Organic Semiconductors and Conductors with tert-Butyl Substituents

Abstract
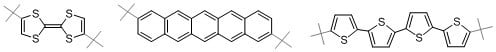
:
1. Introduction

2. Results and Discussion
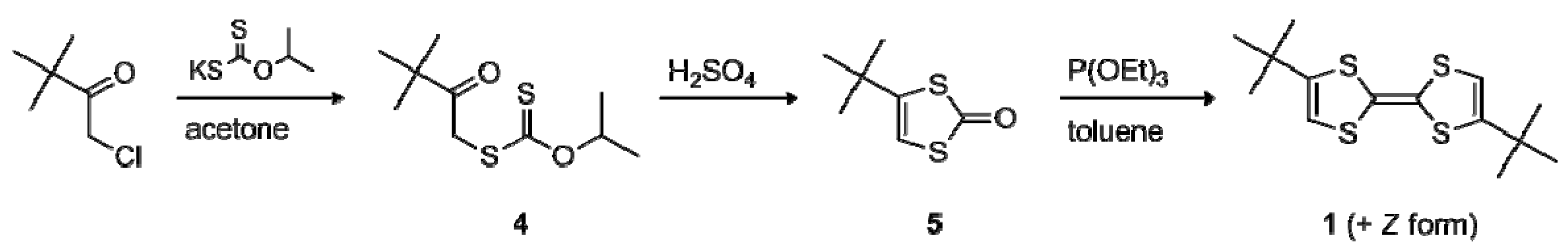
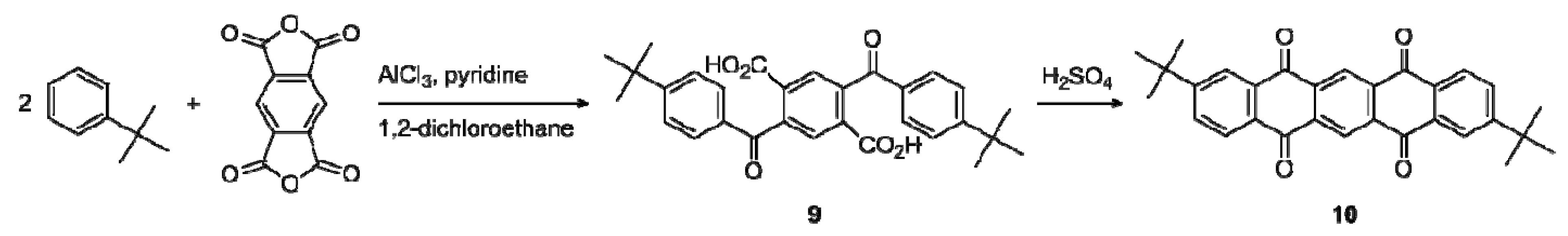
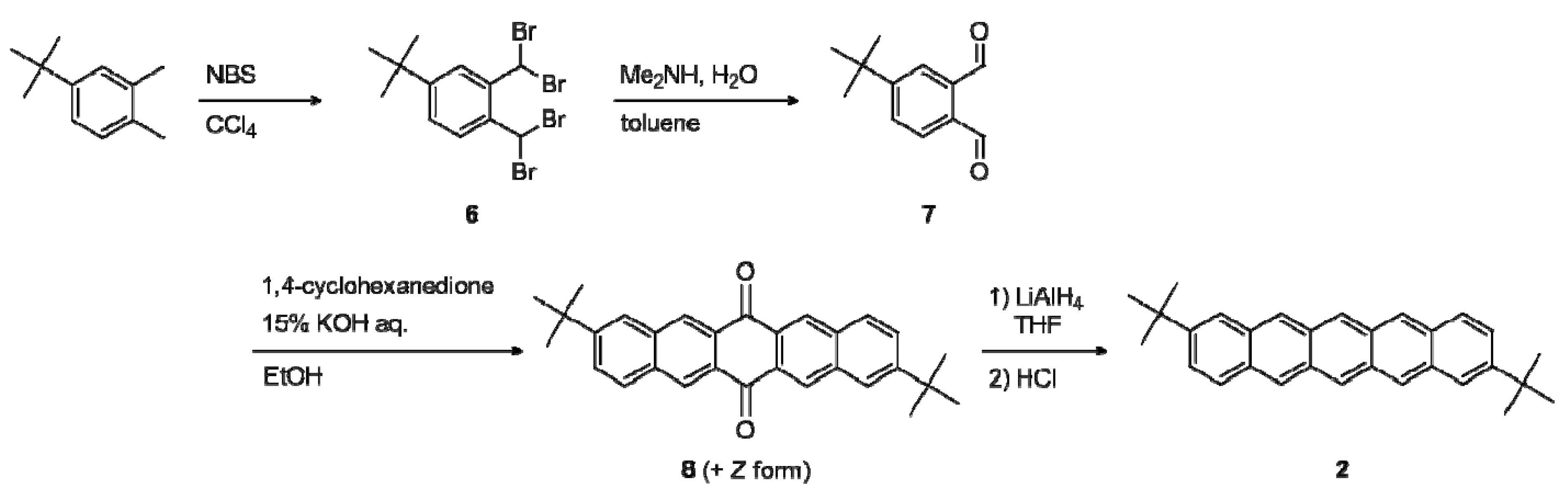
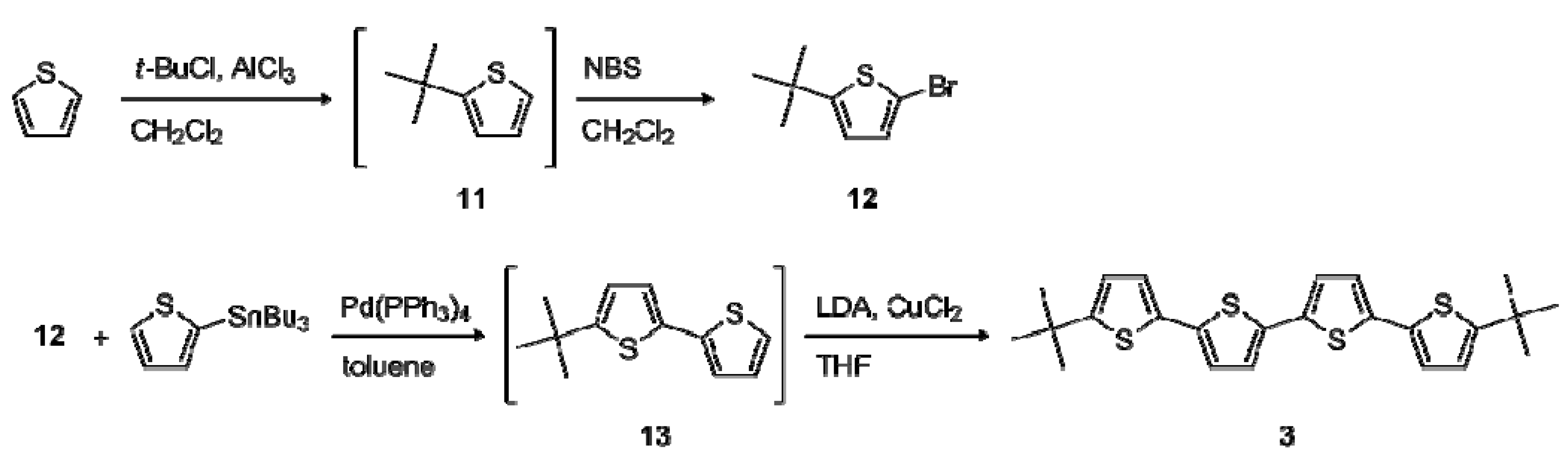
2.1. Synthesis
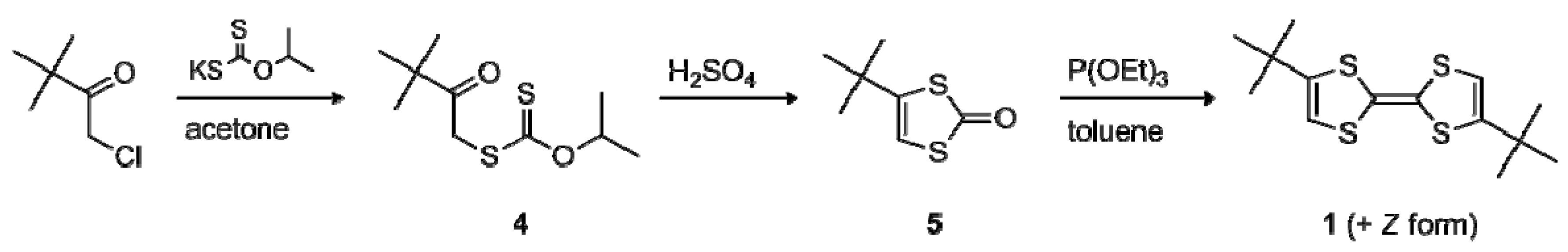
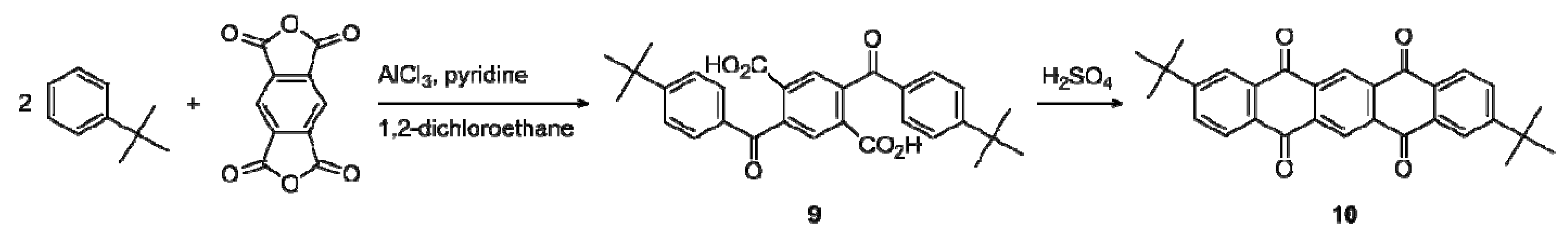
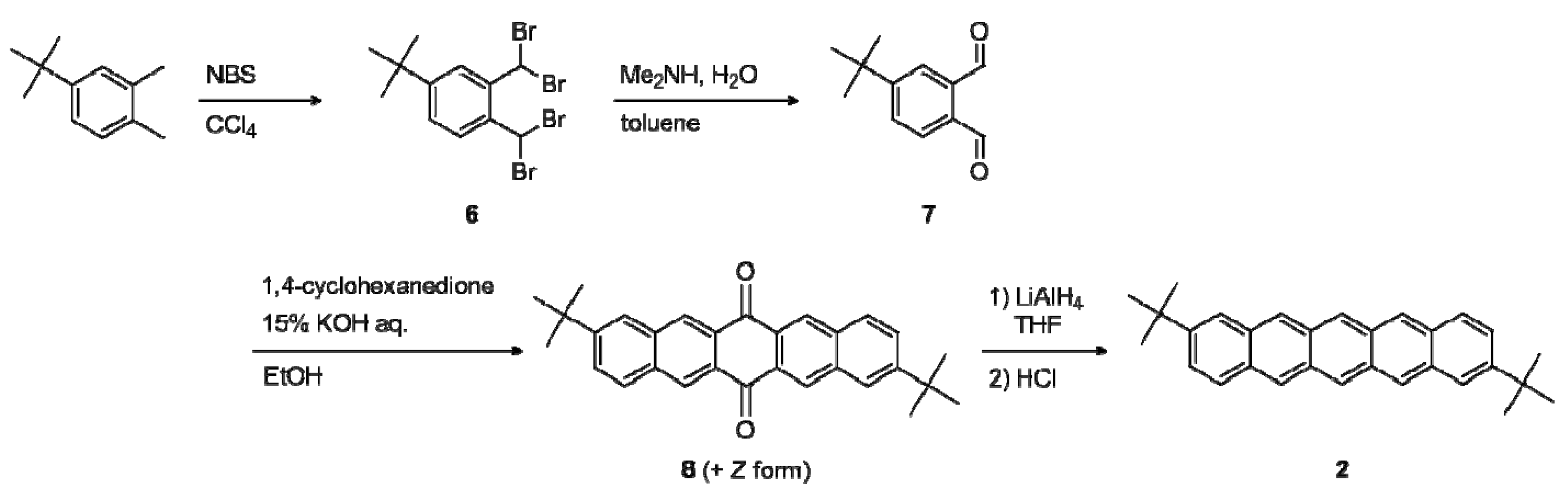
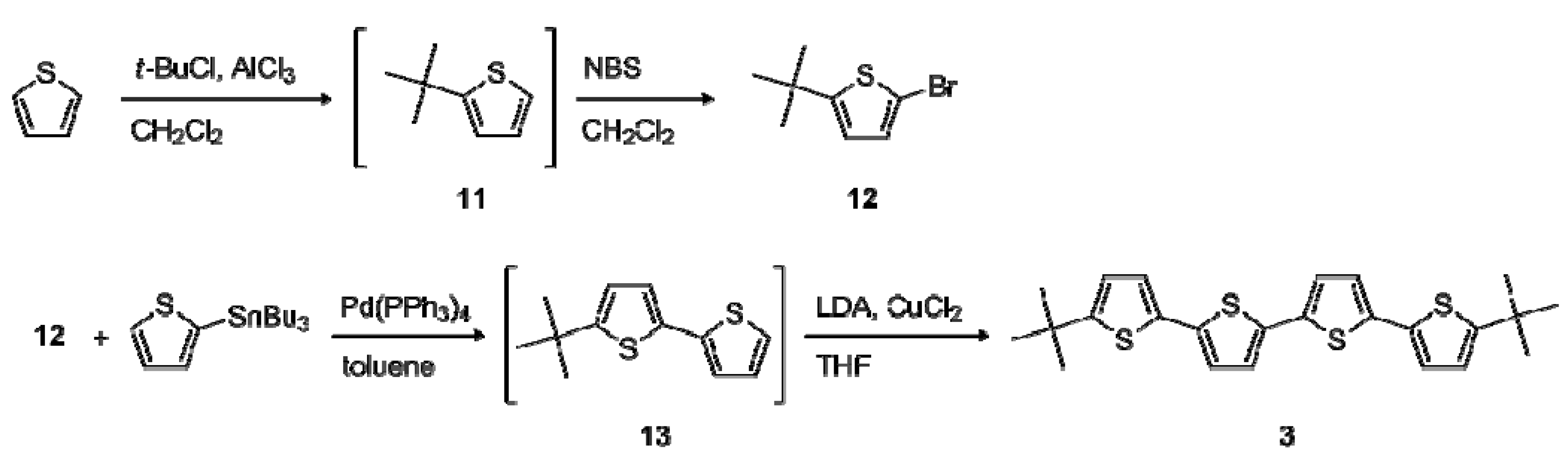
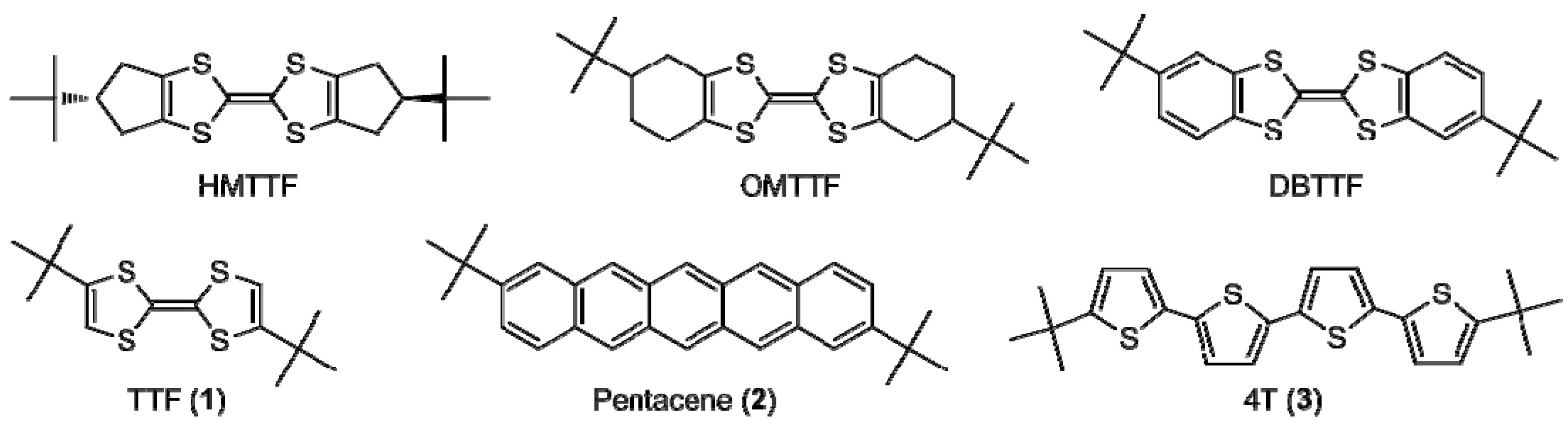
2.2. Electrochemical and Optical Properties

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Experiments | Calculation | |||||
|---|---|---|---|---|---|---|---|
| E1/21 (V) | E1/22 (V) | Gap (eV) | HOMO (eV) | LUMO (eV) | HOMO (eV) | LUMO (eV) | |
| 1 | 0.16 | 0.70 | 3.20 | −4.56 | −1.36 | −4.40 | −0.70 |
| 2 | 0.63 | 1.04 | 2.09 | −5.03 | −2.94 | −4.49 | −2.29 |
| 3 | 0.75 | 0.99 | 2.68 | −5.15 | −2.47 | −4.83 | −1.76 |
| TTF | 0.22 | 0.73 | 3.06 | −4.62 | −1.56 | −4.57 | −0.92 |
| Pentacene | - | - | - | −5.0 b | −3.2 b | −4.61 | −2.40 |
| 4T | 0.78 | - | 2.74 | −5.18 | −2.44 | −4.97 | −1.94 |
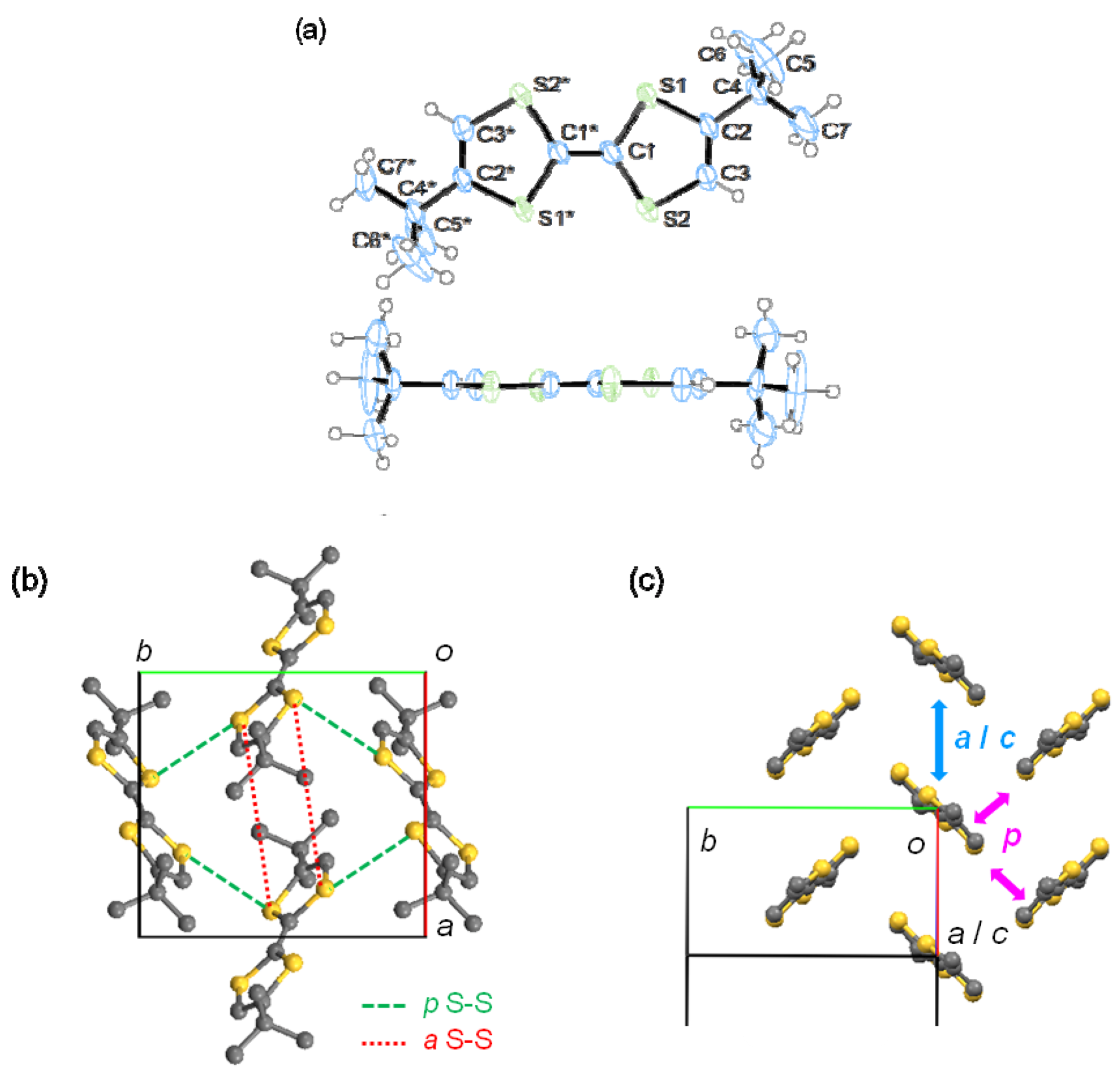
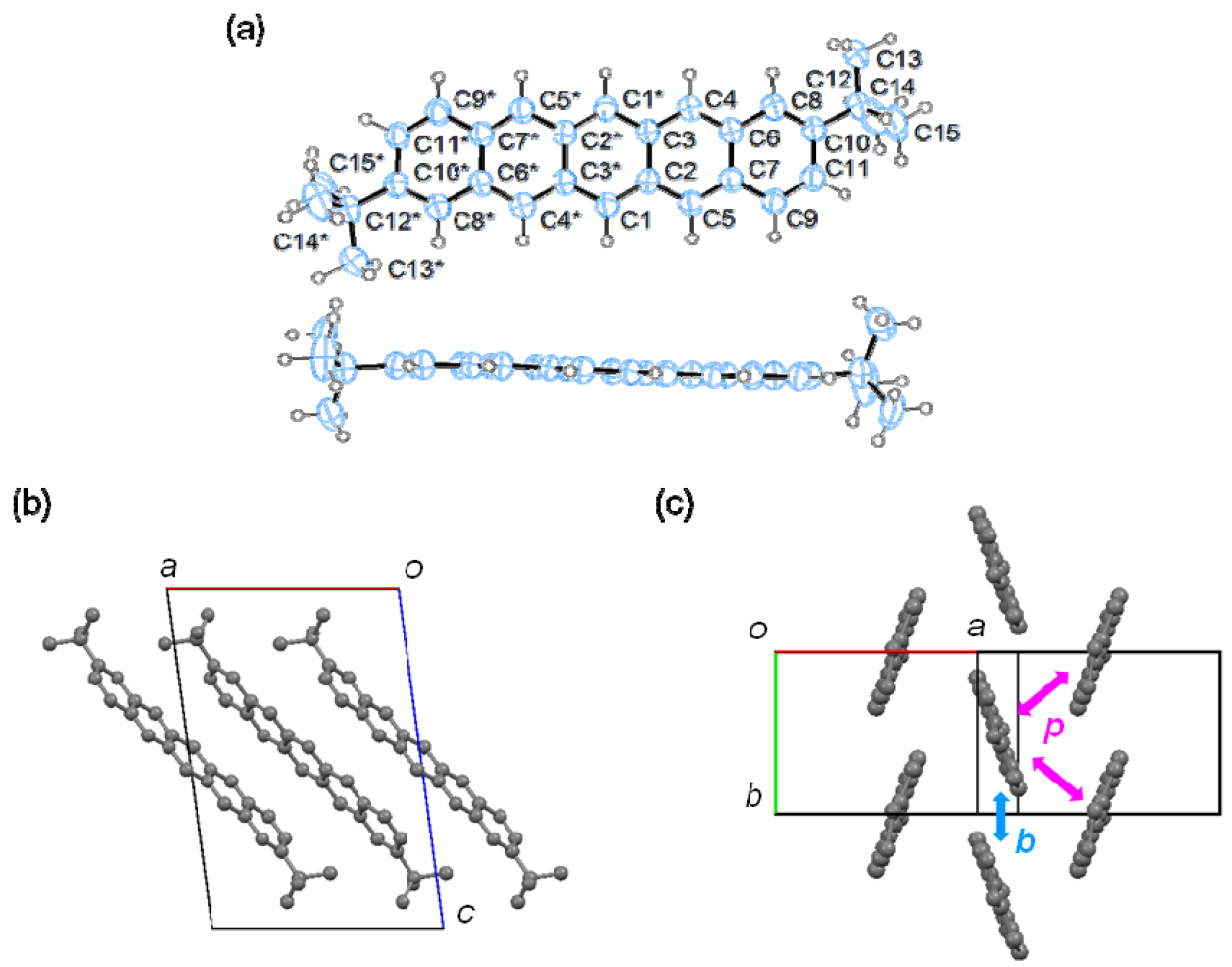
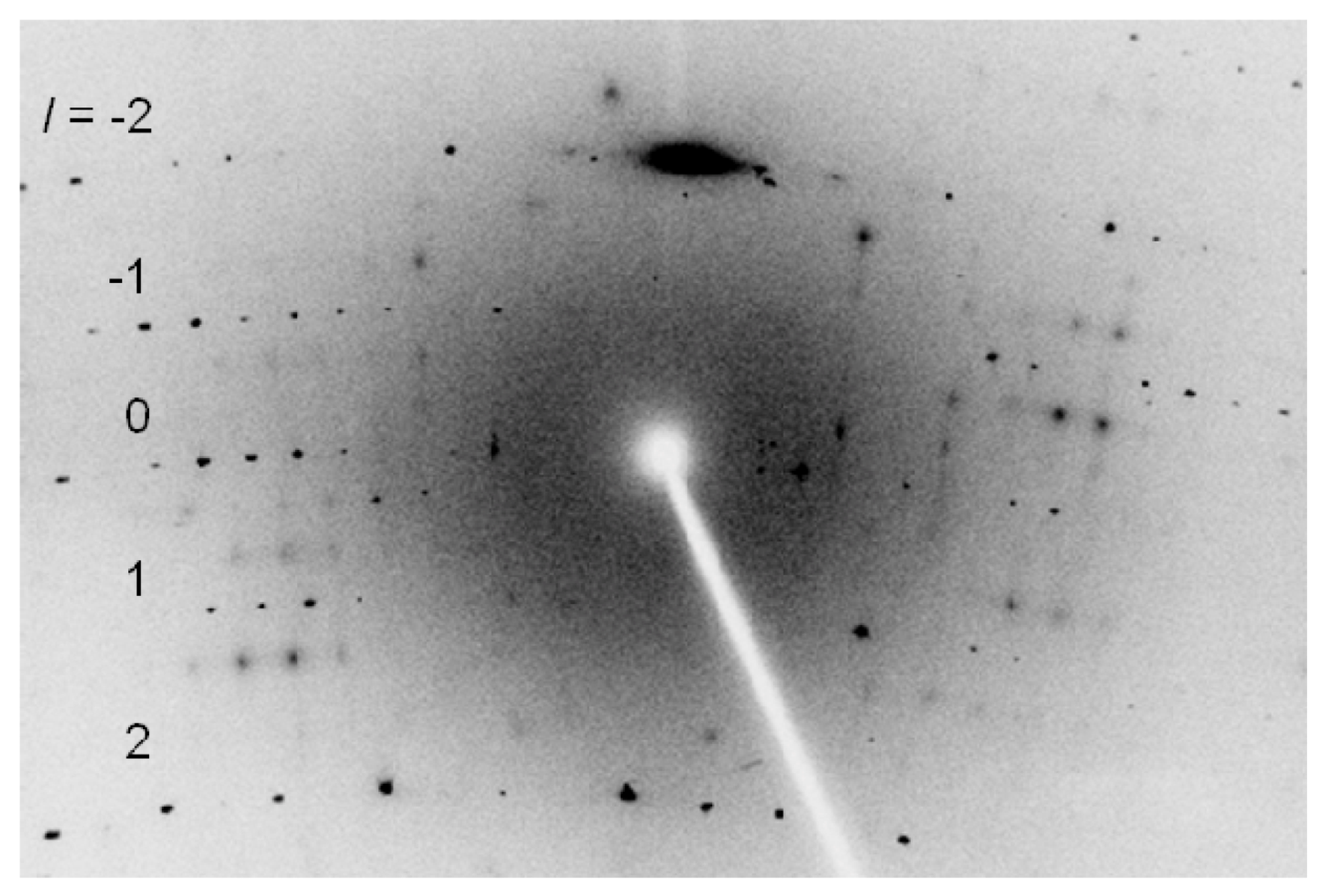
2.3. Single-Crystal X-ray Structure Analysis
| TTF [29] | 1 | Pentacene [30] | 2 | |
|---|---|---|---|---|
| Empirical formula | C2H4S4 | C14H20S4 | C22H14 | C30H30 |
| Formula weight | 204.33 | 316.55 | 278.35 | 390.57 |
| Temperature (K) | 290 | 173 | 293 | 173 |
| Crystal system | monoclinic | monoclinic | triclinic | monoclinic |
| Space group | P21/c | P21/a | P-1 | P21/a |
| α (Å) | 7.352(2) | 9.2639(2) | 6.266(1) | 10.8100(2) |
