable identification of significant transitions that are relevant over longer time scales [51]. The analysis problem arising due to such fast non-Markovian dynamics becomes especially acute for systems consisting of complex molecules with a high degree of conformational flexibility [51]. Due to strong fluctuations of the molecular position and orientation, numerous transitions between crystal-like substates with different number of neighbors, as well as from the crystal-like to the solution-like state can be registered. Consideration and calculation of all of these transition events would lead to strong overestimation of transition rates. The number of such fast, non-Markovian transitions can be reduced by analyzing the data averaged over some time interval, as done by Piana et al. [11,33,35]. However, the resulting transition rates are very sensitive to the choice of the time interval [20,51]. Reily and Briesen paid more attention to the choice of sampling interval and tried to avoid the counting of recrossing events. Namely, the time interval was chosen on the basis of the velocity autocorrelation function (VAF) of solute particles near the equilibrated interface, as well as the VAF of solute particles in the bulk of the crystal slab from the same simulation. Only transitions where the particles remained in the new state for two consecutive time intervals were counted [20]. To improve the estimation of molecular states during crystal growth and dissolution MD simulations, we proposed a new approach [51], based on Kalman filtering [119], making it possible to focus on rare, uncorrelated transition events, i.e., effective dynamics of the Markov chain. The idea is to consider the fluctuations of orientation and position of each crystal molecule as noisy measurements of the “true” (corresponding to the Markovian molecular state) molecular orientation and position and to estimate these “true” values using the Kalman filter algorithm. For the application of a Kalman filter [119], information on the measurement noise variance and the process noise variance is needed. Often, these parameters are just tuned to obtain good filter performance. To avoid this level of arbitrariness, we introduced a scheme to define all filter parameters and thus to provide a way for robust and reliable molecular state definition. According to this scheme, filter parameters as well as tolerance parameters are determined from short preliminary MD simulation, in which the crystal structure still stays stable. The details on the method and its application to processing of MD simulations data can be found in [51]. To analyze nanocrystal MD simulations and calculate rates for individual transition events, like incorporation or dissolution of a solute molecule into/from a particular crystal surface or edge, a scheme to classify all crystal molecules into different edge and surface categories was introduced. There, the neighbor-based approach combined with the geometry-based resolution algorithm is used to identify whether a molecule under consideration belongs to a certain surface or edge from a predefined set of surfaces and edges. Thereby, related faces or edges may be considered to be of the same type (like, e.g., the opposite (00) and (100) faces further referred to as (100) for MD and kMC simulations), as demonstrated in Figure 4.
For each type (e.g., particular edge or surface), rates dependent on the neighbor count can be calculated. Usually, the data points acquired from MD simulations are not distributed over the whole range of the number of neighbors and, thus, need to be interpolated to be used as an input for kMC simulations. Thereby the event count for each specific number of neighbors can be used as the weight for fitting. As the rates for different numbers of neighbors can differ by several orders of magnitude, a logarithmic scale is used. In [40], for each type j, the logarithmic values of the dissolution rates over the corresponding number of neighbors were plotted and approximated with the power law function , where a, b and c are fitting parameters. This function was chosen for fitting, as it is simple and gave a reasonable fit for all types. In this case, the dependence of rates for each type j from the number of neighbors can be rewritten as . Comparing this with the Arrhenius equation , one finds for the activation energy: in the case of . This expression gives us the dependence of activation energy from the number of neighbors x, as well as from their spatial arrangement, varying for different edge and face types, and thus, represented by the use of type-specific coefficients a, b, c [40].
Altogether, for successfully linking of MD and kMC simulations, the whole range of analysis procedures should be performed on MD data, cf. Figure 5.
5. Kinetic Monte Carlo Simulations
A basic n-fold kMC algorithm [120,121] can be employed for the simulation of crystal growth and dissolution. In contrast to MD simulations, each molecule can be represented just by a point on a grid with the arrangement of the lattice sites according to the crystal structure. Moreover, only key growth and dissolution processes are considered for the system. These two simplifications make kMC computationally much less expensive, thus enabling the simulation of larger size and longer time scales compared to MD. For example, the aspirin crystal considered in our kMC studies [40] consisted of about 8.48 million molecules and dissolution was simulated for 7.5 ms, which took only about two days on a single processor. Each grid site in kMC simulations carries the information of whether it is occupied by a crystal molecule or vacant. For each occupied site, it can also be defined how many neighbors it has and to which crystal face or edge it belongs, i.e., its state, based on the applied state identification scheme. For the crystal dissolution simulations in [40], each kMC process corresponded to the removal of one of the crystal sites. All possible kMC processes were grouped into types to distinguish between states of the sites. The dissolution processes from a particular face or edge were considered as belonging to a particular type, cf. Figure 6. The processes within a certain type had different rates depending on the number of neighbors of the specific crystal site. These rates were calculated from MD simulations, as described above.
On each kMC step, first, one of the available types (e.g., dissolution from a particular face or edge) was chosen in proportion to the transition probability obtained from the MD simulations. Then, a particular process within the selected type, i.e., one of the sites for dissolution belonging to the selected face or edge, was chosen using an effective linear selection method [122]. The occupancy of the selected site was then changed to vacant and for all neighbors of the selected site, the state identification scheme was applied to update their states. After updating the system time, the next kMC step could be performed until the maximum number of steps was reached.
In comparison to MD simulations, crystal face displacements could be directly observed in kMC simulations (see Figure 7), and the corresponding rates were calculated. The distance found between the face center calculated on each kMC step and the initial plane was stored during kMC simulations. Displacement rates were obtained from the linear regression of this distance over time. As the Monte Carlo technique is of a stochastic nature, a number of simulations is required to obtain statistically meaningful results. The typical way to determine the required number of replicates is to perform a convergence study by computing the standard deviations. For aspirin crystal dissolution, 25 independent simulations were used for calculation of average face displacement rates, and the relative standard deviations obtained were about 1–2% [40].
The evolution of the whole system is simulated based on the transition rates from MD simulations. Thus, the kMC face displacement rates are obtained at constant concentration (zero concentration in the case of aspirin dissolution simulations) and are only conditionally comparable to the experiment, where the concentration decreases during the crystal growth or rises during the crystal dissolution. To compensate for this, the continuum simulations based on the face displacement rates from kMC simulations can be performed, as described in the next section.
6. Coupling Molecular and Continuum Domains
According to Mullin et al. [4], the kinetics of crystal growth or dissolution of a face j of the crystal can be described by the empirical equation:
where is the face displacement velocity, is a rate constant for a specific temperature T for the respective face, and are the bulk and saturation concentrations, correspondingly, and is the order of the growth process. This equation does not take advection into account, which will be relevant when considering flow conditions, as planned in our future work (see also the Conclusions). Only processes in a stagnant fluid are considered here.
The order of the growth process in the Equation (2) may differ from 1, as this equation accounts not only for integration/disintegration, but also for the diffusion process. However, one can consider diffusion and integration/disintegration as two separate and independent processes. Taking the concentration on the crystal surface instead of the bulk concentration in Equation (2) and setting to 1 would allow one to split integration/disintegration from diffusion process:
For simplicity, we will further consider the case of an aspirin crystal dissolving in a chamber filled with water. With a surface concentration of zero at , the values obtained from combined MD and kMC simulations (cf. Figure 7) can be considered as initial values for the face displacement velocities, as they were obtained at constant zero concentration, provided by the above dummy atoms mechanism in MD simulations:
Thus, we obtain the following expression for time-varying face displacement velocities:
The process of diffusion can be separately covered by the classical diffusion equation known as Fick’s second law:
where D is the diffusion coefficient, the dependent variable c is the concentration in the bulk, the independent variables are the position and time t.
Finding a unique solution requires an initial condition for the concentration in the bulk, as well as boundary conditions on the walls of the chamber and on the crystal surface. For crystal dissolution, the initial bulk concentration can be considered as zero. On the walls of the chamber, the flux in and out of the domain is zero. This is represented by the Neumann boundary condition:
where is the face normal of the walls of the chamber. The boundary condition on the crystal surface is face-specific and can be also represented by the Neumann boundary condition describing the flux over the respective crystal face:
where is the rate constant for the face j directly obtained from kMC simulation (cf. Figure 7), and are the surface and saturation concentrations, is the density and M is the molecular mass of the crystal substance. The diffusion coefficient in Equation (6) as well as the saturation concentration can be easily calculated using MD simulations [40,52]; thus, the equation can be solved, and the face displacement velocities can be evaluated.
