Zintl Salts Ba2P7X (X = Cl, Br, and I): Synthesis, Crystal, and Electronic Structures

Abstract
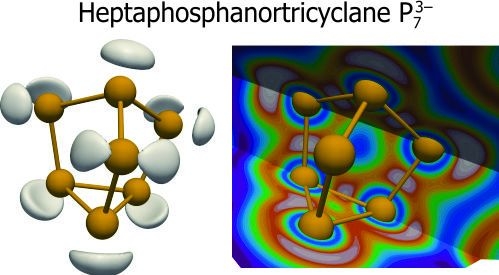
:
1. Introduction
2. Results and Discussion
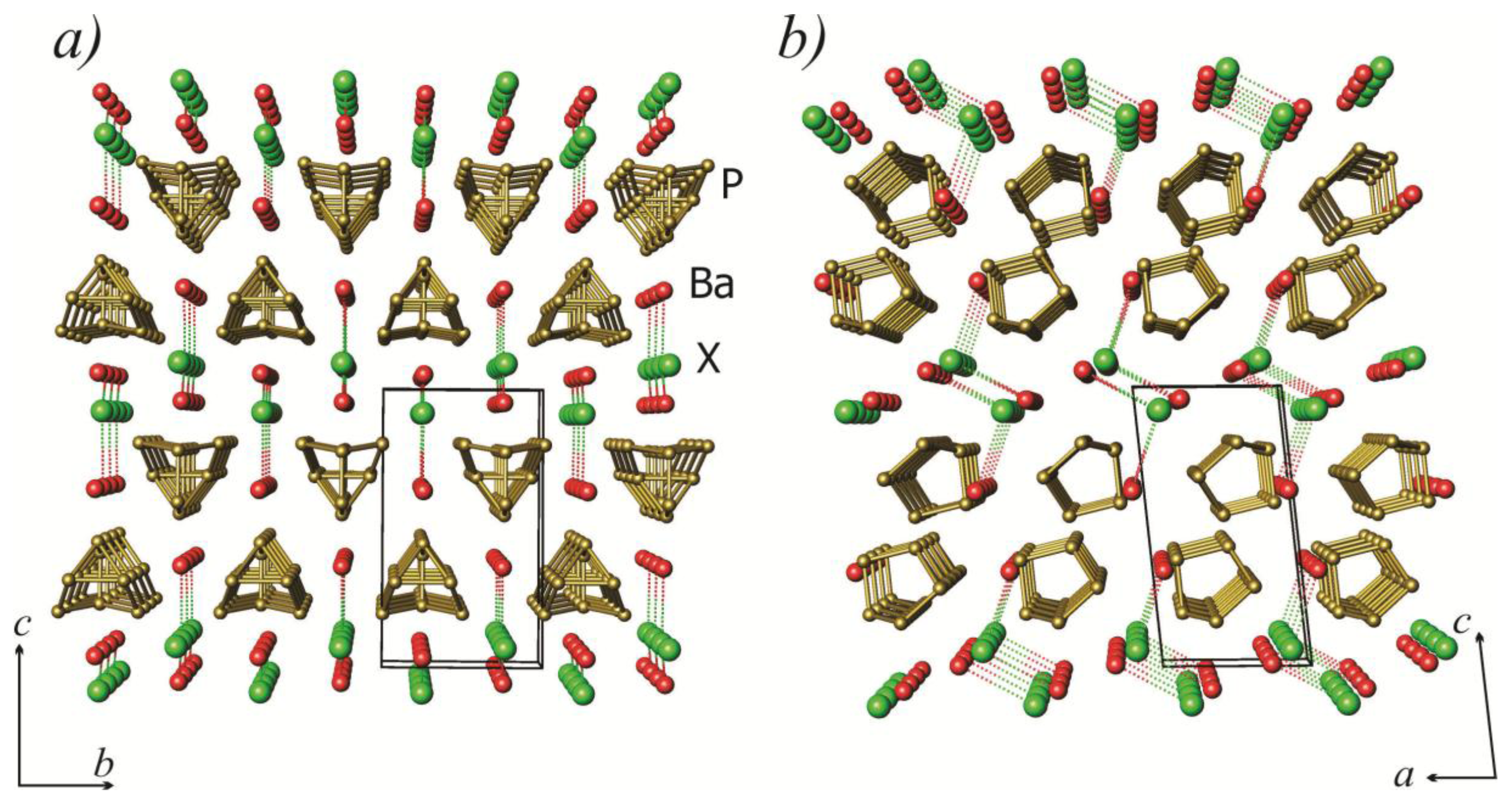
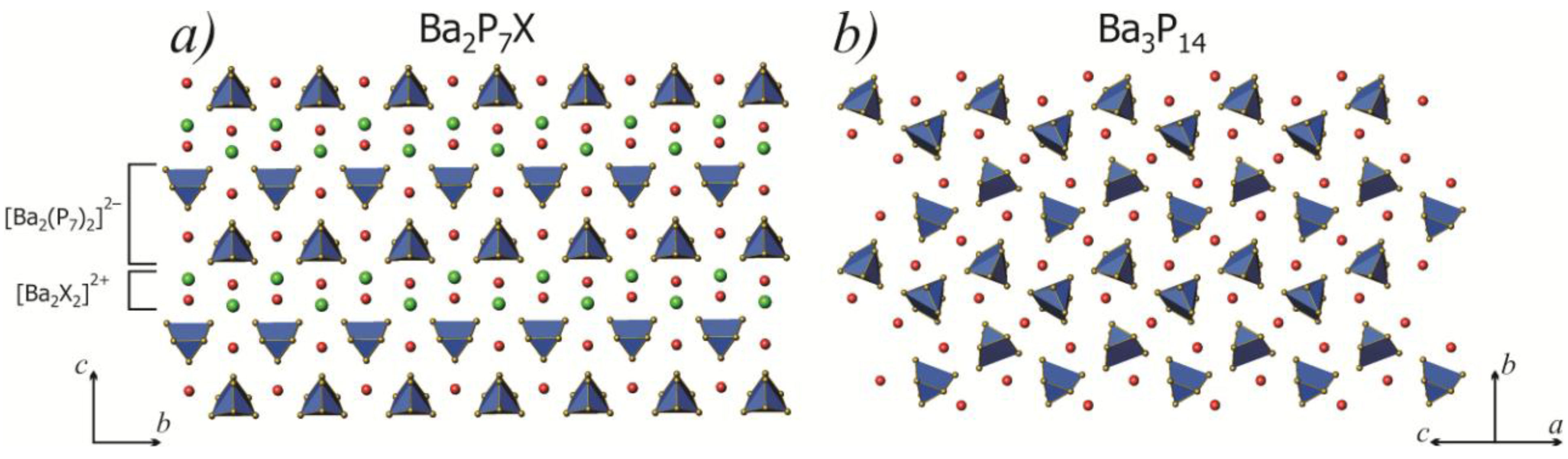
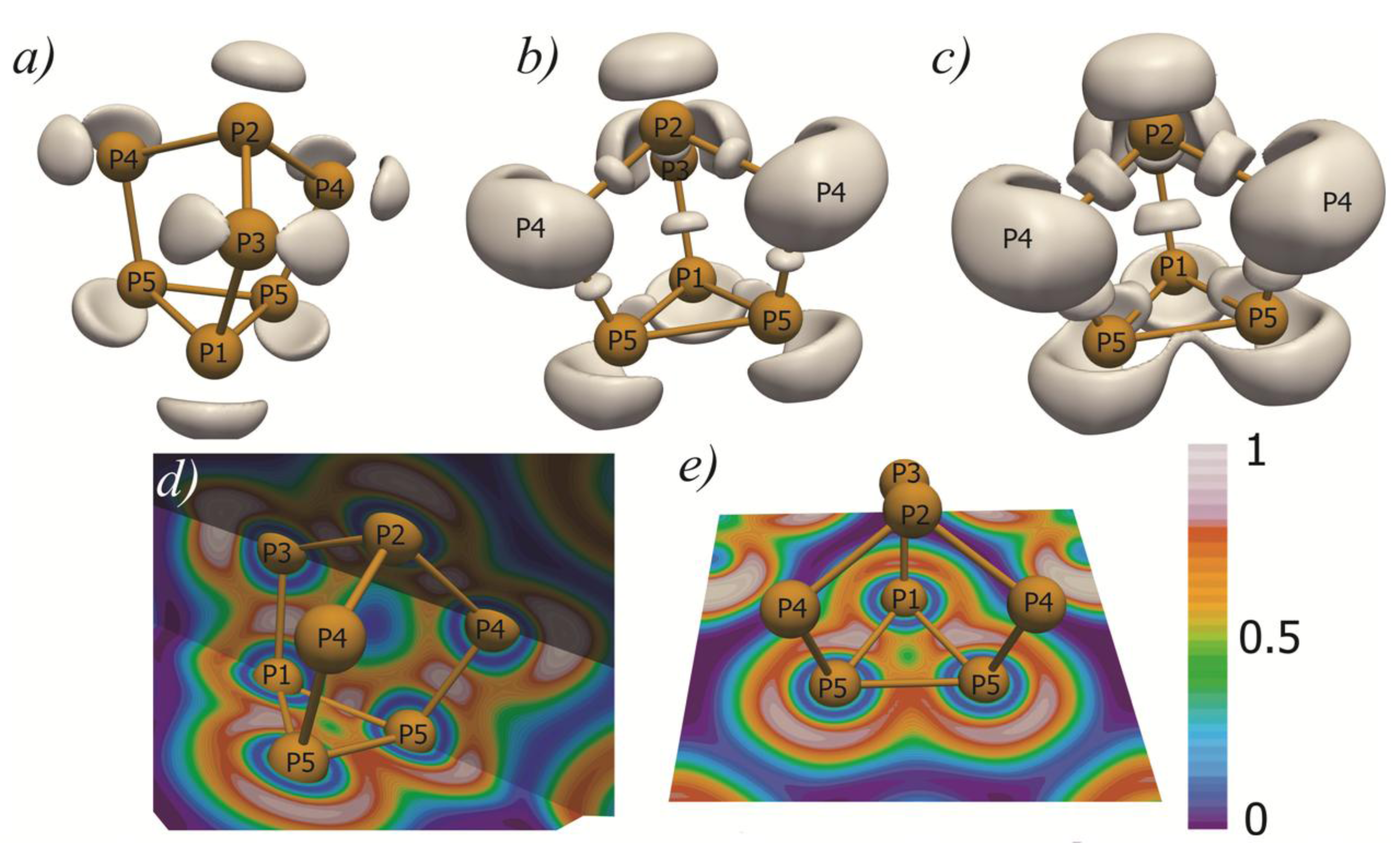
2.1. Crystal Structure

| Composition | Ba2P7Cl | Ba2P7Br | Ba2P7I |
|---|---|---|---|
| CSD number | 426193 | 426194 | 426195 |
| Space Group | P21/m (No. 11) | ||
| Temperature [K] | 90(2) | ||
| λ [Å] | Mo K α, 0.71073 | ||
| a [Å] | 6.329(1) | 6.294(1) | 6.3538(3) |
| b [Å] | 6.805(1) | 6.835(1) | 6.8990(4) |
| c [Å] | 11.709(2) | 11.850(2) | 12.0392(6) |
| β [deg] | 95.267(2) | 95.819(2) | 95.915(1) |
| V [Å3] | 502.2(1) | 507.2(2) | 524.93(5) |
| Z | 2 | ||
| ρ [g cm−3] | 3.485 | 3.742 | 3.912 |
| μ [mm−1] | 9.099 | 12.686 | 11.392 |
| θ [deg.] | 3.23 < θ < 30.66 | 3.25 < θ < 30.57 | 3.22 < θ < 30.58 |
| data/parameters | 1671/55 | 1671/55 | 1733/55 |
| R1 (I > 2 σ(I)) | 0.028 | 0.012 | 0.019 |
| wR2 (I > 2 σ(I)) | 0.047 | 0.025 | 0.037 |
| Goodness-of-fit | 1.087 | 1.202 | 1.117 |