| b (Å) | 4.0181(11) | 9.7388(3) | 7.775(1) | 6.3668(2) |
| c (Å) | 13.901(4) | 9.3381(2) | 14.530(1) | 16.1783(4) |
| α (°) | 90 | 90 | 76.475(4) | 90 |
| β (°) | 101.426(10) | 103.263(2) | 87.682(4) | 97.483(1) |
| γ (°) | 90 | 90 | 84.684(4) | 90 |
| V (Å3) | 402.5(2) | 820.01(4) | 685.15(15) | 1111.24(4) |
| Z | 2 | 2 | 2 | 2 |
| Dcalc (g/cm3) | 1.686 | 1.282 | 1.349 | 1.167 |
| Reflections (unique) | 3571 (926) | 8389 (1501) | 2856 (2684) | 12341 (2033) |
| R1 | 0.028 | 0.0768 | 0.069 | 0.0561 |
| Rw | 0.065 | 0.2215 | 0.179 | 0.1925 |
| GOF | 1.12 | 1.05 | 0.94 | 1.08 |
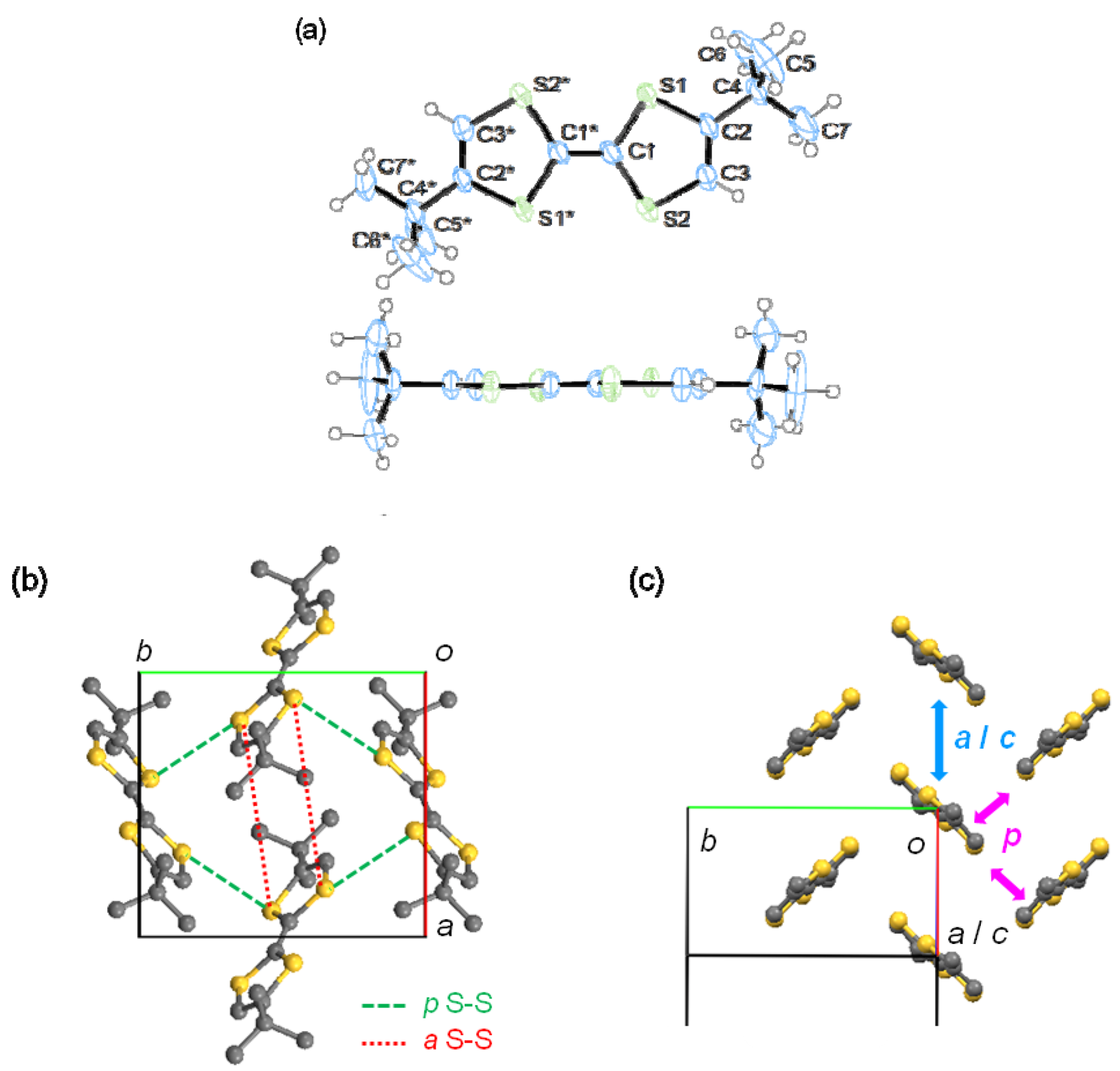

| 1 | t (meV) | D (Å) | S-S (Å) | 2 | t (meV) | D (Å) |
|---|---|---|---|---|---|---|
| a | 1 | 8.1 | 6.59 | b | −23 | 1.2 |
| c | −5 | 5.0 | 5.73 | p | −1 | - |
| p | −11 | - | 3.70 |
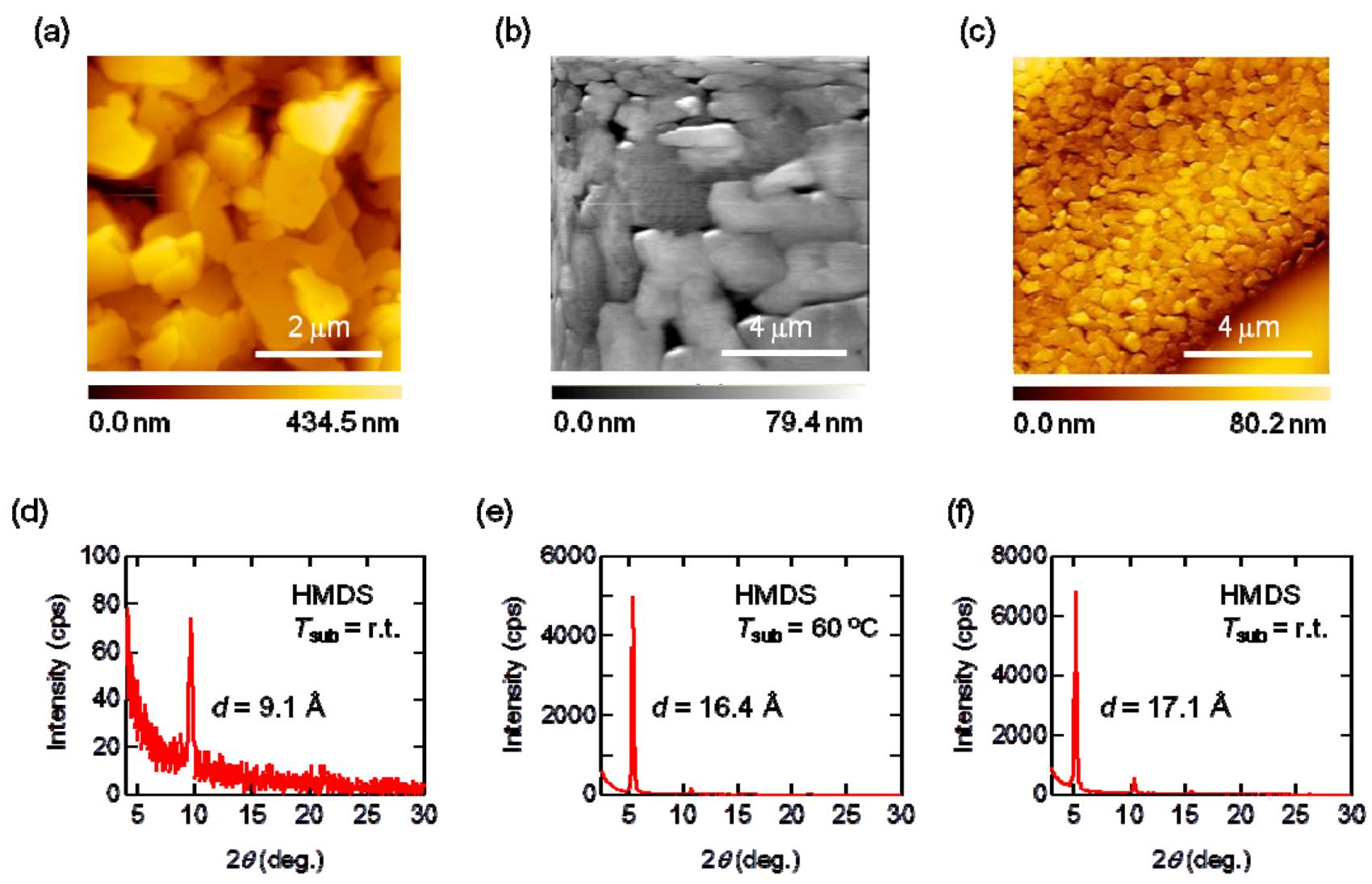
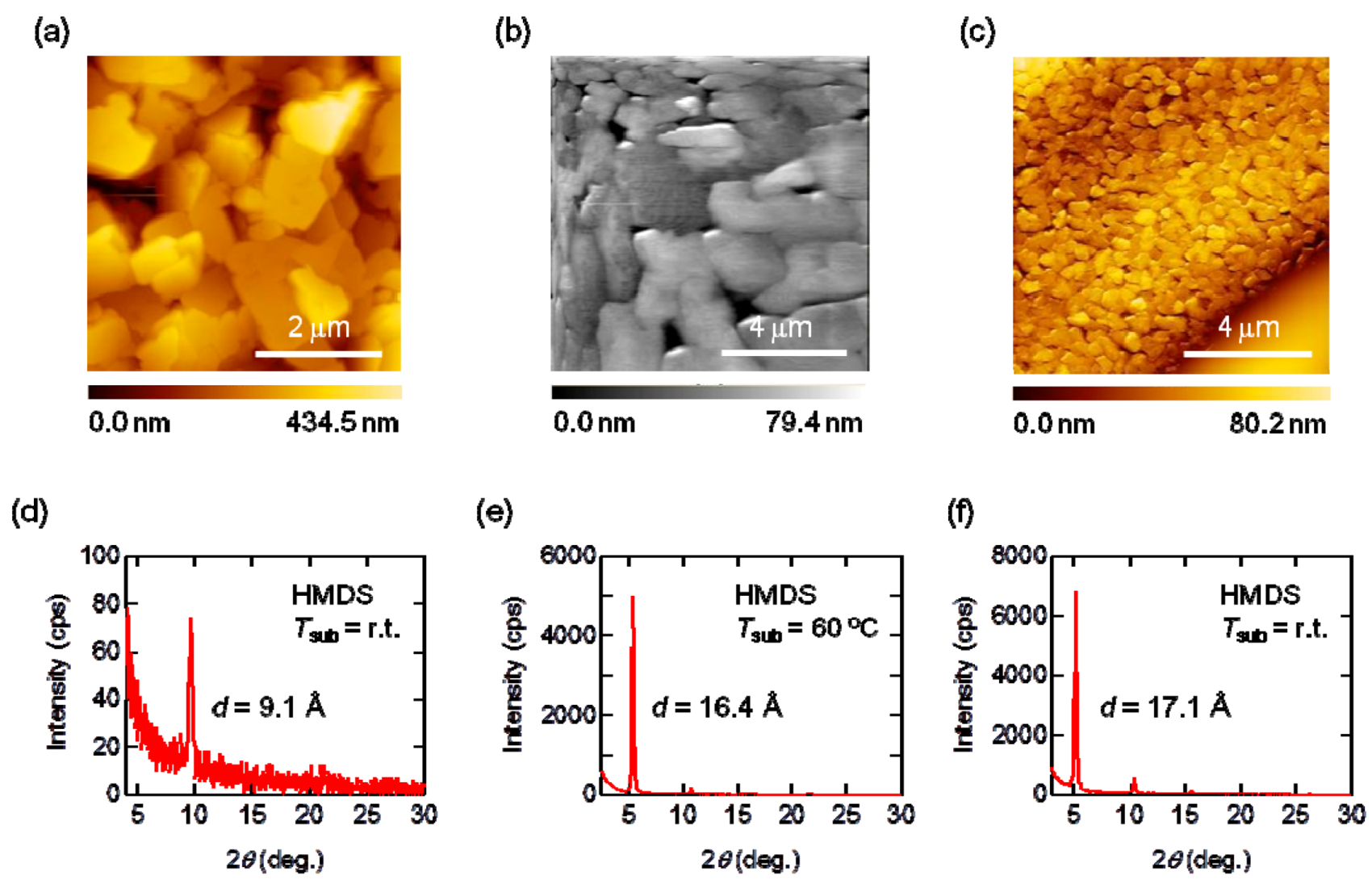
2.4. Thin-Film Properties
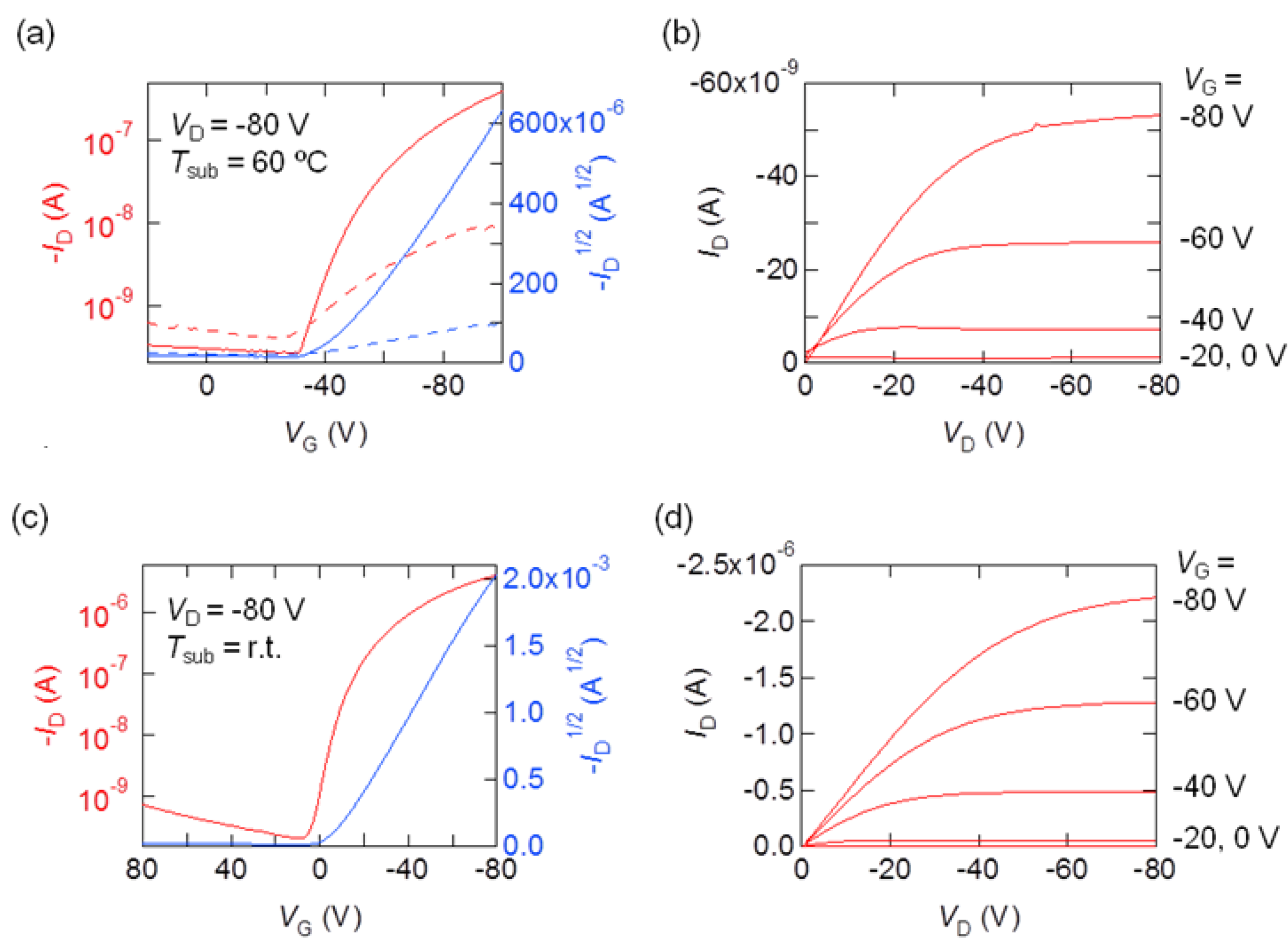
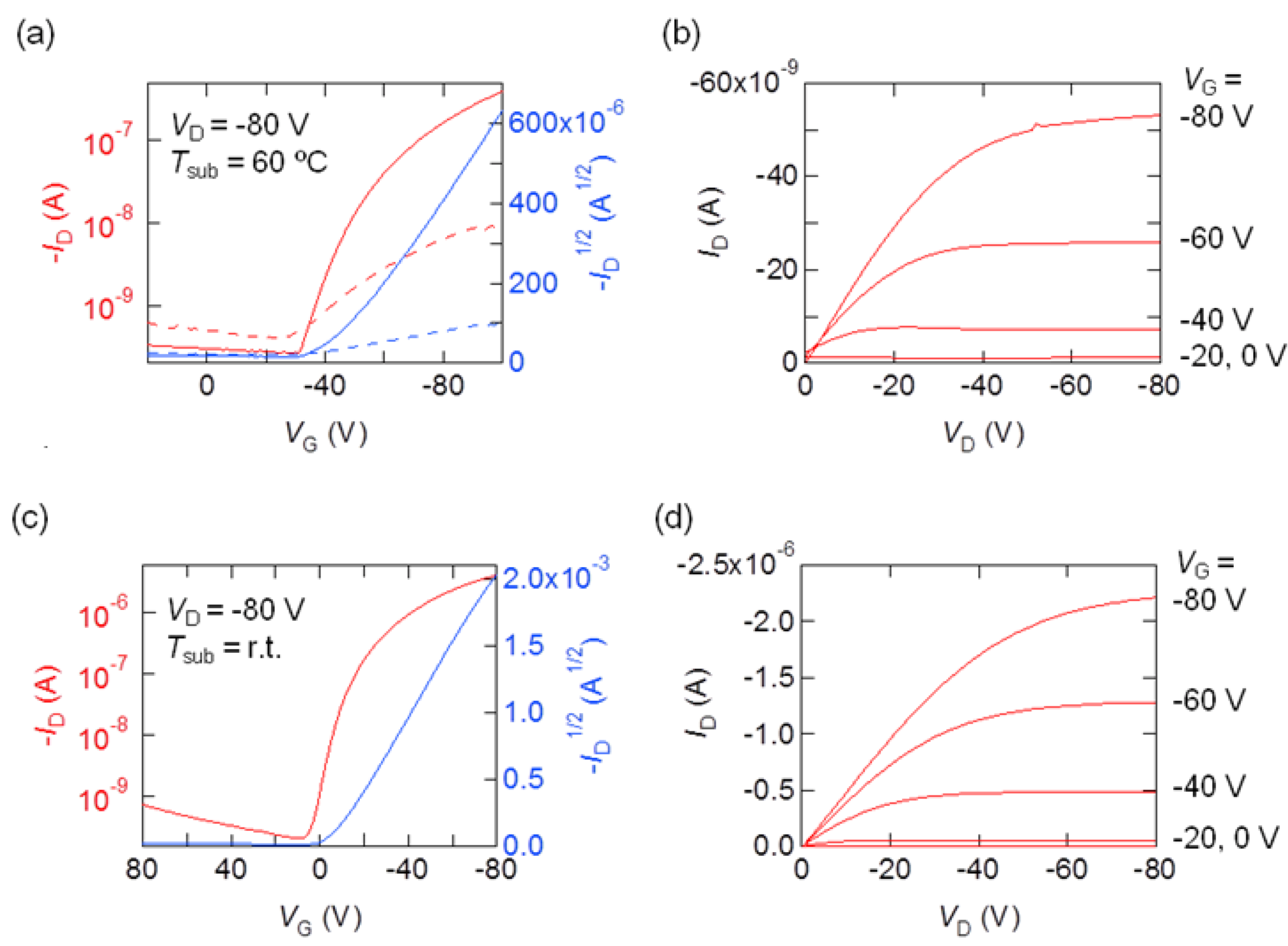
2.5. Transistor Propzerties
| Compounds | SAMs | µ (cm2 V−1 s−1) | Vth (V) | Ion/Ioff | |
|---|---|---|---|---|---|
| 2 | HMDS | r.t. | 2.5 × 10−4 | −36 | 1 × 103 |
| 40 °C | 2.9 × 10−4 | −22 | 2 × 102 | ||
| 60 °C | 2.8 × 10−3 | −44 | 2 × 103 | ||
| after 30 days | 1.3 × 10−5 | −20 | 26 | ||
| 3 | bare | r.t. | 6.3 × 10−3 | −17 | 2 × 104 |
| HMDS | r.t. | 3.5 × 10−3 | −17 | 1 × 104 | |
| 4T | bare | r.t. | 3.5 × 10−3 | −20 | 6 × 103 |
| HMDS | r.t. | 7.8 × 10−3 | −13 | 6 × 104 |
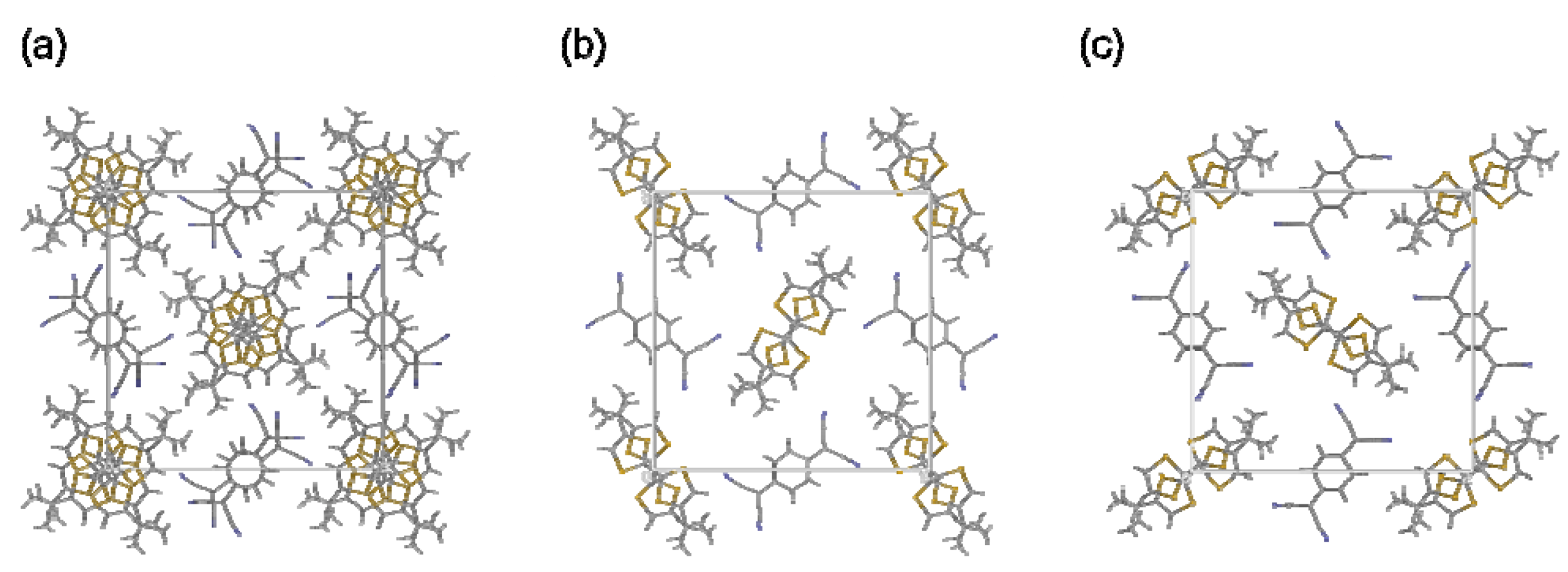
2.6. Charge-Transfer Salt
| (1)(TCNQ) | |
|---|---|
| Empirical formula | C26H24S4N4 |
| Formula weight | 520.74 |
| Temperature (K) | 273 |
