
Bisphthalonitrile with a Disulfide-Based Linker and its Dimethylene Analogue: Comparative Structural Insights


Abstract
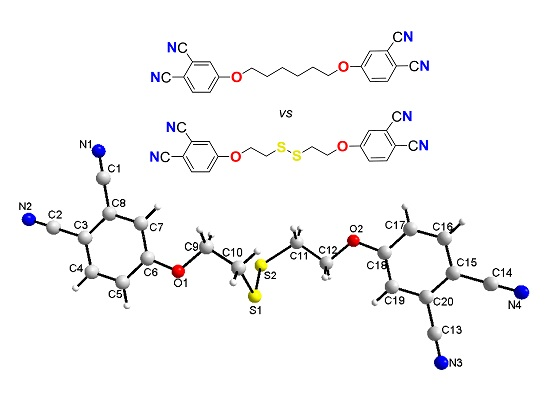
:
1. Introduction
2. Results and Discussion
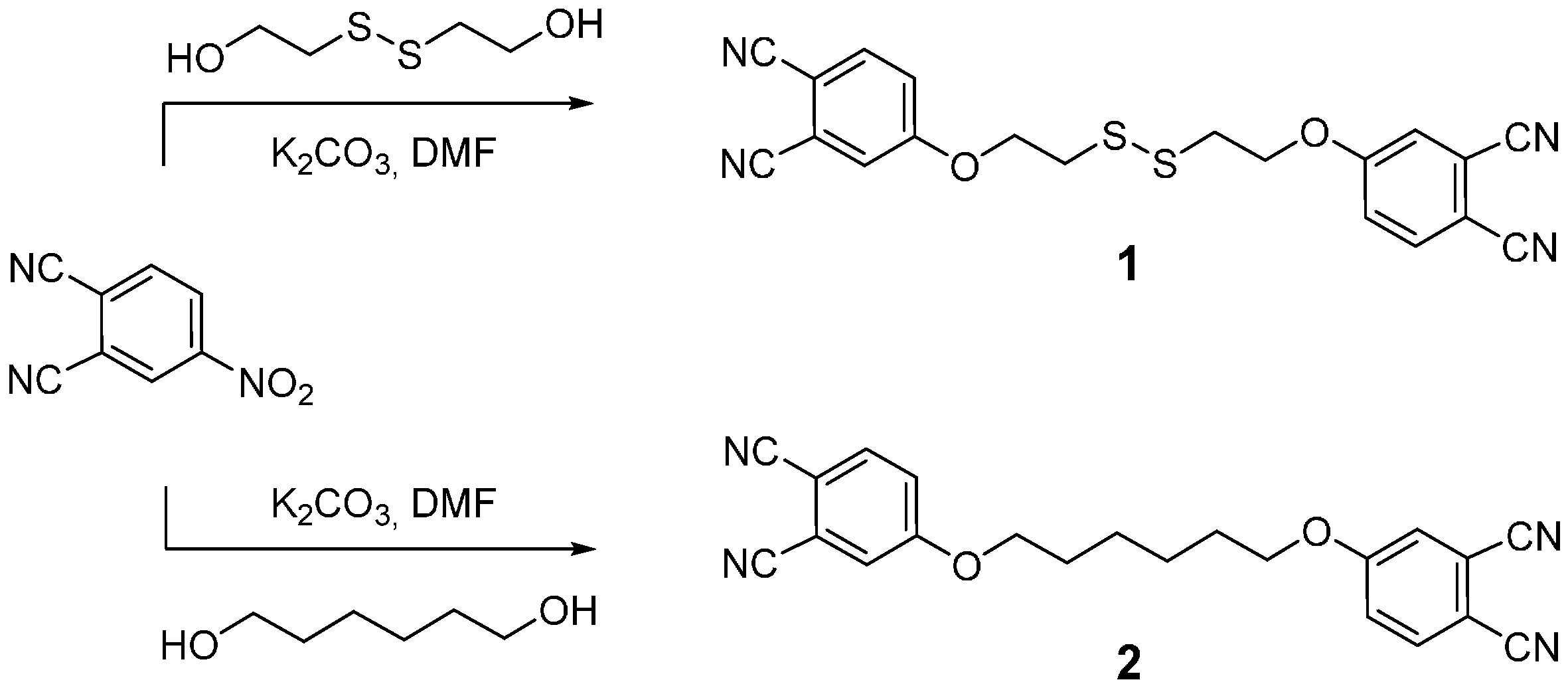
2.1. Synthesis
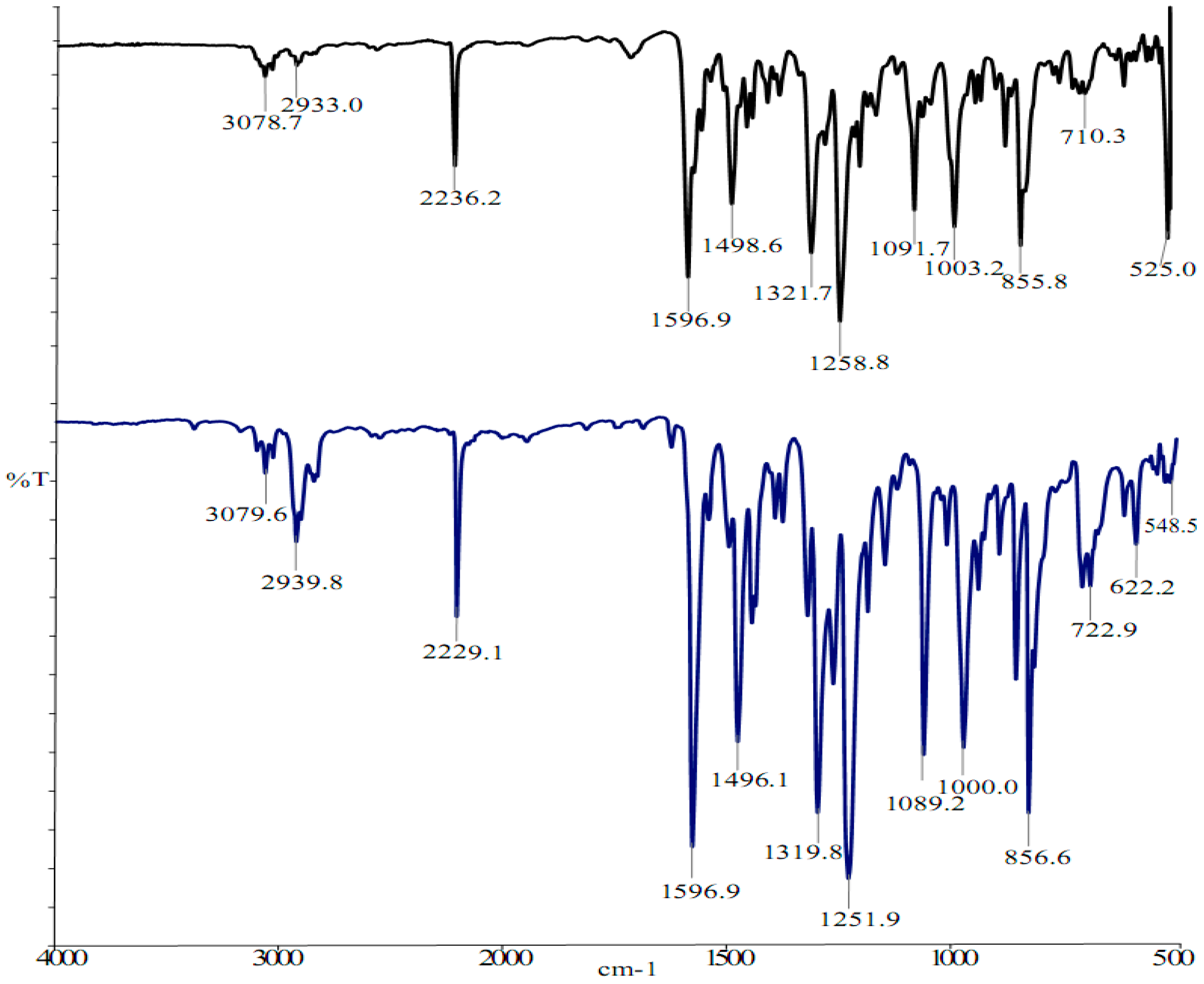
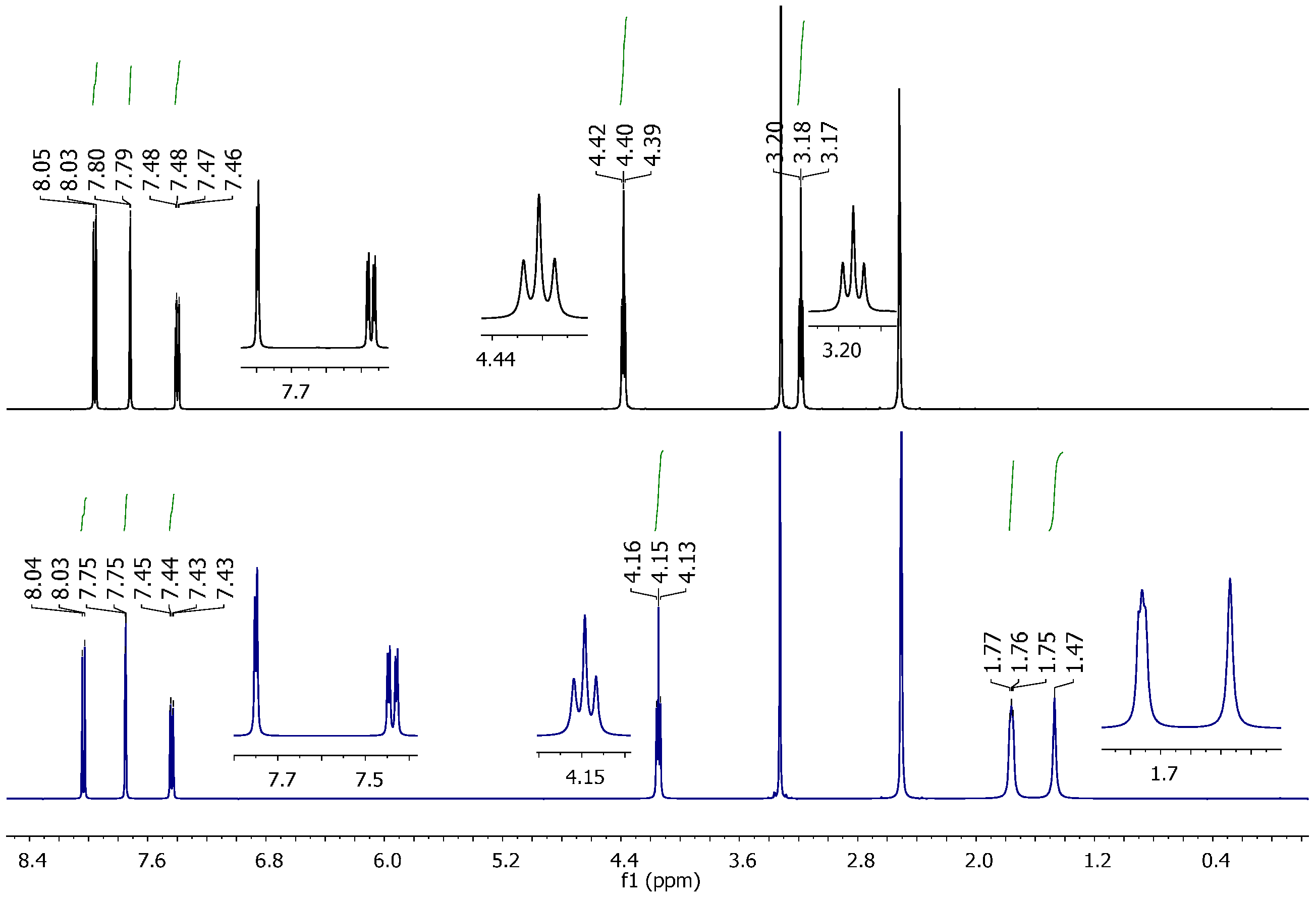
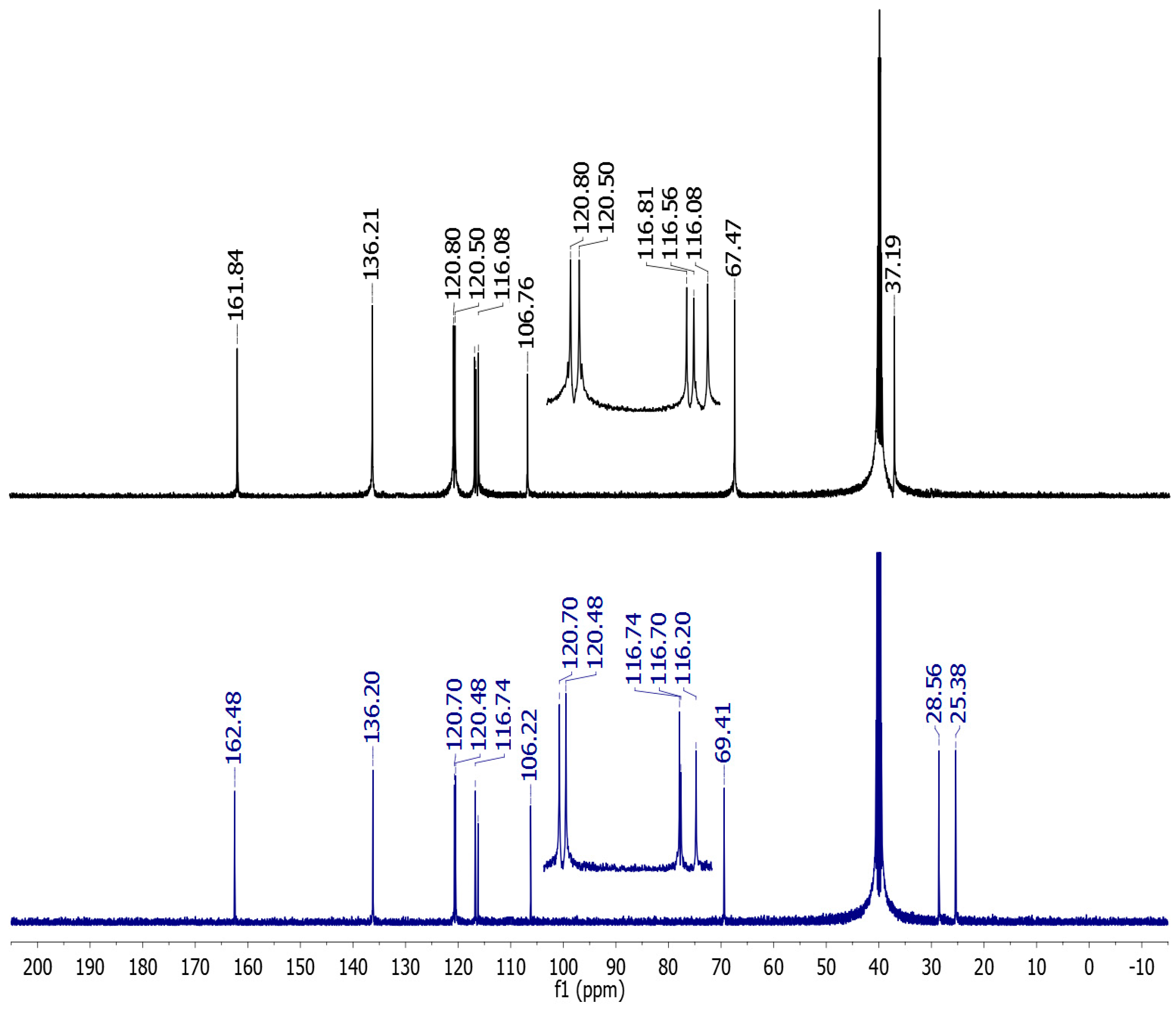
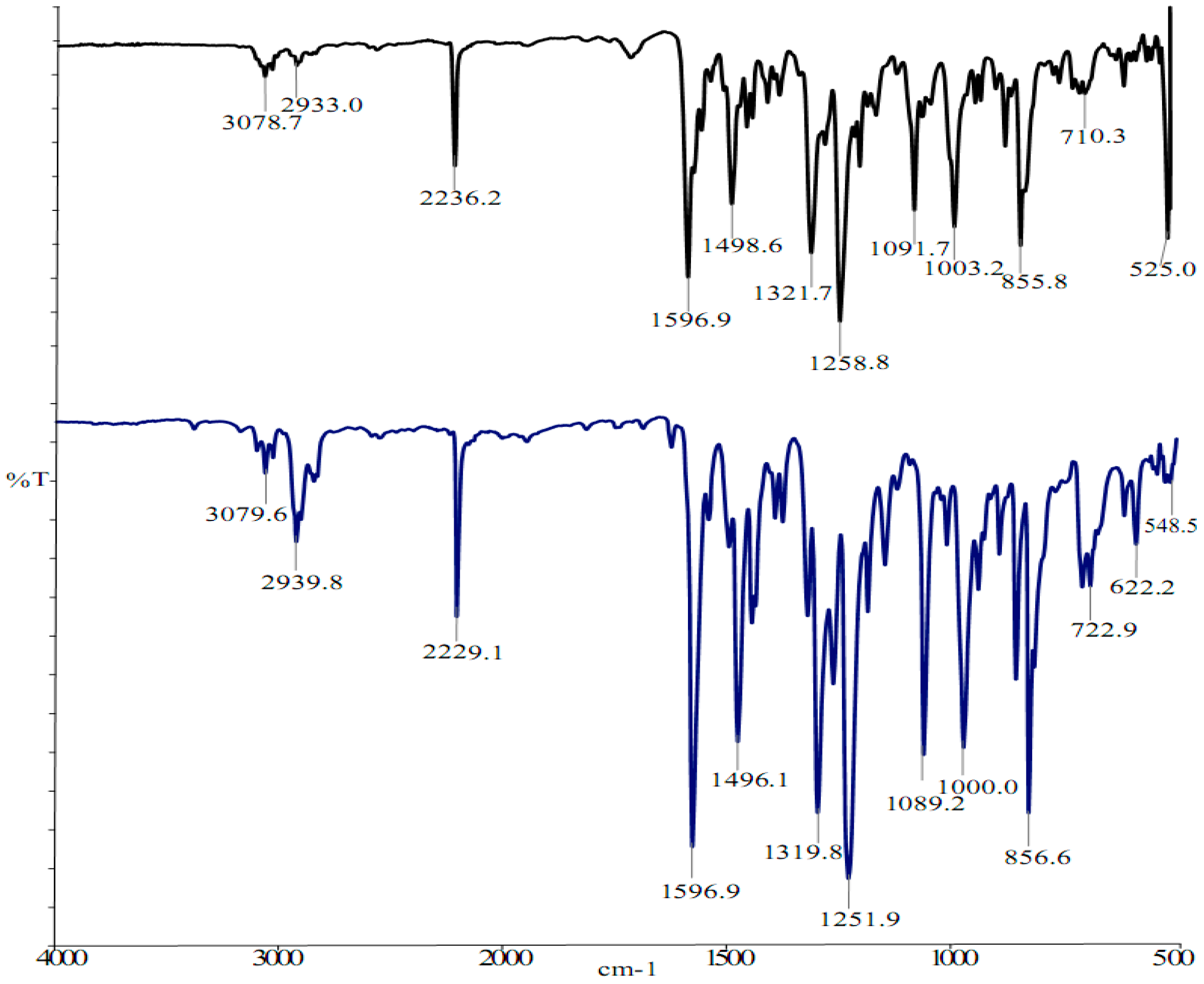
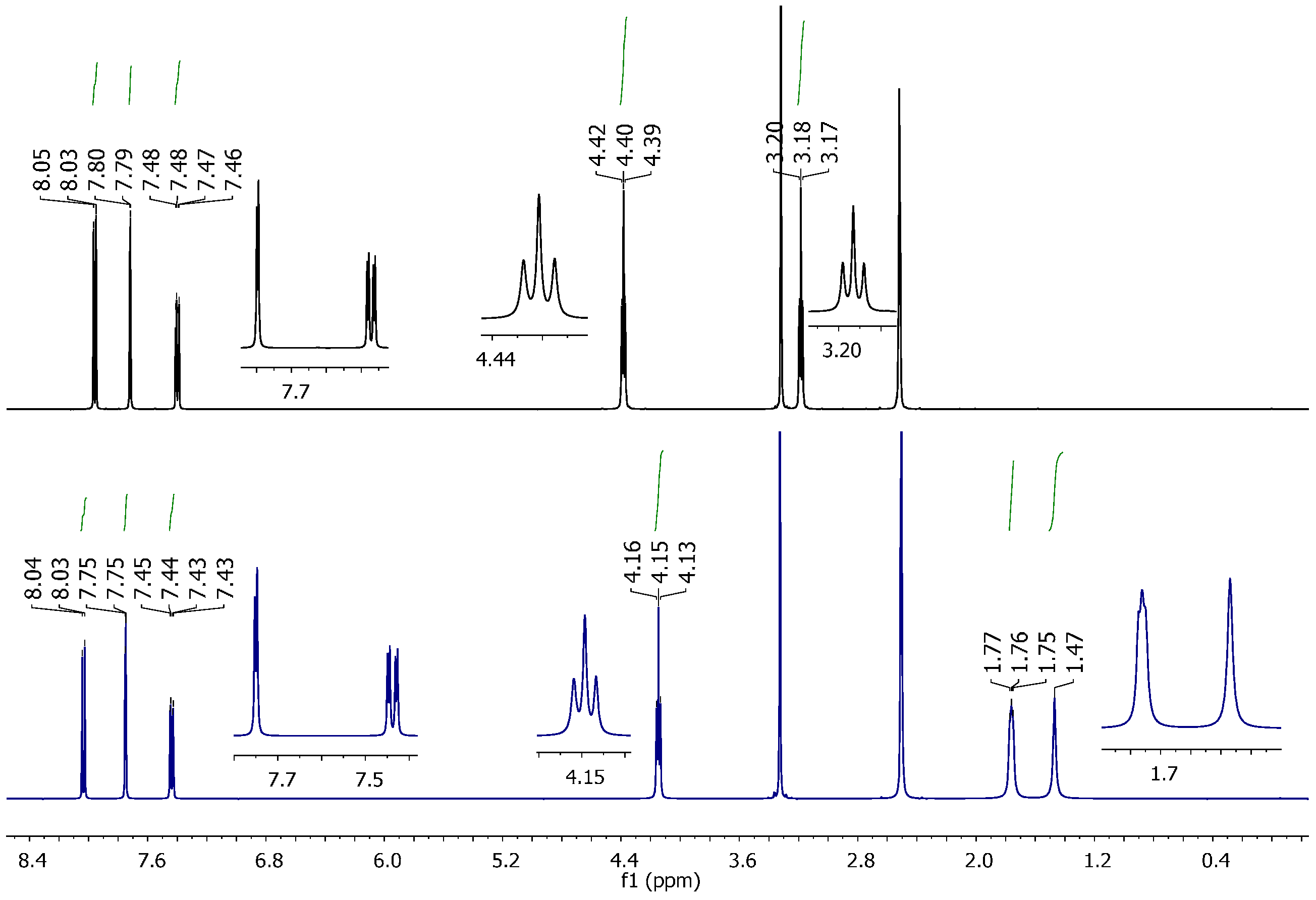
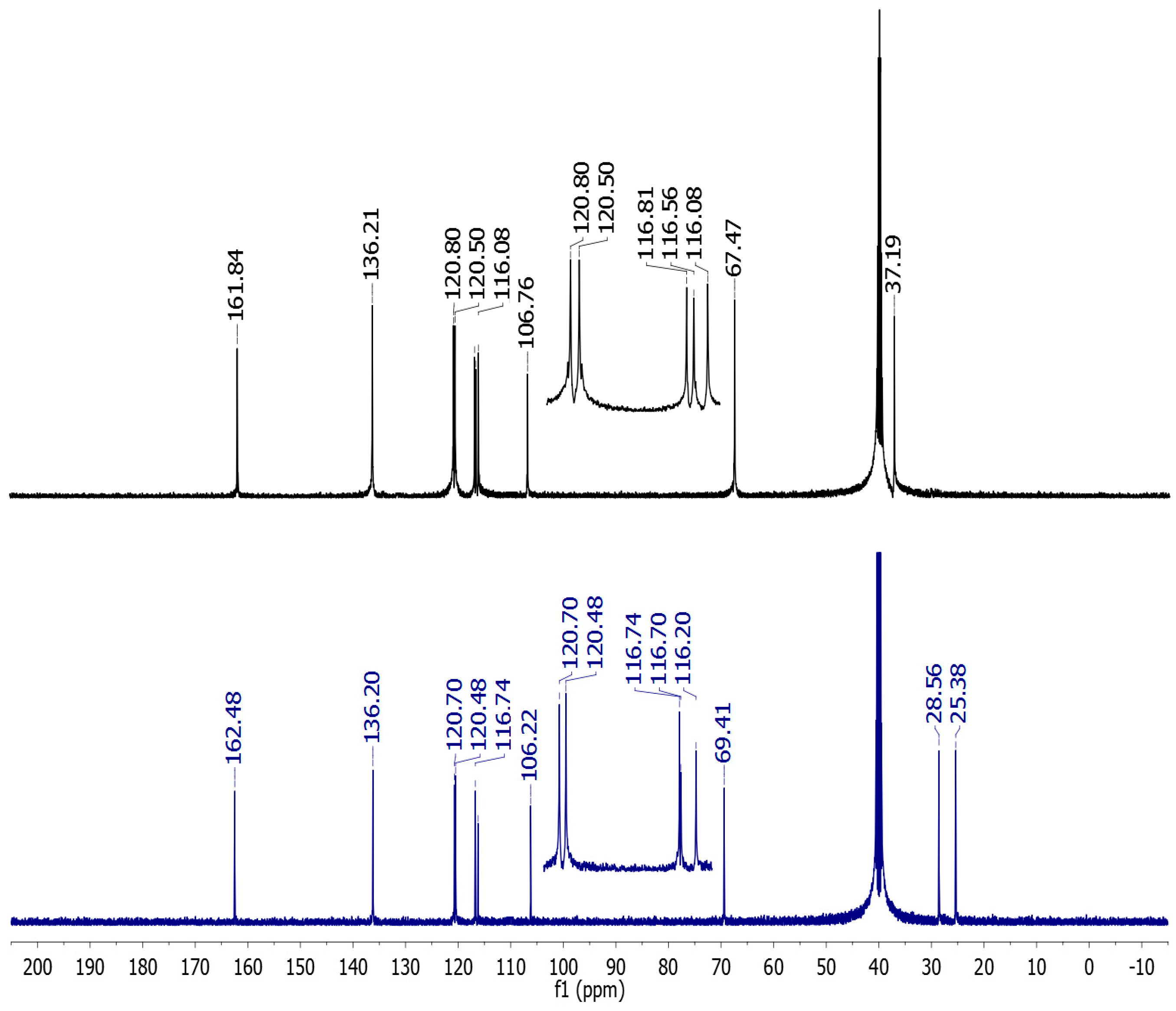
2.2. Vibrational and NMR Spectroscopy
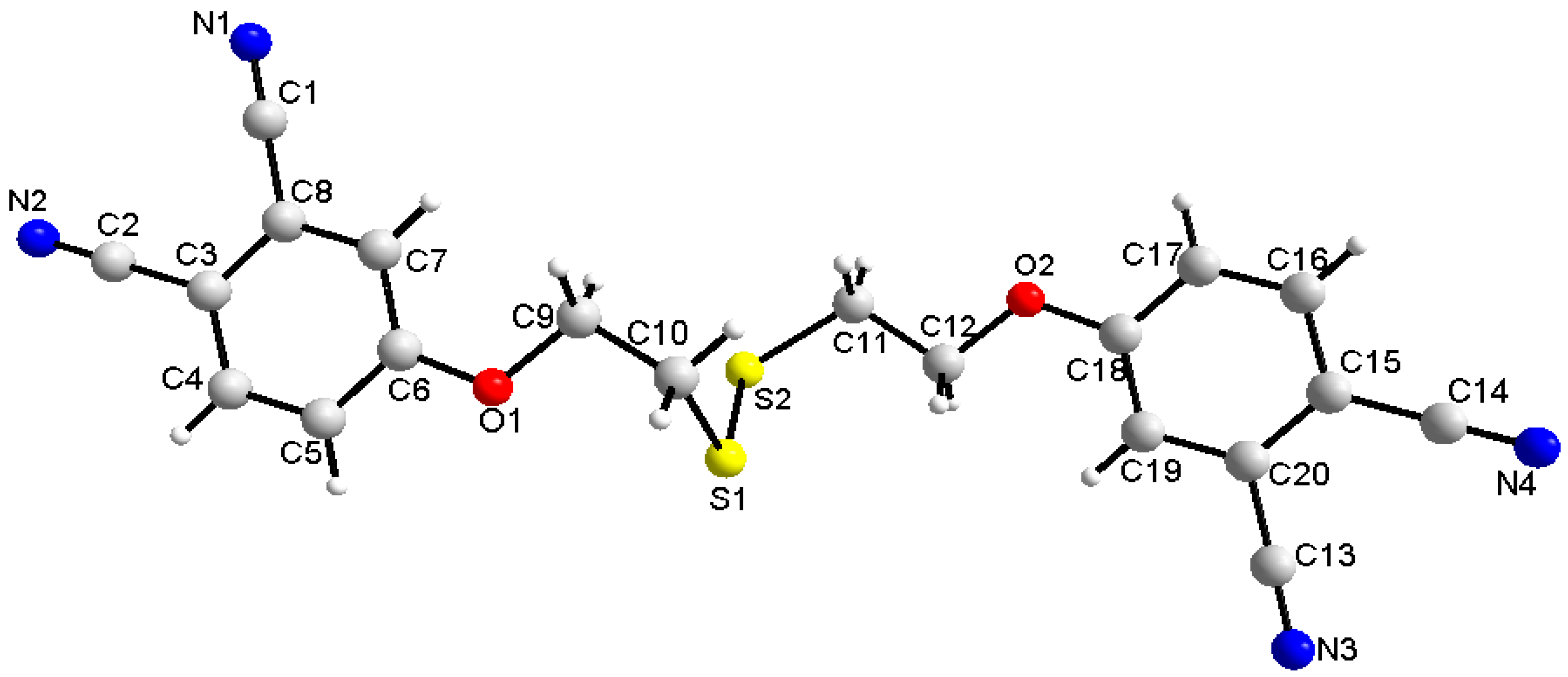
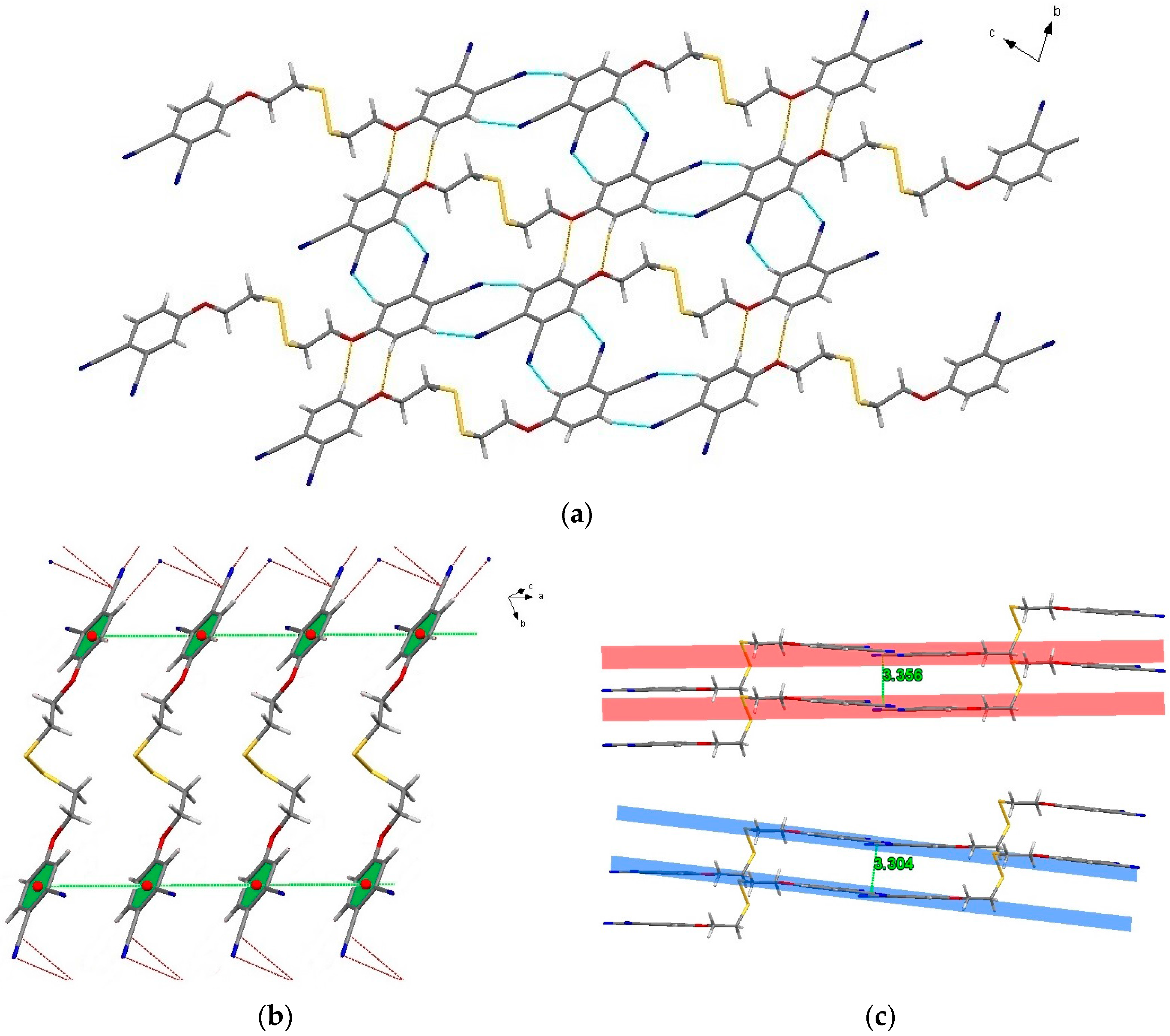
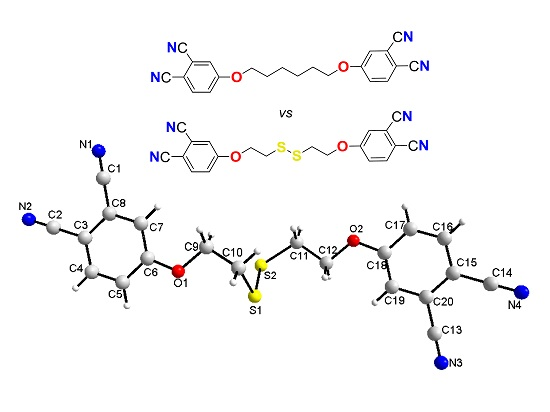
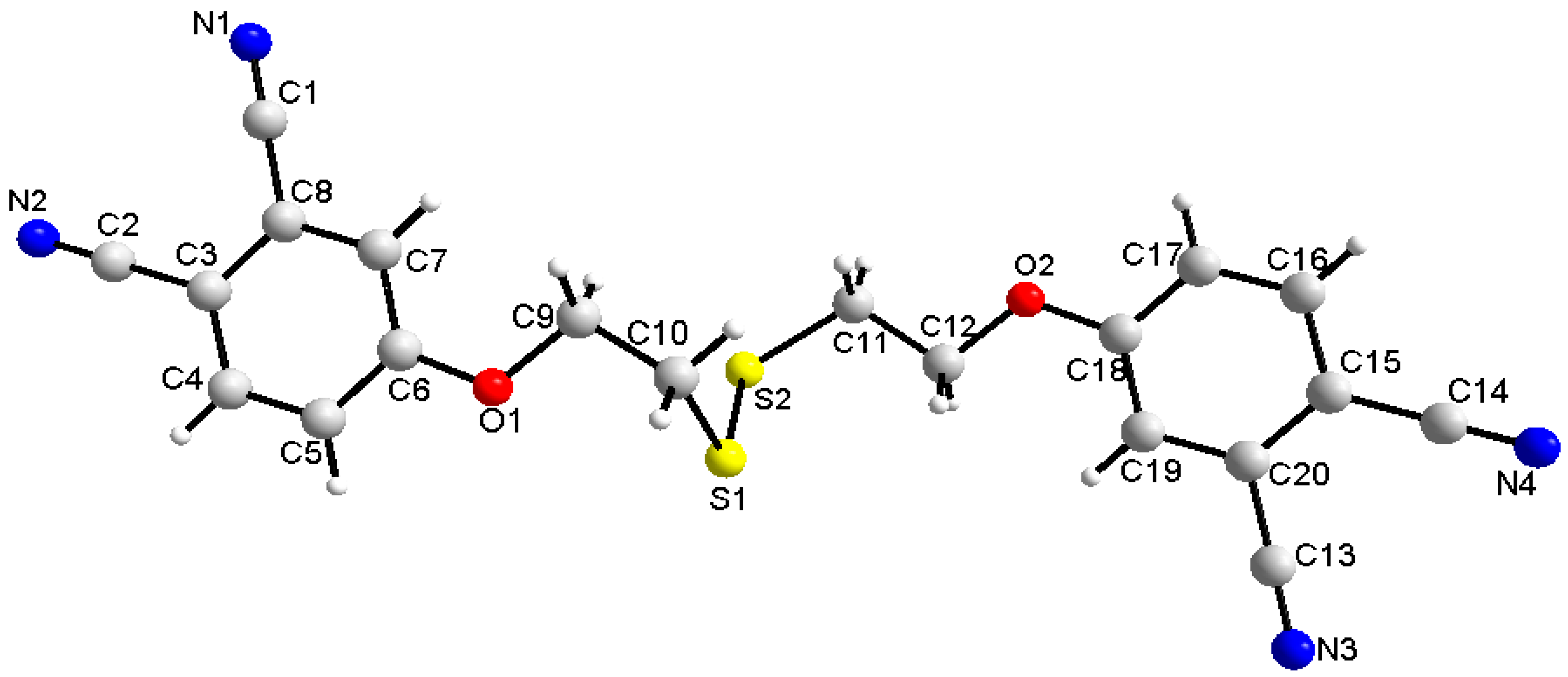
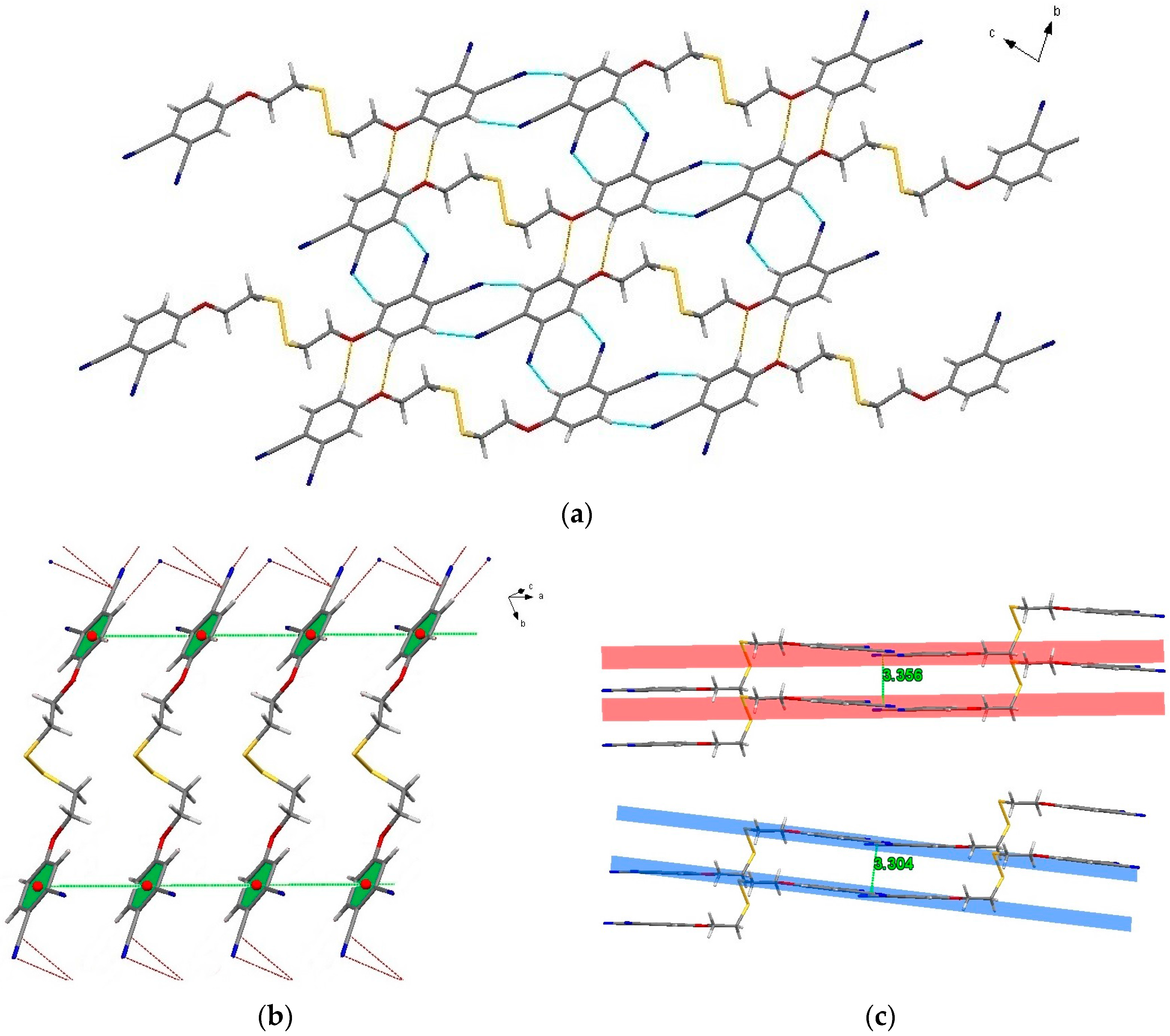
2.3. Single-Crystal Structural Analysis
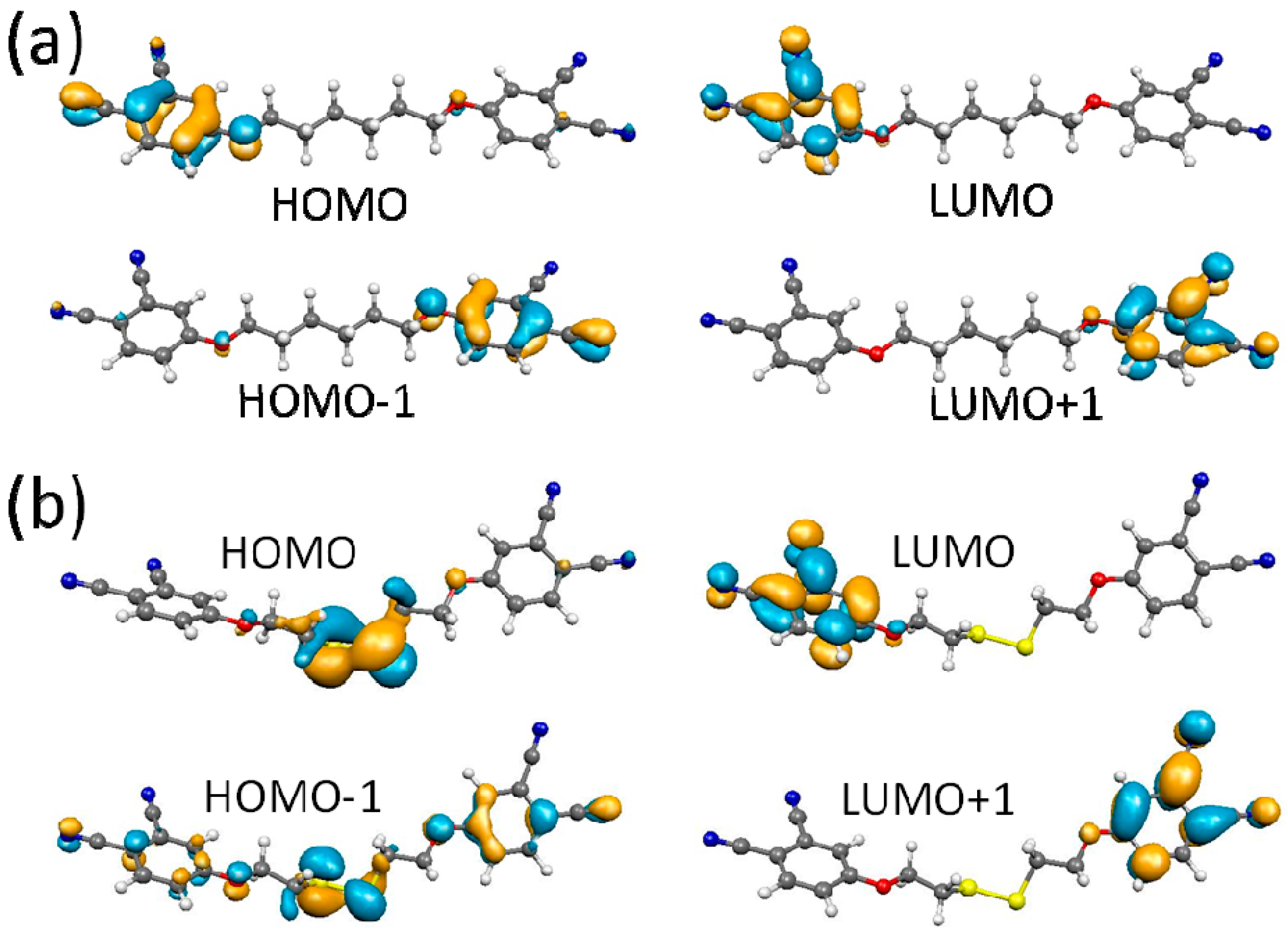
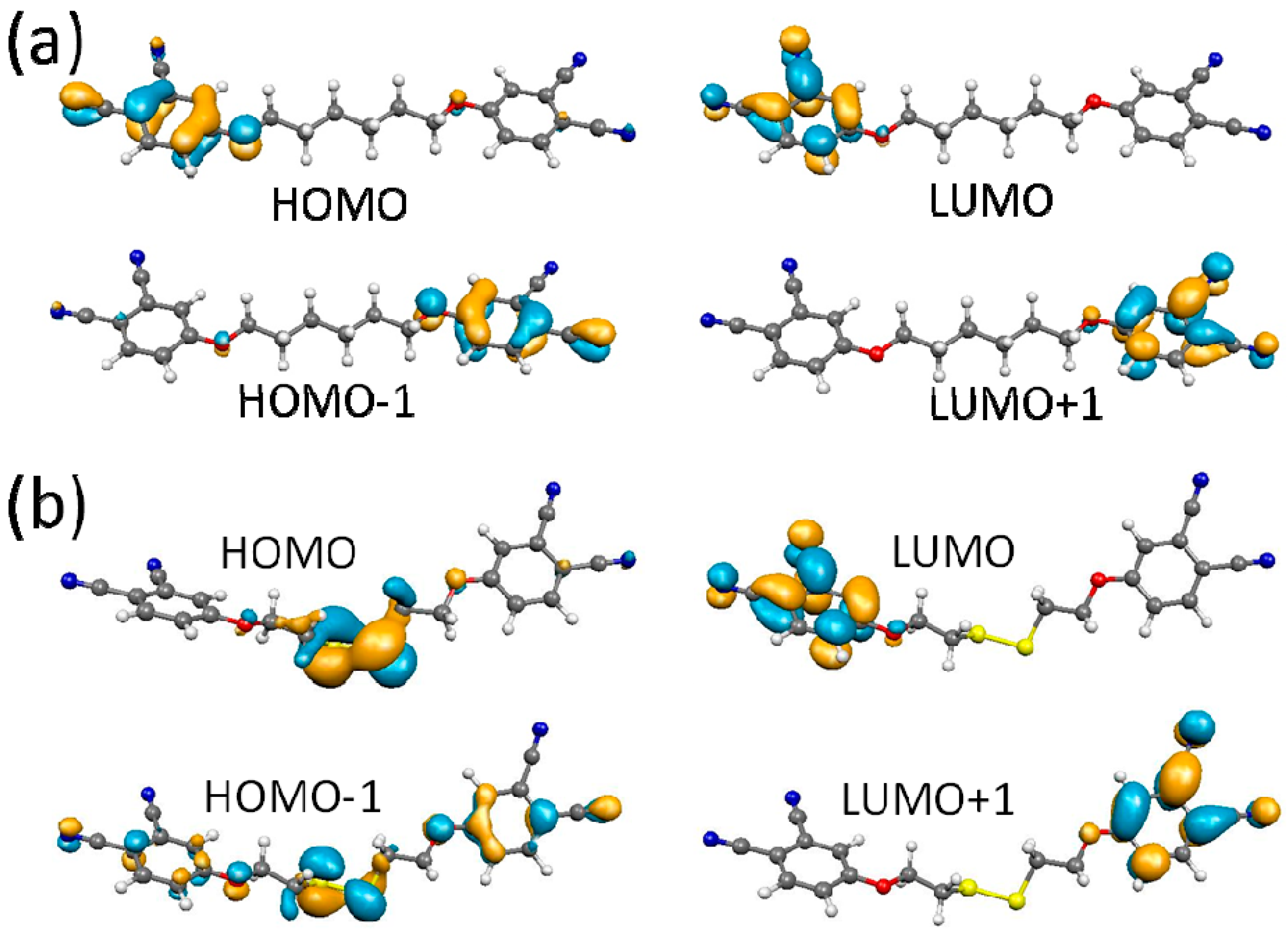
2.4. DFT Calculations
3. Materials and Methods
3.1. Synthesis
3.2. X-Ray Crystallography
3.3. Computational
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Triesscheijn, M.; Baas, P.; Schellens, J.H.M.; Stewart, F.A. Photodynamic therapy in oncology. Oncologist 2006, 11, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Q.; Song, H.T.; Chen, S.P.; Zhang, J.; Niu, G.X.; Liu, X.N. Clinical efficacy of 5-aminolevulinic acid photodynamic therapy in the treatment of moderate to severe facial acne vulgaris. Exp. Ther. Med. 2015, 10, 1194–1198. [Google Scholar] [CrossRef] [PubMed]