| Δ ρmax and Δρmin, e/Å3 | 1.36 and −1.14 | 0.50 and −0.73 | 1.22 and −0.92 |
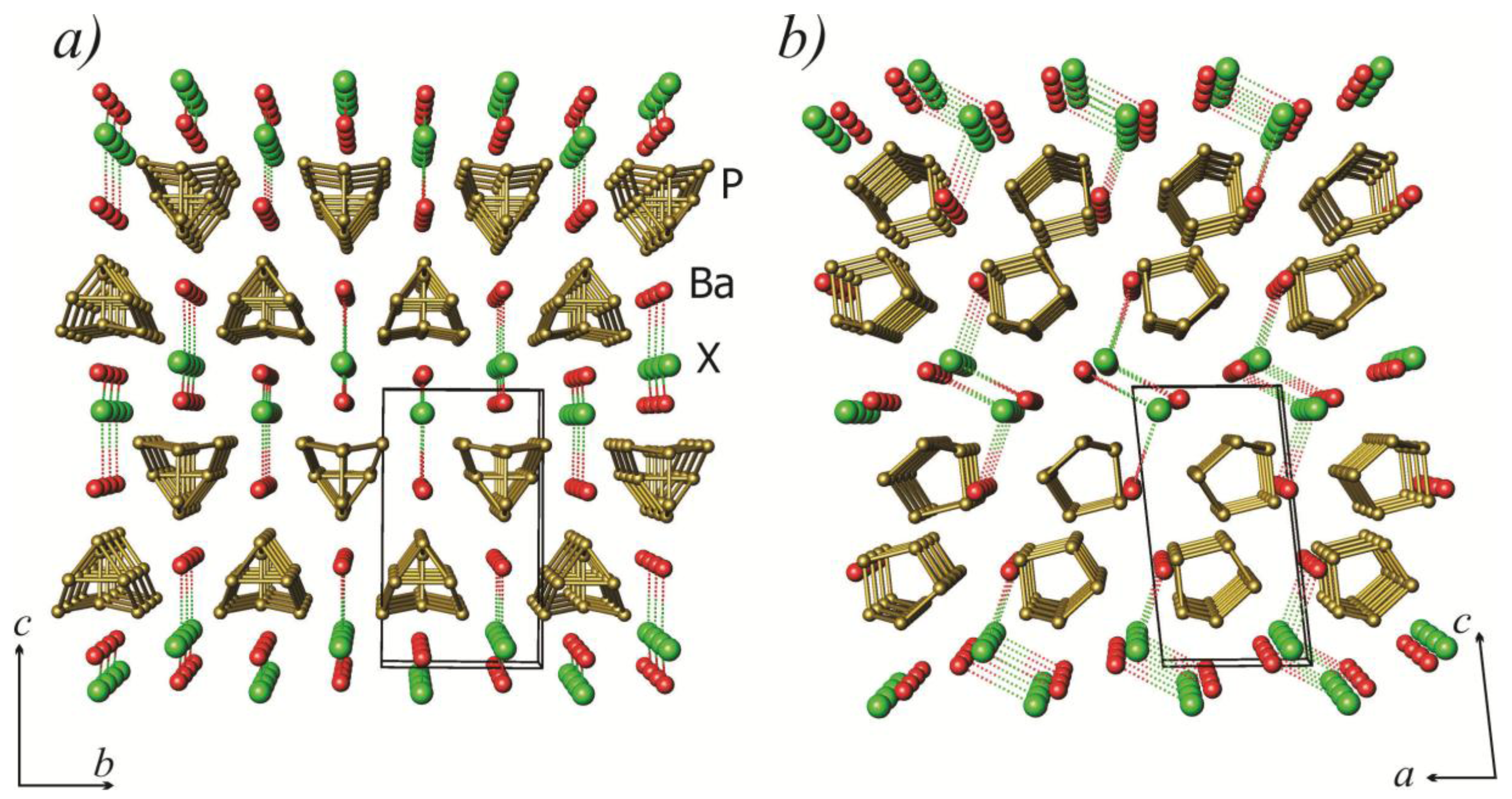


| Atoms | Distances (Å) | Angle Type | Angle (θ) | ||||
|---|---|---|---|---|---|---|---|
| Ba2P7Cl | Ba2P7Br | Ba2P7I | Ba2P7Cl | Ba2P7Br | Ba2P7I | ||
| P1–P3 | 2.156(2) | 2.1558(9) | 2.156(1) | ∠P1–P3–P2 | 97.51(8) | 97.65(3) | 97.77(6) |
| P1–P5 | 2.229(2) | 2.2307(7) | 2.233(1) | ∠P2–P4–P5 | 99.53(7) | 99.53(3) | 99.47(4) |
| P2–P3 | 2.188(2) | 2.1853(9) | 2.188(1) | ∠P1–P5–P4 | 105.44(7) | 105.23(3) | 104.88(5) |
| P2–P4 | 2.170(1) | 2.1706(6) | 2.180(1) | ∠P1–P5–P5 | 58.26(4) | 58.38(1) | 58.41(2) |
| P4–P5 | 2.143(2) | 2.1434(6) | 2.154(1) | ∠P5–P1–P5 | 63.49(7) | 63.24(3) | 63.19(5) |
| P5–P5 | 2.345(2) | 2.339(1) | 2.340(2) | – | – | – | – |
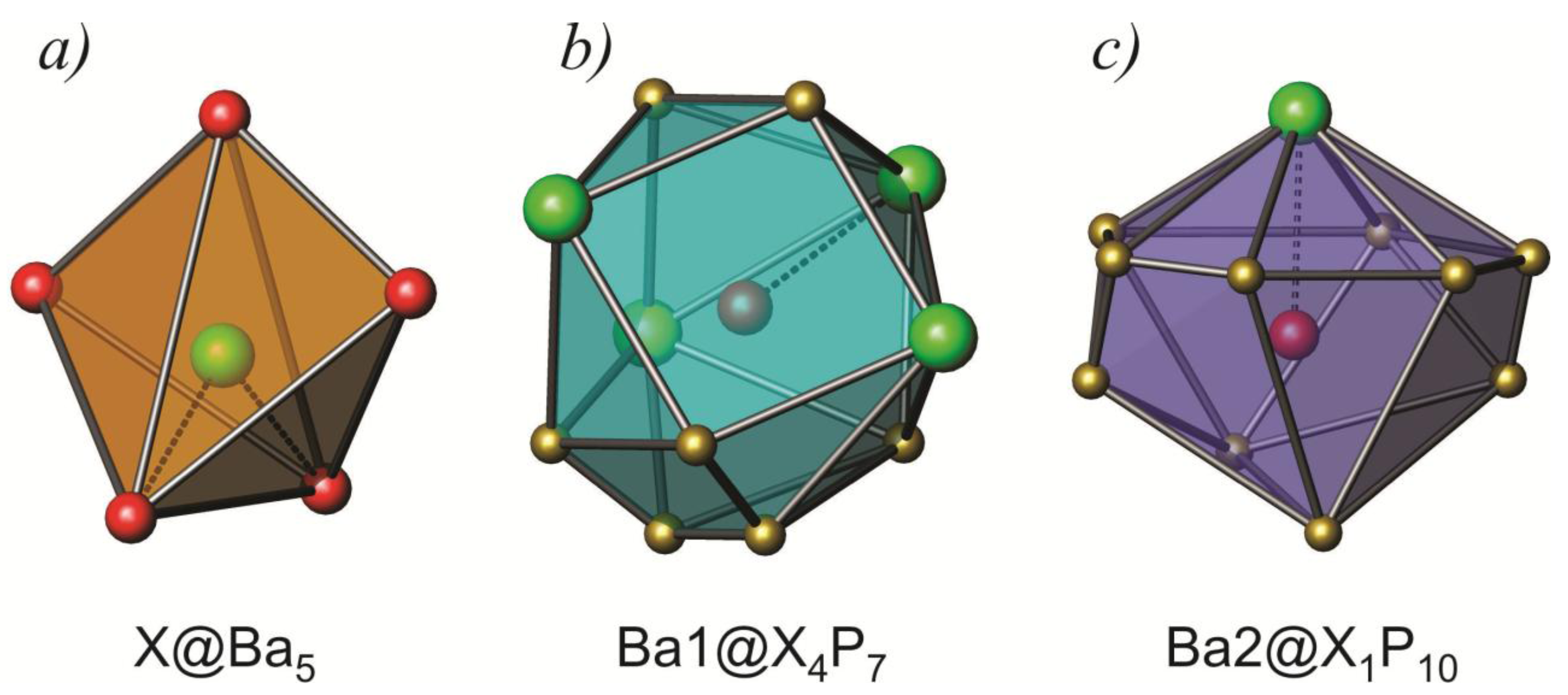
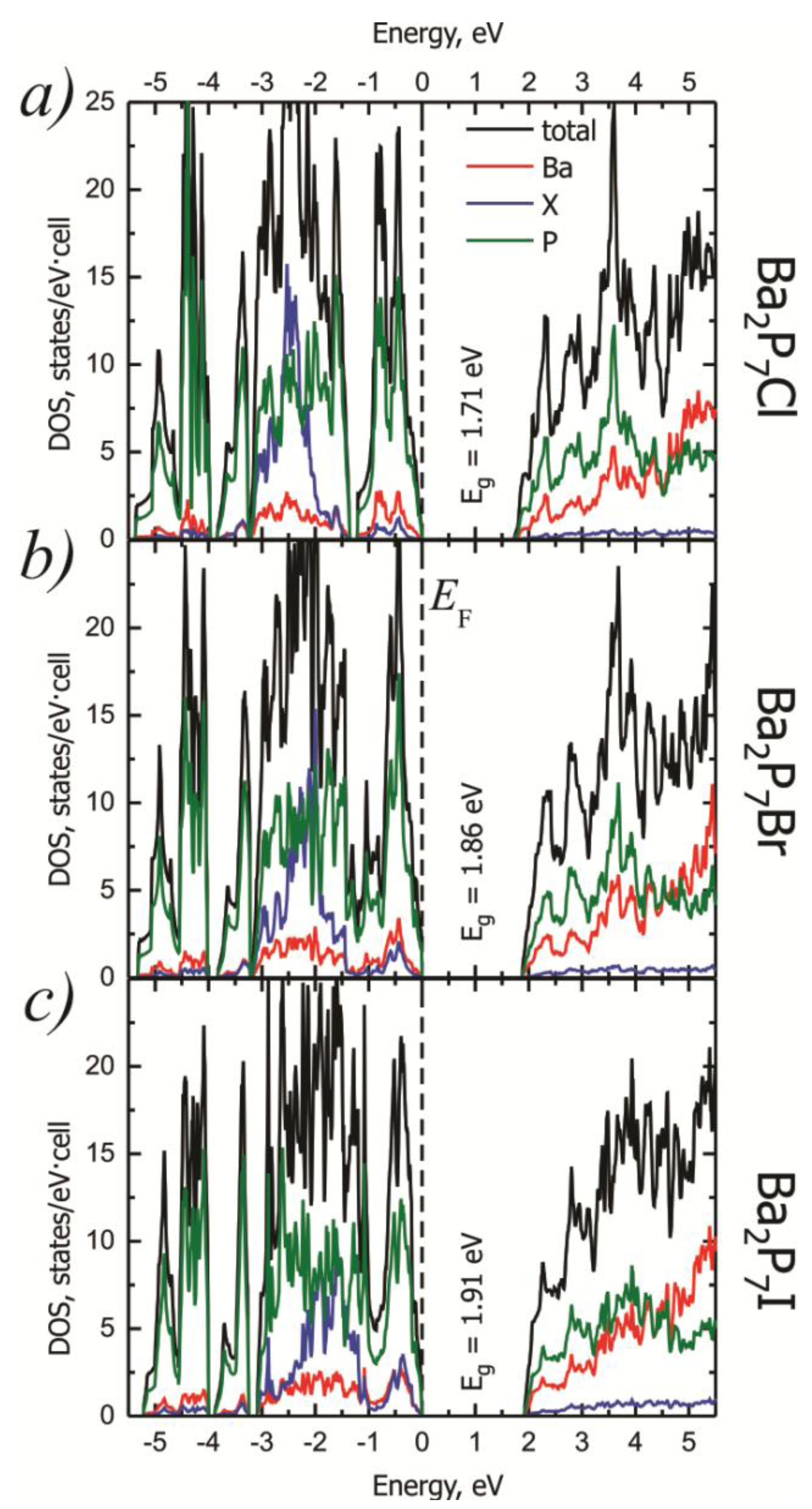
2.2. Electronic Structure
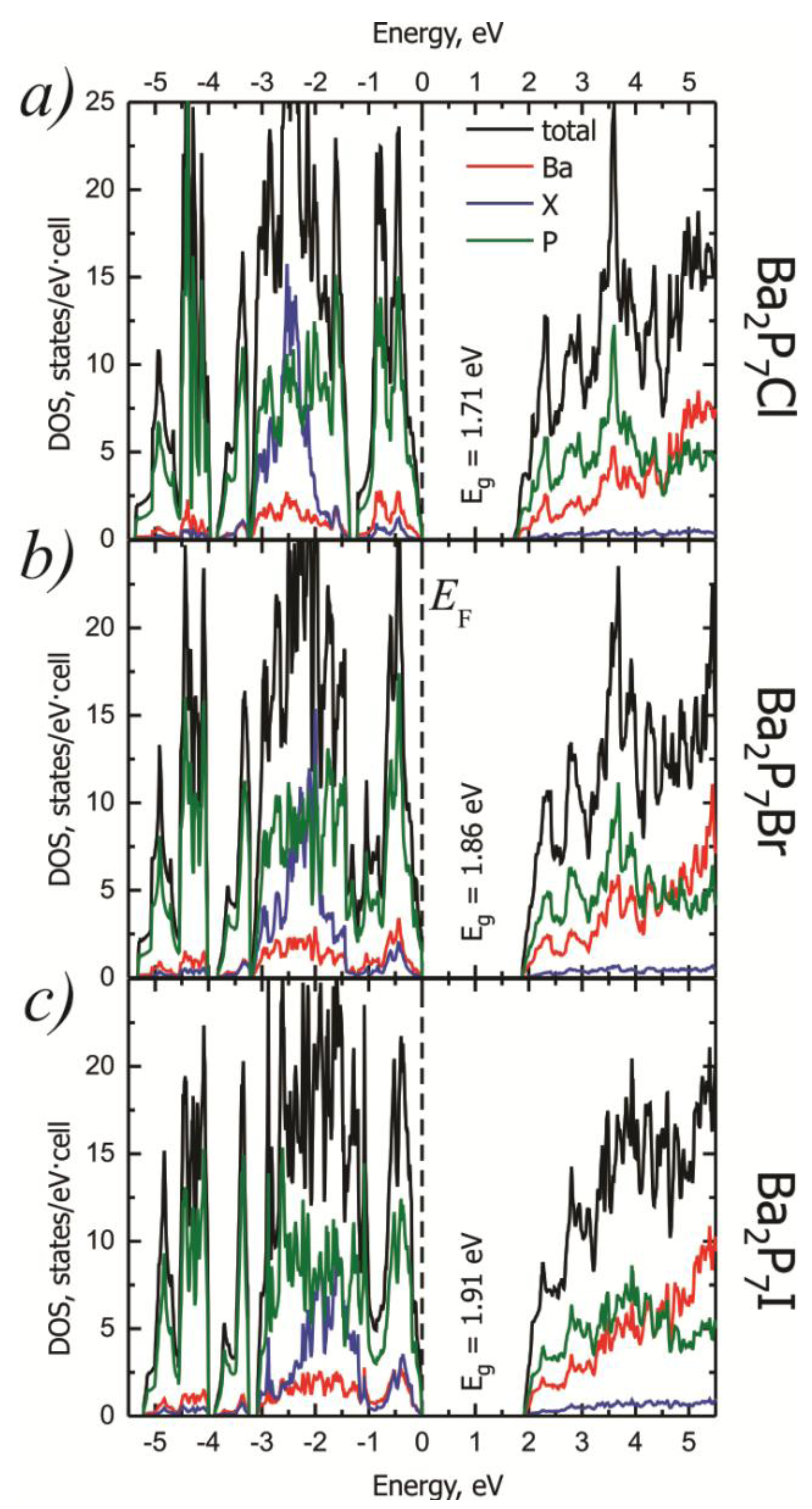

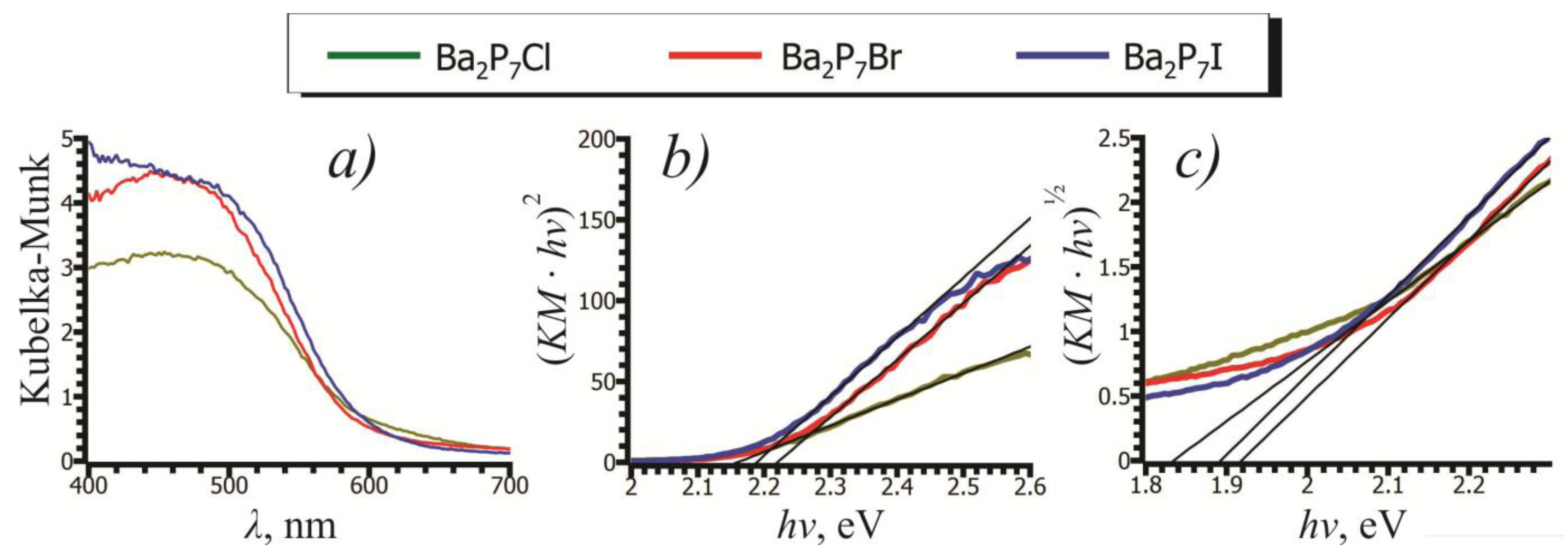
2.3. UV-Vis Spectroscopy
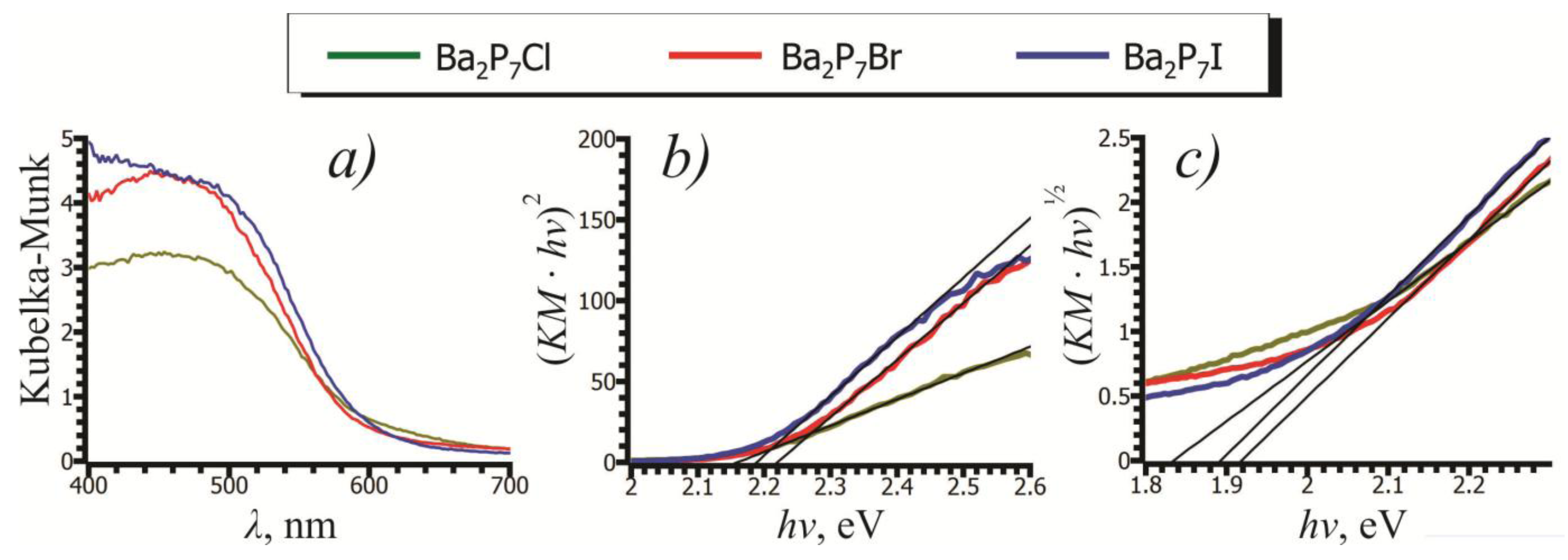
| Composition | Ba2P7Cl | Ba2P7Br | Ba2P7I |
|---|---|---|---|
| Direct Bandgap (eV) | 2.16(3) | 2.20(4) | 2.19(4) |