| Crystal system | tetragonal |
| Space group | P42/mbc |
| a (Å) | 19.7657(7) |
| c (Å) | 6.8345(3) |
| V (Å3) | 2670.1(2) |
| Z | 4 |
| Dcalc (g/cm3) | 1.295 |
| Reflections (unique) | 29583 (1335) |
| R1 | 0.0938 |
| Rw | 0.2868 |
| GOF | 1.08 |
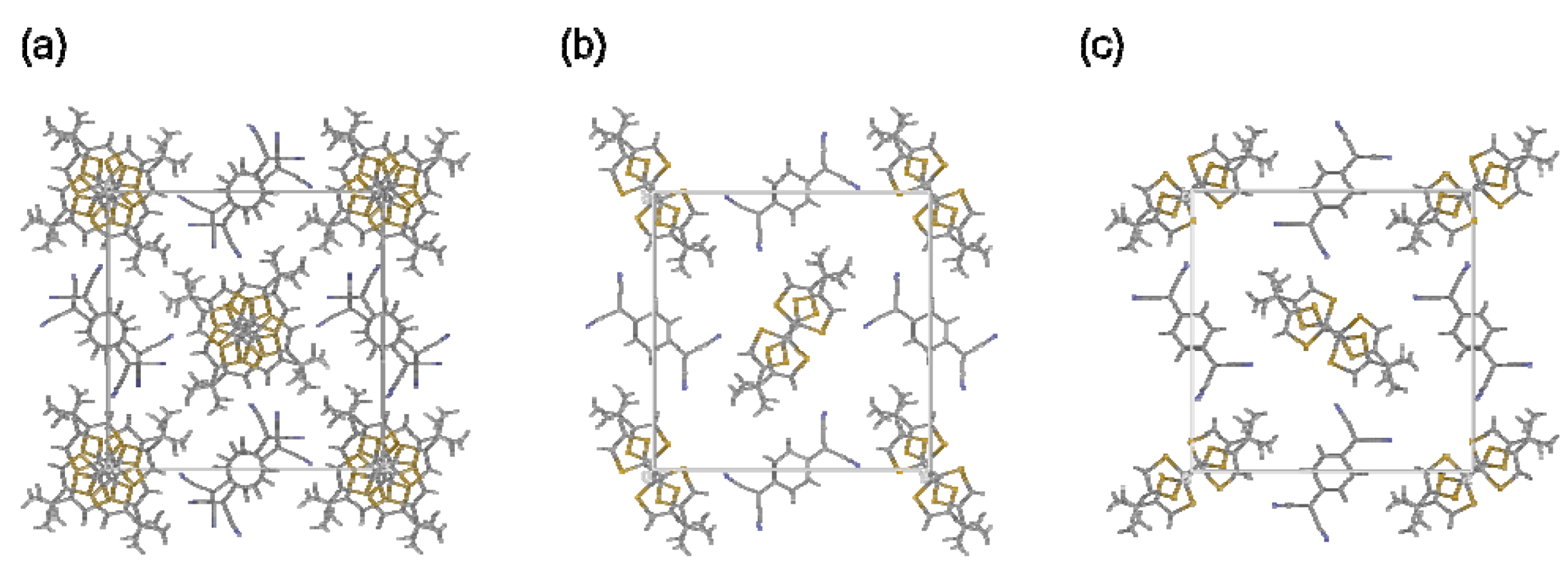
3. Experimental Section
3.1. Synthesis
3.2. Redox Properties
3.3. Photophysical Properties
3.4. Single Crystal Structures and Intermolecular Overlap Integral Calculations
3.5. Device Fabrication
3.6. Conductivity Measurement
4. Conclusions
Acknowledgments
References
- Mori, T. Organic charge-transfer salts and the component molecules in organic transistors. Chem. Lett. 2011, 40, 428–434. [Google Scholar] [CrossRef]
- Kanno, M.; Bando, Y.; Shirahata, T.; Inoue, J.; Wada, H.; Mori, T. Stabilization of organic field-effect transistors in hexamethylenetetrathiafulvalene derivatives substituted by bulky alkyl groups. J. Mater. Chem. 2009, 19, 6548–6555. [Google Scholar] [CrossRef]
- Inoue, J.; Kanno, M.; Ashizawa, M.; Seo, C.; Tanioka, A.; Mori, T. Organic transistors based on octamethylenetetrathiafulvalenes. Chem. Lett. 2010, 39, 538–540. [Google Scholar] [CrossRef]
- Nagakubo, J.; Ashizawa, M.; Kawamoto, T.; Tanioka, A.; Mori, T. Stabilization of organic field-effect transistors by tert-butyl groups in dibenzotetrathiafulvalene derivatives. Phys. Chem. Chem. Phys. 2011, 13, 14370–14377. [Google Scholar]
- Takahashi, Y.; Hasegawa, T.; Horiuchi, S.; Kumai, R.; Tokura, Y.; Saito, G. High mobility organic field-effect transistor based on hexamethylenetetrathiafulvalene with organic metal electrodes. Chem. Mater. 2007, 19, 6382–6384. [Google Scholar] [CrossRef]