- Ideta, R.; Tasaka, F.; Jang, W.D.; Nishiyama, N.; Zhang, G.D.; Harada, A.; Yanagi, Y.; Tamaki, Y.; Aida, T.; Kataoka, K. Nanotechnology-Based Photodynamic Therapy for Neovascular Disease Using a Supramolecular Nanocarrier Loaded with a Dendritic Photosensitizer. Nano Lett. 2005, 5, 2426–2431. [Google Scholar] [CrossRef] [PubMed]
- Maisch, T. Strategies to optimize photosensitizers for photodynamic inactivation of bacteria. J. Photochem. Photobiol. B Biol. 2015, 150, 2–10. [Google Scholar]
- Jiang, Z.; Shao, J.W.; Yang, T.T.; Wang, J.; Jia, L. Pharmaceutical development, composition and quantitative analysis of phthalocyanine as the photosensitizer for cancer photodynamic therapy. J. Pharm. Biomed. Anal. 2014, 87, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Maruani, A.; Savoie, H.; Bryden, F.; Caddick, S.; Boyle, R.; Chudasama, V. Site-selective multi-porphyrin attachment enables the formation of a next-generation antibody-based photodynamic therapeutic. Chem. Commun. 2015, 51, 15304–15307. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Xu, P.; Chen, J.; Chen, H.; Hu, P.; Chen, X.; Lin, L.; Huang, Y.; Zheng, K.; Zhou, S.; et al. Zinc phthalocyanine conjugated with the amino-terminal fragment of urokinase for tumor-targeting photodynamic therapy. Acta Biomater. 2014, 10, 4257–4268. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.D.; Thomas, D.G.T. Biology of Brain Tumour; Martinus Nijhof: Leiden, The Netherlands; Boston, MA, USA, 1986. [Google Scholar]