7. Continuum Simulations and Results
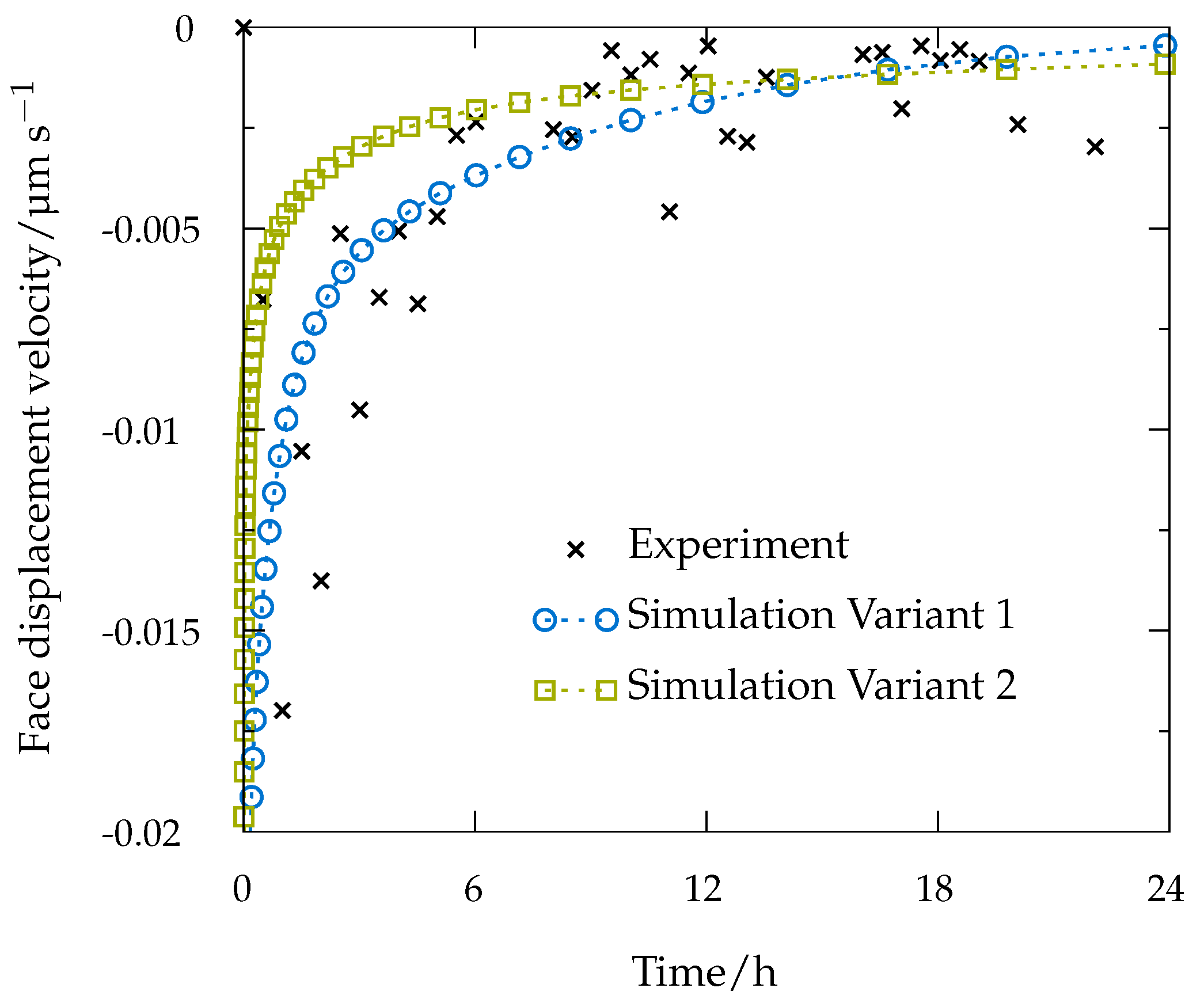
In continuum simulations partial differential equations are solved numerically, e.g., with the finite-element method. Comparing to molecular dynamics and kinetic Monte Carlo simulations, time and size scales can be extended by a large amount, and the simulations can cover typical experimental scales. Thus, a comparison of the respective results yields a good indicator of the simulation accuracy. In [52], a solution of the model Equation (6) with the corresponding boundary conditions (Equations (7) and (8)) was obtained for an aspirin crystal and compared with experimental results obtained using a Jamin-type interferometer [53] with the experimental setup, i.e., the size of the measurement chamber and aspirin crystal, chosen in agreement with the simulation settings. The simulations of the dissolution of an aspirin crystal in the measurement chamber of the interferometric device were performed using COMSOL multiphysics Version 5.0 [123]. Thereby, two different variants were considered: the values for diffusion coefficient D and solubility in model Equations (6)–(8) were either predicted in silico (Variant 1) or set to experimental values known from the literature [52,124] (Variant 2). In the first case the diffusion coefficient was calculated from the averaged mean square displacement of 20 individual MD simulations. The solubility of aspirin in water was calculated with the conductor-like screening model for realistic solvation (COSMO-RS [125]) using commercially available software COSMOtherm (Version C30 Release 15.01., COSMOlogic, Leverkusen, Germany), though it can be also calculated from MD simulations using, e.g., the direct coexistence method or chemical potential route [126], which however could be computationally prohibitive. In continuum simulations, the concentration was evaluated as the average over the whole domain and normalized with the saturation concentration. It is further referred to as relative saturation.
The simulation results yielded a relative saturation of 83% after 24 h of dissolution in the case of predicted parameters (Variant 1) and 67% in the case of experimental model parameters (Variant 2). The relative saturation measured using the Jamin-type interferometer [53] for three individual aspirin crystals with the same crystal dimensions as in simulations was in the range of 55–66% [52]. Thus, the simulation results in Variant 2 only slightly overestimated the experimental data. Overestimation of the final average concentration in the case of predicted model parameters was expected, as the predicted diffusion coefficient was about 20% higher than the known experimental value [52].
The displacement velocity of the (001) face over time was calculated from the total flux of aspirin over the selected crystal surface in simulations:
using as the surface area of the (001) face.
Experimentally, the face displacement velocity was obtained from tracking the surface position over time. For that, the crystal was divided into three segments as described in [52], and experimental results, presented in Figure 8, are given as averaged data points over these segments. Results for both simulations and the experiment exhibit comparably fast dissolution velocities at the beginning, whereas face displacement levels out toward the end. The simulation data obtained with experimental parameters (Variant 2) underestimate the face displacement velocity in the initial hours, while the face displacement velocity almost exactly matches the experimental findings in Variant 1. However, from Figure 8, it can be also seen that the process is diffusion-controlled. A sensitivity analysis performed for all parameters describing the dissolution process: face rate constants obtained from kMC simulations, diffusion coefficient and solubility also revealed a strong diffusion control, and thus, an expressed insensitivity of the simulation results to the actual intrinsic kMC disintegration rates [52].
8. Conclusions and Outlook
Here, we described the multiscale modeling approach for in silico prediction of crystal growth/dissolution rates on the basis of molecular structure only, highlighted the difficulties in the realization of all single steps and illustrated our recent attempts to overcome these on the example of aspirin dissolution. Through a suitable combination of MD, kMC and continuum simulations, we were able to predict the aspirin dissolution rates at tractable computational cost and in good agreement with experimental results [52]. However, the dissolution of aspirin was found to be diffusion-controlled in both the simulation and experiment employing a stagnant fluid. Due to this, the obtained continuum simulation results are not very sensitive to face displacement rates obtained from kMC simulations, but rely mainly on the determined diffusion coefficient. Thus, the validation of the actual multiscale-integration approach is still limited. Additionally, the conditions of a stagnant fluid can generally be considered inadequate for actual oral administration of API crystals, and many reports [127,128,129] suggest that inclusion of hydrodynamic (fluid flow) factors is critical in describing any dissolution processes in the human gastrointestinal tract. The consideration of flow conditions, firstly, would make the study more realistic and, secondly, would allow for a much deeper understanding of the proposed multiscale-integration approach by overcoming the diffusion limitation. Instead of only solving a distributed mass transfer equation, momentum transfer will have to be solved in this case in a coupled manner via a CFD approach. In future work, we will study the dissolution behavior under flow conditions both simulation based and experimentally to validate the scale integration concepts for disintegration-controlled conditions.
The goal to achieve such an assessment on the basis of the molecular structure only has also not yet been met at the other end of the scale. In current aspirin simulations, not only the molecular structure, but also the crystal packing and the crystal shape were used as input. Such information is readily available for model APIs like aspirin. However, this is generally not the case for multicomponent systems, like, e.g., hydrates, salts and co-crystals, which are presently one of the primary strategies pursued to improve the solubility of poorly-soluble drugs [130,131]. The consideration of such systems will also definitely increase the complexity of the state transition description for the MD-kMC integration. In terms of future work, new sophisticated neighbor and orientation definition schemes will be elaborated to expand the scope of the multiscale approach toward multicomponent systems. The state identification scheme mentioned in Section 4 and described in detail in our previous work [40] supports correct assignment of molecules to different face and edge categories only if these are defined by low Miller indices of one and zero. Some complex compounds may, however, require accounting for higher-indexed faces. For that, a new algorithm is currently under development in our group. The proper realization of all protocol steps will enable simplifying the design of crystallizer operations, as well as support decision making in early stages of in silico drug development as it will consider release properties for an API on the basis of the molecular structure only.
Acknowledgments
This work has been supported by the Deutsche Forschungsgemeinschaft (DFG) through Grants BR 2035/4-2 and BR 2035/8-2 and has made use of the computer resources provided by the Leibniz Supercomputing Centre under Grant pr58la. The ScalaLife Competence Center (http://www.scalalife.eu/) is also acknowledged for the assistance with computing resources and the GROMACS code.