- Mas-Torrent, M.; Hadley, P.; Bromley, S.T.; Crivillers, N.; Rovira, C. Single-crystal organic field-effect transistors based on dibenzo-tetrathiafulvalene. Appl. Phys. Lett. 2005, 86, 012110:1–012110:3. [Google Scholar]
- Shibata, K.; Ishikawa, K.; Takezoe, H.; Wada, H.; Mori, T. Contact resistance of dibenzotetrathiafulvalene-based organic transistors with metal and organic electrodes. Appl. Phys. Lett. 2008, 92, 023305:1–023305:3. [Google Scholar]
- Yamada, T.; Hasegawa, T.; Hiraoka, M.; Matsui, H.; Tokura, Y.; Saito, G. Control of film morphology and its effects on subthreshold characteristics in dibenzotetrathiafulvalene organic thin-film transistors. Appl. Phys. Lett. 2008, 92, 233306:1–233306:3. [Google Scholar]
- Dhindsa, A.S.; Bryce, M.R.; Lloyd, J.P.; Petty, M.C.; Kobayashi, K.; Tukada, H. Synthesis of tetrathiafulvalene (TTF) derivatives bearing long alkyl chains. J. Chem. Soc. Chem. Commun. 1988, 1391–1392. [Google Scholar]
- Meng, H.; Bendikov, M.; Mitchell, G.; Helgeson, R.; Wudl, F.; Bao, Z.; Siegrist, T.; Kloc, C.; Chen, C.H. Tetramethylpentacene: Remarkable absence of steric effect on field effect mobility. Adv. Mater. 2003, 15, 1090–1093. [Google Scholar] [CrossRef]
- Okamoto, K.; Kawamura, T.; Sone, M.; Ogino, K. Study on liquid crystallinity in 2,9-dialkylpentacenes. Liq. Cryst. 2007, 34, 1001–1007. [Google Scholar] [CrossRef]
- Kunugi, Y.; Busujima, Y.; Ikari, M.; Okamoto, K.; Ogino, K. Organic field-effect transistors based on 2,9-disubstituted pentacene. ECS Trans. 2008, 16, 273–282. [Google Scholar]
- Perepichka, I.F.; Perepichka, D.F. Handbook of Thiophene-based Materials; Wiley: Chichester, UK, 2009. [Google Scholar]
