Structural Aspects of the Bechgaard and Fabre Salts: An Update

Laboratoire de Physique des Solides, Université Paris-sud, CNRS, UMR 8502, Bâtiment 510, Orsay Cedex F-91405, France
Crystals 2012, 2(2), 466-520; https://doi.org/10.3390/cryst2020466
Submission received: 20 March 2012
/
Revised: 19 April 2012
/
Accepted: 20 April 2012
/
Published: 21 May 2012
(This article belongs to the Special Issue Molecular Conductors)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:We review structural aspects of the Bechgaard and Fabre salts in relationship with their electronic, magnetic and superconducting properties. We emphasize the role of bond and charge modulations of the quarter filled organic stack in the various instabilities and ground states exhibited by these salts. A special consideration is also devoted to the influence of anions and methyl groups in these processes. In particular we point out the importance of the anions in achieving the inter-stack coupling by either direct or indirect (via the polarization of the methyl group cavities) interactions with the donors. In this framework we discuss the role of anions and methyl group disorders in the inhibition of the divergence of the high temperature bond order wave instability of the Bechgaard salts. We analyze the modulation in the magnetic ground states by considering explicitly the coupling of the magnetization with structural degrees of freedom. We consider the role of the anions and methyl groups in stabilizing the charge ordering pattern in the Fabre salts. We also discuss the spin-Peierls transition of the Fabre salts in relation with the charge ordering transition and the adiabaticity of the phonon field. We review the anion ordering transitions by considering more particularly the influence of the ordering process on the electronic structure and on the ground states which results. In this framework we show that the texture of the anion ordered structure has direct consequences on the superconducting properties of (TMTSF)2ClO4. Finally we conclude on the essential implication of the structural degrees of freedom on the generic phase diagram of the Bechgaard and Fabre salts.
1. Introduction
For several decades the synthesis of new classes of materials has triggered original fields of research where the discovery of new electronic states of matter has increasingly enriched the number of fundamental concepts in solid state physics while opening new routes for potential applications. This has been the case, to name a few examples, of oxides and carbon based compounds including conducting polymers. In this respect, the study of organic conductors since the beginning of the 1970s has largely contributed to this revival [1,2,3].
Organic conductors presenting a regular array of quasi-planar organic molecules, developing a preferential overlap of their pπ HOMO in one or two crystallographic directions, provide very anisotropic materials exhibiting a relatively simple electronic structure. The originality of these materials relies on the following facts:
- (i) Their pπ electron bandwidth is comparable to the electron–electron Coulomb repulsions which, being weakly screened, decrease slowly with the intermolecular separation.
- (ii) Their lattice is soft and quite compressible, which leads to rapid change of the electronic properties under pressure or uniaxial stress, while providing a sizeable coupling between the electronic and structural degrees of freedom.
- (iii) Their crystal array is quite perfect, which allows a fine control of the perturbation of physical properties by defects intentionally introduced in the structure.
If one restricts to quasi one-dimensional (1D) systems, the organic conductors provide classes of materials which realize a very large panel of different physical situations. The charge transfer salts of the TTF-TCNQ family are composed of segregated stacks of donors (D: TTF) and acceptors (A: TCNQ) with generally an incommensurate charge transfer between the two metallic 1D subsystems. These salts are subject to a charge density wave (CDW) instability which triggers a Peierls insulating ground state [1]. The 2:1 A2Y or D2X salts, where Y or X is a monovalent cationic or anionic entity, form quasi-1D quarter filled electronic systems built exclusively on acceptors, A, or donors, D. These “less screened” metallic systems generally experience quite strong Coulomb repulsions driving high temperature charge localization. This localization, accompanied by a spin charge decoupling, leaves the spin degrees of freedom active for a low temperature anti-ferromagnetic (AF) or spin-Peierls (SP) instability. Among these 2:1 series considerable studies have been devoted to the so-called Fabre and Bechgaard salts built with TMTTF and TMTSF donors respectively. The Fabre salts show charge localization, spin-charge decoupling and prototypal examples of AF and SP ground states while the Bechgaard salts exhibit a Fermi surface (FS) nesting driven spin density wave (SDW) ground state. The suppression of the SDW ground state under pressure restores the metallic state which is thus subject to superconductivity [1,2]. To complete this survey of different classes of organic 1D system, let us briefly mention very original 2:1 series of quasi-1D organic systems based on acceptors incorporating a metal transition element M. The corresponding salts, in presence of perylene or of TTF based donor, achieve segregated metallic and magnetic stacks [Per2-M(mnt)2 series [4]] or spin ladders [(DT-TTF)2-M(mnt)2 series [5]]. Finally, let us mention conducting and even superconducting 2:1 (M(dmit)2)2Y or charge transfer TTF(M(dmit)2)2 systems [2], where the electronic structure of the M(dmit)2 acceptor, composed of dmit side groups weakly coupled through a central M atom, involves both HOMO and LUMO electronic states [6]. These highly original conducting salts, which are not considered here, deserve a special review in their own right.
This present review focuses on structural aspects of Bechgaard and Fabre salts and their relationship with electronic, magnetic and superconducting properties. It will cover the most recent findings concerning the structural instabilities exhibited by these salts, and will present quantitative analyses of earlier data. In this respect this paper updates our 15 year old reviews [7,8]. The review is organized as follows: The structure of the Bechgaard and Fabre salts is presented in Section 2 with special consideration of entities achieving a subtle coupling to the electronic degrees of freedom. The underlying bond order wave (BOW) instability, always present in these salts, is quantitatively analyzed in Section 3. The AF and SDW magnetic ground states are described in Section 4, which also includes the often ignored coupling between magnetic and structural order parameters. Section 5 considers charge ordering (CO) effects in the Fabre salts with a special investigation of the role of the anions and methyl groups in the stabilization of the CO ground state. Section 6 is devoted to the SP transition and its coupling with the CO. Section 7 will review the most recent findings concerning the influence of the anion ordering (AO) transitions on the electronic structures. Finally we conclude this review by summarizing the influence of the structural degrees of freedom on the generic phase diagram of the Fabre and Bechgaard salts.
2. Basic Aspects of the Crystallographic Structure in Relationship with Electronic Properties
2.1. Structural Arrangement
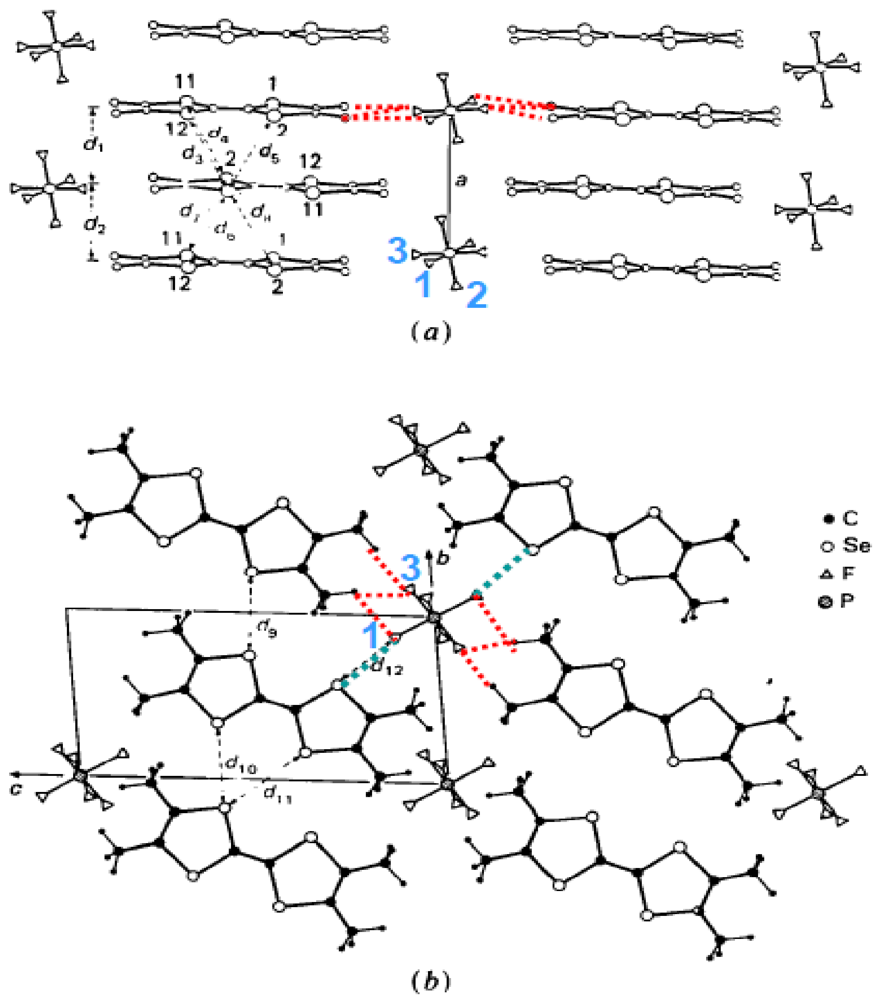
As shown in Figure 1, Bechgaard and Fabre salts are made of a zig-zag stacking of TMTSF and TMTTF molecules arranged nearly perpendicularly to the 1D direction, a. In the short direction of the molecule, stacks, shifted from each other along b, form (a, b) donor layers. In the long direction of the molecule, out of phase first neighboring zig-zag stacks delimit methyl group cavities which are filled by the anions X. This packing leads to an alternation of layers of well coupled donors with anions along c. Two aspects of the structure at ambient conditions are noticeable: (i) the slight dimerization of the zig-zag stack; and (ii) the presence of a sizeable thermal motion of the anion X in its cavity [9]. These salts crystallize in the triclinic space group P-1 with the inversion centers located in between the molecules and in the center of the methyl group cavities. If one ignores the slight stack dimerization and the location of the anions close to the shortest zig-zag bond, the structure has an underlying monoclinic C2/m symmetry (a: binary axis, b’ = 2b-a, c) tending to promote a 21 screw axis symmetry in the stack direction [8]. In this respect it is interesting to remark that both Bechgaard and Fabre salts, with X = PF6, undergo, at 5.5GPa and 8.5GPa respectively, a first order phase transition to a monoclinic (a: binary axis, b’, c) structure [10]. In the (a, b) donor layer, this discontinuous structural change could be achieved by the relative shift, along a, of first neighboring stacks.
Figure 1.
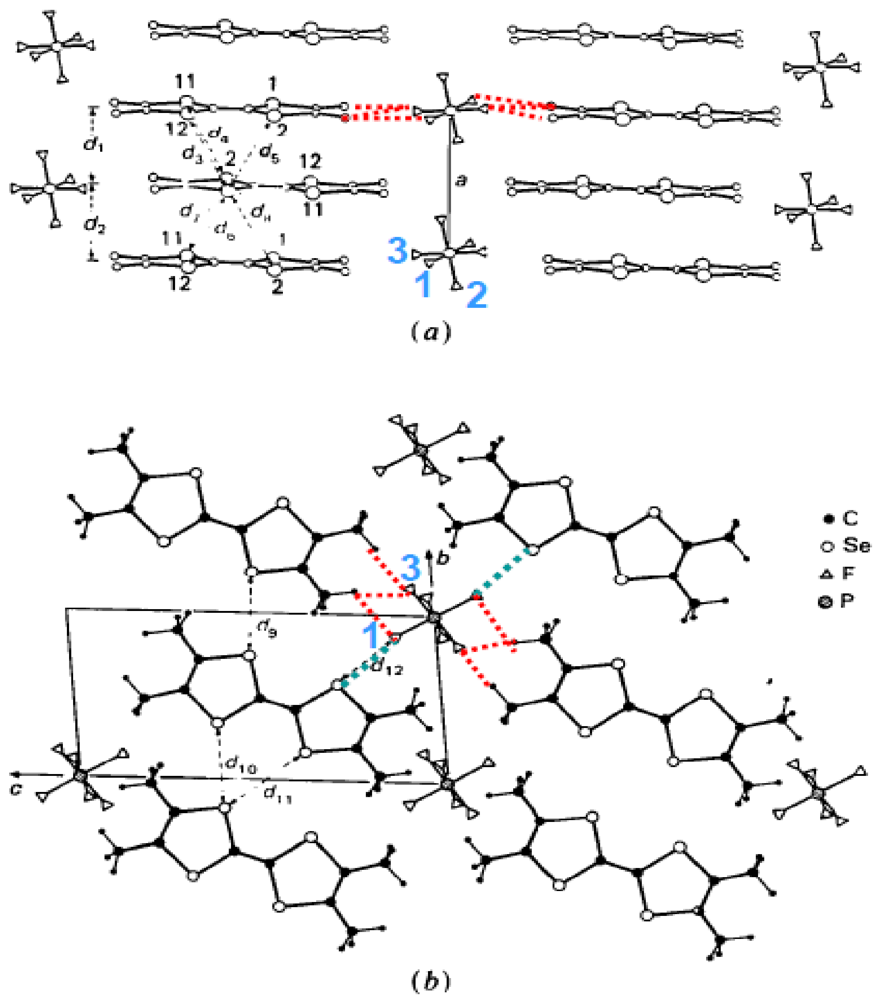
Structure of (TMTSF)2PF6: (a) Projection in the (a, [011]) plane; (b) Projection along a. The F atoms are labeled. In (b) the methyl groups located in the low temperature neutron scattering structural refinement [11] are shown. The shortest F(1)-Se distances, F(1)-H and F(3)-H bonds are indicated by the blue and red dotted lines respectively.
Figure 1.
Structure of (TMTSF)2PF6: (a) Projection in the (a, [011]) plane; (b) Projection along a. The F atoms are labeled. In (b) the methyl groups located in the low temperature neutron scattering structural refinement [11] are shown. The shortest F(1)-Se distances, F(1)-H and F(3)-H bonds are indicated by the blue and red dotted lines respectively.
2.2. Dimerization of the Stack
In a 1D description of the quarter filled interacting electron gas, the dimerization of the zig-zag stack activates an umklapp 4kF = a* electron–electron scattering term which, in presence of sizeable Coulomb repulsions, facilitates the charge localization (kF is the Fermi wave vector of the quarter filled 1D electron gas) [12]. The strength of this umklapp process depends of the amplitude of the dimerization gap 2ΔD which opens at ±a*/2 in the band structure well below the Fermi level. ΔD amounts to the difference of transfer integrals (in presence of the Hartree anion potential) on the two inequivalent bonds of the zig-zag. ΔD calculated by the DFT differs from ΔD calculated by the Extended Hűckel Theory (EHT) which does not take into account the anion potential. For example in (TMTSF)2ClO4 at room temperature (RT) DFT gives ΔD = 26 meV [13], while EHT gives ΔD = 37–54 meV (see Table 4.3 in [2]). Another complication arises if one considers the interstack coupling: the inter-chain transfer integrals lead to a significant variation of ΔD along b*. In (TMTSF)2ClO4ΔD varies from ~50meV in (a*/2, 0) to ~0meV in (a*/2, b*/2) [13] at RT. ΔD decreases upon cooling and under pressure, while ΔD increases from the TMTSF’salts to the TMTTF’salts. ΔD is controlled by the shape and volume of the anion which deforms the donor packing via its interaction with the terminal methyl groups.
2.3. Anion Cavities
Structural effects associated to anions require a special attention because the anions are located in soft cavities delimited by the methyl groups of the TMTSF or the TMTTF molecules. The anions fit these cavities in a more or less compact manner depending upon the volume and shape of the anion [14,15,16]. The anions were considered in earlier studies as spacers acting on the inter-layer distance as does the pressure [17]. In fact recent works summarized in this review show that their role is more subtle than it was previously believed. First, the anions are located in centro-symmetric cavities. This location causes no problem for centro-symmetric anions such as octahedron MF6 (M = P, As, Sb) or sphere (Br), but it implies at RT the existence of an orientation disorder for tetrahedral (ReO4, ClO4, BF4), triangular (NO3) or asymmetric linear (SCN) anions. Second, each anion experiences quite a symmetric environment from its six first neighbor donors [18] due to the six closest methyl groups. Among them, four groups are located in the (b’, c*) plane perpendicular to the stacking axis, while the two remaining groups, located in the (a, c*) plane, are significantly tilted with respect to a. These six methyl groups delimit three sets of three-fold symmetry axis close to the symmetry axis of the octahedral anions. The four planar methyl groups delimit two sets of two-fold symmetry axis close to the symmetry axis of the tetrahedral anions [18]. Third, the F or O outer atoms of the anion form weak H-bonds with the closest methyl groups, especially at low temperatures when thermal disorder is reduced [19]. The 4K neutron scattering structural refinement [11], which allows precisely locating the H of the methyl groups of (TMTSF)2PF6, shows the formation of 6 H-bonds with F(1) and F(3) of the PF6 (red dotted lines in Figure 1). All these H-bonds are located in the plane perpendicular to the stacking axis a. In addition, the anion develops short contact distances with the Se of the closest donors. Figure 1 shows (blue dotted lines) the two short Se…F(1) contacts established in (TMTSF)2PF6. The anion and thermal dependences of the geometry of the S...F interactions have been analyzed in (TMTTF)2PF6, AsF6 and SbF6 [20]. Note that F(2), which presents the strongest disorder [11], is not involved in any interactions. Any change of the shape of the methyl group cavity, caused by an elastic deformation of the donor stack for example, should react on the location of the anions and vice versa.
These two kinds of short contacts provide respectively indirect (via the polarization of the H-bond network and of the intra-molecular σ bonds) [21] and direct (via the Se or S atoms) attractive interactions between the anion and the π holes cloud located in the inner part of the donor. In this respect infrared measurements provided evidence of a considerable electron-molecular-vibration coupling of the methyl groups of both TMTSF and TMTTF with the charges on the molecules [22]. These interactions are involved in the inter-chain coupling mechanism (see Section 3.1.2) and for this reason play a key role in the stabilization of various ground states exhibited by Bechgaard and Fabre salts such as the AO transition involving non centro-symmetric anions (see Section 7), the CO transition breaking the inversion symmetry (see Section 5) and the SP transition subject to an important magneto-elastic coupling (see Section 6).
2.4. Anion and Methyl Group Disorders
The location of anions in methyl group cavities provide flexibility in the structure due to the soft interface provided by the methyl groups between the anions and the core of the donors and the incomplete fit of the anion in the volume delimited by these methyl groups [14,15,16]. This soft environment tolerates the presence of disordered anions in the structure even for centro-symmetrical anions [9]. At ambient conditions, the non centro-symmetrical tetrahedral anions are disordered in Bechgaard and Fabre salts while they are ordered in others structurally related 2:1 salts, such as (BEDT-TTF)2ReO4, (t-TTF)2X with X = ClO4 and BF4 and (DMtTTF)2ClO4, built with donors which do not provide a methyl group environment at the anion.
The lattice softness is revealed by thermal expansion measurements. First, linear expansion coefficients, αi, are one order of magnitude larger in the PF6 salt than in the Br salt where the anion better fits the volume of its methyl group cavity [23]. Second, in salts incorporating PF6 or AsF6 anions, the rate of variation of αi upon heating becomes negative at high temperature [24]. This finding points out peculiar lattice dynamics, possibly due to “free” rotation and/or translation of the anions in their cavities. This effect, which is observed both for TMTTF and TMTSF salts, is mostly pronounced along the c* direction where layers of donors and anions alternate [23,24,25].