Author Contributions
All authors jointly contributed to devising the presented methodology; M.G. performed the simulations; E.E. elaborated and developed analysis tools to link MD and kMC techniques; M.G. and E.E. developed kMC code; H.B. initiated and supervised the investigations; E.E. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Weeks, J.; Gilmer, G.H. Advances in Chemical Physics; Chapter Dynamics of Crystal Growth; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2007; Volume 40, pp. 157–228. [Google Scholar]
- Dhanaraj, G.; Byrappa, K.; Prasad, V.; Dudley, M. (Eds.) Springer Handbook of Crystal Growth; Springer: Heidelberg, Germany, 2010. [Google Scholar]
- Nishinaga, T. (Ed.) Handbook of Crystal Growth. Fundamentals: Thermodynamics and Kinetics; Elsevier: Amsterdam, The Netherland, 2014. [Google Scholar]
- Mullin, J. Crystallization: Third Edition; Butterworth-Heinemann: London, UK, 1997. [Google Scholar]
- Hurle, D. (Ed.) Handbook of Crystal Growth; Fundamentals. (a): Thermodynamics and Kinetics; (b): Transport and Stability; North Holland Elsevier Science Publishers: Amsterdam, The Netherlands, 1993; Volume 1. [Google Scholar]
- Dove, P.; Han, N. Kinetics of Mineral Dissolution and Growth as Reciprocal Microscopic Surface Processes Across Chemical Driving Force. Perspectives on Inorganic, Organic and Biological Crystal Growth: From Fundamentals to Applications Directions. Am. Inst. Phys. Conf. Ser. 2007, 916, 215–234. [Google Scholar]
- Clark, J.N.; Ihli, J.; Schenk, A.S.; Kim, Y.Y.; Kulak, A.N.; Campbell, J.M.; Nisbet, G.; Meldrum, F.C.; Robinson, I.K. Three-Dimensional Imaging of Dislocation Propagation During Crystal Growth and Dissolution. Nat. Mater. 2015, 14, 780–784. [Google Scholar] [CrossRef] [PubMed]
- Veesler, S.; Puel, F. Handbook of Crystal Growth. Fundamentals: Thermodynamics and Kinetics; Part A, Chapter Crystallization of Pharmaceutical Crystals; Elsevier: Amsterdam, The Netherland, 2014; Volume I, pp. 915–951. [Google Scholar]
- Derby, J.J.; Chelikowsky, J.R.; Sinno, T.; Dai, B.; Kwon, Y.I.; Lun, L.; Pandy, A.; Yeckel, A. Large-Scale Numerical Modeling of Melt and Solution Crystal Growth. AIP Conf. Proc. 2007, 916, 139–158. [Google Scholar]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Oxford University Press Inc.: New York, NY, USA, 1989. [Google Scholar]
- Piana, S.; Gale, J.D. Understanding the Barriers to Crystal Growth: Dynamical Simulation of the Dissolution and Growth of Urea from Aqueous Solution. J. Am. Chem. Soc. 2005, 127, 1975–1982. [Google Scholar] [CrossRef] [PubMed]
- Salvalaglio, M.; Vetter, T.; Giberti, F.; Mazzotti, M.; Parrinello, M. Uncovering Molecular Details of Urea Crystal Growth in the Presence of Additives. J. Am. Chem. Soc. 2012, 134, 17221–17233. [Google Scholar] [CrossRef] [PubMed]
- Salvalaglio, M.; Vetter, T.; Mazzotti, M.; Parrinello, M. Controlling and Predicting Crystal Shapes: The Case of Urea. Angew. Chem. Int. Ed. 2013, 52, 13369–13372. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Briesen, H. Molecular dynamics simulations of glycine crystal-solution interface. J. Chem. Phys. 2009, 131, 184705. [Google Scholar] [CrossRef] [PubMed]
- Gnanasambandam, S.; Rajagopalan, R. Growth Morphology of α-Glycine Crystals in Solution Environments: An Extended Interface Structure Analysis. CrystEngComm 2010, 12, 1740–1749. [Google Scholar] [CrossRef]
- Cheong, D.W.; Boon, Y.D. Comparative Study of Force Fields for Molecular Dynamics Simulations of α-Glycine Crystal Growth from Solution. Cryst. Growth Des. 2010, 10, 5146–5158. [Google Scholar] [CrossRef]
- Parks, C.; Koswara, A.; Tung, H.H.; Nere, N.K.; Bordawekar, S.; Nagy, Z.K.; Ramkrishna, D. Nanocrystal Dissolution Kinetics and Solubility Increase Prediction from Molecular Dynamics: The Case of α-, β-, and γ-Glycine. Mol. Pharm. 2017, 14, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Volkov, I.; Cieplak, M.; Koplik, J.; Banavar, J.R. Molecular Dynamics Simulations of Crystallization of Hard Spheres. Phys. Rev. E 2002, 66, 061401. [Google Scholar] [CrossRef] [PubMed]
- Lemarchand, C.A. Molecular dynamics simulations of a hard sphere crystal and reaction-like mechanism for homogeneous melting. J. Chem. Phys. 2012, 136, 234505. [Google Scholar] [CrossRef] [PubMed]
- Reilly, A.; Briesen, H. Modeling crystal growth from solution with molecular dynamics simulations: Approaches to transition rate constants. J. Chem. Phys. 2012, 136, 034704. [Google Scholar] [CrossRef] [PubMed]
- Gruhn, T.; Monson, P.A. Molecular dynamics simulations of hard sphere solidification at constant pressure. Phys. Rev. E 2001, 64, 061703. [Google Scholar] [CrossRef] [PubMed]
- Mandal, T.; Huang, W.; Mecca, J.M.; Getchell, A.; Porter, W.W.; Larson, R.G. A framework for multi-scale simulation of crystal growth in the presence of polymers. Soft Matter 2017, 13, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Mandal, T.; Marson, R.L.; Larson, R.G. Coarse-grained modeling of crystal growth and polymorphism of a model pharmaceutical molecule. Soft Matter 2016, 12, 8246–8255. [Google Scholar] [CrossRef] [PubMed]
- Ingólfsson, H.I.; Lopez, C.A.; Uusitalo, J.J.; de Jong, D.H.; Gopal, S.M.; Periole, X.; Marrink, S.J. The power of coarse graining in biomolecular simulations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 225–248. [Google Scholar] [CrossRef] [PubMed]
- Kästner, J. Umbrella sampling. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 932–942. [Google Scholar] [CrossRef]
- Quigley, D.; Rodger, P. A metadynamics-based approach to sampling crystallisation events. Mol. Simul. 2009, 35, 613–623. [Google Scholar] [CrossRef]
- Allen, R.J.; Valeriani, C.; ten Wolde, P.R. Forward flux sampling for rare event simulations. J. Phys. Condens. Matter 2009, 21, 463102. [Google Scholar] [CrossRef] [PubMed]
- Sosso, G.C.; Chen, J.; Cox, S.J.; Fitzner, M.; Pedevilla, P.; Zen, A.; Michaelides, A. Crystal Nucleation in Liquids: Open Questions and Future Challenges in Molecular Dynamics Simulations. Chem. Rev. 2016, 116, 7078–7116. [Google Scholar] [CrossRef] [PubMed]
- Dogan, B.; Schneider, J.; Reuter, K. In Silico Prediction of Dissolution Rates of Pharmaceutical Ingredients. Chem. Phys. Lett. 2016, 662, 52–55. [Google Scholar] [CrossRef]
- Schneider, J.; Zheng, C.; Reuter, K. Thermodynamics of Surface Defects at the Aspirin/Water Interface. J. Chem. Phys. 2014, 141, 124702. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Reuter, K. Efficient Calculation of Microscopic Dissolution Rate Constants: The Aspirin-Water Interface. J. Phys. Chem. Lett. 2014, 5, 3859–3862. [Google Scholar] [CrossRef] [PubMed]
- Burton, W.K.; Cabrera, N.; Frank, F.C. The Growth of Crystals and the Equilibrium Structure of their Surfaces. Philos. Trans. R. Soc. 1951, 243, 299–358. [Google Scholar] [CrossRef]
- Piana, S.; Reyhani, M.; Gale, J.D. Simulating micrometre-scale crystal growth from solution. Nature 2005, 483, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Reilly, A.; Briesen, H. A detailed kinetic Monte Carlo study of growth from solution using MD-derived rate constants. J. Cryst. Growth 2012, 354, 34–43. [Google Scholar] [CrossRef]
- Piana, S.; Gale, J.D. Three-dimensional kinetic Monte Carlo simulation of crystal growth from solution. J. Cryst. Growth 2006, 294, 46–52. [Google Scholar] [CrossRef]
- Kurganskaya, I.; Luttge, A. Kinetic Monte Carlo Simulations of Silicate Dissolution: Model Complexity and Parametrization. J. Phys. Chem. C 2013, 117, 24894–24906. [Google Scholar] [CrossRef]
- Stack, A.G.; Raiteri, P.; Gale, J.D. Accurate Rates of the Complex Mechanisms for Growth and Dissolution of Minerals Using a Combination of Rare-Event Theories. J. Am. Chem. Soc. 2012, 134, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; Reischl, B.; Spijker, P.; Holmberg, N.; Laasonen, K.; Foster, A.S. Ab initio Kinetic Monte Carlo simulations of dissolution at the NaCl-water interface. Phys. Chem. Chem. Phys. 2014, 16, 22545–22554. [Google Scholar] [CrossRef] [PubMed]
- Dkhissi, A.; Estéve, A.; Mastail, C.; Olivier, S.; Mazaleyrat, G.; Jeloaica, L.; Rouhani, M.D. Multiscale Modeling of the Atomic Layer Deposition of HfO2 Thin Film Grown on Silicon: How to Deal with a Kinetic Monte Carlo Procedure. J. Chem. Theory Comput. 2008, 4, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Elts, E.; Greiner, M.; Briesen, H. Predicting Dissolution Kinetics for Active Pharmaceutical Ingredients on the Basis of Their Molecular Structures. Cryst. Growth Des. 2016, 16, 4154–4164. [Google Scholar] [CrossRef]