- Oliva, M.M.; Casado, J.; Navarrete, J.T.L.; Patchkovskii, S.; Goodson, T.; Harpham, M.R.; de Melo, J.S.S.; Amir, E.; Rozen, S. Do [all]-S, S′-Dioxide Oligothiophenes show electronic and optical properties of oligoenes and/or of oligothiophenes? J. Am. Chem. Soc. 2010, 132, 6231–6242. [Google Scholar]
- Alberola, A.; Bosch-Navarro, C.; Gavina, P.; Tatay, S. Synthesis of brominatedtetrathiafulvalenes via phosphite-mediated cross-coupling. Synth. Met. 2010, 160, 1797–1799. [Google Scholar] [CrossRef]
- Fieser, L.F. Organic Synthesses Collected Volume I; Gilman, H., Ed.; Wiley: New York, NY, USA, 1941; pp. 517–519. [Google Scholar]
- Fieser, L.F. Organic Experiments; D.C. Heath: Lexington, MA, USA, 1965; pp. 195–200. [Google Scholar]
- Bénard, C.P.; Geng, Z.; Heuft, M.A.; Van Crey, K.; Fallis, A.G. Double diels-alder strategies to Soluble 2,9- and 2,9,6,13-Tetraethynylpentacenes, Photolytic [4+4] Cycloadditions, and pentacene crystal packing. J. Org. Chem. 2007, 72, 7229–7236. [Google Scholar]
- Okamoto, T.; Reese, C.; Senatore, M.L.; Tang, M.L.; Jiang, Y.; Parkin, S.R.; Bao, Z. 2,9-Dibromopentacene: Synthesis and the role of substituent and symmetry on solid-state order. Synth. Met. 2010, 160, 2447–2451. [Google Scholar]
- Vets, N.; Smet, M.; Dehaen, W. Synthesis and thermolysis of a Diels Alder adduct of pentacene and thiophosgene. Tetrahedron Lett. 2004, 45, 7287–7289. [Google Scholar] [CrossRef]
- Belen’kii, L.I.; Yakubov, A.P. Stable heteroareniumions–VIII some transformations of alkylthiophenium ions and new synthesis of 2-t-butylthiophene. Tetrahedron 1984, 40, 2471–2477. [Google Scholar] [CrossRef]