The enhanced values of Debye Waller factors obtained in structural refinements show the presence of an atomic disorder for both the methyl groups and the outer atoms of the anions. An orientation disorder is expected for non centro-symmetrical anions located in a centro-symmetric cavity. This disorder is removed at a well defined AO transition which will be the object of Section 7. Below we shall consider the case of centro-symmetrical octahedral anions such as PF6. At RT, F atoms exhibit a sizeable thermal motion but the finding of well defined maxima in electron-density map, obtained in the structural refinement of (TMTSF)2PF6 [9], show that there is no free rotation of the centro-symmetrical anion. At 20K, the anion disorder is almost completely suppressed especially for F(1) and F(3) atoms forming H bonds and a linkage with the Se (see Figure 1) [11]. However neutron refinements show that methyl groups sustain a certain disorder even at 4K [11], probably due to quantum-mechanical tunneling. NMR studies show that PF6 disorder is established upon heating above ~70K in (TMTSF)2PF6 [26]. More recent NMR studies performed in (TMTTF)2SbF6 [27,28] show, by the measurement of different activation energies in the molecular motion, that in fact anion disorder is progressively set upon heating. A detailed analysis [29] of the Debye Waller factor of (TMTSF)2AsF6 at 125K suggests that the disorder could take place between two well defined orientations of the AsF6 anion in its cavity. Several activation energies also govern the motion of the methyl groups in TMTSF salts [30,31,32,33].
Thermal variation of the principal directions of the dilatation tensor of (TMTSF)2PF6 [11] suggests a lattice modification around 60–50K which could be related to the “freezing out” of the PF6 disorder previously considered. More precisely, the thermal variation of the lattice expansion in the direction i divided by T, αi/T, or more likely the volume expansion divided by T, (Σiαi)/T, is proportional to the entropy derivative ∂S/∂T [34]. α┴/T taken from the data of [24] exhibits broad maxima around 30–40 K in (TMTSF)2PF6 before dropping to zero at lower temperature [34]. This finding implies that below these maxima a net decrease of the “lattice” entropy, probably due to the vanishing of the structural disorder, should occur. The best way to reduce simultaneously the PF6 and methyl group disorders is to link these two entities, as suggested from methyl protons NMR [32]. These findings have important consequences on the density wave instabilities which will be considered in Section 3.
The PF6 anion disorder could be even more significantly reduced by a well defined thermodynamic transformation recalling the AO transition (see Section 7). This could be achieved by an orientation ordering transition achieving a well defined orientation at the anion which oscillates between several positions in its methyl group cavity at high temperature [29]. Such a transition has been observed in (EDO-TTF)2PF6 [35] where the orientation ordering of PF6, accompanied by its shift, stabilizes a 3D pattern of CO and BOW distortions of the EDO-TTF stack. All these effects lead to a unit cell doubling which causes the 2kF MI transition of (EDO-TTF)2PF6. In P-1 triclinic (TMTSF)2PF6, where there is no cell doubling (above TSDW), the only possible symmetry breaking operation while keeping the translation symmetry will be the loss of inversion centers as for the CO transition of the (TMTTF)2X’s (see Section 5). However neutron scattering structural refinements of (TMTSF)2PF6 performed at 4K and 20K [11] do not provide any evidence of the loss of the inversion centers (Note that this also implies that PF6 does not shift from the centre of its methyl group cavity).
3. The Bond Order Wave Instability in the Bechgaard and Fabre Salts
3.1. The Interplay between 2kF BOW and SDW Instabilities in (TMTSF)2PF6
3.1.1. Quantitative Analysis of the 1D 2kF BOW Instability
All charge transfer salts of the TTF-TCNQ family exhibit a divergent 2kF CDW instability which, when coupled to the lattice degrees of freedom, drives a low temperature TP (~30–50 K) metal-insulator (MI) transition [1,36]. In TTF-TCNQ the electron-phonon coupling of the 1D electron gas with the lattice modes leads to the formation above TP of a Kohn anomaly in a transverse acoustic (TA) branch [37] which softening at TP leads to a static modulation of the intra-stack bond distances. This stabilizes a 2kF BOW ground state below TP. Such instability of a 1D metal with respect to a 2kF periodic lattice distortion (PLD) was first proposed by Peierls in 1955.
Above TP the 1D BOW structural instability gives also rise to critical fluctuations consisting, in reciprocal space, in a planar X-ray diffuse scattering (or diffuse lines on X-ray patterns) at the wave vector Q = G + q, where G is a reciprocal lattice wave vector and q ~ 2kF. The intensity, I(Q), of this diffuse scattering can be expressed in the form [36,38]:
I(Q)= │F(Q)│2 <│uq│2> (1)
In expression (1) uq is the atomic displacement (of the TA critical phonon mode at q in TTF-TCNQ for example) and F(Q) is the structure factor of the modulation. In the classical limit when the thermal energy kBT is larger than the critical phonon energy, ħΩ, one has:
<│uq│2 >≈ kBTχ(q) (2)
In expression (2) χ(q) is the CDW or BOW response function of the electron-phonon coupled system. For a standard Peierls chain χ(q) is peaked at the 2kF wave vector. In the regime of 1D fluctuations χ(q) has a Lorentzian dependence in q:
χ(q) = χ(2kF)/[1 + ξ2(q − 2kF)2] (3)
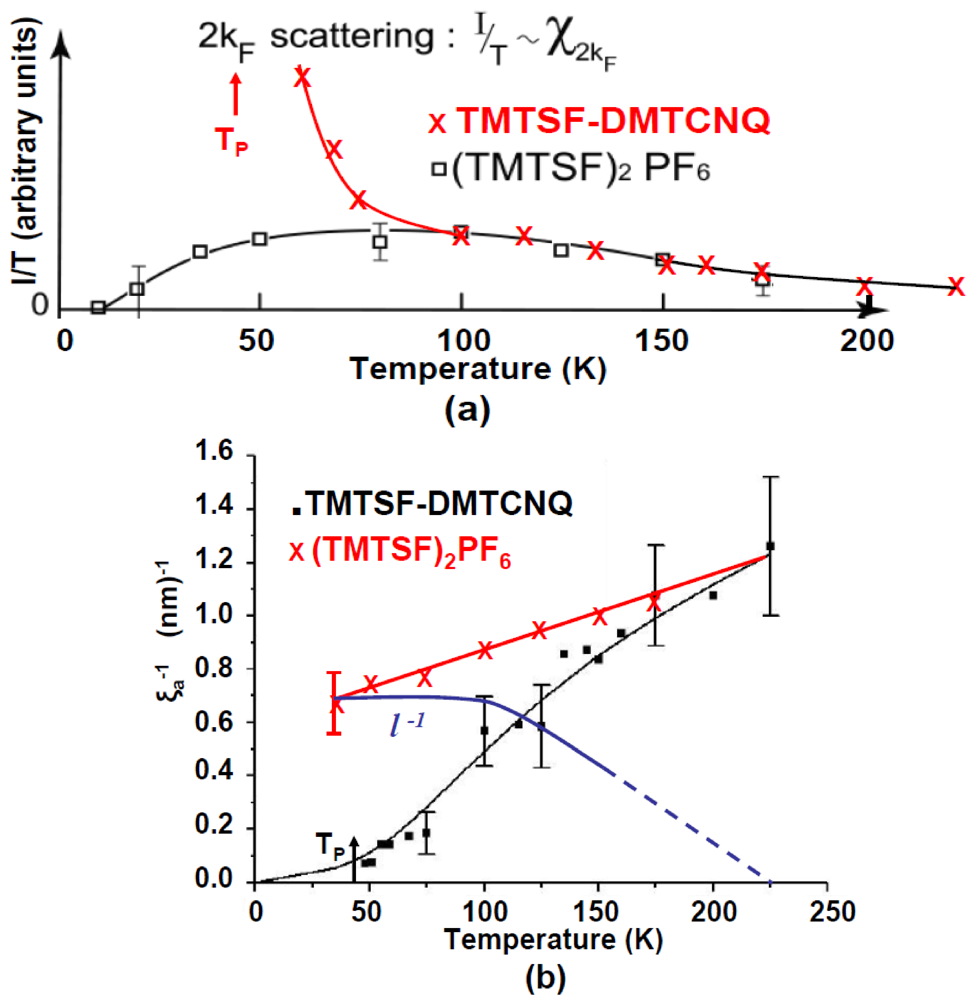
In expression (3) the correlation length of the intra-chain structural fluctuations, ξ, is given by the inverse half width at half maximum (HWHM) of χ(q). In the case of a Peierls instability (case of TMTSF-DMTCNQ considered below in Figure 2) both χ(2kF) and ξ diverge at TP.
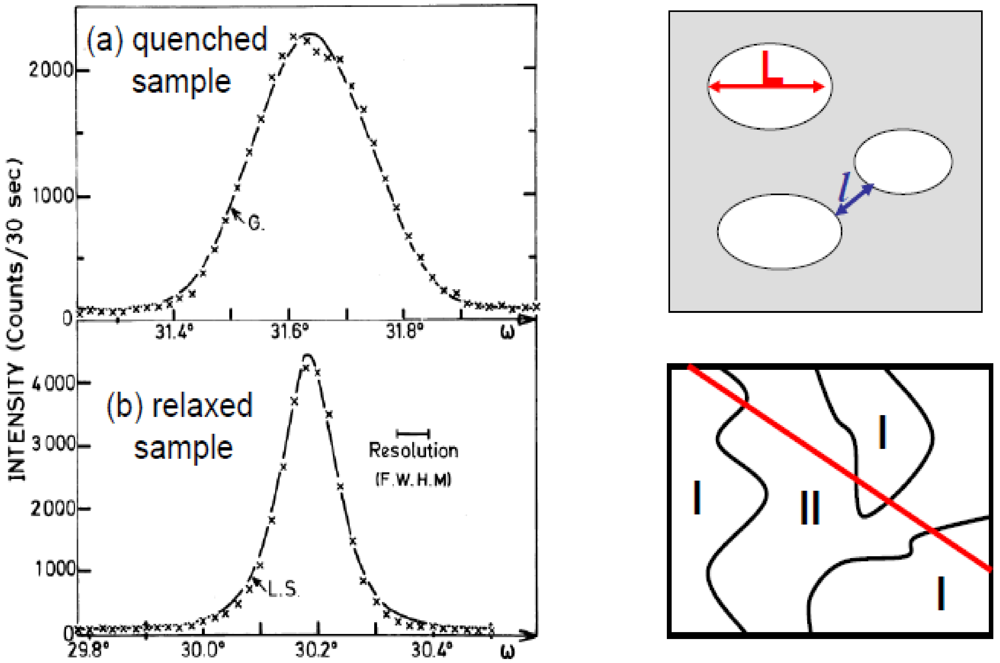
The surprise of the investigation of the Bechgaard salts was the observation in (TMTSF)2AsF6 [39] and PF6 [40] of X-ray diffuse lines, corresponding to the 1D 2kF BOW instability previously described, whose intensity I(Q) progressively vanishes upon cooling below ~50K. Below ~30K these structural fluctuations are no longer observable. The MI transition, which occurs at TMI = 12K in these salts, is thus not due to the setting of a Peierls ground state. The MI is caused by the establishment of a 2kF SDW order [1,2] by a mechanism similar to the one proposed by Slater in 1951 [41]. In this mechanism the divergence of 2kF SDW (AF in the model of Slater) correlations give rise to a 2kF periodic exchange potential which opens a gap at the Fermi level in the 1D band structure and thus drives the electronic system to an insulating ground state (here the 2kF periodic exchange potential plays the role of the 2kF PLD of the Peierls transition).
The reason for the low temperature vanishing of the 2kF BOW instability in the Bechgaard salts has never been elucidated. To this end, we compare below the 2kF BOW instability of (TMTSF)2PF6 with the 2kF BOW instability of TMTSF-DMTCNQ which diverges at the TP = 42 K Peierls transition [36]. This comparison is all the more justified because, both in (TMTSF)2PF6 and in TMTSF-DMTCNQ, the 2kF BOW instability develops on the same TMTSF stack which presents similar electronic characteristics in the two salts (same bandwidth, same band filling and comparable dimensionless Luttinger parameter Kρ~0.25 [8]).
Figure 2 quantitatively compares the thermal dependence of the 1D 2kF BOW fluctuations of (TMTSF)2PF6 [40] and of TMTSF-DMTCNQ [36,42]. Part (a) shows the 2kF susceptibility, χ(2kF), given by I(Q)/T in the expressions (1) + (2), while part (b) shows the inverse intra-chain correlation length ξa−1, given by expression (3). Above ~100K χ(2kF) behaves similarly in the two salts. Below 100K χ(2kF) of TMTSF-DMTCNQ grows rapidly and diverges at TP while χ(2kF) of (TMTSF)2PF6 saturates before decreasing below ~50K. Above ~150K ξa−1 of TMTSF-DMTCNQ and of (TMTSF)2PF6 are comparable. Below this temperature ξa−1 of TMTSF-DMTCNQ drops rapidly to reach zero at TP while ξa−1 of (TMTSF)2PF6 decreases very slowly.
The thermal dependence of ξa of TMTSF-DMTCNQ (named ξ/T below) has been quantitatively analyzed in [42] using exact calculations of the fluctuations of the Peierls chain (black continuous line of Figure 2b). Upon cooling, ξ/T is governed by the diverging growth of intra-chain electron-hole pair correlations associated with the increase of the thermal life time of the individual electron-hole pairs. In (TMTSF)2PF6 the saturation in the growth of ξa upon cooling means that the low temperature divergence of ξ/T expected for a standard Peierls chain is capped by a cut off length. We associate at this cut off length a “mean free path” l = v*τ, where τ corresponds to a non thermal lifetime of the electron-hole pair, and where v* is the “charge velocity” depending of the microscopic parameters of the TMTSF electron gas (v* amounts to ~vF/2 in TMTSF-DMTCNQ [42]). By adding these two independent contributions in quadrature:
ξa−2= ξ/T−2 + l−2 (4)
l−1 can be obtained if one assumes that ξ/T−1 is given by ξa−1 measured in TMTSF-DMTCNQ. Figure 2b gives the thermal dependence of l−1, deduced from expression (4), for (TMTSF)2PF6. l−1 increases upon cooling and saturates below 100K, as does χ(2kF). The life time τ smoothes the q dependence of the CDW response function near 2kF, with the result to kill the low temperature divergence of both ξa and χ(2kF).
Figure 2.
Thermal dependence of (a) the 2kF BOW susceptibility, χ(2kF); and (b) the inverse intra-chain correlation length ξa−1 in (TMTSF)2PF6 and in TMTSF-DMTCNQ. In (b) the thermal dependence of the inverse mean free path l−1 of the electron-hole pairs of (TMTSF)2PF6, given by (4), is given by the blue line.
Figure 2.
Thermal dependence of (a) the 2kF BOW susceptibility, χ(2kF); and (b) the inverse intra-chain correlation length ξa−1 in (TMTSF)2PF6 and in TMTSF-DMTCNQ. In (b) the thermal dependence of the inverse mean free path l−1 of the electron-hole pairs of (TMTSF)2PF6, given by (4), is given by the blue line.

l saturates at about 14 Å, which corresponds to one wave length 2a of the 2kF BOW. From this length one deduces an inverse electron-hole life time ħ/τ = ħv*l−1 of 43meV. This value is one order of magnitude larger than the phonon frequency, ħΩ = 5 meV, of the transverse acoustic branch which bears the Kohn anomaly in TTF-TCNQ [37]. As Ωτ << 1 the 2kF BOW dynamics of (TMTSF)2PF6 should occur in the anti-adiabatic limit and thus a Kohn anomaly should not form in the phonon spectrum. In this regime the dynamics is achieved by the critical growth of quasi-static 2kF fluctuations which are visualized in reciprocal space by the presence of 2kF X-ray diffuse lines above 30 K.
The life time τ of the electron-hole pairs could be caused by the orientation disorder of the anion. The change of orientation of the anion in its cavity should break both the short F…Se contact distance and the weak F…H-CH2 bonds (see Section 2.4). This will respectively modulate directly and indirectly (via the polarization of the H bonds and of the σ intra-molecular electrons [21]) the Hartree potential experienced by the π holes. These fluctuations of the Hartree potential should provide new scattering mechanisms of holes and electrons at the Fermi level, resulting in either an incoherent inter-stack charge transfer and/or intra-stack backward and forward scatterings. The inter-stack transfer and the backward scattering change the direction of the kF wave vector of the electron or of the hole of a pre-formed 2kF electron-hole pair. This change breaks the pairing, and thus reduces the lifetime τ of the electron-hole pairs.
Using the value at saturation of l, the FS should be broadened by ħ/τ~43 meV. This lifetime broadening amounts to the warping of the Fermi surface due to coherent interchain hopping (2t'b = 44meV from the transverse plasma edge measurement at 25 K [22]). Thus, the warping of the FS should be smeared out by this lifetime effect on all the temperature range of observation of the 2kF BOW diffuse lines. In this framework it has been suggested from the absence of Drude absorption in intra-chain optical reflectance of (TMTSF)2ClO4 that the single particle transport is diffusive in all directions and temperatures and that the high dc conductivity in chain direction is due to collective sliding CDW [43]. Similar features are observed in (TMTSF)2PF6 [44], but the optical spectrum has been interpreted differently [45].
Note that this lifetime effect should similarly affect the 2kF SDW fluctuations also driven by the divergence of the electron-hole response function of the correlated electron gas. In particular a reduced SDW correlation length, similarly to ξ(T) of the 2kF BOW fluctuations reported in Figure 2b, should control the spatial dependence of the 2kF SDW fluctuations. Such SDW fluctuations have been probed by NMR [46] which however does not give any information on their spatial extent.
The TN = 12 K SDW transition of (TMTSF)2PF6 is achieved by a FS nesting process (see next section). However FS nesting should provide an efficient inter-chain coupling between density waves only if the FS is warped. Thus the setting of the inter-chain coupling requires a net sharpening of the FS upon cooling which can be achieved only if the extra scattering processes due to disorder vanish.
Evidence of a vanishing of the structural disorder comes from the thermal decrease of lattice entropy below ~30–40 K ascribed to the anchorage of the anions to the methyl groups (see Section 2.4). The cooperative locking of PF6 due to the formation of H-bonds with the methyl groups could explain the stiffening of the lattice revealed by the hardening of sound velocity below ~ 45 K in (TMTSF)2PF6 [47]. Interestingly, a similar hardening of sound velocity is observed at the 24 K AO transition of (TMTSF)2ClO4 when the orientation ordering of the ClO4 is accompanied by its linkage to the methyl groups (see section 7.3) [48]. This scenario is sustained by microwave transport measurements showing a dimension crossover in the charge transport at about the same temperature, TX ~ 35 K [49,50], at which the disorder vanishes.