- Boetker, J.P.; Rantanen, J.; Rades, T.; Müllertz, A.; Østergaard, J.; Jensen, H. A New Approach to Dissolution Testing by UV Imaging and Finite Element Simulations. Pharm. Res. 2013, 30, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Haddish-Berhane, N.; Nyquist, C.; Haghighi, K.; Corvalan, C.; Keshavarzian, A.; Campanella, O.; Rickus, J.; Farhadi, A. A multi-scale stochastic drug release model for polymer-coated targeted drug delivery systems. J. Control. Release 2006, 110, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Bai, G.; Armenante, P.M. Hydrodynamic, mass transfer, and dissolution effects induced by tablet location during dissolution testing. J. Pharm. Sci. 2009, 98, 1511–1531. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, G.; Galdi, I.; Barba, A.A. Controlled release from hydrogel-based solid matrices. A model accounting for water up-take, swelling and erosion. Int. J. Pharm. 2011, 407, 78–86. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, D.M.; Healy, A.M.; Corrigan, O.I. Towards determining appropriate hydrodynamic conditions for in vitro in vivo correlations using computational fluid dynamics. Eur. J. Pharm. Sci. 2009, 37, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Boetker, J.; Raijada, D.; Aho, J.; Khorasani, M.; Søgaard, S.V.; Arnfast, L.; Bohr, A.; Edinger, M.; Water, J.J.; Rantanen, J. In silico product design of pharmaceuticals. Asian J. Pharm. Sci. 2016, 11, 492–499. [Google Scholar] [CrossRef]
- Kindgen, S.; Rach, R.; Nawroth, T.; Abrahamsson, B.; Langguth, P. A Novel Disintegration Tester for Solid Dosage Forms Enabling Adjustable Hydrodynamics. J. Pharm. Sci. 2016, 105, 2402–2409. [Google Scholar] [CrossRef] [PubMed]
- Haddish-Berhane, N.; Rickus, J.L.; Haghighi, K. The role of multiscale computational approaches for rational design of conventional and nanoparticle oral drug delivery systems. Int. J. Nanomed. 2007, 2, 315–331. [Google Scholar]
- Greiner, M.; Elts, E.; Schneider, J.; Reuter, K.; Briesen, H. Dissolution study of active pharmaceutical ingredients using molecular dynamics simulations with classical force fields. J. Cryst. Growth 2014, 405, 122–130. [Google Scholar] [CrossRef]
- Greiner, M.; Elts, E.; Briesen, H. Insights into Pharmaceutical Nanocrystal Dissolution: A Molecular Dynamics Simulation Study on Aspirin. Mol. Pharm. 2014, 11, 3009–3016. [Google Scholar] [CrossRef] [PubMed]
- Elts, E.; Greiner, M.; Briesen, H. Data Filtering for Effective Analysis of Crystal-Solution Interface Molecular Dynamics Simulations. J. Chem. Theory Comput. 2014, 10, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Greiner, M.; Choscz, C.; Eder, C.; Elts, E.; Briesen, H. Multiscale modeling of aspirin dissolution: From molecular resolution to experimental scales of time and size. CrystEngComm 2016, 18, 5302–5312. [Google Scholar] [CrossRef]
- Eder, C.; Choscz, C.; Müller, V.; Briesen, H. Jamin-interferometer-setup for the determination of concentration and temperature dependent face-specific crystal growth rates from a single experiment. J. Cryst. Growth 2015, 426, 255–264. [Google Scholar] [CrossRef]
- Sundaralingam, M.; Jensen, L.H. Refinement of the structure of salicylic acid. Acta Crystallogr. 1965, 18, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Payne, R.S.; Rowe, R.C.; Roberts, R.J.; Charlton, M.H.; Docherty, R. Potential polymorphs of aspirin. J. Comput. Chem. 1999, 20, 262–273. [Google Scholar] [CrossRef]
- Bond, A.; Boese, R.; Desiraju, G. On the Polymorphism of Aspirin: Crystalline Aspirin as Intergrowths of Two “Polymorphic” Domains. Angew. Chem. Int. Ed. 2007, 46, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Hammond, R.B.; Pencheva, K.; Ramachandran, V.; Roberts, K.J. Application of Grid-Based Molecular Methods for Modeling Solvent-Dependent Crystal Growth Morphology: Aspirin Crystallized from Aqueous Ethanolic Solution. Cryst. Growth Des. 2007, 7, 1571–1574. [Google Scholar] [CrossRef]
- Hammond, R.B.; Pencheva, K.; Roberts, K.J. A Structural-Kinetic Approach to Model Face-Specific Solution/Crystal Surface Energy Associated with the Crystallization of Acetyl Salicylic Acid from Supersaturated Aqueous/Ethanol Solution. Cryst. Growth Des. 2006, 6, 1324–1334. [Google Scholar] [CrossRef]
- Kim, Y.; Matsumoto, M.; Machida, K. Specific Surface Energies and Dissolution Behavior of Aspirin Crystal. Chem. Pharm. Bull. 1985, 33, 4125–4131. [Google Scholar] [CrossRef]
- Danesh, A.; Connell, S.D.; Davies, M.C.; Roberts, C.J.; Tendler, S.J.B.; Williams, P.M.; Wilkins, M.J. An in situ dissolution study of aspirin crystal planes (100) and (001) by atomic force microscopy. Pharm. Res. 2001, 18, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Woodley, S.M.; Catlow, R. Crystal structure prediction from first principles. Nat. Mater. 2008, 7, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Day, G. Computational Pharmaceutical Solid State Chemistry; Chapter Advances in Crystal Structure Prediction and Applications to Pharmaceutical Materials; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 87–115. ISBN 978-1-11-870068-6. [Google Scholar]
- Price, S. Predicting crystal structures of organic compounds. Chem. Soc. Rev. 2014, 43, 2098–2111. [Google Scholar] [CrossRef] [PubMed]
- Reilly, A.M.; Cooper, R.I.; Adjiman, C.S.; Bhattacharya, S.; Boese, A.D.; Brandenburg, J.G.; Bygrave, P.J.; Bylsma, R.; Campbell, J.E.; Car, R.; et al. Report on the Sixth Blind Test of Organic Crystal-Structure Prediction Methods. Acta Crystallogr. Sect. B 2016, 72, 439–459. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.P.; Day, G.M. Which conformations make stable crystal structures? Mapping crystalline molecular geometries to the conformational energy landscape. Chem. Sci. 2014, 5, 3173–3182. [Google Scholar] [CrossRef]
- Nývlt, J.; Ulrich, J. Addmixtures in Crystallization; VCH: Weinheim, Germany, 1995. [Google Scholar]
- Wulff, G. Velocity of growth and dissolution of crystal faces. Z. Kristallogr. 1901, 34, 449–530. [Google Scholar]
- Rogal, J.; Reuter, K.; Scheffler, M. Thermodynamic stability of PdO surfaces. Phys. Rev. B 2004, 69, 075421. [Google Scholar] [CrossRef]
- Ouyang, R.; Liu, J.X.; Li, W.X. Atomistic Theory of Ostwald Ripening and Disintegration of Supported Metal Particles under Reaction Conditions. J. Am. Chem. Soc. 2013, 135, 1760–1771. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mota, M.; Rieger, M.; Reuter, K. Ab Initio Prediction of the Equilibrium Shape of Supported Ag Nanoparticles on α-Al2O3(0001). J. Catal. 2015, 321, 1–6. [Google Scholar] [CrossRef]
- Lovette, M.A.; Robben Browning, A.; Griffin, D.W.; Sizemore, J.P.; Snyder, R.C.; Doherty, M.F. Crystal Shape Engineering. Ind. Eng. Chem. Res. 2008, 47, 9812–9833. [Google Scholar] [CrossRef]
- Kuvadia, Z.B.; Doherty, M.F. Spiral growth model for faceted crystals of non-centrosymmetric organic molecules grown from solution. Cryst. Growth Des. 2011, 11, 2780–2802. [Google Scholar] [CrossRef]
- Mayerson, A.S. (Ed.) Handbook of industrial Crystallization; Butterworth-Heinemann: Newton, MA, USA, 2002. [Google Scholar]
- Bravais, A. Etudes Crystallographiques; Academie des Sciences: Paris, France, 1913. [Google Scholar]
- Friedel, G. Etudes sur la loi de Bravais. Bull. Soc. Fr. Miner. 1907, 30, 326–455. [Google Scholar]
- Donnay, J.D.H.; Harker, D. A new law of crystal morphology extending the law of Bravais. Am. Miner. 1937, 22, 446–467. [Google Scholar]
- Hartman, P.; Bennema, P. The attachment energy as a habit controlling factor. I. Theoretical considerations. J. Cryst. Growth 1980, 49, 145–156. [Google Scholar] [CrossRef]
- Rai, B. (Ed.) Molecular Modeling for the Design of Novel Performance Chemicals and Materials; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Li, J.; Doherty, M.F. Steady State Morphologies of Paracetamol Crystal from Different Solvents. Cryst. Growth Des. 2017, 17, 659–670. [Google Scholar] [CrossRef]
- Tilbury, C.J.; Green, D.A.; Marshall, W.J.; Doherty, M.F. Predicting the Effect of Solvent on the Crystal Habit of Small Organic Molecules. Cryst. Growth Des. 2016, 16, 2590–2604. [Google Scholar] [CrossRef]
- Shim, H.M.; Koo, K.K. Prediction of Growth Habit of β-Cyclotetramethylene-tetranitramine Crystals by the First-Principles Models. Cryst. Growth Des. 2015, 15, 3983–3991. [Google Scholar] [CrossRef]