- Jiang, X.J.; Lo, P.C.; Tsang, Y.M.; Yeung, S.L.; Fong, W.P.; Ng, D.K.P. Phthalocyanine-Polyamine Conjugates as pH-Controlled Photosensitizers for Photodynamic Therapy. Chem. Eur. J. 2010, 16, 4777–4783. [Google Scholar] [CrossRef] [PubMed]
- Isık, M.; Guliyev, R.; Kolemen, S.; Altay, Y.; Senturk, B.; Tekinay, T.; Akkaya, E.U. Designing an Intracellular Fluorescent Probe for Glutathione: Two Modulation Sites for Selective Signal Transduction. Org. Lett. 2014, 16, 3260–3263. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, D.; Lange, N.; Chobaz-Peclat, V.; Zuluaga, M.F.; Gurny, R.; van den Bergh, H.; Busso, N. Thrombin-sensitive dual fluorescence imaging and therapeutic agent for detection and treatment of synovial inflammation in murine rheumatoid arthritis. J. Control. Release 2012, 163, 178–186. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Lo, P.-C.; Ng, D.K.P.A. Glutathione-Activated Phthalocyanine-Based Photosensitizer for Photodynamic Therapy. Chem. Eur. J. 2014, 20, 6241–6245. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.T.F.; Jiang, X.-J.; Ng, D.K.P.; Lo, P.-C. A disulfide-linked conjugate of ferrocenyl chalcone and silicon (IV) phthalocyanine as an activatable photosensitiser. Chem. Commun. 2013, 49, 4274–4276. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.T.F.; Lo, P.-C.; Jiang, X.-J.; Wang, Q.; Ng, D.K.P. A Dual Activatable Photosensitizer toward Targeted Photodynamic Therapy. J. Med. Chem. 2014, 57, 4088–4097. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.L.; Cho, H.; Li, L.; Kang, H.C.; Huh, K.M. Biarmed Poly (ethylene glycol)-(pheophorbide a)2 Conjugate as a Bioactivatable Delivery Carrier for Photodynamic Therapy. Biomacromolecules 2014, 15, 2224–2234. [Google Scholar] [CrossRef] [PubMed]