Note that if the transverse coupling t'b is only considered, the warping of the FS and thus the 1D-2D spatial deconfinement of the electron gas should be effective at a larger TX ~ t'b/π ~ 80 K. However if the confinement due to electron-electron repulsions is considered the effective t'b is reduced and it is estimated [51] that TX can be depressed below 50K. On the basis of available experimental arguments the generic phase diagram of [52] was built with TX ~ 80 K. However the expected development of inter-chain correlations below TX ~ 80 K are neither detected in the 2kF BOW X-ray diffuse scattering experiments nor in the thermal dependence of the 2kF SDW fluctuations measured by NMR [46]. All these features show that the nature of the dimension crossover and of the deconfinement transition is unclear in the Bechgaard salts and that the value of the crossover temperature TX remains debated in the literature [45,51,53]. Our finding of additional effects due to anion disorder adds new elements at the discussion.
(TMTSF)2PF6 exhibits below 200-150K critical 1D SDW fluctuations, probed by T1−1 NMR measurements [46]. These fluctuations occur in the same temperature range where the 1D 2kF BOW fluctuations are also detected, which means that at high temperature (TMTSF)2PF6 exhibits both 2kF SDW and 2kF BOW instabilities. Below T* ~ 30 K inter-chain 2kF SDW critical fluctuations, due to the FS nesting process, develop [45] and the 2kF BOW fluctuations are no longer detected. However the precise reason of the vanishing of the BOW fluctuations below T* requires clarifications which will be the object of the next section.
3.1.2. Interchain Coupling between the 2kF Density Waves and the Vanishing of the Anion Disorder
It is now well established that the SDW transition of (TMTSF)2PF6 is due to a FS nesting because the interchain component qb ~ 0.20 ± 0.05b* [54] - 0.24 ± 0.03b* [55] of the SDW order, obtained by NMR, is close to the best nesting wave vector ~0.3b* of its FS determined from the 4 K structure [56]. In addition to this FS nesting process, inter-chain Coulomb interaction, g1┴, provides also an efficient coupling mechanism between CDW [1]. The inter-chain coupling g1┴ achieves the 3D ordering of 2kF CDW/BOW in charge transfer Peierls systems like TTF-TCNQ and TMTSF-DMTCNQ [36,38].
However there is the possibility for the Bechgaard salts, and as previously suggested for the Fabre salts [57], of conflicting interchain coupling mechanisms if Coulomb interactions, g1┴, and FS nesting tend to promote different b* transverse periodicities: g1┴ (possibly mediated by the anion shift–see below) favoring qb = 1/2b*, as observed for the AO and SP modulations (see Section 7 and Section 6), while the nesting of the FS favoring qb ~ 1/4b* as discussed above.
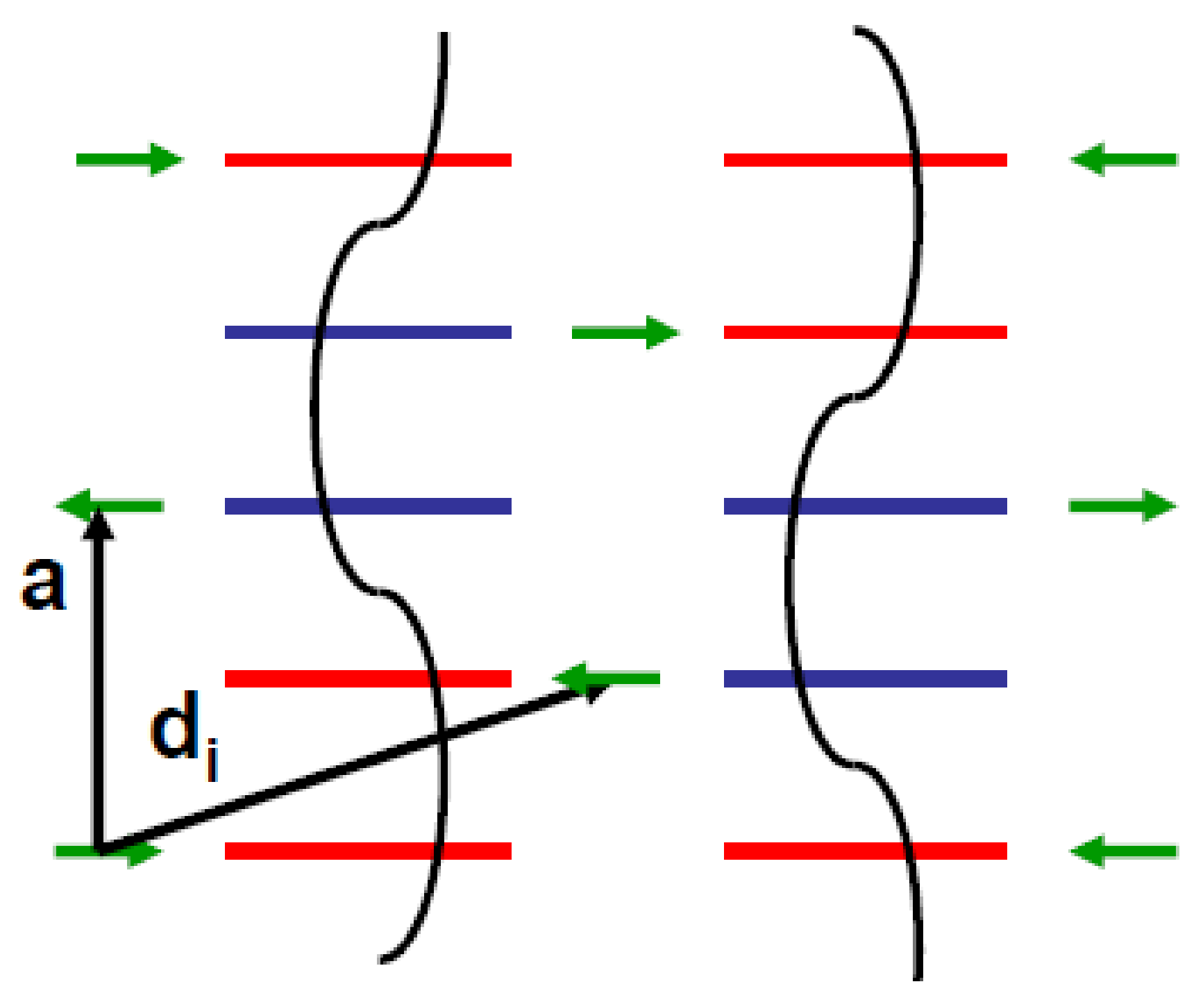
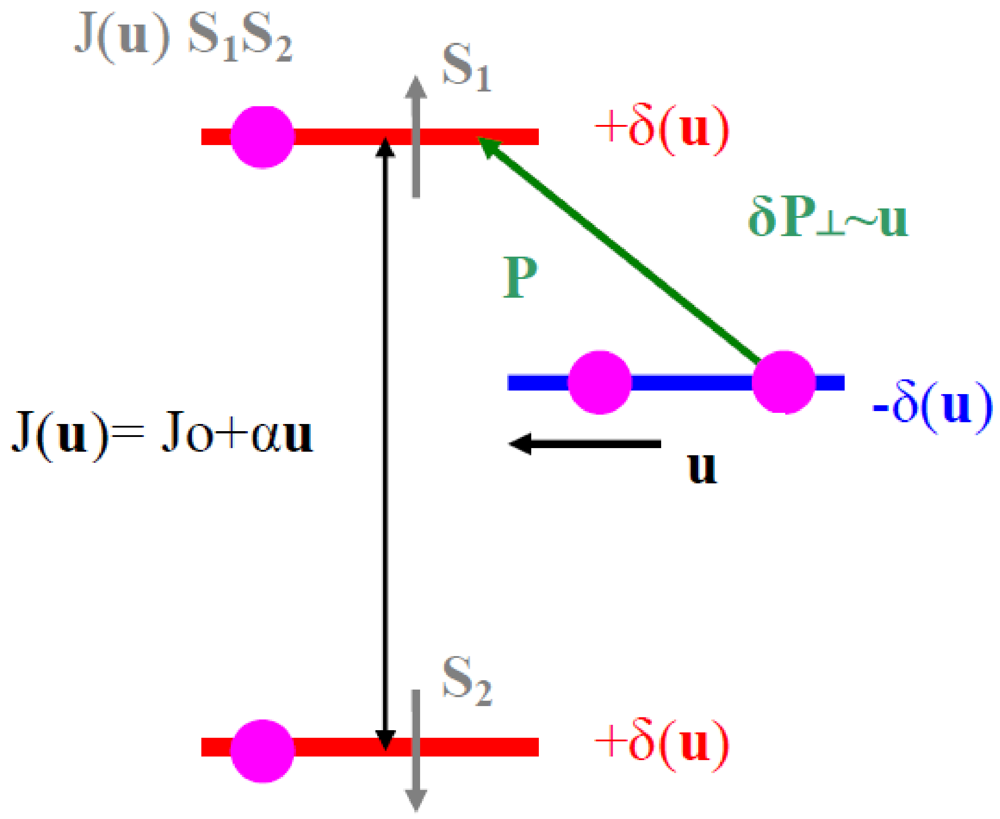
In all the superstructures achieving a transverse order between BOW, such as those formed at the (1/2, 1/2, 1/2) AO transition [58,59] (see Section 7.2), the shift of the anion from the inversion center seems necessary to stabilize the 3D pattern of 2kF BOW in the Bechgaard and Fabre salts. In these superstructures:
- - The (2kF)−1 periodicity is achieved in stack direction by repeating periodically two neighboring maxima of hole concentration with two neighboring minima of hole concentration.
- - The anion shift allows to achieve a 3D coupling between neighboring 2kF CDW by locking a maxima of the 2kFπ hole CDW on the molecule towards which the anion approaches and concomitantly a minimum of π hole density on the molecule that the anion leaves.
- - By combining these two features, the 3D CDW pattern requires a synchronous staggered shift of near-neighbor anions along a, along the d1 = a − b + c direction (which achieves the coupling of anions with the closest Se atom of the donor) and along the d2 = a + b + c direction (which achieves the coupling of the anions with the methyl groups and the polarization of σ bonds of the donor).
Figure 3 resumes this synchronous CDW – anion shift pattern.
Figure 3.
Schematic representation in the (a, di) plane of the array of 2kF CDW (in black) coupled to anion shifts (in green). Donors with an excess (defect) of hole are shown in red (blue).
Figure 3.
Schematic representation in the (a, di) plane of the array of 2kF CDW (in black) coupled to anion shifts (in green). Donors with an excess (defect) of hole are shown in red (blue).

In this model the CDW wave vector q allowing such π phase shifts between the anion displacements is given by:

The relationships (5) lead to ± qb + qc = 0 (mod.1) which has two solutions: qb = qc = 1/2 and qb = qc = 0. The first solution (1/2, 1/2, 1/2) corresponds to the 2kF BOW/AO modulation, qAO, (see Section 7.2) and the SP modulation, qSP (see Section 6). The second solution (1/2, 0, 0) corresponds to the 2kF BOW/AO modulation in the NO3 salts (see Section 7.5). As usual the 2kF BOW has a phase shift of π/2 with the 2kF CDW. In [60] this model is used to derive the anion shift pattern setting the 4kF CDW or CO.
In this picture the suppression of the 2kF BOW at low temperature could be explained by the impossibility to establish an inter-chain coupling between CDW via the anion shift. The absence of a low temperature anion shift, ascribed the lock-in of the PF6 to the methyl groups via the formation of H-bonds, was previously discussed in Section 2.4 and Section 3.1.1.
3.2. Evolution of the 2kF BOW Instability along the TMTSF-TMTTF Series
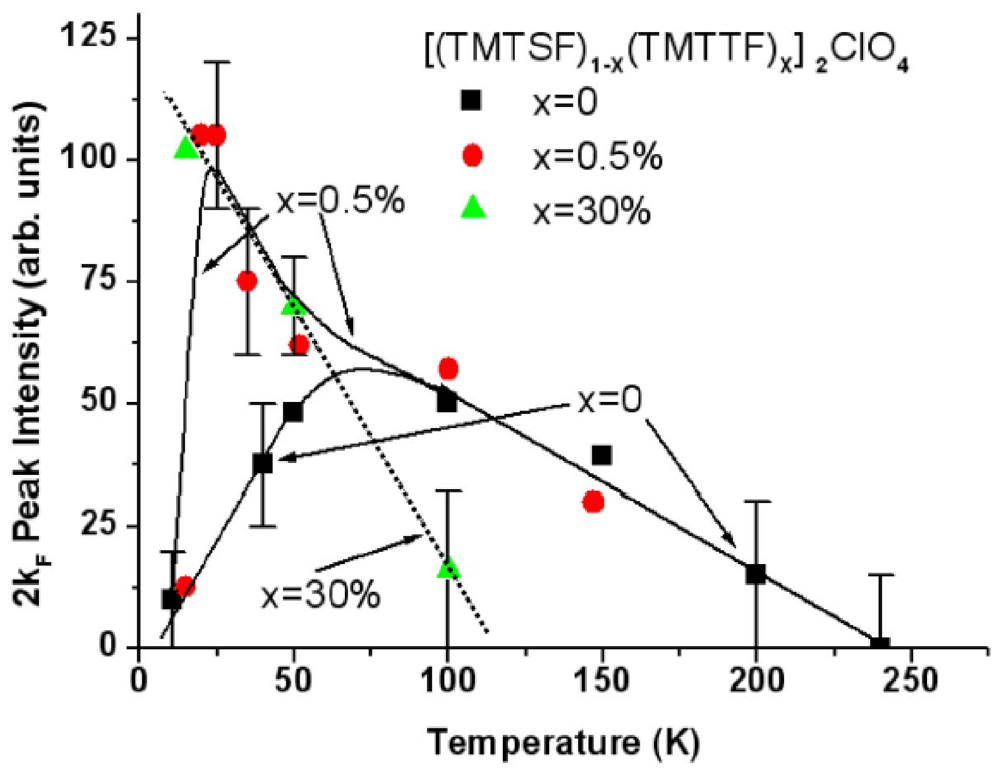
The 1D 2kF BOW instability of (TMTSF)2ClO4 [40] behaves as the one of (TMTSF)2PF6 and AsF6. Figure 4 reports the thermal dependence of the 2kF peak intensity, I(2kF) given by expression (1), of the X-ray diffuse scattering from (TMTSF)2ClO4. In this salt the low temperature vanishing of I(2kF) has to be associated with the 3D critical growth below ~40 K of the (0, 1/2, 0) AO structural instability promoting the uniform ClO4 ordering and anion shift in stack direction (see Section 7.3) [61]. Figure 4 shows surprisingly that the thermal dependence of the 2kF BOW fluctuations of (TMTSF)2ClO4 is strongly modified upon substitution by the TMTTF [62].
Figure 4.
Thermal dependence of the 2kF peak intensity, I(2kF), in the solid solution [(TMTSF)1−x(TMTTF)x]2ClO4 for x= 0 (black squares), 0.5% (red circles) and 30% (green triangles) of TMTTF deduced from the data reported in [40] and [62].
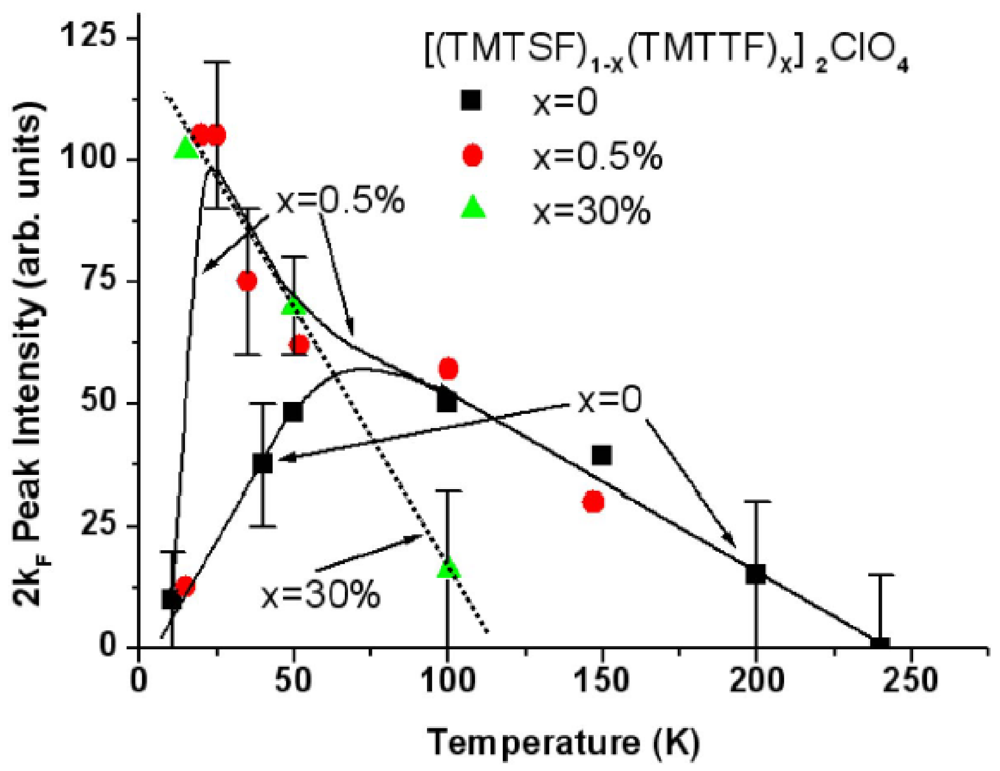
A low temperature divergence of I(2kF) appears for very small amount of TMTTF substituent. For x = 0.5% this additional divergence pushes down at lower temperatures the drop of I(2kF) which remains due to the competing AO instability as in pure (TMTSF)2ClO4. The thermal behavior of I(2kF) in the x = 0.5% salt resembles the one observed (see Figure 2 in [63]) in the (TMDTDSF)2PF6 iso-structural salt built on a donor hybrid between the TMTSF and TMTTF molecules. However in (TMDTDSF)2PF6 the drop of I(2kF) below 20 K is not due to an AO transition but, as for (TMTSF)2PF6, to the stabilization of a SDW ground state below 7 K. Note that because of the orientation disorder of the TMDTDSF molecule in the structure [64], I(2kF) does not vanish completely at low temperature in (TMDTDSF)2PF6 so that a 1D SP short range order (on ξa~25 Å) still coexists with the 2kF SDW modulation.
Only the low temperature divergence of I(2kF) remains in the x = 30% salt below 100K. This low temperature divergence of the 2kF diffuse scattering intensity recalls the divergence of the SP fluctuations observed in (TMTTF)2Br and PF6 below 80 K and 60 K respectively [65].
In TMTTF salts the occurrence of a low temperature divergence of I(2kF) coincides with the presence of a high temperature charge localization [51,52] which leaves the spin degrees of freedom available for a low temperature SP or AF instability. This charge localization manifests below a temperature, Tρ [66], higher than the onset temperature of the critical growth of the SP fluctuations. In the x = 30% salt the 2kF fluctuations detected below the spin-charge separation temperature Tρ [67] are thus the structural fingerprint of an incipient SP instability which fully diverges in (TMTTF)2PF6 .