| Indirect Bandgap (eV) | 1.83(2) | 1.92(2) | 1.89(2) |
| Calculated Bandgap (eV) | 1.71 | 1.86 | 1.91 |
3. Experimental Section
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Pöttgen, R.; Hönle, W.; von Schnering, H.G. Phosphides: Solid State Chemistry. In Encyclopedia of Inorganic Chemistry, 2nd ed.; King, R.B., Ed.; Wiley: Chichester, UK, 2005; volume 8, pp. 4255–4308. [Google Scholar]
- Von Schnering, H.G.; Hoenle, W. Bridging chasms with polyphosphides. Chem. Rev. 1988, 88, 243–273. [Google Scholar] [CrossRef]
- Dahlmann, W.; von Schnering, H.G. Die Polyphosphide SrP3 und Ba3P14. Naturwissenschaften 1973, 60, 429–429. [Google Scholar] [CrossRef]
- Manriquez, V.; Hönle, W.; von Schnering, H.G. Trilithiumheptaphosphid Li3P7: Darstellung, Struktur und Eigenschaften. Z. Anorg. Allg. Chem. 1986, 539, 95–109. [Google Scholar] [CrossRef]
- Scharfe, S.; Kraus, F.; Stegmaier, S.; Schier, A.; Fässler, T.F. Zintl ions, cage compounds, and intermetalloid clusters of group 14 and group 15 elements. Angew. Chem. Int. Ed. 2011, 50, 3630–3670. [Google Scholar] [CrossRef]