- Zhang, C.; Ji, C.; Li, H.; Zhou, Y.; Xu, J.; Xu, R.; Li, J.; Luo, Y. Occupancy Model for Predicting the Crystal Morphologies Influenced by Solvents and Temperature, and Its Application to Nitroamine Explosives. Cryst. Growth. Des. 2013, 13, 282–290. [Google Scholar] [CrossRef]
- Yang, X.; Qian, G.; Zhang, X.; Duan, X.; Zhou, X. Effects of Solvent and Impurities on Crystal Morphology of Zinc Lactate Trihydrate. Chin. J. Chem. Eng. 2014, 22, 221–226. [Google Scholar] [CrossRef]
- Wilson, C.C. Interesting proton behaviour in molecular structures. Variable temperature neutron diffraction and ab initio study of acetylsalicylic acid: Characterising librational motions and comparing protons in different hydrogen bonding potentials. New J. Chem. 2002, 26, 1733–1739. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins Struct. Funct. Bioinf. 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general AMBER force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam, a Fast Force Field Generation Tool For Small Organic Molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Da Silva Sousa, A.; Alan, W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Schüttelkopf, A.W.; van Aalten, D.M.F. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Lemkul, J.A.; Allen, W.J.; Bevan, D.R. Practical Considerations for Building GROMOS-Compatible Small-Molecule Topologies. J. Chem. Inf. Model. 2010, 50, 2221–2235. [Google Scholar] [CrossRef] [PubMed]
- Hawtin, R.; Quigley, D.; Rodger, P. Gas hydrate nucleation and cage formation at a water/methane interface. Phys. Chem. Chem. Phys. 2008, 10, 4853–4864. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Kusalik, P.G. Explorations of gas hydrate crystal growth by molecular simulations. Chem. Phys. Lett. 2010, 494, 123–133. [Google Scholar] [CrossRef]
- Jacobson, L.C.; Matsumoto, M.; Molinero, V. Order parameters for the multistep crystallization of clathrate hydrates. J. Chem. Phys. 2011, 135, 074501. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Olsen, K.W. Molecular Dynamics of Drug Crystal Dissolution: Simulation of Acetaminophen Form I in Water. Mol. Pharm. 2013, 10, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Wen, H.; Li, T.; Morris, K.R.; Park, K. Dissolution Study on Aspirin and α-Glycine Crystals. J. Phys. Chem. B 2004, 108, 11219–11227. [Google Scholar] [CrossRef]
- Wedelkind, J.; Reguera, D.; Strey, R. Finite-size effects in simulations of nucleation. J. Chem. Phys. 2006, 125, 214505. [Google Scholar] [CrossRef] [PubMed]
- Salvalaglio, M.; Mazzotti, M.; Parrinello, M. Urea homogeneous nucleation mechanism is solvent dependent. Faraday Discuss. 2015, 179, 291–307. [Google Scholar] [CrossRef] [PubMed]
- Grossier, R.; Veesler, S. Reaching One Single and Stable Critical Cluster through Finite-Sized Systems. Cryst. Growth Des. 2009, 9, 1917–1922. [Google Scholar] [CrossRef]
- Perego, C.; Salvalaglio, M.; Parrinello, M. Molecular dynamics simulations of solutions at constant chemical potential. J. Chem. Phys. 2015, 142, 144113. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, N.E.R.; Vorselaars, B.; Quigley, D.; Peters, B. Nucleation of NaCl from Aqueous Solution: Critical Sizes, Ion-Attachment Kinetics, and Rates. J. Am. Chem. Soc. 2015, 137, 13352–13361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anwar, J.; Zahn, D. Uncovering Molecular Processes in Crystal Nucleation and Growth by Using Molecular Simulation. Angew. Chem. Int. Ed. 2011, 50, 1996–2013. [Google Scholar] [CrossRef] [PubMed]
- Steinhardt, P.J.; Nelson, D.R.; Ronchetti, M. Bond-orientational order in liquids and glasses. Phys. Rev. B 1983, 28, 784–805. [Google Scholar] [CrossRef]
- Lechner, W.; Dellago, C. Accurate determination of crystal structures based on averaged local bond order parameters. J. Chem. Phys. 2008, 129, 114707. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, R.; Trout, B.L. Nucleation of Hexagonal Ice (Ih) in Liquid Water. J. Am. Chem. Soc. 2003, 125, 7743–7747. [Google Scholar] [CrossRef] [PubMed]
- Brukhno, A.; Anwar, J.; Davidchack, R.; Handel, R. Challenges in molecular simulation of homogeneous ice nucleation. J. Phys. Condens. Matter 2008, 20, 494243. [Google Scholar] [CrossRef]
- Leyssale, J.M.; Delhommelle, J.; Millot, C. Reorganization and Growth of Metastable α-N2 Critical Nuclei into Stable β-N2 Crystals. J. Am. Chem. Soc. 2004, 126, 12286–12287. [Google Scholar] [CrossRef] [PubMed]
- Mettes, J.A.; Keith, J.B.; McClurg, R.B. Molecular crystal global phase diagrams. I. Method of construction. Acta Crystallogr. Sect. A Found. Crystallogr. 2004, 60, 621–636. [Google Scholar] [CrossRef] [PubMed]
- Zahn, D. Atomistic Mechanisms of Phase Separation and Formation of Solid Solutions: Model Studies of NaCl, NaCl-NaF, and Na(Cl1−xBrx) Crystallization from Melt. J. Phys. Chem. B 2007, 111, 5249–5253. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Bartell, L. Analysis of Orientational Order in Molecular Clusters. A Molecular Dynamics Study. J. Phys. Chem. 1993, 97, 13544–13549. [Google Scholar] [CrossRef]
- Kinney, K.E.; Xu, S.; Bartell, L.S. Molecular Dynamics Study of the Freezing of Clusters of Chalcogen Hexafluorides. J. Phys. Chem. 1996, 100, 6935–6941. [Google Scholar] [CrossRef]
- Santiso, E.E.; Trout, B.L. A general set of order parameters for molecular crystals. J. Chem. Phys. 2011, 134, 064109. [Google Scholar] [CrossRef] [PubMed]
- Reuter, K. First-Principles Kinetic Monte Carlo Simulations for Heterogeneous Catalysis: Concepts, Status, and Frontiers. In Modeling and Simulation of Heterogeneous Catalytic Reactions: From the Molecular Process to the Technical System; Deutschmann, O., Ed.; Wiley-VCH: Weinheim, Germany, 2011; pp. 71–111. [Google Scholar]
- Kalman, R.E. A New Approach to Linear Filtering and Prediction Problems. J. Basic Eng. 1960, 82, 35–45. [Google Scholar] [CrossRef]
- Bortz, A.B.; Kalos, M.H.; Lebowitz, J.L. A new algorithm for Monte Carlo simulation of Ising spin systems. J. Comput. Phys. 1975, 17, 10–18. [Google Scholar] [CrossRef]
- Chatterjee, A.; Vlachos, D. An overview of spatial microscopic and accelerated kinetic Monte Carlo methods. J. Comput. Aided Mater. Des. 2007, 14, 253–308. [Google Scholar] [CrossRef]
- Burghaus, U.; Stephan, J.; Vattuone, L.; Rogowska, J. A Practical Guide to Kinetic Monte Carlo Simulations and Classical Molecular Dynamics Simulations; Nova Science Publishers, Inc.: New York, NY, USA, 2006. [Google Scholar]
- Comsol AB. COMSOL Multiphysics User’S Guide; Comsol AB: Stockholm, Sweden, 2014. [Google Scholar]
- Edwards, L.J. The Dissolution and Diffusion of Aspirin In Aqueous Media. Trans. Faraday Soc. 1951, 47, 1191–1210. [Google Scholar] [CrossRef]
- Klamt, A. Conductor-like Screening Model for Real Solvent: A New Approach to the Quantitative Calculation of Solvation Phenomena. J. Phys. Chem. A 1995, 99, 2224–2235. [Google Scholar] [CrossRef]
- Espinosa, J.R.; Young, J.M.; Jiang, H.; Gupta, D.; Vega, C.; Sanz, E.; Debenedetti, P.G.; Panagiotopoulos, A.Z. On the calculation of solubilities via direct coexistence simulations: Investigation of NaCl aqueous solutions and Lennard-Jones binary mixtures. J. Chem. Phys. 2016, 145, 154111. [Google Scholar] [CrossRef] [PubMed]
- Sugano, K. Theoretical comparison of hydrodynamic diffusion layer models used for dissolution simulation in drug discovery and development. Int. J. Pharm. 2008, 363, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Southard, M. Control of Poorly Soluble Drug Dissolution in Conditions Simulating the Gastrointestinal Tract Flow. 1. Effect of Tablet Geometry in Buffered Medium. J. Pharm. Sci. 1996, 85, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Southard, M.Z. Control of Poorly Soluble Drug Dissolution in Conditions Simulating the Gastrointestinal Tract Flow. 2. Cocompression of Drugs with Buffers. J. Pharm. Sci. 1997, 86, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Sikarra, D.; Shukla, V.; Kharia, A.; Chatterjee, D. Techniques for solubility enhancement of poorly soluble drugs: An overview. J. Med. Pharm. Allied Sci. 2012, 1, 1–22. [Google Scholar]
- Tiwle, R.; Ajazuddin; Giri, T.; Tripathi, D.; Jain, V.; Alexander, A. An exhaustive review on solubility enhancement for hydrophobic compounds by possible applications of novel techniques. Trends Appl. Sci. Res. 2012, 7, 596–619. [Google Scholar]
Figure 1.