- Tuncel, S.; Trivella, A.; Atilla, D.; Bennis, K.; Savoie, H.; Albrieux, F.; Delort, L.; Billard, H.; Dubois, V.; Ahsen, V.; et al. Assessing the Dual Activity of a Chalcone—Phthalocyanine Conjugate: Design, Synthesis, and Antivascular and Photodynamic Properties. Mol. Pharm. 2013, 10, 3706–3716. [Google Scholar] [CrossRef] [PubMed]
- Işci, Ü.; Beyreis, M.; Tortik, N.; Topal, S.Z.; MGlueck, M.; Ahsen, V.; Dumoulin, F.; Kiesslich, T.; Plaetzer, K. Methylsulfonyl Zn phthalocyanine: A polyvalent and powerfulhydrophobic photosensitizer with a wide spectrum of photodynamicapplications. Photodiagn. Photodyn. Ther. 2016, 13, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Lafont, D.; Zorlu, Y.; Savoie, H.; Albrieux, F.; Ahsen, V.; Boyle, R.W.; Dumoulin, F. Monoglycoconjugated Phthalocyanines. Effect of Sugar and Linkage on Photodynamic Activity. Photodiagn. Photodyn. Ther. 2013, 10, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Yu Tolbin, A.; Tomilova, L.G.; Zefirov, N.S. Bi- and polynuclear phthalocyanines: Synthesis and study of physicochemical properties. Russ. Chem. Rev. 2008, 77, 435–449. [Google Scholar] [CrossRef]
- Makhseed, S.; Machacek, M.; Alfadly, W.; Tuhl, A.; Vinodh, M.; Simunek, T.; Novakova, V.; Kubat, P.; Rudolf, E.; Zimcik, P. Water-soluble non-aggregating zinc phthalocyanine and in vitro studies for photodynamic therapy. Chem. Commun. 2013, 49, 11149–11151. [Google Scholar] [CrossRef] [PubMed]