- Shatruk, M.M.; Kovnir, K.A.; Shevelkov, A.V.; Popovkin, B.A. Ag3SnP7: A polyphosphide with a unique (P7) chain and a novel Ag3Sn heterocluster. Angew. Chem. Int. Ed. 2000, 39, 2508–2509. [Google Scholar] [CrossRef]
- Lange, S.; Sebastian, C.P.; Zhang, L.; Eckert, H.; Nilges, T. Ag3SnCuP10: [Ag3Sn] tetrahedra embedded between adamantane-type [P10] cages. Inorg. Chem. 2006, 45, 5878–5885. [Google Scholar] [CrossRef]
- Lange, S.; Bawohl, M.; Weihrich, R.; Nilges, T. Mineralization routes to polyphosphides: Cu2P20 and Cu5InP16. Angew. Chem. Int. Ed. 2008, 47, 5654–5657. [Google Scholar] [CrossRef]
- Dewalsky, M.V.; Jeitschko, W.; Wortmann, U. The metallic polyphosphide titanium nickel phosphide (Ti2NiP5). Chem. Mater. 1991, 3, 316–319. [Google Scholar] [CrossRef]
- Eisenmann, B.; Rößler, U. Ein Erdalkalimetallpolyphosphid ungewöhnlicher Zusammensetzung: Die Kristallstruktur von Ba5P9. Z. Anorg. Allg. Chem. 2003, 629, 459–462. [Google Scholar] [CrossRef]
- Chen, X.; Zhu, L.; Yamanaka, S. High-pressure synthesis and structural characterization of three new polyphosphides, α-SrP3, BaP8, and LaP5. J. Solid State Chem. 2003, 173, 449–455. [Google Scholar] [CrossRef]