Protocol for in silico prediction of crystal growth/dissolution rates on the basis of the molecular structure only (case scenario: aspirin).
Figure 1.
Protocol for in silico prediction of crystal growth/dissolution rates on the basis of the molecular structure only (case scenario: aspirin).
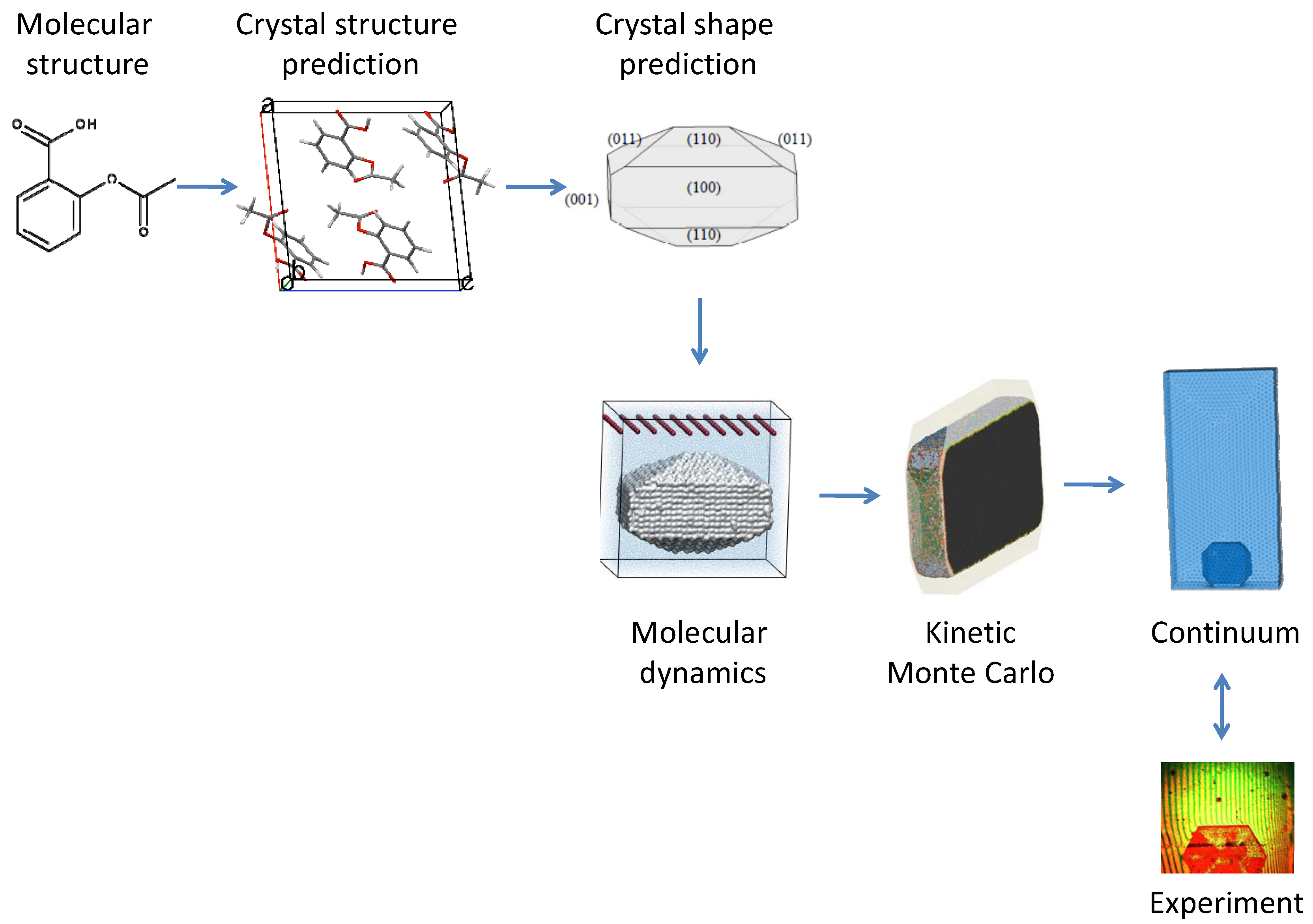
Figure 2.
Representations of aspirin crystal morphology in MD simulations: (a) Starting configuration of the nanocrystal in its experimental shape, (b) starting configuration with the (010) face and (c) block structure revealing only the (100), (010), and (001) faces.
Figure 2.
Representations of aspirin crystal morphology in MD simulations: (a) Starting configuration of the nanocrystal in its experimental shape, (b) starting configuration with the (010) face and (c) block structure revealing only the (100), (010), and (001) faces.
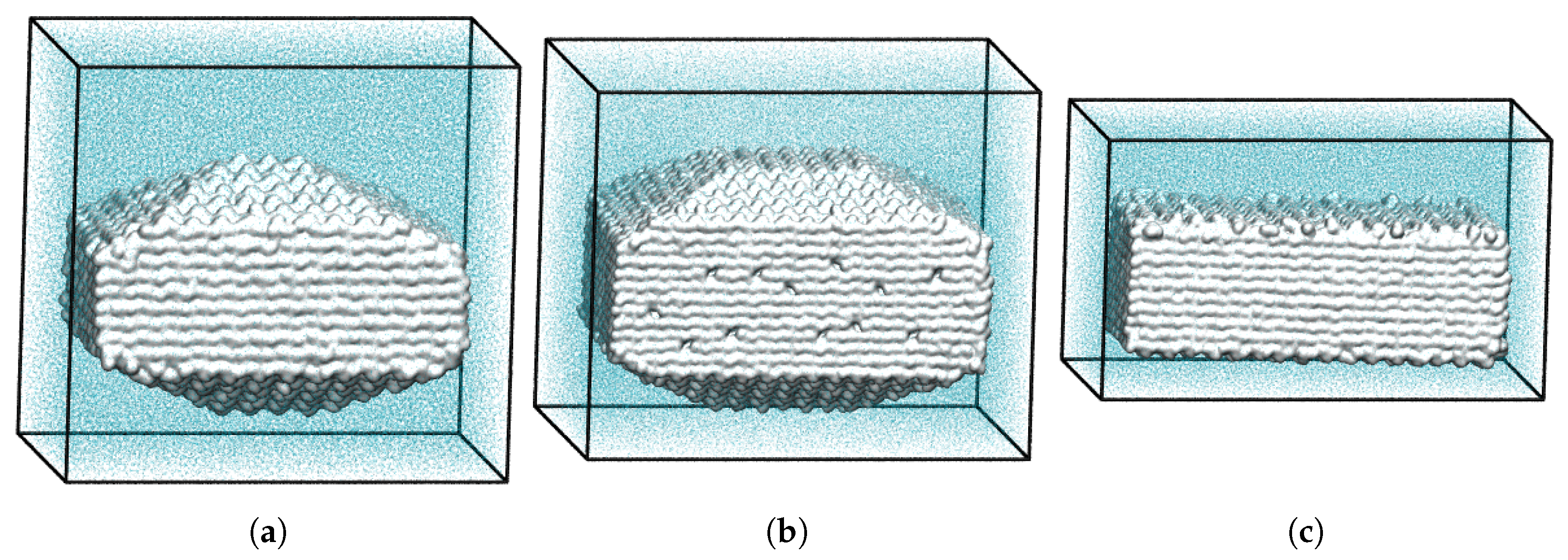
Figure 3.
Sticky dummy atoms (red points above the nanocrystal) in action: (a) initial and (b) final snapshots from MD simulation of aspirin nanocrystal in its experimental form with crystal-like molecules plotted as white surface, liquid-like molecules in blue and adsorbed molecules in green. Water molecules are not shown for clarity.
Figure 3.
Sticky dummy atoms (red points above the nanocrystal) in action: (a) initial and (b) final snapshots from MD simulation of aspirin nanocrystal in its experimental form with crystal-like molecules plotted as white surface, liquid-like molecules in blue and adsorbed molecules in green. Water molecules are not shown for clarity.