- Köç, M.; Zorlu, Y.; İşci, Ü.; Berber, S.; Ahsen, V.; Dumoulin, F. A library of dimeric and trimeric phthalonitriles linked by a single aromatic ring: Comparative structural and DFT investigation. CrystEngComm 2016, 18, 1416–1426. [Google Scholar] [CrossRef]
- Sinnokrot, M.O.; Valeev, E.F.; Sherrill, C.D. Estimates of the Ab Initio Limit for π−π Interactions: The Benzene Dimer. J. Am. Chem. Soc. 2002, 124, 10887–10893. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R.; Gavezzotti, A. From molecular to crystal structure; polynuclear aromatic hydrocarbons. J. Chem. Soc. Chem. Comm. 1989, 621–623. [Google Scholar] [CrossRef]
- Castineiras, A.; Sicilia-Zafra, A.G.; Gonzalez-Perez, J.M.; Choquesillo-Lazarte, D.; Niclos-Gutierrez, J. Intramolecular “Aryl-Metal Chelate Ring” π,π-Interactions as Structural Evidence for Metalloaromaticity in (Aromatic α,α‘-Diimine)-Copper(II) Chelates: Molecular and Crystal Structure of Aqua(1,10-phenanthroline) (2-benzylmalonato)copper(II) Three-hydrate. J. Inorg. Chem. 2002, 41, 6956–6958. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Uchimaru, T.; Sugawara, K.; Mikami, M. Energy profile of the interconversion path between T-shape and slipped-parallel benzene dimers. J. Chem. Phys. 2002, 117, 11216–11221. [Google Scholar] [CrossRef]
- Ekineker, G.; Pilet, G.; Berber, S.; Ahsen, V.; Dumoulin, F.; Önal, E. Spectroscopic and structural properties of bisphthalonitriles with O/S/SO2 grafting: Comparative theoretical and experimental studies. J. Mol. Struct. 2016, 1123, 261–270. [Google Scholar] [CrossRef]