In conclusion the [(TMTSF)1−x(TMTTF)x]2ClO4 solid solution illustrates quite well when x increases that the evolution from a 2kF BOW instability, which vanishes at low temperature, to a divergent SP instability follows the growth of the 4kF charge localization
3.3. The Spin-Peierls Instability
(TMTTF)2PF6 develops 2kF SP critical fluctuations below about 60 K (taken as the mean field transition temperature of the SP transition, TSPMF). The critical nature of the SP fluctuations is assessed by the divergent growth of the SP susceptibility, χ(2kF) [7,8,65], and of the intra-chain correlation length, ξSP; both quantities being defined by expression (3). The divergence is achieved at TSP ~ 17 K, temperature at which a long range (1/2, 1/2, 1/2) SP stack tetramerization occurs [68].
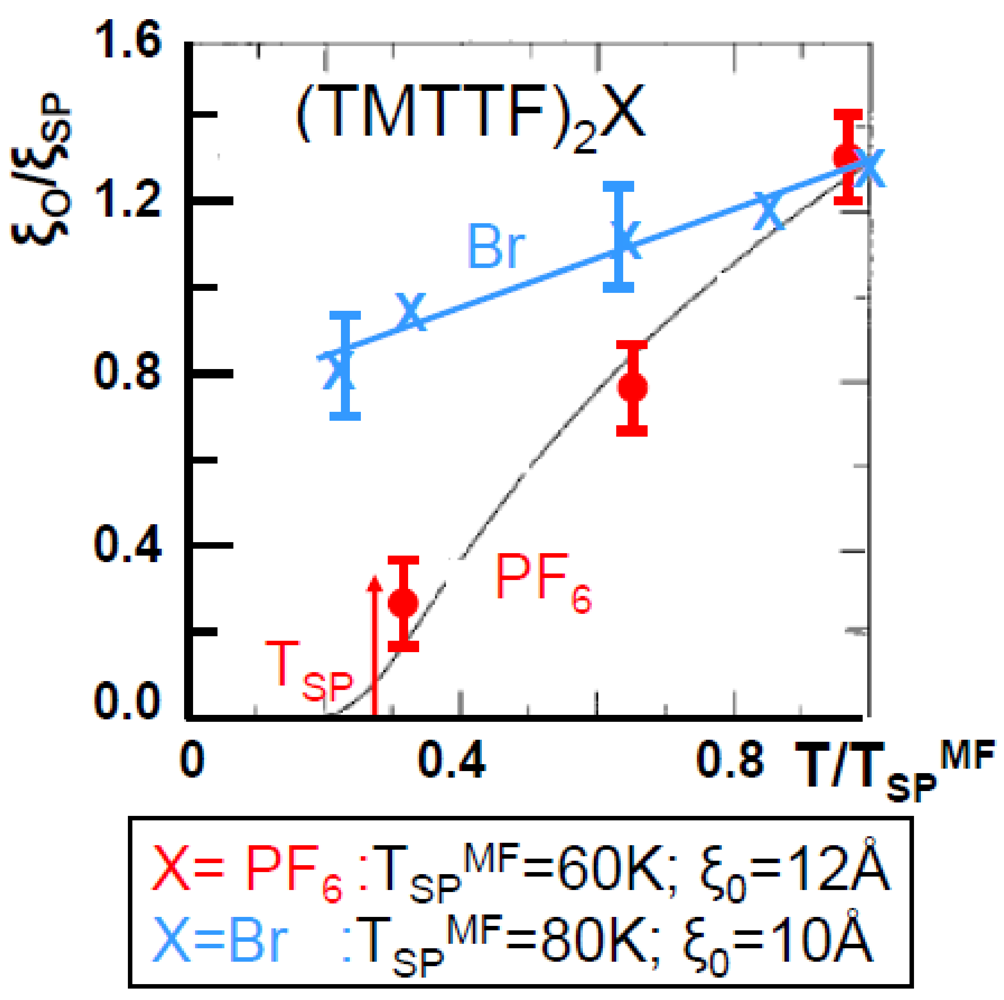
Figure 5 compares the thermal dependence of ξSP−1 of (TMTTF)2PF6 and of (TMTTF)2Br. It clearly shows that the SP fluctuations of (TMTTF)2Br do not diverge. Thus Figure 5 resembles for the SP fluctuations of (TMTTF)2Br at Figure 2b for the 2kF BOW fluctuations of (TMTSF)2PF6. We shall consider at the end of this section this aspect of the SP fluctuations of (TMTTF)2Br.
Figure 5.
Thermal dependence of the inverse intra-chain correlation length ξSP−1 of the SP instability of (TMTTF)2PF6 (red dots) and of (TMTTF)2Br (blue crosses). TSPMF and ξ0 are indicated in the lower panel for each salt. The continuous line for the PF6 salt is ξSP−1 calculated [69] for the weakly localized SP chain.
Figure 5.
Thermal dependence of the inverse intra-chain correlation length ξSP−1 of the SP instability of (TMTTF)2PF6 (red dots) and of (TMTTF)2Br (blue crosses). TSPMF and ξ0 are indicated in the lower panel for each salt. The continuous line for the PF6 salt is ξSP−1 calculated [69] for the weakly localized SP chain.
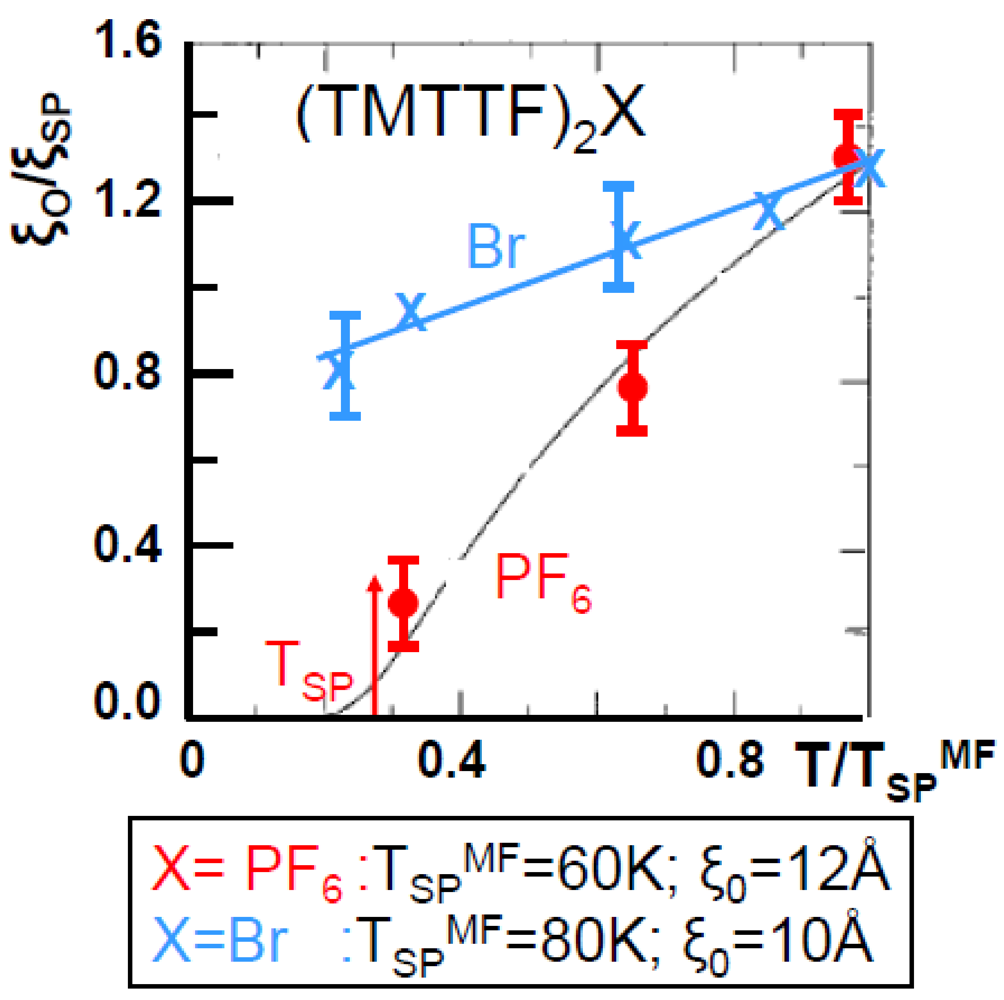
The thermal dependence of ξSP−1 of (TMTTF)2PF6 can be well accounted for by the calculation of the fluctuations of the weakly localized SP chain [69]. From this calculation one deduces that the SP coherence length ξ0 amounts to 12 Å. This value is very close to ξ0 ~ 10 Å found for the SP instability of (BCPTTF)2PF6 [7]. From the relationship linking ξ0 to TSPMF:
ξ0= ħvσ/πkBTSPMF (6)
one gets a “spin velocity” ħvσ ~ 0.2 eVÅ in (TMTTF)2PF6. A similar value ħvσ = 0.27 eVÅ is obtained in (BCPTTF)2PF6 where TSPMF ~ 100 K. For this last compound which can be described by the Heisenberg Hamiltonian coupling well localized spins one gets, with ħvσ = πJa/2, an AF exchange coupling J = 280K which is close to J = 330 K deduced from the fit of the thermal dependence of the spin susceptibility of (BCPTTF)2PF6 [70]. If the same analysis is performed for (TMTTF)2PF6, where the holes are less localized than in (BCPTTF)2PF6, one gets, using the above quoted ħvσ value, J = 210 K, which is twice smaller than J = 420K obtained from the fit of the thermal dependence of the spin susceptibility [71]. A similar discrepancy is obtained for (TMTTF)2Br where expression (6) leads, with ξ0 ~ 10 Å and TSPMF ~ 80 K, to a J value of 230 K twice smaller than J = 500 K obtained from the fit of the thermal dependence of the spin susceptibility [71]. It thus appears that the analysis using the Heisenberg Hamiltonian, strictly valid for localized spins, does not hold for the TMTTF salts where the holes bearing the spins are too delocalized.
Let us remark that in the TMTTF salts kBTSPMF amounts to the energy of the transverse acoustic phonon mode, ħΩ~5meV, which controls the dynamics of the SP instability. With kBTSPMF ~ ħΩ, the SP transition should occur in the adiabatic limit with however a strongly damped soft mode (see Figure 4b in [72]). This is consistent with the adiabatic limit used in [69] for the calculation of ξSP-1 shown in Figure 5. However with kBTSPMF ~ ħΩ, (TMTTF)2PF6 should not be too far from the adiabatic-antiadiabatic crossover. As TSPMF decreases when the size of the anion increases, the AsF6 salts could be located in the anti-adiabatic regime and the SbF6 salt in the gapless regime where the SP ground state cannot be stabilized (see Section 6.2 and the analysis performed in [73]). The location of the SbF6 salt in the gapless regime agrees with the non detection of SP X-ray diffuse scattering fluctuations in this salt. Due to the absence of a SP instability the SbF6 salt undergoes an AF transition. The SCN salt behaves similarly. However the mutual exclusion between AF and SP instabilities is not general among the TMTTF salts because the Br salt exhibits below 80 K quasi-1D SP fluctuations which coexist with AF fluctuations whose divergence [74] stabilizes below TN = 13 K the AF ground state.
As previously mentioned, the thermal dependence of the SP fluctuations of (TMTTF)2Br resembles those of the 2kF BOW fluctuations of (TMTSF)2PF6. Figure 5 shows that ξSP reaches ~13 Å before the vanishing of the SP fluctuations, a similar value of ξBOW (14 Å) was found in (TMTSF)2PF6 (Figure 2b). The vanishing of the SP fluctuations in the Br salt could have the same origin as for the BOW fluctuations of (TMTSF)2PF6. However there is no low temperature structural refinement in the Br salt allowing sustaining this assertion. Such a structure would be all the more desirable than thermal expansion measurements [23] show that the lattice (as well as electronic properties, see Section 4.1) exhibits unusual features just above TN. Upon cooling below 18 K αc* abruptly drops, exhibits a negative maximum around 17 K, then gently increases towards zero. These low temperature thermal expansion anomalies, which recall glassy phenomena involving ethylene groups in (BEDT-TTF)2X [3], could be also associated to the freezing of the disorder of methyl groups of the TMTTF.
4. The Magnetic Ground States of the Bechgaard and Fabre Salts
At ambient pressure (TMTTF)2Br undergoes a commensurate AF order at TN = 13 K while (TMTSF)2PF6 undergoes an incommensurate SDW order at TSDW = 12 K. Pressure studies show that the AF ground state evolves into a SDW ground state when the electron localization (i.e., Tρ), occurring on the TMTTF side of the generic phase diagram drops [51,52]. This section will show that, because of the presence of a sizeable magneto-elastic coupling, the magnetic ground states of the Fabre and Bechgaard salts, which involve also structural degrees of freedom [65], are quite a bit more complex than those expected for a simple magnetic transition. This magneto-elastic coupling is also responsible of the occurrence of SP transitions which will be considered in Section 6.
4.1. The AF Phase of (TMTTF)2Br and the Pre-Transitional Anomalies
AF order which develops below a 2nd order phase transition in (TMTTF)2Br stabilizes a qAF = (1/2, 1/4, ?) commensurate magnetic modulation [75]. The magnetic order is accompanied by a structural modulation at the qS = (1, 1/2, ?) reduced wave vector (the component 1a*, corresponding to the 4kF wave vector, means that the superstructure reflections are detected in H odd layers of Bragg reflections [65]). As qS = 2qAF the structural modulation should result from a “magneto-elastic” coupling which will be considered below.
The coupling between the structural and AF order parameters is analyzed in Annex A.1 using the simplest Landau development of the free energy. Its minimization gives two solutions shown in Figure 6.
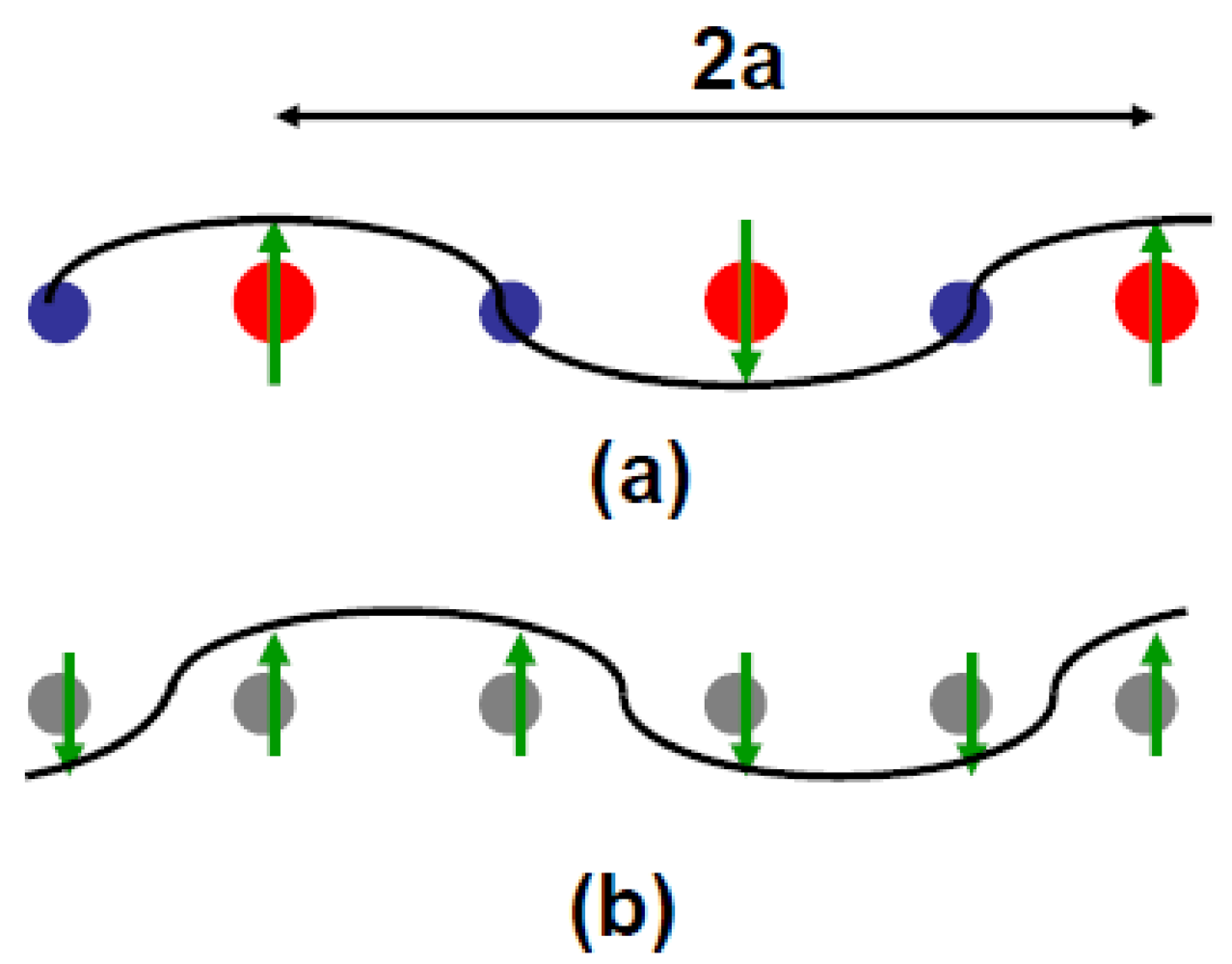
Figure 6.
(a) Mixed AF–CO order where the hole rich molecule bears the spin and the hole poor molecule is a node of the magnetization wave; (b) AF modulation where each site bears magnetization and the same charge. In (b) the nodes of the magnetization wave are located on one bond out of two. The spin direction is given by the sign of η(q) defined in Annex A.1. In (a), the charge rich/poor (+ρ/–ρ) sites are represented by the red/blue circles of a different size. The dimerization of the stack is ignored in this figure.
Figure 6.
(a) Mixed AF–CO order where the hole rich molecule bears the spin and the hole poor molecule is a node of the magnetization wave; (b) AF modulation where each site bears magnetization and the same charge. In (b) the nodes of the magnetization wave are located on one bond out of two. The spin direction is given by the sign of η(q) defined in Annex A.1. In (a), the charge rich/poor (+ρ/–ρ) sites are represented by the red/blue circles of a different size. The dimerization of the stack is ignored in this figure.