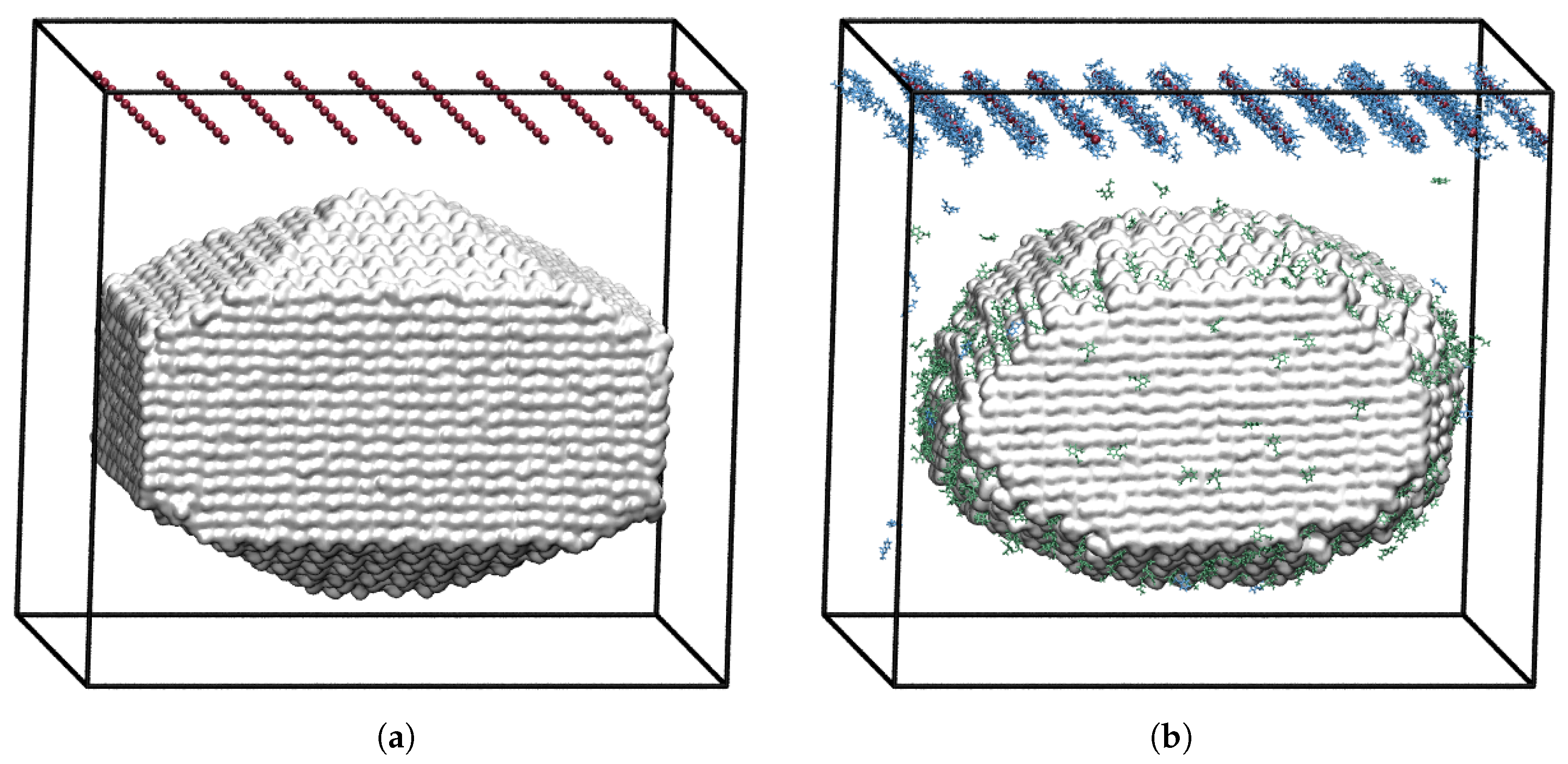
Figure 4.
Result of the application of state classification scheme for an aspirin nanocrystal at the beginning of MD simulation. The molecules located at flat faces are gray for the (100) face, blue for the (001) face, light blue for the (011) face and light gray for the (110) face. Molecules located on edges formed by the intersection of single-indexed faces are indicated in light green, and purple is used to show molecules on the edges formed by the intersection of double-indexed faces. Orange indicates molecules on the edges formed by single- and double-indexed faces. Water and sticky dummy atoms are not shown. Reprinted with permission from [40]. Copyright 2016 American Chemical Society.
Figure 4.
Result of the application of state classification scheme for an aspirin nanocrystal at the beginning of MD simulation. The molecules located at flat faces are gray for the (100) face, blue for the (001) face, light blue for the (011) face and light gray for the (110) face. Molecules located on edges formed by the intersection of single-indexed faces are indicated in light green, and purple is used to show molecules on the edges formed by the intersection of double-indexed faces. Orange indicates molecules on the edges formed by single- and double-indexed faces. Water and sticky dummy atoms are not shown. Reprinted with permission from [40]. Copyright 2016 American Chemical Society.

Figure 5.
Linking MD and kMC approaches.
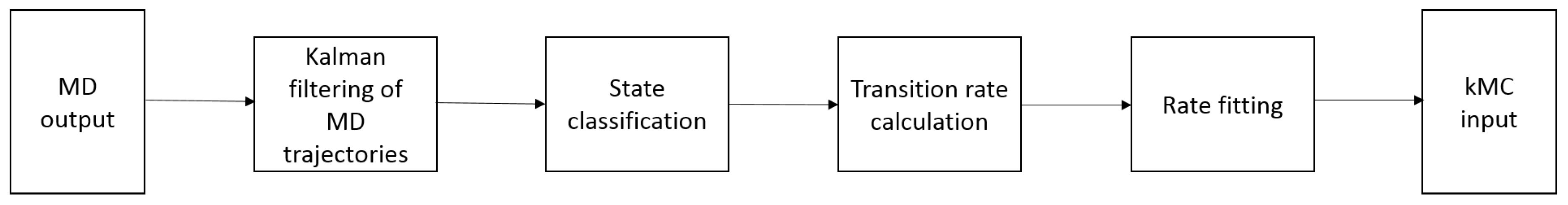
Figure 6.
Representation of aspirin crystal in kMC simulations. The molecules located at flat faces are gray for the (100) face, blue for the (001) face, light blue for the (011) face and light gray for the (110) face. Molecules located on edges formed by the intersection of single-indexed faces are indicated in light green, and purple is used to show molecules on the edges formed by the intersection of double-indexed faces. Orange indicates molecules on the edges formed by single- and double-indexed faces.
Figure 6.
Representation of aspirin crystal in kMC simulations. The molecules located at flat faces are gray for the (100) face, blue for the (001) face, light blue for the (011) face and light gray for the (110) face. Molecules located on edges formed by the intersection of single-indexed faces are indicated in light green, and purple is used to show molecules on the edges formed by the intersection of double-indexed faces. Orange indicates molecules on the edges formed by single- and double-indexed faces.
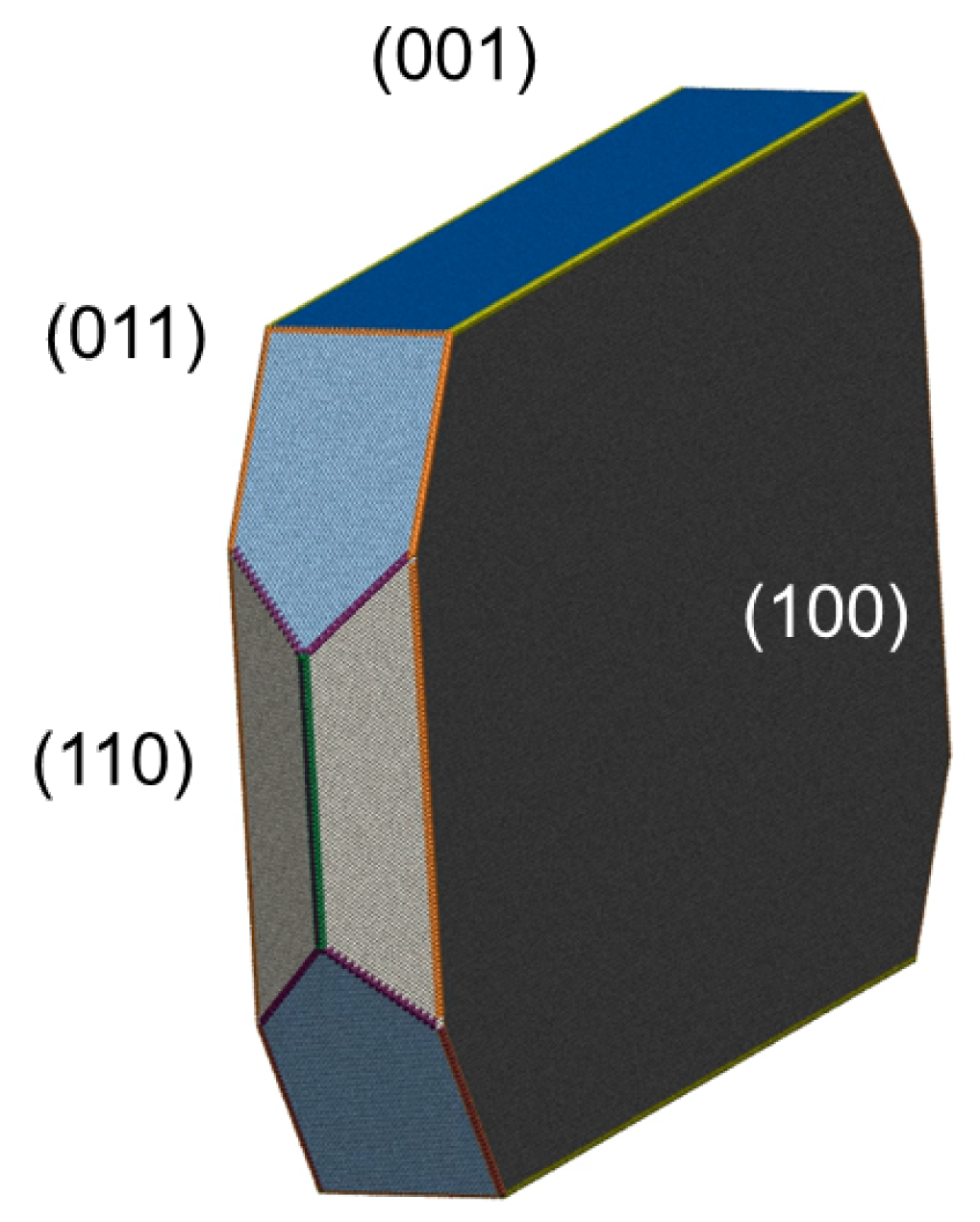
Figure 7.