- Zorlu, Y.; Taşkıran, D.T.; Tarakçı, D.K.; Kumru, U.; Alpugan, S.; İşçi, Ü.; Dumoulin, F.; Ahsen, V. Dihydroxylated Alkylsulfanyl Phthalonitriles. J. Chem. Crystallogr. 2014, 44, 337–345. [Google Scholar] [CrossRef]
- Zeng, W.-L.; Xin, C.-H. Synthesis and Crystal Structure of 4-(2-Isopropyl-5-methylcyclohexyloxy) phthalonitrile. Asian J. Chem. 2013, 25, 2067–2069. [Google Scholar]
- Janczak, J.; Kubiak, R. 1,2-Dicyanobenzene. A Precursor of Phthalocyanines. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1995, 51, 1399–1401. [Google Scholar] [CrossRef]
- Zorlu, Y.; İşci, Ü.; Hirel, C.; Dumoulin, F.; Ahsen, V. Phthalonitriles Functionalized for Click Chemistry. Design, Synthesis and Structural Characterization. J. Chem. Crystallogr. 2013, 43, 636–645. [Google Scholar] [CrossRef]
- Chi, Y.J.; Tan, A.L.; Wimmer, F.L.; Mirza, A.H.; Young, D.J.; Ng, S.W.; Tiekink, E.R.T. 4-(Prop-2-yn-1-yloxy) benzene-1,2-dicarbonitrile. Acta Cryst. Sec. E 2012, 68, o2293–o2294. [Google Scholar]
- Young, J.G.; Onyebuagu, W. Synthesis and characterization of di-disubstituted phthalocyanines. J. Org. Chem. 1990, 55, 2155–2159. [Google Scholar] [CrossRef]
- Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals, 2nd ed.; Pergamon Press: Oxford, UK, 1989. [Google Scholar]
- Bruker. APEX2; Bruker AXS Inc.: Madison, WI, USA, 2008. [Google Scholar]