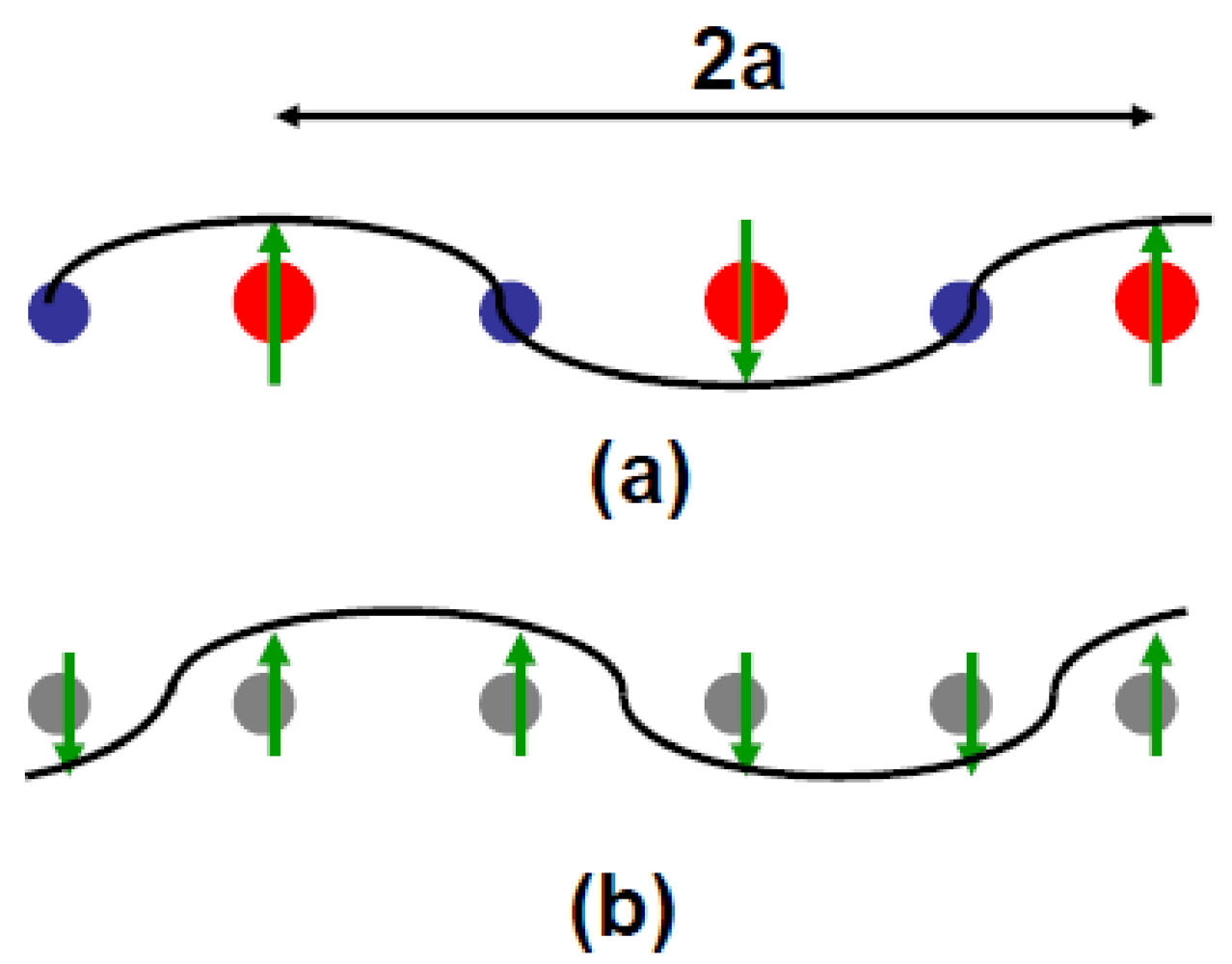
Solution (a) corresponds to an AF order with one site out of two bearing the magnetization, while solution (b) gives an AF order where all the sites bear the magnetization. In the Landau theory of Annex A.1 only solution (a) gains energy by setting a 4kF structural modulation. NMR lineshape analysis [75] better agrees with the spin configuration shown in (a). In the schematic representation of Figure 5a the presence of a spin on one site out of two is accompanied by an excess of hole on the magnetic site and a defect of hole on the non magnetic site. This leads to the formation of a 4kF CDW or CO [76]. CO should be accompanied by a 4kF internal deformation wave of the TMTTF molecules in phase with the charge density per molecule. The CO pattern could be also stabilized by a displacement wave of the Br towards the molecule bearing the excess of hole (if there is no H-bonds blocking the anion shift), recalling the one found in the structural refinement of δ-(EDT-TTF-CONMe2)2Br in its CO ground state [77]. As previously mentioned in Section 3.3, (TMTTF)2Br exhibits unusual features few degrees above TN. In addition to the lattice thermal expansion anomaly previously reported, (TMTTF)2Br exhibits below T* ~ 18–22 K anomalous electronic properties:
- - The thermal dependence of the electrical conductivity exhibits, depending on the measurements, either a kink at T* or a slope anomaly at T*, or even a plateau below T* (to my knowledge the T* anomaly was first reported in Figure 1 of [78]).
- - A broadening of the NMR spectra, interpreted as being due to non-homogeneities in the electronic states, is observed below T* [80].
(TMTTF)2Br bears some resemblance with (TMTTF)2SCN which also stabilizes the same modulations. (TMTTF)2SCN (see Section 7.4) sets at TCO = 160K an anti-ferroelectric (1, 1/2, 1/2) CO superstructure followed at TN = 7 K by a qAF = (1/2, 1/4, ?) magnetic order while in (TMTTF)2Br both modulations are established at the same 13K transition. This analogy is better revealed by dielectric permittivity measurements showing in both salts upon cooling an increase of dielectric constant, probably due to the growth of CO ferroelectric segments on individual stacks, followed by an abrupt drop of the dielectric constant ~20K above TCO in (TMTTF)2SCN [81] and below ~T* in (TMTTF)2Br [82], probably when the inter-chain anti-ferroelectric coupling develops. 3D CO local order should induce the charge non homogeneities revealed by NMR [80] below T* in (TMTTF)2Br. These charge non homogeneities could trigger local random Br displacements, possibly inducing methyl group disorder, which could cause the lattice expansion anomaly observed along the c* direction [23].
4.2. The SDW Phase of (TMTSF)2PF6
(TMTSF)2PF6 stabilizes below TSDW = 12 K an incommensurate qSDW = (1/2, ~1/4, ?) magnetic modulation [54,55] stabilized by the nesting of its FS [56] and where the exchange field due to the intra-site coulomb repulsion U opens a full gap at the Fermi energy. However the phase transition of (TMTSF)2PF6 exhibits additional features. First, the 2kF SDW order is stabilized by a first order transition [83], while the Peierls or Slater transition caused by the simple divergence of CDW/BOW or SDW electron-hole response functions is of second order [1,2]. It follows that the order parameter, the magnetization [71] or the square root of the 2kF satellite intensity [65] (see below), does not continuously vanish to zero at TSDW. There is also a jump of electrical conductivity at the MI transition. Second, a “specific heat” anomaly is observed at TSDW in thermal expansion measurements [25]; a feature not expected for a pure electronic SDW transition. Third, very weak X-ray satellite reflections (but too intense to be due to magnetic scattering) are observed below TSDW at the reduced qSDW (2kF CDW reflections) and 2qSDW (4kF CDW reflections) wave vectors [7,65,84].
Thus the ground state of (TMTSF)2PF6 must be described with 3 order parameters related to the 2kF SDW, 2kF CDW and 4kF CDW modulations. A simplified analysis of the phase transition is given Annex A.2 in the frame work of the Landau development of the free energy. This simple analysis shows, in agreement with experimental observations, that for an attractive enough coupling between the 2kF SDW and 2kF CDW order parameters a mixed SDW-CDW modulated ground state can be directly obtained through a first order transition.
The stabilization of a first order phase transition particularly requires that the Landau coefficients:
- - a1, associated to the 2kF SDW order parameter, changes of sign near TSDW, which means that the 2kF SDW susceptibility, a1−1, should diverge. Such a divergence is detected by NMR [46].
- - a2, associated to the 2kF CDW order parameter, should be very small, which means that the system should present incipient 2kF CDW instability. This is the case above ~30K for the 2kF BOW previously discussed in Section 3.1.
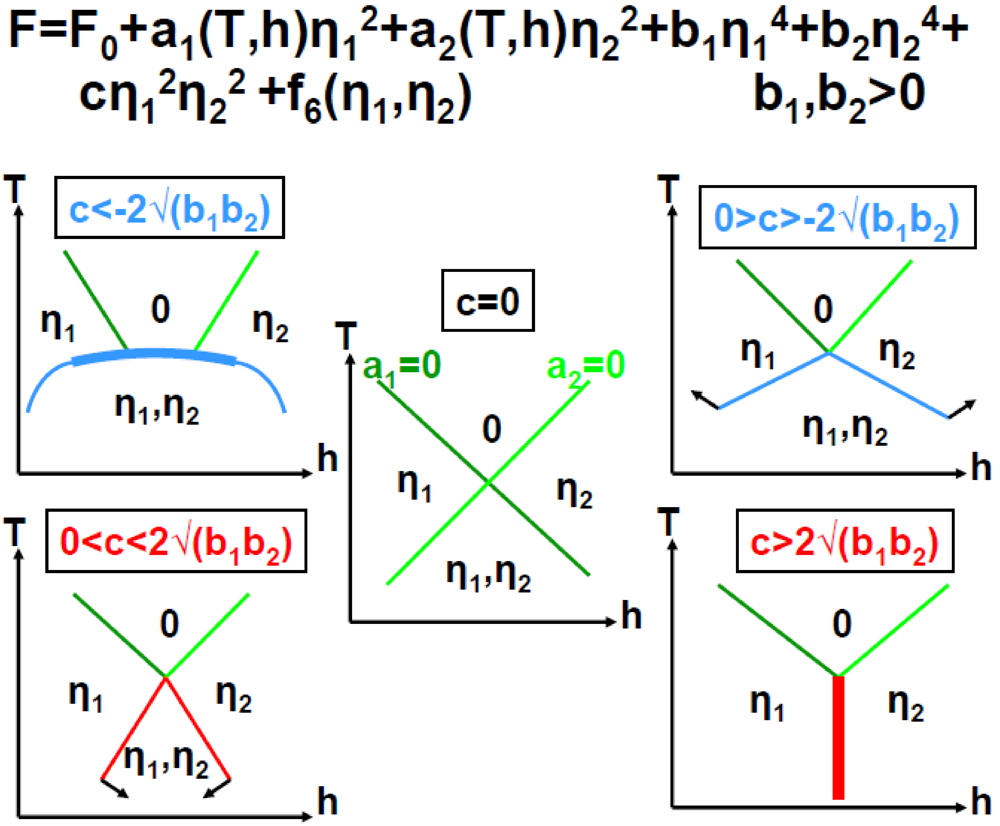
These conditions together with additional requirements given in Annex A.2, lead to phase diagrams shown in Figure 7.
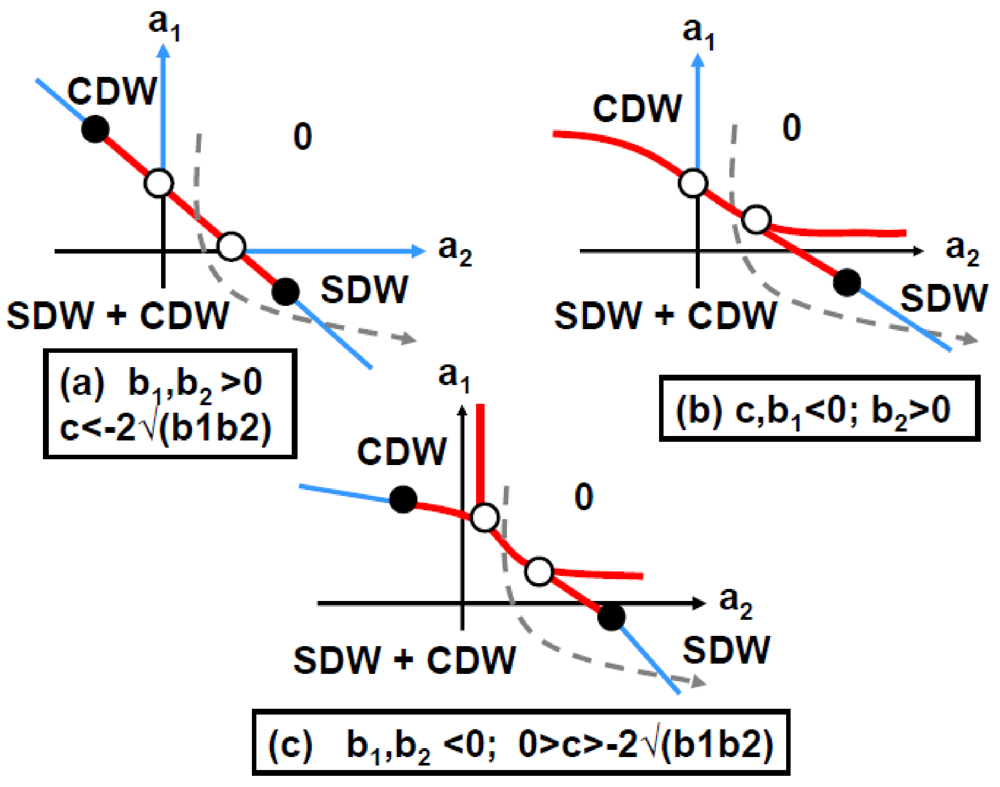
Figure 7.
2kF SDW/CDW phase diagrams in function of the Landau coefficients a1 and a2 for an attractive interaction, c, between the 2kF SDW and 2kF CDW order parameters (adapted from Figure 3 in [85]). (a) Corresponds to c < −2√(b1b2) and b1,b2 > 0 (case also displayed in Figure A.1); (b) Corresponds to c, b1 < 0 and b2 > 0; (c) Corresponds to 0 > c > −2√(b1b2) and b1, b2 < 0. The notations are defined in Annex A.2. Thin (thick) lines represent 2nd (1st) order transition lines. White dots indicate merging points between transition lines and black dots represent tricritical points. In each phase diagram the grey interrupted lines show a trajectory starting from the uniform phase “0”, crossing, after a 1st order transition, the SDW + CDW phase to finally reach, after a 2nd order transition, a pure SDW phase.
Figure 7.
2kF SDW/CDW phase diagrams in function of the Landau coefficients a1 and a2 for an attractive interaction, c, between the 2kF SDW and 2kF CDW order parameters (adapted from Figure 3 in [85]). (a) Corresponds to c < −2√(b1b2) and b1,b2 > 0 (case also displayed in Figure A.1); (b) Corresponds to c, b1 < 0 and b2 > 0; (c) Corresponds to 0 > c > −2√(b1b2) and b1, b2 < 0. The notations are defined in Annex A.2. Thin (thick) lines represent 2nd (1st) order transition lines. White dots indicate merging points between transition lines and black dots represent tricritical points. In each phase diagram the grey interrupted lines show a trajectory starting from the uniform phase “0”, crossing, after a 1st order transition, the SDW + CDW phase to finally reach, after a 2nd order transition, a pure SDW phase.
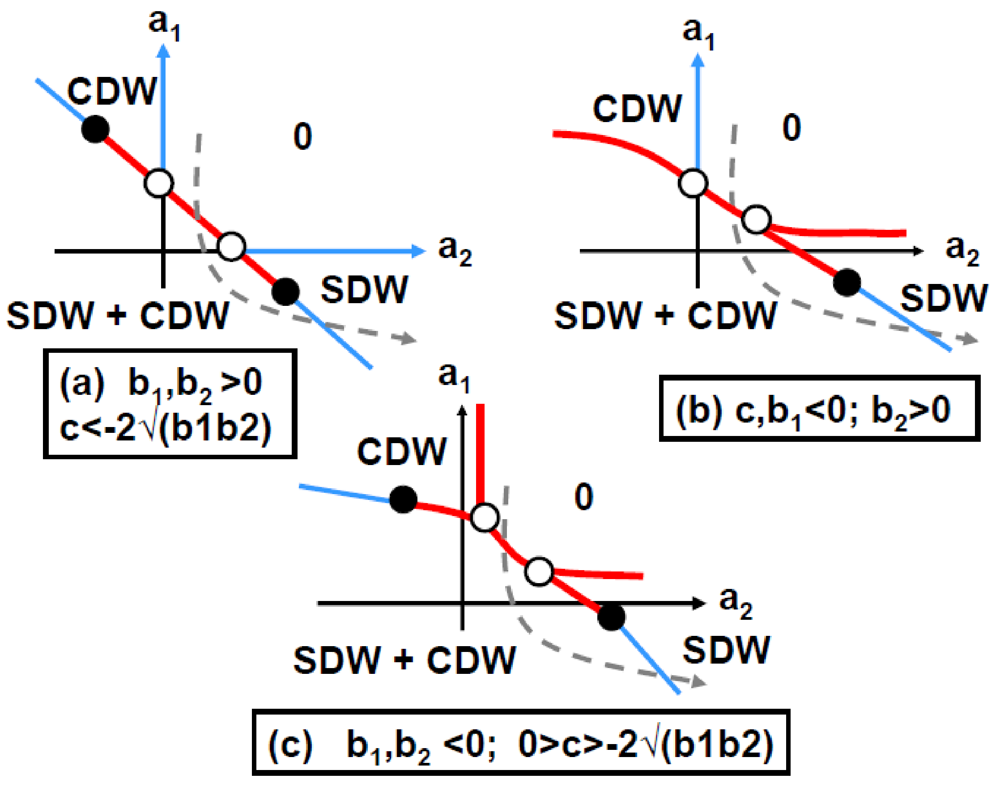
The phase diagrams shown in Figure 7 leave the possibility (interrupted lines) of an additional transition at lower temperature towards a pure 2kF SDW phase. This transition could occur in (TMTSF)2PF6 at ~3.5 K, temperature at which earlier NMR measurements [86,87] exhibit anomalies. Consistently with the occurrence of a pure SDW sub-phase, a vanishing of the 2kF CDW satellite reflection intensity is observed below ~3.5 K [84].
The finding of a mixed 2kF SDW/CDW ground state implies that both 2kF SDW and 2kF CDW response functions should be strong. Let us first consider the electronic phase diagram. Recent calculation [88,89] shows that, in addition to the FS nesting process tending to promote, in presence of sizeable intra-chain coulomb repulsions, the 2kF SDW ground state, the inter-chain backward repulsive coulomb interaction g1┴ previously considered in Section 3.1.2 can in parallel stabilize a 2kF CDW ground state for strong enough g1┴. These calculations show that with both kinds of inter-chain coupling there is a proximity between the 2kF SDW and 2kF CDW phases, and that, even if the SDW ground state is stabilized, sizeable 2kF CDW electronic correlations remain above the transition temperature. Interestingly when the FS nesting breaking effects destabilize the insulating density wave ground state, strong g1┴ tends to stabilize triplet f superconductivity instead of the singlet d superconductivity occurring for small g1┴ (for a recent review of the interplay between superconductivity and 2kF density waves see [90]).
However in the present case, already discussed in Section 3.1.2, g1┴ stabilizes a different periodicity as does the FS nesting, a feature not considered in the theory performed in [88,89]. This competition together with the lock-in of the anion destabilizes the 2kF BOW as seen in Section 3.1. However the CDW counterpart of 2kF instability could remain active and contribute to the ground state modulation if the 3D ordering of CDW is achieved by the same FS nesting mechanism as for the 2kF SDW. As g1┴ is not activated below 30K, the structural modulation observed below TSDW must not be of the BOW type (The absence of BOW structural features explains why the 2kF satellite reflections are of very weak intensity). For this reason it was proposed in [7] and [65] that the weak satellite reflections detected below TSDW could correspond to the X-ray scattering by the electronic 2kF CDW. Eventually a molecular distortion could follow the setting of the 2kF CDW (see below).