Face displacements from kMC simulations.
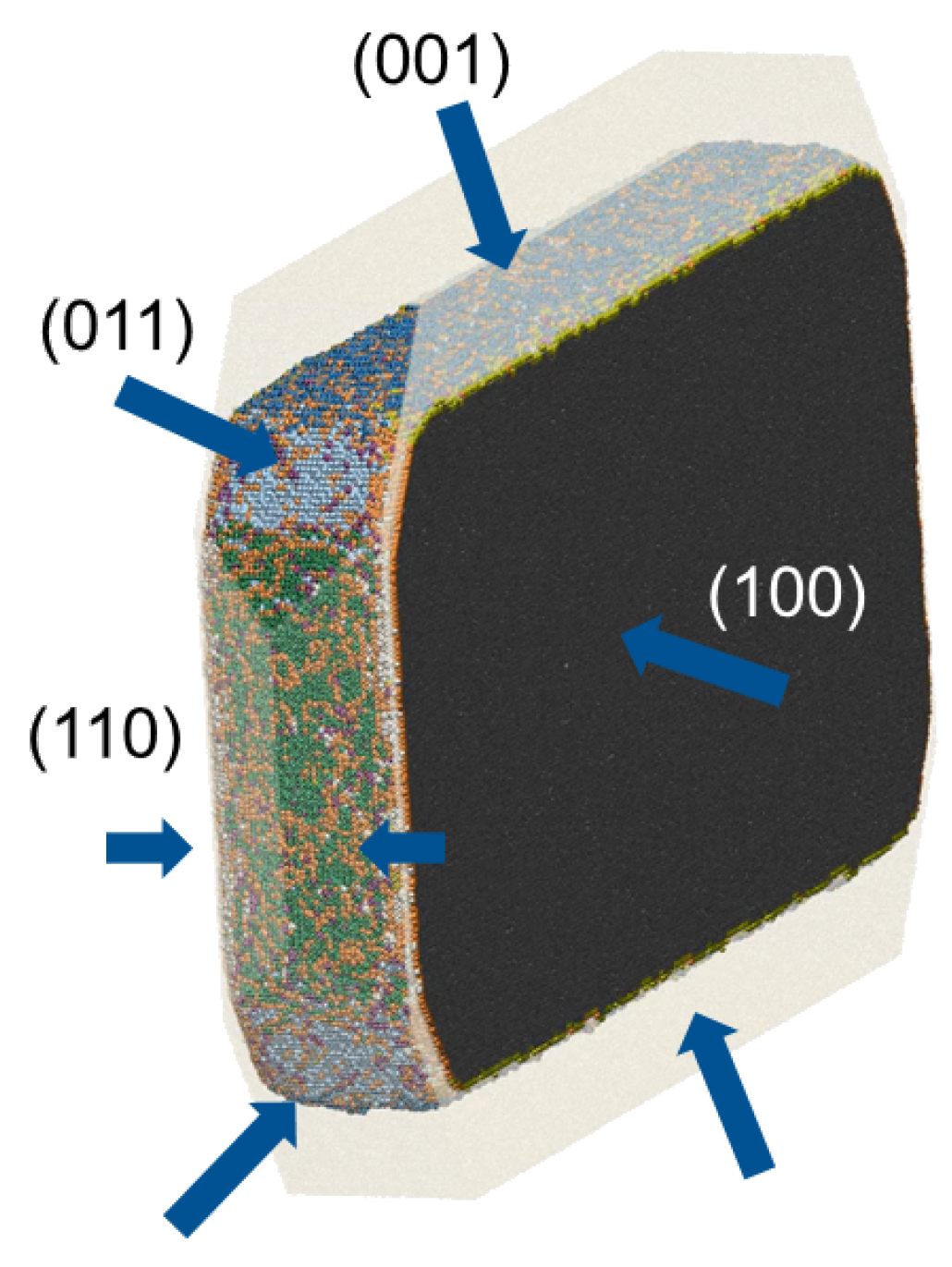
Figure 8.
Face displacement velocity for the (001) face of aspirin crystal as obtained in the experiment and calculated from continuum simulations based on kMC face rate constants and (Variant 1) the predicted in silico values for the diffusion coefficient and solubility or (Variant 2) experimental values for the diffusion coefficient and solubility.
Figure 8.
Face displacement velocity for the (001) face of aspirin crystal as obtained in the experiment and calculated from continuum simulations based on kMC face rate constants and (Variant 1) the predicted in silico values for the diffusion coefficient and solubility or (Variant 2) experimental values for the diffusion coefficient and solubility.
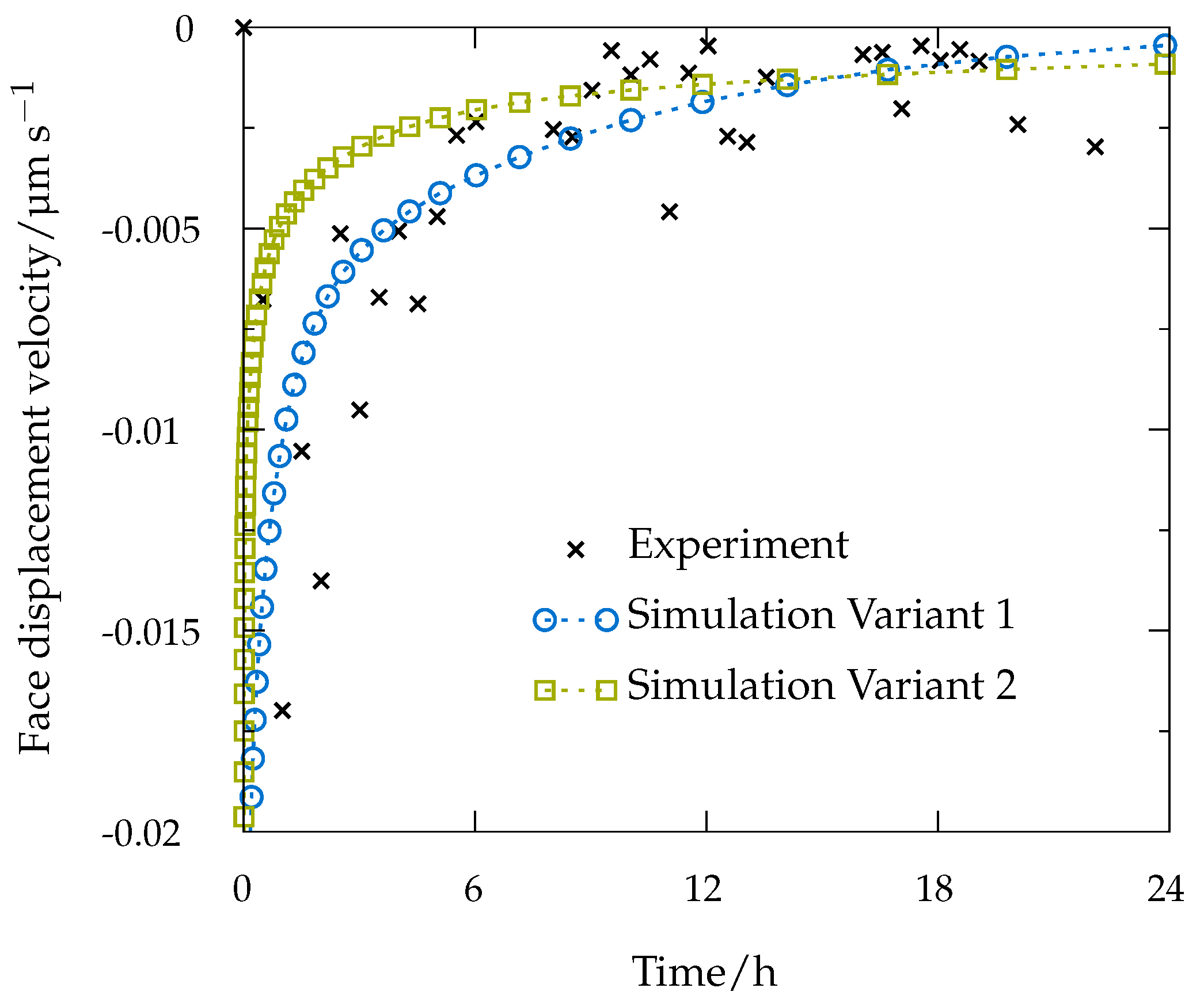
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Elts, E.; Greiner, M.; Briesen, H. In Silico Prediction of Growth and Dissolution Rates for Organic Molecular Crystals: A Multiscale Approach. Crystals 2017, 7, 288. https://doi.org/10.3390/cryst7100288
AMA Style
Elts E, Greiner M, Briesen H. In Silico Prediction of Growth and Dissolution Rates for Organic Molecular Crystals: A Multiscale Approach. Crystals. 2017; 7(10):288. https://doi.org/10.3390/cryst7100288
Chicago/Turabian StyleElts, Ekaterina, Maximilian Greiner, and Heiko Briesen. 2017. "In Silico Prediction of Growth and Dissolution Rates for Organic Molecular Crystals: A Multiscale Approach" Crystals 7, no. 10: 288. https://doi.org/10.3390/cryst7100288
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.