- Bruker. Multi-Scan SADABS (V2014/5); Bruker AXS Inc.: Madison, WI, USA, 2014. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745–2779. [Google Scholar] [CrossRef]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef]
- Kleinman, L.; Bylander, D.M. Efficacious Form for Model Pseudopotentials. Phys. Rev. Lett. 1982, 48, 1425–1428. [Google Scholar] [CrossRef]
- Valiev, M.; Bylaska, E.J.; Govind, N.; Kowalski, K.; Straatsma, T.P.; van Dam, H.J.J.; Wang, D.; Nieplocha, J.; Apra, E.; Windus, T.L.; et al. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 2010, 181, 1477–1489. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical Formula | C20H14N4O2S2 |
|---|---|
| Formula weight | 406.4780 |
| Temperature (K) | 299 |
| Crystal system | Triclinic |
| Space group | P 1 |
| a (Å) | 5.3047(3) |
| b (Å) | 9.4281(5) |
| c (Å) | 10.3733(6) |
| β(°) | 79.443(4) |
| Volume (Å3) | 488.84(5) |
| Z | 1 |
| λ (Mo Kα)(Å) | 0.71073 |
| Crystal shape | Block |
| Crystal colour | Colourless |
| Crystal size (mm3) | 0.359 × 0.159 × 0.116 |
| ρ (calc, g/cm3) | 1.381 |
| Absorption coeff. (mm−1) | 0.296 |
| F(000) | 210.0 |
| Reflections collected | 7730 |
| Independent reflections | 3697 |
| Rint (merging R value) | 0.0213 |
| Absorption correction | multi-scan |
| Data/Restrains/Parameters | 3697/3/253 |
| R/Rw (F > 3σF) | 0.0353/0.0874 |
| Goodness-of-fit on F | 1.031 |
| Δρmin/Δρmax (e-.Å−3) | −0.143/0.265 |
| CCDC number | 1061336 |
| D–H···A | Symmetry | d(D–H) | d(H···A) | d(D–H···A) | D–H···A |
|---|---|---|---|---|---|
| C5–H5....O2 | −1 + x, 1 + y, z | 0.9300 | 2.6200 | 3.547(4) | 171.800 |