- Pinault, T.; Chérioux, F.; Therrien, B.; Süss-Fink, G. An iterative strategy for the synthesis of oligothiophenes by catalytic cross-coupling reactions. Heteroatom Chem. 2004, 15, 121–126. [Google Scholar] [CrossRef]
- Kagan, J.; Arora, S.K.; Prakash, I.; Üstünol, A. The synthesis of 2,2′:5′,3″-terthiophene. Heterocycles 1983, 20, 1341–1345. [Google Scholar]
- Tang, M.L.; Reichardt, A.D.; Miyaki, N.; Stoltenberg, R.M.; Bao, Z. Ambipolar, high performance, acene-based organic thin film transistors. J. Am. Chem. Soc. 2008, 130, 6064–6065. [Google Scholar]
- Tang, M.L.; Reichardt, A.D.; Wei, P.; Bao, Z. Correlating carrier type with frontier molecular orbital energy levels in organic thin film transistors of functionalized acene derivatives. J. Am. Chem. Soc. 2009, 131, 5264–5273. [Google Scholar]
- Meng, H.; Zheng, L.; Lovinger, A.J.; Wand, B.-C.; Patten, P.G.V.; Bao, Z. Oligofluorene−thiophene derivatives as high-performance semiconductors for organic thin film transistors. Chem. Mater. 2003, 15, 1778–1787. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Yasuda, T.; Goto, T.; Fujita, K.; Tsutsui, T. Ambipolar carrier transport in polycrystalline pentacene thin-film transistors. Mol. Cryst. Liq. Cryst. 2006, 444, 219–224. [Google Scholar] [CrossRef]
- Batsanov, A.S. Tetrathiafulvalene revisited. Acta Crystallogr. 2006, C62, o501–o504. [Google Scholar]
- Mattheus, C.C.; Dros, A.B.; Baas, J.; Meetsma, A.; de Boer, J.L.; Palstra, T.M. Polymorphism in pentacene. Acta Crystallogr. 2001, C57, 939–941. [Google Scholar]
- Mori, T.; Kobayashi, A.; Sasaki, Y.; Kobayashi, H.; Saito, G.; Inokuchi, H. The intermolecular interaction of tetrathiafulvalene and bis(ethylenedithio)tetrathiafulvalene in organic metals. calculation of orbital overlaps and models of energy-band structures. Bull. Chem. Soc. Jpn. 1984, 57, 627–633. [Google Scholar] [CrossRef]