- Eschen, M.; Jeitschko, W. Au2PbP2, Au2TlP2, and Au2HgP2: Ternary gold polyphosphides with lead, thallium, and mercury in the oxidation state zero. J. Solid State Chem. 2002, 165, 238–246. [Google Scholar] [CrossRef]
- Kovnir, K.; Stockert, U.; Budnyk, S.; Prots, Y.; Baitinger, M.; Paschen, S.; Shevelkov, A.V.; Grin, Y. Introducing a magnetic guest to a tetrel-free clathrate: Synthesis, structure, and properties of EuxBa8−xCu16P30 (0 ≤ x ≤ 1.5). Inorg. Chem. 2011, 50, 10387–10396. [Google Scholar] [CrossRef]
- Fulmer, J.; Kaseman, D.C.; Dolyniuk, J.; Lee, K.; Sen, S.; Kovnir, K. BaAu2P4: Layered Zintl Polyphosphide with Infinite
![Crystals 03 00431 i001]() Chains. Inorg. Chem. 2013, 52, 7061–7067. [Google Scholar] [CrossRef]
Chains. Inorg. Chem. 2013, 52, 7061–7067. [Google Scholar] [CrossRef] - Kraus, F.; Korber, N. The Chemical Bond in Polyphosphides: Crystal Structures, the Electron Localization Function, and a New View of Aromaticity in P42− and P5−. Chem. A Eur. J. 2005, 11, 5945–5959. [Google Scholar] [CrossRef]