The separation between pure 2kF SDW and 2kF CDW ground states implies that there is a natural repulsion between the associated density waves, which means that the bare coupling term ν introduced in the Landau free energy of Annex A.2 is repulsive. However if these two density waves are simultaneously stabilized in the same phase, the free energy is minimized if the 2kF SDW and 2kF CDW are in phase quadrature (ω = π/2 phase shift). This corresponds also to a π/2 phase shift between the spin ↑ and spin ↓ components of the SDW and of the electronic CDW [8,65].
The “specific heat” anomaly observed at TSDW in thermal expansion measurements [25] shows that the electronic CDW should have a structural counterpart. This counterpart is not a BOW modulation (see Section 3.1). However if the CDW consists primarily in a modulation of the hole occupancy on the molecules, one expects that molecular deformations should follow the electronic density wave by elongating the TMTSF hole rich molecules and contracting the TMTSF electron rich molecules. This should lead to a modification of intra-molecular vibration modes in Raman and/or infrared spectra of (TMTSF)2PF6 below TSDW. The electronic CDW could also trigger “phase phonons” which should be observable in reflectance measurements. These latter effects apparently have been detected below TSDW in the parent (TMTSF)2SbF6 compound [91]. However there are very few optical studies performed below TSDW in (TMTSF)2X’s to assess this statement.
The coupling between the 2kF SDW and the 4kF CDW could be achieved by a “magneto-elastic” effect similar to the one considered in (TMTTF)2Br. By analogy with the findings of Annex A.1, this coupling should lead to an increase of hole concentration on the site having the larger magnetization. However as the intensity of the 4kF CDW reflections of (TMTSF)2PF6 is much weaker than the one of (TMTTF)2Br, the lattice distortion which eventually accompanies the 4kF CDW of (TMTSF)2PF6 must be much smaller than the 4kF distortion of (TMTTF)2Br. This difference could be explained by the incommensurate nature of the modulation (leading to the loss of umklapp effects) and the smaller amplitude of modulation [a reduction of the magnetization by a factor 1.75 (0.08μB for (TMTSF)2PF6 versus 0.14μB for (TMTTF)2Br [71]) reduces, according to (A.2), the amplitude of the distortion by a factor 3 and the 4kF reflection intensity by a factor 9]. Whatever its microscopic origin, the coupling between the 2kF SDW and the lattice remains appreciable in the density wave ground state because optical studies [92] show that the SDW condensate presents a large dynamical mass enhancement, which is however several times smaller than the mass enhancement of a CDW condensate.
5. The Charge Ordering Transition in the Fabre Salts
5.1. Basic Features: The Historical Scenario
The first indication of the now well established symmetry breaking CO phase transition was revealed by anomalies in conductivity measurements performed in the (TMTTF)2X salts with X = PF6, AsF6 and SbF6 and their solid solutions [93]. The most striking finding was the observation of a MI transition at TCO = 154 K in the SbF6 salt, recalling the one previously observed at 160 K in the SCN salt [66]. Although this transition was correctly interpreted as due to a 4kF electronic localization phenomenon, no lattice symmetry breaking could be detected at TCO in this preliminary study, in the difference of the SCN salt [94]. For this reason this transition was labeled “structureless” in later studies [95] which also revealed an anomaly at TCO in the thermal dependence of the thermo-power (possibly due to a gap opening) of these salts and similar anomalies in the ReO4 salt well above its AO transition. During the following year two important, but often ignored studies, revealed a dielectric divergence [81] and a few percent Young modulus softening [96] at TCO. The symmetry breaking consisting of a loss of all the inversion centers at TCO was only revealed 15 years later by the NMR observation of a charge differentiation between the two TMTTF molecules of the unit cell [97] providing the first evidence of the occurrence of a charge disproportion below TCO. Charge disproportion between molecules was soon confirmed by the observation of split intra-molecular vibration modes below TCO [98,99]. This charge disproportion together with the incipient stack dimerization leads to a stack dielectric polarization and, as there is no cell doubling (at the difference of the SCN salt), to the establishment of electronic ferroelectricity. This was sustained by accurate measurements of the dielectric divergence at TCO [100].
Although it was suspected in these first works that the anions should control the CO transition [93] or even should be displaced with respect to donors at TCO[38], several attempts to find a structural modification at TCO were unsuccessful [7,101,102,103]. The reason, found recently [104], is that minute irradiation defects created by X-ray beams in laboratory diffraction conditions kill the CO. It was only recently that the finding [24] of a lattice thermal expansion anomaly at TCO provides convincing evidences of a structural counterpart at the electronic ferroelectricity. Then evidence of a tiny structural modification was found from the detection of weak (<15%) variations at TCO of the intensity of several main Bragg reflections using neutron diffraction on (TMTTF)2PF6 deuterated powders [21,105]. These quite small structural modifications explain why earlier attempts to solve the (TMTTF)2PF6 and AsF6 structures on hydrogenated crystals in the P1 space group from conventional neutron scattering data collection at 4K were unsuccessful [106]. Recently the low temperature non centro-symmetric P1 structure of (TMTTF)2PF6 has been assessed using high energy X-ray beam from synchrotron radiation [107]. The main result of this study was the finding of intra-molecular deformations associated to charge disproportion. However no sizeable change of the F-S contact distance could be detected at TCO.
5.2. Structural Ingredients of the CO Transition
From a general point of view, three main structural features are expected at the CO transition of the Fabre salts [108]:
- - an internal deformation of the TMTTF molecules following their charge occupancy,
- - a deformation of the methyl group cavities associated to the loss of inversion symmetry,
- - a shift of the anions from the inversion centers.
The first feature, expected from the splitting of intra-molecular mode frequencies at TCO [98,99,109], has been recently detected [107]. Although the differentiation of the two molecules of the unit cell already suppresses the inversions centers, a deformation of the methyl group cavities and/or a shift of the anions should also contribute to stabilize the non centro-symmetric structure. In this case both the polarization of the methyl groups and/or the shift of the anions provide the efficient couplings with the π cloud [21,110] necessary to stabilize the CO ground state [111].
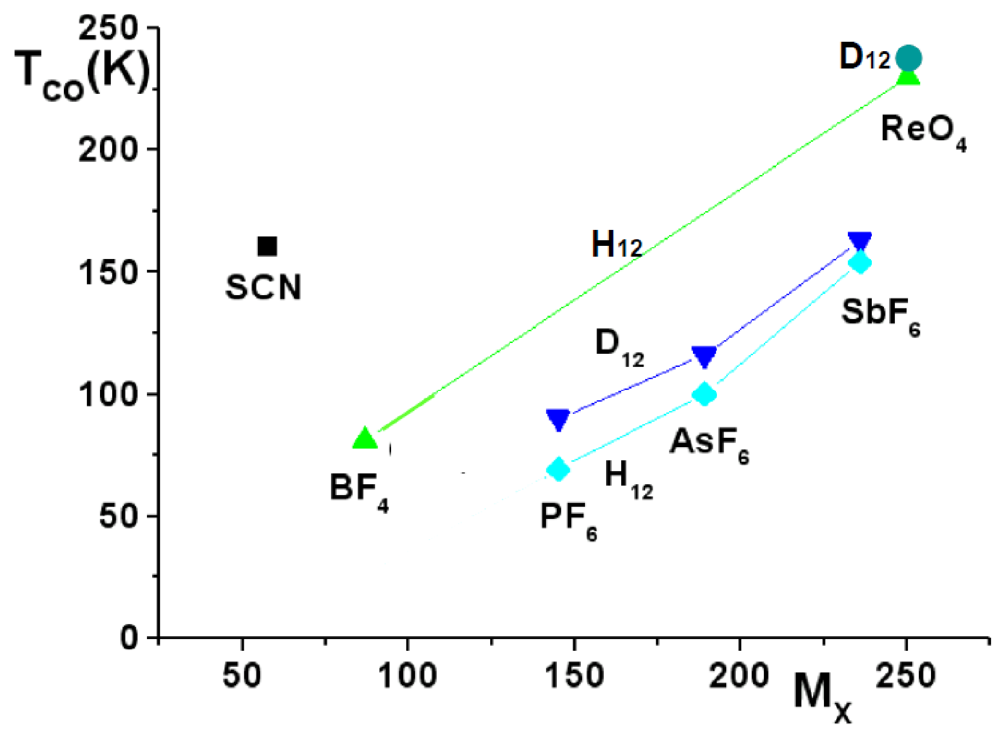
Figure 8.
TCO in function of the anion (labeled by its mass MX) in hydrogenated (H12) and deuterated (D12) (TMTTF)2X (adapted from [112]). The continuous lines connect TCO of H12 and D12 salts with anions of same symmetry.
Figure 8.
TCO in function of the anion (labeled by its mass MX) in hydrogenated (H12) and deuterated (D12) (TMTTF)2X (adapted from [112]). The continuous lines connect TCO of H12 and D12 salts with anions of same symmetry.
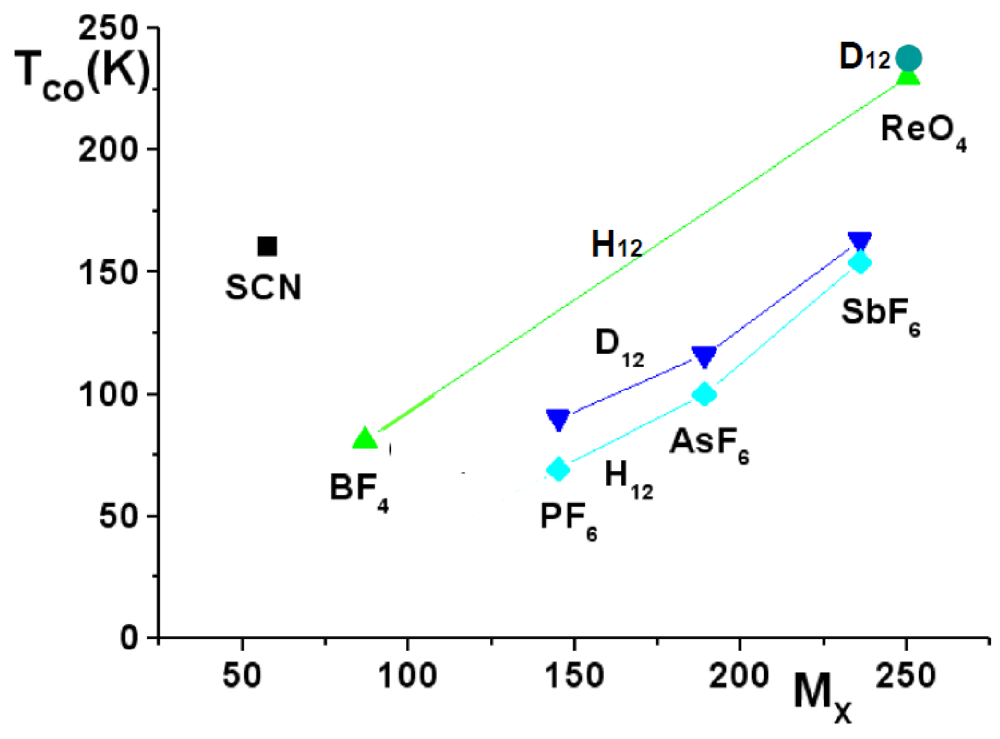
The influence of the anions in the CO process is assessed by the observation that:
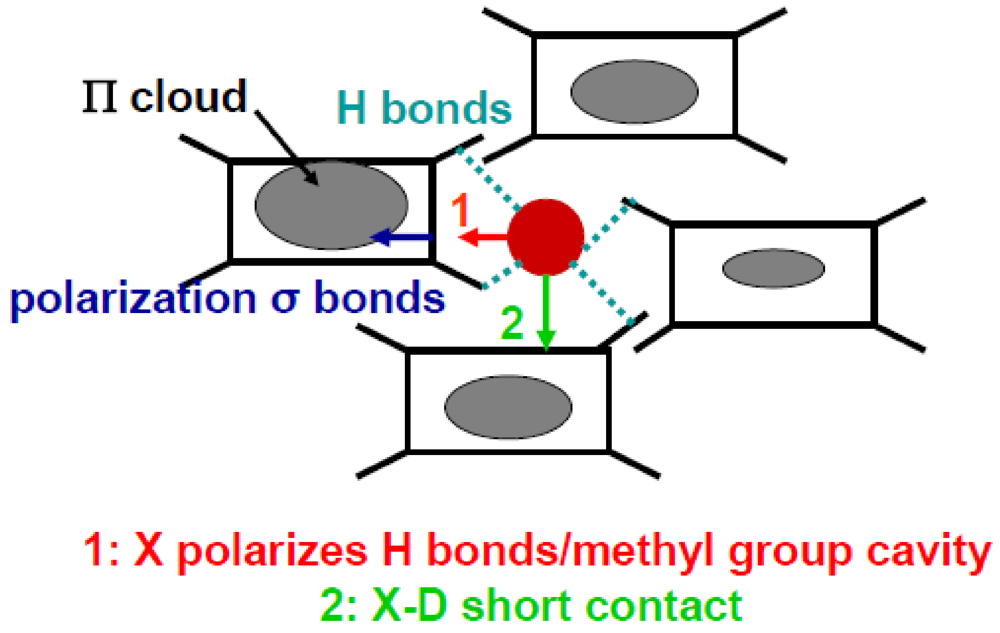
Figure 9 shows two different processes where the shift of the anion could lead to a disproportion of charge on the TMTTF. In Process 2 of Figure 9, the anion moves towards the S of a TMTTF where the shortening of the anion –S contact distance below TCO enhances directly the π hole density of the TMTTF in the vicinity of the anion. In Process 1 of Figure 9, the anion moves inside its methyl group cavity and deforms it. This deformation polarizes the H-bond network. The H-bond polarization induces a displacement of charge in the σ bonds connected to the H-bonds. This shift of σ electrons towards the center of the TMTTF stabilizes the excess of π holes [21].
Figure 9.
Schematic illustration of the two types of anion shifts. Process 1 (in red): displacement of the anion inside the methyl group cavity which polarizes the H-bond network and the σ electron skeleton of the TMTTF. Process 2 (in green): shift of the anion X towards the Se/S of a donor D establishing a short X-D contact distance. The modulation of the density of π holes is indicated for Process 1.
Figure 9.
Schematic illustration of the two types of anion shifts. Process 1 (in red): displacement of the anion inside the methyl group cavity which polarizes the H-bond network and the σ electron skeleton of the TMTTF. Process 2 (in green): shift of the anion X towards the Se/S of a donor D establishing a short X-D contact distance. The modulation of the density of π holes is indicated for Process 1.
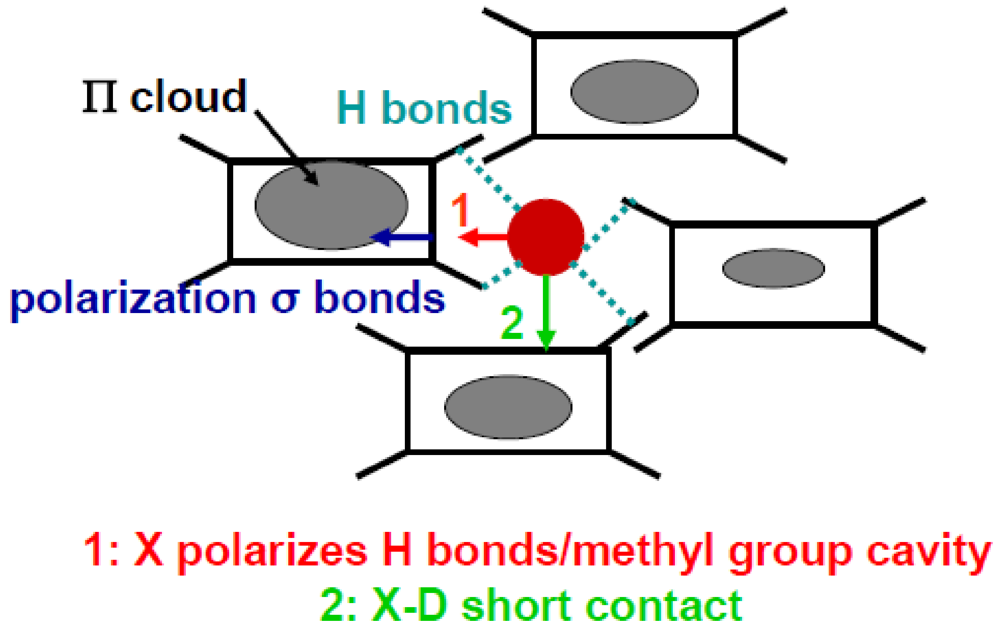
Both direct process 2 and indirect process1 stabilize an excess of π hole on the TMTTF towards which the anion moves. However the excess of hole is not localized on the same molecule because the direction of displacement of the anion is different in the two processes. However as the recent structural refinement [107] does not provide evidence of a sizeable change of the anion -S short contact distance below TCO in the PF6 salt, the stabilization of the CO pattern probably occurs via the deformation of the methyl group cavity in Process 1. Such an assertion is sustained by the following observations:
- - TCO increases when, with the larger size (and polarizability) of octahedral and tetrahedral anions, the contact between the anion and the periphery of the cavity delimited by the methyl groups is strengthened,
- - Far infrared measurements show there is a modification of the coupling between the methyl groups and the anions at the CO transition [109].
Interactions via the methyl groups could also explain why strong TCO lattice parameter anomalies are observed in the (b', c*) plane containing the H-bond network (see Figure 1); the strongest anomaly being along c* [24]. In addition the easy squeezing of the soft methyl group cavity under pressure explains simply, via the blockade of the anion in its cavity, the rapid decrease of TCO under pressure [116]. Until now there is no direct evidence of the deformation of the methyl group cavity at the CO transition. This deformation should be better revealed by neutron diffraction more sensitive to H or D positions.
An eventual shift of the anion in the CO phase does not mean that the anion orientation disorder is completely removed at TCO in the TMTTF salts. 19F NMR studies of (TMTTF)2SbF6 show the contrary [27,28]. Evidence of the decoupling between CO and orientation order of the anion is provided by the observation of successive CO and AO transitions in the ReO4 (TCO = 230 K and TAO = 154 K) and BF4 (TCO = 83 K and TAO = 40 K) salts [117].
Process 1 in Figure 9 which achieves an anion shift inside the methyl group cavity is structurally different from Process 2 which achieves an anion shift towards the S of the donor as found at the (1/2, 1/2, 1/2) AO transition (see Section 7.2). It is thus possible that these two types of nearly perpendicular anion displacements could be successively active in the CO and (1/2, 1/2, 1/2) AO transitions of the TMTTF salts with the ReO4 and BF4 tetrahedral anions. This decoupling allows to simply understand that the (1/2, 1/2, 1/2) AO transition, which does not substantially activate the H-bond network, is less sensitive to deuteration [114] and irradiation defects [118].
In this picture TMTTF salts incorporating small anions, such as ClO4 and NO3 which do not establish important interactions with the methyl groups [14,110] should not exhibit the CO transition. This seems to be the case for the ClO4 salt [113]. In the opposite situation where an anion perfectly fits the methyl group cavity there is no room for an anion shift inside this cavity and thus no CO transition is expected. This could be the case of the Br salt. This better fit, which also occurs under pressure with the squeezing of the methyl group cavities, can be taken as responsible of the rapid drop of TCO, as observed for example in pressurized AsF6 salt [116].