- Kojima, H.; Mori, T. Dihedral angle dependence of transfer integrals in organic semiconductors with herringbone structures. Bull. Chem. Soc. Jpn. 2011, 84, 1049–1056. [Google Scholar] [CrossRef]
- Campbell, R.B.; Robertson, J.M.; Trotter, J. The crystal and molecular structure of pentacene. Acta Crystallogr. 1961, 14, 705–711. [Google Scholar] [CrossRef]
- Aoyagi, I.; Katsuhara, M.; Mori, T. Syntheses and structures of highly solublebis(ethylenedithio)tetrathiafulvalene molecules with alkyl chains. Sci. Technol. Adv. Mater. 2004, 5, 443–447. [Google Scholar] [CrossRef]
- Shitzkovsky, S.; Weger, M.; Gutfreund, H. Band structure of TTF-TCNQ. J. Phys. (Paris) 1978, 39, 711–717. [Google Scholar]
- Kunugi, Y.; Takimiya, K.; Yamane, K.; Yamashita, K.; Aso, Y.; Otsubo, T. Organic field-effect transistor using oligoselenophene as an active layer. Chem. Mater. 2003, 15, 6–7. [Google Scholar] [CrossRef]
- Takimiya, K.; Kunugi, Y.; Toyoshima, Y.; Otsubo, T. 2,6-Diarylnaphtho[1,8-bc:5,4-b′c′]dithiophenes as new high-performance semiconductors for organic field-effect transistors. J. Am. Chem. Soc. 2005, 127, 3605–3612. [Google Scholar] [CrossRef]
- Scott, B.A.; La Placa, S.J.; Torrance, J.B.; Silverman, B.D.; Welber, B.J. The crystal chemistry of organic metals. Composition, structure, and stability in the tetrathiafulvalenium-halide systems. J. Am. Chem. Soc. 1977, 99, 6631–6639. [Google Scholar] [CrossRef]
- Fourmigué, M.; Reinheimer, E.W.; Dunbar, K.R.; Auban-Senzier, P.; Pasquier, C.; Coulon, C. A series of strongly one-dimensional organic metals with strictly uniform stacks: (o-DMTTF)2X (X = Cl, Br, I). Dalton Trans. 2008, 4652–4658. [Google Scholar]