- He, H.; Tyson, C.; Bobev, S. New compounds with [As7]3− clusters: Synthesis and crystal structures of the Zintl phases Cs2NaAs7, Cs4ZnAs14 and Cs4CdAs14. Crystals 2011, 1, 87–98. [Google Scholar] [CrossRef]
- Knapp, C.M.; Large, J.S.; Rees, N.H.; Goicoechea, J.M. A versatile salt-metathesis route to heteroatomic clusters derived from phosphorus and arsenic Zintl anions. Dalton Trans. 2011, 40, 735–745. [Google Scholar] [CrossRef]
- Hirschle, C.; Röhr, C. Darstellung und Kristallstruktur der Bekannten Zintl-Phasen Cs3Sb7 und Cs4Sb2. Z. Anorg. Allg. Chem. 2000, 626, 1992–1998. [Google Scholar] [CrossRef]
- Kauzlarich, S.M. Chemistry, Structure, and Bonding of Zintl Phases and Ion; John Wiley and Sons Ltd.: New York, NY, USA, 1996. [Google Scholar]
- Miller, G.J.; Schmidt, M.W.; Wang, F.; You, T.S. Quantitative advances in the Zintl-Klemm formalism. Struct. Bond. 2011, 139, 1–55. [Google Scholar]
- Von Schnering, H.G.; Menge, G. Dibariumheptaphosphidchlorid Ba2P7Cl, eine Verbindung mit dem polycyclischen Anion P73−. Z. Anorg. Allg. Chem. 1981, 481, 33–40. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Savin, A.; Jepsen, O.; Flad, J.; Anderson, O.K.; Preuß, H.; von Schnering, H.G. Electron localization in solid-state structures of the elements: The diamond structure. Angew. Chem. Int. Ed. 1992, 31, 187–188. [Google Scholar] [CrossRef]
- Savin, A.; Nesper, R.; Wengert, S.; Fässler, T.F. ELF: The electron localization function. Angew. Chem. Int. Ed. 1997, 36, 1808–1832. [Google Scholar] [CrossRef]
- Kovnir, K.; Kolen’ko, Y.V.; Baranov, A.I.; Neira, I.S.; Sobolev, A.V.; Yoshimura, M.; Presniakov, I.A.; Shevelkov, A.V. Sn4As3 revisited: Solvothermal synthesis and crystal and electronic structure. J. Solid State Chem. 2009, 182, 630–639. [Google Scholar] [CrossRef]
- Morales, A.; Mora, E.; Pal, U. Use of diffuse reflectance spectroscopy for optical characterization of un-supported nanostructures. Rev. Mex. Fis. S. 2007, 53, 18–22. [Google Scholar]
- Nobbs, J.H. Kubelka—Munk theory and the prediction of reflectance. Rev. Prog. Color. Relat. Top. 1985, 15, 66–75. [Google Scholar] [CrossRef]
- SMART and SAINT. Bruker AXS Inc.: Madison, WI, USA, 2007.