In this framework the (0, 1/2, 1/2) anti-ferroelectric CO transition of (TMTTF)2SCN, which coincides with the AO transition of the SCN [94], is singular. In this salt the small SCN anion, which has no real linkage with the methyl groups [110], can only strengthen its contacts with one donor out of two via the AO shift [103]. In this respect the wave vector of the AO superstructure of (TMTTF)2SCN, which simultaneously stabilizes the CO, is different from the wave vectors stabilized at the others AO and CO transitions (see Section 7). The AO/CO transition of the SCN salt which does not directly involve the methyl groups is consistently weakly sensitive to X-ray irradiation damage.
Because of the involvement of the fragile methyl groups in the stabilization of the CO pattern, the ferroelectric ground state is subject to defects. Defects break the long range ferroelectric order into domains forming local clusters of polarization. This leads to a frequency dependant dielectric permittivity which does not really diverge at a well defined TCO. Such features, recalling those of dielectric relaxors, are found in (TMTTF)2PF6 at ambient pressure and in (TMTTF)2SbF6 under pressure near its transformation to a (local) SP ground state [117]. The domain walls nucleated by these defects are described by charge soliton excitations of the CO ground state [119].
Electronic ferroelectricity [100,119] occurs in others organic systems [120]. Among them let us mention the 2D organic salt α–(BEDT-TTF)2I3 where ferroelectricity appears at the 135 K MI transition [121]. The interesting aspect of this phase transition is that, analogously to TMTTF salts, the charge rearrangement on type A BEDT-TTF molecules (called CO for this reason) is triggered by the deformation of the I3 sublattice which thus modifies of the H-bond network between I3 and the ethylene groups of the BEDT-TTF [122]. This mechanism, similarly to Process 1 in Figure 9, modulates the density of π holes on the BEDT-TTF.
5.3. The Underlying CO Instability
The CO transition observed in the Fabre salts is just a manifestation of the general tendency of organic conductors to form a Wigner lattice of localized charges because of the presence of long range intermolecular Coulomb repulsions (for a recent review see [73]). As the lattice is soft such an electronic instability is coupled to the lattice where it drives a 4kF CDW which also, in presence of an electron-phonon coupling with the acoustic modes, leads to the formation of a 4kF BOW [123,36]. However in quarter filled band systems, one has to distinguish between two different types of charge localization phenomena [73,76]:
- - charge localization on the bonds, stabilizes a 4kF BOW or Dimer Mott (DM) ground state,
- - charge localization on the sites, stabilizes a 4kF CDW or CO ground state.
For uniform quarter filled band systems, these two ground states have different inversion symmetry. However the TMTTF salts are more complex because with a stack being already dimerized (see Section 2.2) the divergence of the 4kF BOW instability is killed (the static dimerization itself gives rise to a charge localization [12] “visualized” by the development of an activated conductivity below Tρ ~ 200K higher than TCO). Thus the only symmetry breaking instability remaining in the dimerized salts is the CO if the stacks are not too strongly dimerized [73,119].
Ferroelectric CO is not announced by easily detectable pre-transitional structural fluctuations (the pretransitional structural fluctuations at the anti-ferroelectic CO transition of (TMTTF)2SCN are also barely detectable). Also the ferroelectric instability associated to CO appears to be mean-field because the dielectric susceptibility divergence follows a Curie Weiss behavior on a large (~30 K) temperature range above TCO [100,117]. This means that CO in the TMTTF’s is mostly announced by a regime of 3D fluctuations which isotropy reflects the importance of the inter-chain Coulomb coupling. In contrast, regular (i.e., non dimerized) quarter filled systems such as (DIDCNQI)2Ag [124] or (o-DMTTF)2X [125] exhibit a sizeable regime of 1D 4kF BOW fluctuations above the “CO” transition which in fact presents a mixed CO/DM character.
6. The Spin-Peierls Transition in the Fabre Salts
6.1. Basic Features
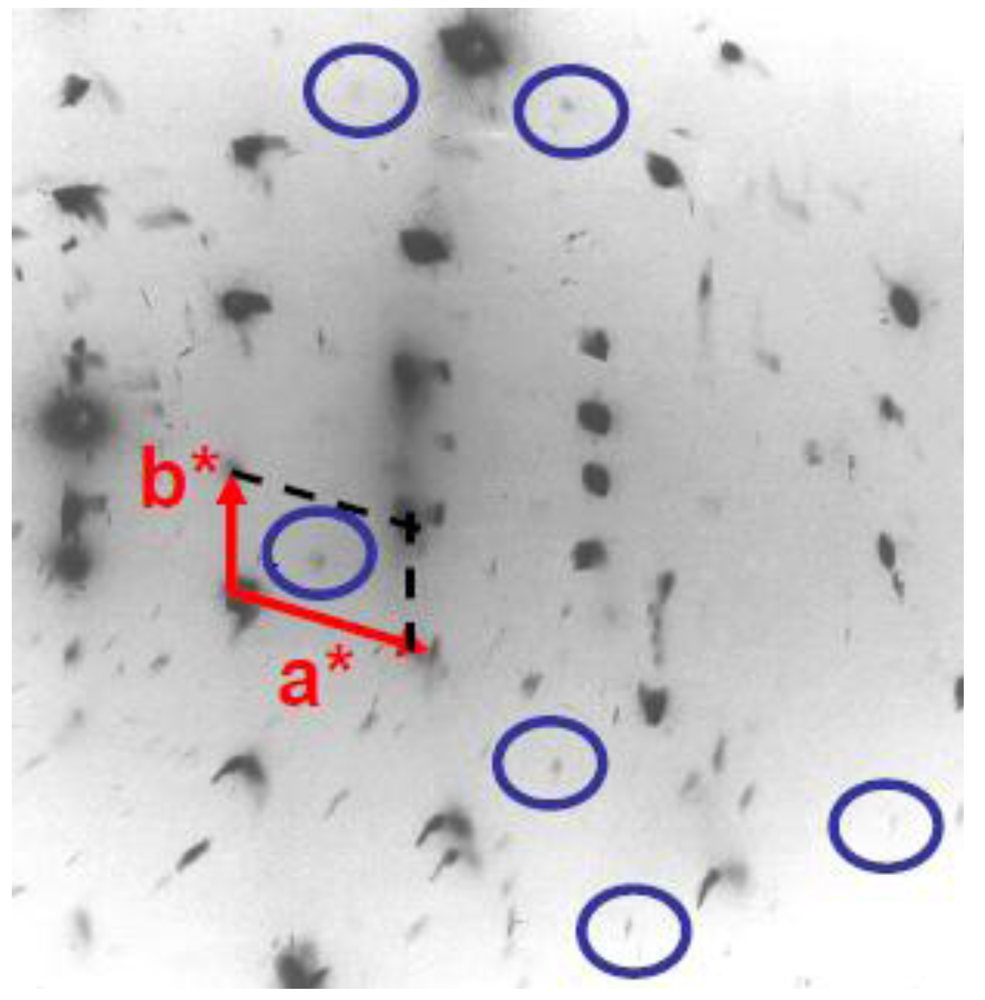
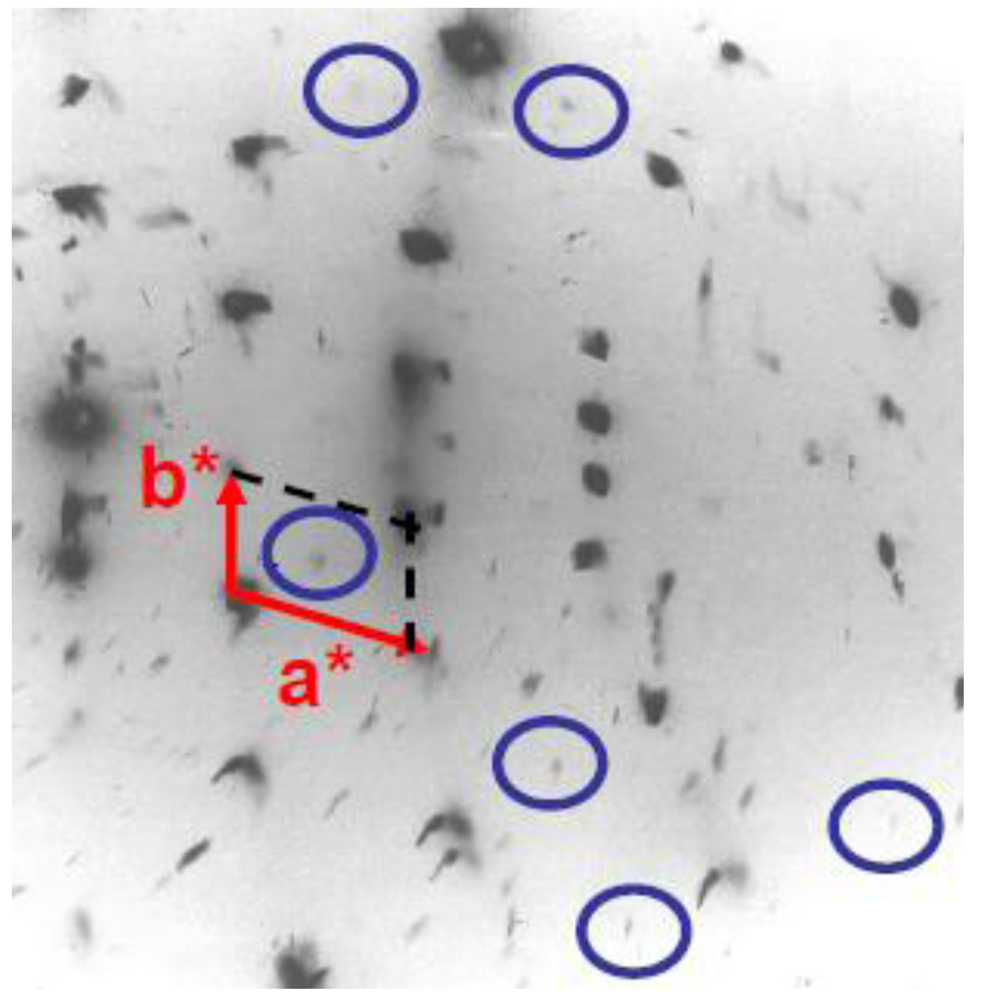
The SP transition of (TMTTF)2PF6 is characterized by the appearance below TSP ~ 17 K of superlattice reflections of very weak intensity at the (1/2, 1/2, ?) reduced reciprocal position according to earlier X-ray investigations [40] (see Figure 10). The third component 1/2c* has been determined by a recent neutron scattering investigation [68].
Figure 10.
X-ray diffuse scattering pattern from (TMTTF)2PF6 at 10 K in its SP ground state showing inside the blue circles very weak (1/2, 1/2, ?) superlattice spots. The (a*, b*) unit cell is shown (original data of the study reported in [40]).
Figure 10.
X-ray diffuse scattering pattern from (TMTTF)2PF6 at 10 K in its SP ground state showing inside the blue circles very weak (1/2, 1/2, ?) superlattice spots. The (a*, b*) unit cell is shown (original data of the study reported in [40]).
At ambient pressure the SP transition of the TMTTF’s occurs in the CO phase. The SP order is rapidly destroyed by X-ray irradiation as for the CO transition. This irradiation sensitivity explains that recent structural studies of the SP order have been performed with neutron scattering. The structure of the SP ground state has not yet been determined, but it should present some analogies with the (1/2, 1/2, 1/2) AO superstructure [58,59]. Thus it is expected that the stack tetramerization, corresponding to a dimerization of stack of dimers, each dimer bearing a spin ½, will be accompanied by synchronized shift of the anions towards donors [68]. The involvement of the anions in the SP instability is assessed by the observation of a TSP critical divergence of the 75As NMR T1−1 in (TMTTF)2AsF6 [126,127]. In this process the anion shift should tune the interchain coupling between the individual stack tetramerization, as discussed in Section 3.1.2. The 3D inter-chain coupling regime should occur in the near vicinity of TSP because the observation of precursor X-ray diffuse scattering lines on a large temperature range above TSP shows that the SP instability is basically 1D (see Section 3.3).
The temperature TSP at which the (1/2, 1/2, 1/2) superlattice reflections are observed coincides with the temperature at which a singlet gap develops in the spin susceptibility [112]. At TSP lattice expansion measurements exhibit a lambda type anomaly which coincides with the specific heat anomaly [128]. The value of the singlet-triplet gap in controversial in the literature which mixes calculated values, using approximate theories [129,130], and measured quantities [74,112]. These last measurements have been confirmed by a direct determination, using inelastic neutron scattering, of the SP gap in the spin excitation spectrum [131]. The SP wave vector is the same in the TMTTF salts and in the isostructural BCPTTF salts [7,70]. BCPTTF)2PF6 and AsF6, which does not undergo a CO transition, have a twice higher TSP than (TMTTF)2PF6 and AsF6.
6.2. Competition with the Charge Ordering
NMR measurements show that there is coexistence between CO and SP pairing in (TMTTF)2PF6 and AsF6. However the observation of an opposite variation of TCO and TSP under pressure shows that there is repulsion between the order parameters associated at the CO and SP transitions [116]. In this context, the real part of the microwave dielectric function, ε', which increases by ~10% in the SP phase, shows that the gap of charge (i.e., CO order parameter) decreases at the SP transition [132]. This competition is analyzed in Annex A.3 in the framework of the Landau theory. In particular Landau development predicts the behavior of TSP in function of TCO. This dependence, obtained with the restrictions outlined in Annex A3 and shown in Figure 11, accounts for the experimental results.
Figure 11.
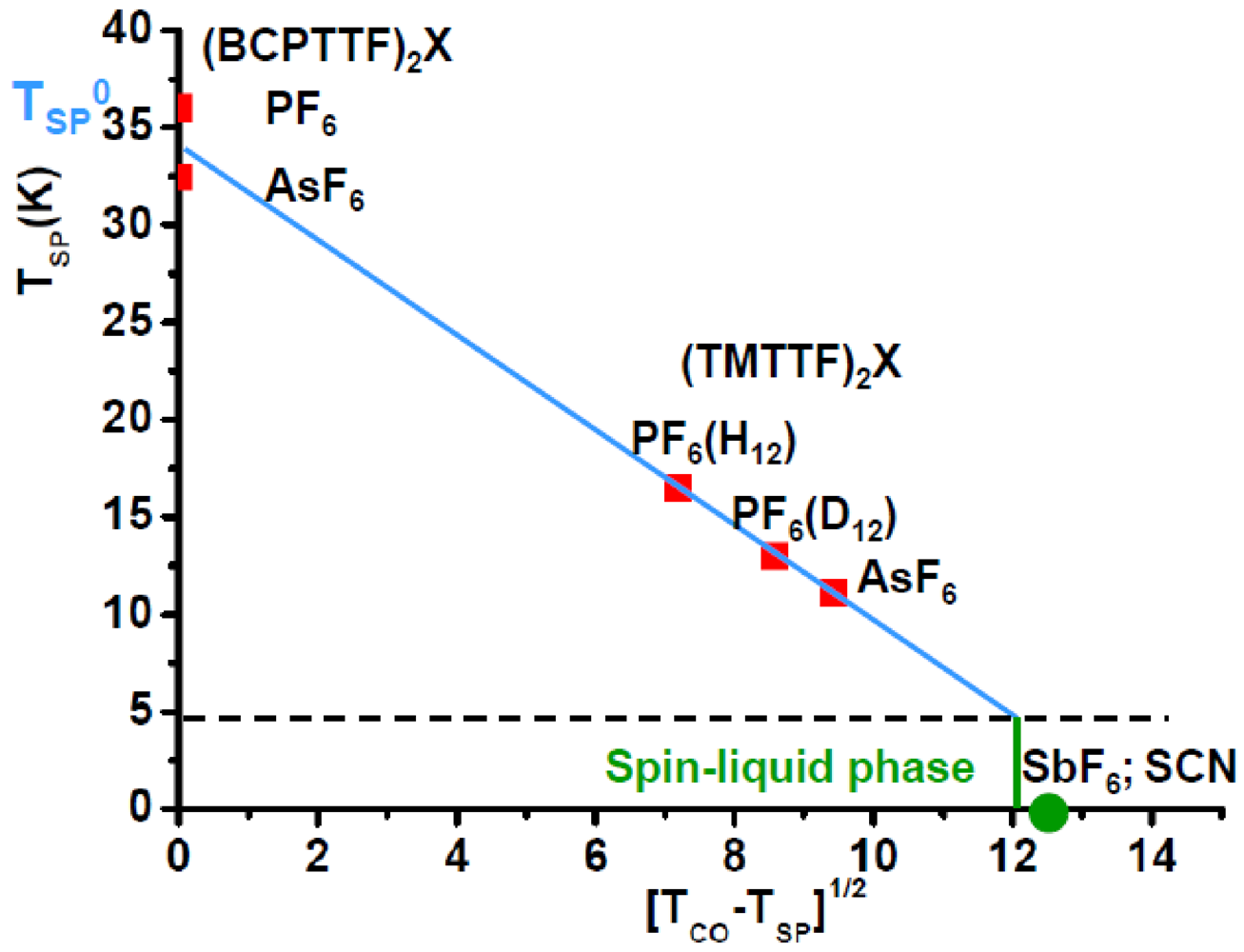
Spin-Peierls critical temperature, TSP, of (TMTTF)2X versus √(TCO −TSP), as given by the expression (A12). TSP0 of (BCPTTF)2X, which does not undergo a CO, is also given, as well as √(TCO) for the SbF6 and SCN salts having no SP ground state. The spin-liquid phase occurring for TSP < 5 K (see text) is indicated.
Figure 11.
Spin-Peierls critical temperature, TSP, of (TMTTF)2X versus √(TCO −TSP), as given by the expression (A12). TSP0 of (BCPTTF)2X, which does not undergo a CO, is also given, as well as √(TCO) for the SbF6 and SCN salts having no SP ground state. The spin-liquid phase occurring for TSP < 5 K (see text) is indicated.

Figure 11 shows that TSP of (BCPTTF)2PF6 and AsF6 is consistent with the absence of CO (in that case the SP transition occurs at TSP0 defined Annex A3). Figure 11 shows also that the SP transition vanishes for TCO ~ 190 K or more likely for ~ 150 K if one assumes that for TSP < 5 K the spin-liquid phase is stabilized (see below). 150 K is close to TCO of (TMTTF)2SbF6 and SCN which do not exhibit a SP ground state.
To describe completely the SP phase diagram of the TMTTF salts, one has to consider explicitly the non-adiabatic effect of the phonon field (see Section 3.3). These effects have already been discussed in [73] with the main conclusions being:
- - SP transition does not occur in (TMTTF)2SbF6 and SCN because the spin-phonon coupling is smaller than the zero-point lattice fluctuations (this condition corresponds to TSPMF < Ω/4 ~ 15 K, or TSP < 5 K because in the PF6 and AsF6 salts TSP ~ TSPMF/3). These salts are thus located in the spin-liquid phase outlined in Figure 11phase the inter-chain exchange coupling stabilizes a low temperature (TN ~ 7K) AF ground state.
- - SP transitions of (BCPTTF)2PF6 and AsF6 occur in the adiabatic limit.