- Yamamoto, H.M.; Kosaka, Y.; Maeda, R.; Yamaura, J.; Nakano, A.; Nakamura, T.; Kato, R. Supramolecular insulating networks sheathing conducting nanowires based on organic radical cations. ACS Nano 2008, 2, 143–155. [Google Scholar] [CrossRef]
- Chappell, J.S.; Bloch, A.N.; Bryden, W.A.; Maxfield, M.; Poehler, T.O.; Cowan, D.O. Degree of charge transfer in organic conductors by infrared absorption spectroscopy. J. Am. Chem. Soc. 1981, 103, 2442–2443. [Google Scholar]
- Kagoshima, S.; Ishiguro, T.; Anzai, H. X-ray scattering study of phonon anomalies and superstructures in TTF-TCNQ. J. Phys. Soc. Jpn. 1976, 41, 2061–2071. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. ActaCrystallogr. 2008, A64, 112–122. [Google Scholar]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Burla, M.C.; Polidori, G.; Camalli, M. SIR92—A program for automatic solution of crystal structures by direct methods. J. Appl. Crystallogr. 1994, 27, 435. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Higashino, T.; Akiyama, Y.; Kojima, H.; Kawamoto, T.; Mori, T. Organic Semiconductors and Conductors with tert-Butyl Substituents. Crystals 2012, 2, 1222-1238. https://doi.org/10.3390/cryst2031222
Higashino T, Akiyama Y, Kojima H, Kawamoto T, Mori T. Organic Semiconductors and Conductors with tert-Butyl Substituents. Crystals. 2012; 2(3):1222-1238. https://doi.org/10.3390/cryst2031222
Chicago/Turabian StyleHigashino, Toshiki, Yuto Akiyama, Hirotaka Kojima, Tadashi Kawamoto, and Takehiko Mori. 2012. "Organic Semiconductors and Conductors with tert-Butyl Substituents" Crystals 2, no. 3: 1222-1238. https://doi.org/10.3390/cryst2031222