| C7–H7....N3 | 1 + x, 1 + y, −1 + z | 0.9300 | 2.4900 | 3.390(5) | 163.200 |
| C9–H9B....S2 | x, y, z | 0.9700 | 2.9100 | 3.401(4) | 112.700 |
| C17–H17....O1 | 1 + x, −1 + y, z | 0.9300 | 2.6300 | 3.553(4) | 172.300 |
| C19–H19....N1 | −1 + x, −1 + y, 1 + z | 0.9300 | 2.5000 | 3.398(5) | 161.500 |
| C13–N3 | 1.137(5) |
| C14–N4 | 1.138(5) |
| C6–O1 | 1.352(4) |
| O1–C9 | 1.438(4) |
| C10–S1 | 1.812(4) |
| S1–S2 | 2.029(3) |
| S2–C11 | 1.817(3) |
| C12–O2 | 1.433(4) |
| C18–O2 | 1.357(4) |
| C5–C6–C7 | 119.6(3) |
| C5–C6–O1 | 116.3(3) |
| C6–O1–C9 | 118.2(3) |
| C10–S1–S2 | 103.9(4) |
| S1–S2–C11 | 103.4(2) |
| C12–O2–C18 | 119.0(2) |
| C20–C15–C16 | 118.9(3) |
| C20–C15–C14 | 121.4(3) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alpugan, S.; Ekineker, G.; Ahsen, V.; Berber, S.; Önal, E.; Dumoulin, F. Bisphthalonitrile with a Disulfide-Based Linker and its Dimethylene Analogue: Comparative Structural Insights. Crystals 2016, 6, 89. https://doi.org/10.3390/cryst6080089
Alpugan S, Ekineker G, Ahsen V, Berber S, Önal E, Dumoulin F. Bisphthalonitrile with a Disulfide-Based Linker and its Dimethylene Analogue: Comparative Structural Insights. Crystals. 2016; 6(8):89. https://doi.org/10.3390/cryst6080089
Chicago/Turabian StyleAlpugan, Serkan, Gülçin Ekineker, Vefa Ahsen, Savaş Berber, Emel Önal, and Fabienne Dumoulin. 2016. "Bisphthalonitrile with a Disulfide-Based Linker and its Dimethylene Analogue: Comparative Structural Insights" Crystals 6, no. 8: 89. https://doi.org/10.3390/cryst6080089