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Jepsen, O.; Burkhardt, A.; Andersen, O.K. The Program TB-LMTO-ASA, Version 4.7; Max-Planck-Institut für Festkörperforschung: Stuttgart, Germany, 1999. [Google Scholar]
- Barth, U.V.; Hedin, L. A local exchange-correlation potential for the spin polarized case. J. Phys. C Solid State Phys. 1972, 5. [Google Scholar] [CrossRef]
- Paraview: Parallel visualization application, version 3.8.1 64 bit. Available online: http://paraview.org.
- Baranov, A.I. Visualization Plug-in for ParaView, Version 3.4.11; Springer: Dresden, Germany, 2012. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dolyniuk, J.-A.; Kovnir, K. Zintl Salts Ba2P7X (X = Cl, Br, and I): Synthesis, Crystal, and Electronic Structures. Crystals 2013, 3, 431-442. https://doi.org/10.3390/cryst3030431
Dolyniuk J-A, Kovnir K. Zintl Salts Ba2P7X (X = Cl, Br, and I): Synthesis, Crystal, and Electronic Structures. Crystals. 2013; 3(3):431-442. https://doi.org/10.3390/cryst3030431
Chicago/Turabian StyleDolyniuk, Juli-Anna, and Kirill Kovnir. 2013. "Zintl Salts Ba2P7X (X = Cl, Br, and I): Synthesis, Crystal, and Electronic Structures" Crystals 3, no. 3: 431-442. https://doi.org/10.3390/cryst3030431