- - SP transitions of (TMTTF)2PF6 and AsF6 are located between the adiabatic limit and the spin-liquid phase, close to the crossover line between the adiabatic and anti-adiabatic regimes.
The microscopic origin of the competition between CO and SP instabilities relies on the fact that CO, which localizes the holes every second neighbor molecules, establishes also a reduced AF exchange integral, J, between the spin of the holes. With a spin-phonon coupling following the decrease of J, TSPMF (i.e. the SP instability) decreases [73].
The weakening of the SP instability by the CO is just the manifestation of the repulsion existing between 4kF CDW (or CO) and 2kF BOW (or SP) instabilities in quarter filled band systems: the next neighbour Coulomb repulsion V1 which promotes CO in quarter filled band systems kills in the same time the divergence of the 2kF BOW response function [135,136]. In other words CO induces hetero-polar charge configurations which destabilize the modulation of bond distances in the 2kF Peierls mechanism. This was clearly shown in the study of the [(TMTSF)1−x (TMTTF)x]2ReO4 solid solution [137,138]. For x = 0.5 the alternate stacking of TMTSF and TMTTF induces a chemical CO. In its presence it is observed a sizeable decrease of TAO of the (1/2, 1/2, 1/2) AO transition which involves the formation of 2kF BOW on the organic stacks (see Section 7.2).
6.3. The Vanishing of the SP Phase under Pressure
TSP increases when TCO decreases under pressure [116]. At higher pressure when the CO vanishes instead of reaching TSP0 at a value comparable to TSP measured in the BCPTTF’s, TSP further decreases. In the PF6 salts TSP even exhibits a singular depression around Pc ~ 9 kbar before entering the AF phase [139]. This behavior suggests the existence of a quantum critical point where the normal phase is further stabilized around Pc.
A possible explanation could be, that with TMTTF salts being close to the crossover boundary between the adiabatic and anti-adiabatic regime, the hardening of the phonon mode under pressure will shift the SP instability to the anti-adiabatic regime followed by the spin-liquid regime around Pc. In the anti-adiabatic SP regime the enhancement of quantum fluctuations should strongly reduce the amplitude of 1D SP gap. In this scenario the suppression of the SP phase should occur through a critical point due to the enhancement of spin-liquid fluctuations. Then above Pc the 3D AF ground state will be stabilized by the inter-chain exchange coupling. If the succession of ground states is described by the generic phase diagram [52], AF pressurized (TMTTF)2PF6 should be identical to AF ambient pressure (TMTTF)2Br.
In order to assess the development of a pressure induced non-adiabaticity one must prove that the amplitude of the 1D SP gap decreases under pressure as a result of the enhanced quantum fluctuations. However, as it is observed that TSPMF (linearly related to the mean field 1D SP gap when the phonon fluctuations are neglected) slightly increases from (TMTTF)2PF6 to (TMTTF)2Br (see Figure 5), the opposite situation seems to occur. In addition in order to sustain the non-adiabatic scenario one should have a considerable phonon frequency hardening (by a factor ~4) to stabilize the spin-liquid phase in pressurized (TMTTF)2PF6. This requires a phonon frequency as high as Ω > 4TSPMF, which seems unrealistic for an acoustic mode. The difficulty with this scenario is that SP fluctuations are still detected in AF (TMTTF)2Br, at the difference of (TMTTF)2SbF6 where it was stated in Section 6.2 that the AF ground state is caused by the enhanced non-adiabaticity of the phonon field which in the same time kills the SP fluctuations.
Another scenario could be that the squeezing of the methyl group cavities under pressure blocks the shift of the PF6, the necessary ingredient setting the interchain coupling between the individual SP stack tetramerization. This is corroborated by a recent determination of the structure of pressurized (TMTTF)2PF6 [140] showing a sizeable contraction of the methyl group cavity. The mechanism of blockade of the anion in its methyl group cavity explains also that only a short range SP order is stabilized in pressurized (TMTTF)2SbF6 [27] when both the CO and AF phases are suppressed above 0.5 GPa.
7. The Anion Ordering Transitions
7.1. Basic Features
In Fabre and Bechgaard salts incorporating non-centrosymmetric anions, the anion orientation is disordered at RT. However for entropy reasons, the anions order upon cooling. The ordering is achieved at a well defined phase transition showing that the AO leads to a symmetry breaking. The AO transition stabilizes a superstructure which generally doubles the RT lattice periodicity in one or several directions [7]. The AO transition can be of 2nd [(0, 1/2, 0), (1/2, 0, 0) and (0, 1/2, 1/2) AO] or 1st order [(1/2, 1/2, 1/2) AO] depending on the strength of the coupling of the AO process at the elastic lattice degrees of freedom The transition not only consists of an orientation ordering of the anion inside its methyl group cavity, which in itself breaks its inversion symmetry, but also of a concomitant deformation of this cavity associated with an anion shift. This shift can be either towards selected methyl groups as in the (0, 1/2, 0) AO transition of (TMTSF)2ClO4 (direction 1 in Figure 9) [141] or towards the S or Se atom of a given donor as in the (1/2, 1/2, 1/2) AO transition of (TMTSF)2ReO4 (direction 2 in Figure 9) [58,59]. The deformation of the methyl group cavities can be achieved by a cooperative PLD of the organic stacks. In the case of the (1/2, 1/2, 1/2) AO transition this corresponds to a 2kF BOW distortion of the stacks. In (TMTSF)2ReO4 the 2kF PLD leads to a MI transition [142]. Even if the coupling of the PLD with the 1D electron gas provides the necessary energy gain to open a gap at the Fermi level, the (1/2, 1/2, 1/2) AO transition is not achieved by a standard Peierls instability because the associated pretransitional fluctuations are 3D and not 1D [143]. This means that the anions located in between the stacks trigger by their ordering the 2kF stack deformation while providing the necessary inter-chain coupling to stabilize the AO insulating ground state. This differs of a conventional Peierls transition where the driving force is due to the individual stack instability towards the formation of a 2kF PLD. Indeed the electron-anion coupling displayed in Figure 9 plays in the AO transition the role played by the intra-stack electron-phonon coupling in the Peierls transition.
These few examples, as well as the case of (TMTTF)2SCN already considered Section 5.2, show that important changes of the electronic properties generally accompany the AO transition. AO transitions lead to substantial modifications of the electronic structure [144,145] and of the electronic ground states [146,147] of the Bechgaard and Fabre salts. These structural-electronic coupled properties will be revisited below in light of the most recent findings.
7.2. The (1/2, 1/2, 1/2) AO Transition of Tetrahedral Anions
Among salts undergoing an (1/2, 1/2, 1/2) AO transition, several cases must be distinguished:
- - TMTSF salts where the AO transition coincides with a MI transition.
- - TMTTF salts where 4kF charge localization takes place above the AO transition.
- - Salts with dipolar anions which incorporate additional degrees of freedom.
7.2.1. TMTSF Salts
ReO4 and BF4 salts undergo 1st order AO/MI transitions, at TAO = 176 K and 36 K respectively, which open a sizeable energy gap of ~0.2eV [142]. These MI transitions cannot be classified as standard Peierls transitions because these latter are 2nd order transitions and open much smaller energy gaps. For these reasons it has been suggested [148] that these AO transitions should be understood in the framework of a strong-coupling theory where the anion shift towards the Se of the closest TMTSF [58,59,149] is the essential ingredient. In this picture the anion shift, associated to its orientation ordering, localizes a pπ hole on the molecule towards which the anion establishes a short contact. In this mechanism the magnitude of the energy gap should reflect the staggered Hartree anion potential experienced by the pπ holes. However structural refinements of (TMTSF)2ReO4 [59] and BF4 [149] superstructures do not exhibit an appreciable internal deformation of the TMTSF revealing the formation of a sizeable 2kF CDW induced by the direct Coulomb coupling with the anions. Structural refinements [58,59] show that the 2kF BOW stack distortion consists mainly in a displacement wave of an amplitude ~0.07 Å (essentially directed along c) of dimers, composed of the two inequivalent donors below TAO, which alternates in stack direction by inversion symmetry in order to set the 2kF periodicity. However tight binding band calculations [149,150] considering only the 2kF stack distortion cannot account for the entire value of the energy gap. Thus it should be interesting to perform a DFT calculation of the electronic structure which fully incorporates the shifted and ordered anions.
The AO transition of the ReO4 salt occurs in the temperature range above TX where the warping of the FS is not relevant, but this is probably not the case for the BF4 salt. Under pressure it is observed that the MI transition of the ReO4 salt disappears [151] when the superstructure periodicity changes from (1/2, 1/2, 1/2), to (0, 1/2, 1/2) through a 1st order phase transition [152]. When, with the setting of the (0, 1/2, 1/2) superstructure, the metallic state is restored pressurized (TMTSF)2ReO4 becomes a superconductor with a TS as high as 1.5K [149]–1.7 K [153] in the coexistence region of the two AO superstructures. The (0, 1/2, 1/2) superstructure, occurring at a TAO of 240 K at 1.7 GPa, could be stabilized at ambient temperature around 2.5 GPa. The squeezing of the methyl group cavities under pressure is certainly responsible for the locking of the orientation of the anions in the (0, 1/2, 1/2) superstructure. A refinement of this superstructure would be desirable.
7.2.2. TMTTF Salts
Although presenting structural distortions of same nature as in TMTSF salts, the (1/2, 1/2, 1/2) AO ground state of the TMTTF salts exhibit different electronic properties. At atmospheric pressure, TAO occurs in the 4kF charge localization regime (i.e. charge localization in the TMTTF dimers below Tρ). TAO occurs even below the CO transition (TCO < Tρ) in the ReO4 and BF4 salts. TCO and TAO are quite decoupled in these salts, which means, as already discussed in Section 5.2, that the anions, remaining disordered below TCO, undergo a new shift towards the S of the TMTTF when they order at TAO.
Structural refinement below TAO [103] shows that (TMTTF)2ReO4 exhibits both:
- - a sizeable 2kF CDW with an amplitude of 0.25 electrons estimated from the bond lengths of the two inequivalent TMTTF of the dimer, and which must add to a CO amplitude of 0.17 electrons estimated by NMR between TCO and TAO [154],
- - a sizeable 2kF BOW achieving a quite large dimerization of the stack of dimers with substantially different inter-dimer distances of 3.9 Å and 3.75 Å; the intra-dimer distance being of 3.7 Å.
Indeed a significant increase of the activation energy of the conductivity [113,117] and a rapid drop of spin susceptibility [118,130] follow the 2kF AO structural distortion.
The AO transition of (TMTTF)2BF4 is quantitatively different from the AO transition of the ReO4 salt because there is no appreciable change of the activation energy of the conductivity [66,113] and only a weak drop of spin susceptibility [118,130] at TAO. This should be due to the presence of a very weak 2kF PLD of the stack below TAO. In this picture the slight increase of conductivity observed at TAO [113] can be explained by an increase of carrier mobility due to the reduction of the electron scattering by the AO. This finding is very surprising because a large gap of charge opens at the AO transition of (TMTSF)2BF4. A possible explanation could be that the high temperature CO of (TMTTF)2BF4 kills substantially the 2kF BOW/SP response of the stack at the staggered AO potential (see the end of Section 6.2), leaving the stack nearly unaffected by the AO transition. In this case the AO transition should consist in a simple ordering of the BF4.
Similar features [66,113,118,130] are observed at the AO transition of (TMTTF)2ClO4 where the structural refinement performed below TAO [155] shows that only very weak anion shift and stack tetramerization accompany the AO process. The AO transition of (TMTTF)2ClO4 thus corresponds to the simple ordering of well decoupled ClO4, a finding in agreement with a measured entropy of transition of kBln2 per formula unit [66].
7.2.3. Salts with Dipolar Tetrahedral Anions
Bechgaard and Fabre salts incorporate also non symmetrical tetrahedral anions such as PF2O2 and FSO3 which possess an electric dipolar momentum. These anions provide an additional degree of freedom in the structure related to the orientation of their electric dipoles.
(TMTSF)2PF2O2 exhibits two competitive instabilities at ambient pressure which stabilize after a 2nd order transition the (1/2, ±1/4, 0) superstructure followed a few degrees below by a 1st order transition towards the more “conventional” (1/2, 1/2, 1/2) superstructure [156]. It is not known if the dipole of the anion is ordered in these superstructures. Note that the (1/2, ±1/4, 0) superstructure does not fulfill all the conditions (5) minimizing the interchain coupling via the anion shift.
(TMTTF)2FSO3 undergoes a 1st order transition at 58K which stabilizes the (1/2, 1/2, 1/2) AO superstructure. However at the difference of the (F-1) AO superstructure of (TMTTF)2ReO4, which contains two identical tetramerized stacks per unit cell related by the F centering (and inversion) symmetry, the superstructure of (TMTTF)2FSO3 contains two different stacks which breaks the F symmetry [155]. The ordered FSO3 anions located between the stacks form ferroelectric chains. This removes also the inversion symmetry which relates the two donor stacks in (TMTTF)2ReO4 and lowers the space group symmetry to P1.
(TMTSF)2FSO3 exhibits also at 87.5K and atmospheric pressure a strongly 1st order MI transition which stabilizes the (1/2, 1/2, 1/2) AO superstructure [157], which, because of the ferroelectric order of the FSO3 [158], is probably non centro-symmetric as the TMTTF analogue. Under pressure the MI is removed and above ~0.5GPa (TMTSF)2FSO3 becomes a superconductor with a TS as high as 3 K [159,160]. Preliminary X-ray diffraction measurements [158] show that in the pressure range of observation of superconductivity the (1/2, 1/2, 1/2) superstructure is transformed into the (0, 0, 1/2) superstructure, which itself transforms into the (0, 1/2, 0) superstructure at higher pressure. Recent transport measurements provide evidence that the (1/2, 1/2, 1/2) AO transition splits into several phases under pressure [160]. NMR studies [161] suggest that this splitting reveals the succession upon cooling of the ordering of the FSO3 without ordering of the F-sites then of the ordering of the F-sites (i.e. ordering of electric dipoles).
7.3. The (0, 1/2, 0) AO Transition of (TMTSF)2ClO4
7.3.1. Basic Aspects of the AO Superstructure and of Its Electronic Structure
The AO transition of the (TMTSF)2ClO4 is significantly different from the AO transition of other salts of tetrahedral anions. For example in (TMTTF)2ClO4, the ClO4 adopts a staggered order in all directions without important interaction of the ClO4 with the organic stacks (see Section 7.2.2). In (TMTSF)2ClO4, the ClO4 adopts a staggered order in the b direction and a uniform order in a and c directions [61]. Surprisingly, the ordering of the ClO4 is accompanied by its shift which increases the closest Se-O distance [115] and a de-centering of the ClO4 inside its methyl group cavity [141]. This de-centering reinforces H-bonds of the ClO4 with the methyl groups of the closest TMTSF (TMTSF A) and form loose contacts with the methyl groups of the other TMTSF (TMTSF B) of the cavity. The uniform shift of anions along a, thus stabilizes two types A and B of TMTSF stacks per unit cell.
The polarization of the methyl group cavities by the anion shift causes a charge transfer from stack A to stack B via Process 1 shown in Figure 9. TMTSF A towards which the anion moves becomes hole rich while TMTSF B which has looser contacts becomes hole poor [21]. The charge transfer from A to B molecules drives an incommensurate band filling [147]. EHT [141] and more recent DFT [162] band structure calculations estimate this charge transfer at ~0.025 electrons per TMTSF. In a 1D picture, this deviation from the quarter band filling should kill the 4kF = a* umklapp electron-electron scattering terms which tend to promote the Mott-Hubbard gap.
Another important consequence of the AO transition is that TMTSF A towards which the ClO4 move is deformed (chair-like conformation), probably because of the constraints exercised by the H-bonds, while unconstrained TMTSF B remains planar [141]. These different molecular deformations achieve different HOMO energies for A and B molecules (separated by ~0.1 eV [141,162]). Also the non equivalent stacks A and B become differently dimerized.
The band structure and the FS of the AO superstructure have been calculated using EHT [141] and DFT [162,163]. All these calculations give an open FS composed of four quasi-1D sheets. The sheets originate from the “bonding” and “anti-bonding” combinations of the dimeric electronic structure of A and B stacks. Both the A-B inter-stack transfer integrals and the difference of site energies (including the HOMO energy separation plus the staggered anion potential) contribute to the warping and splitting of the FS. DFT calculation [162,163] shows that the anion gap has no significant effect on the FS shape and that the main contribution at the FS splitting comes from the difference of HOMO energies between the A and B donors. However the warping of the FS differs with the method of calculation [141,163]. In spite of this problem the assumption frequently done in the literature of a high temperature band structure only split by the staggered anion potential [144,145] is not valid to interpret the data obtained below TAO.
In addition, as revealed by the most recent DFT calculations [162] and in contradiction with the earlier EHT calculation [141], the “bonding” and “anti-bonding” combinations of the dimeric electronic structure of A and B stacks lead to differently warped quasi-1D sheets that do not nest “perfectly” on each other. This could explain why the SDW ground state is not stabilized in the AO superstructure. This means also that the placement of (TMTSF)2ClO4 in the generic phase diagram built on octahedral anions [52] is ambiguous because the vanishing of the SDW ground state in AO ClO4 salt is not due to the enhanced warping of the FS, as it occurs under pressure in salts with octahedral anions such as PF6, but to the reconstruction of the band structure. A proof is that in quenched ClO4 salt (see below), where the band structure is not reconstructed by the AO process, the SDW ground state is kept. Another ambiguity with the placement of relaxed (TMTSF)2ClO4 in the generic phase diagram is that superconductivity occurs for an electronic structure substantially different of the pressurized (TMTSF)2PF6 one.
To summarize these recent findings, AO opens space for superconductivity in (TMTSF)2ClO4 by killing the SDW with the reconstruction of the electronic structure. AO also kills the tendency to form a 4kF commensurate Mott-Hubbard gap by achieving an inter-stack electronic charge transfer.
7.3.2. Texture of the AO Phase
By its FS reconstruction AO controls the low temperature electronic properties of TMTSF)2ClO4. In particular the tuning between SDW and superconductivity which results from this electronic structure modification is revealed using the slow kinetics required to orient the anions. The slow kinetics is due to the fact that AO occurs at a temperature TAO = 24 K much lower than the barrier height (~240 K) necessary to overcome to change the orientation of the ClO4. The achievement of the AO phase thus requires a very slow cooling rate. In that case relaxed (TMTSF)2ClO4 becomes a superconductor with a TS ~ 1.2 K [1]. For faster cooling rates the AO is incomplete and (TMTSF)2ClO4 undergoes a SDW order, due to the FS nesting, at a TSDW increasing with the cooling rate and capped to ~ 6.5 K for the most rapidly cooled (quenched) samples [2]. Although it is often stated that a very slow cooling rate achieves a perfect AO and that a very rapid cooling rate leaves the anions disordered, high resolution X-ray measurements [164] show that this assertion is incorrect: there is no true long range AO even in the best relaxed samples and a local AO is always present even in the best quenched samples. Also the cooling rate modifies progressively the fraction of ordered anions and the texture of the AO pattern, as we shall see below.
It is interesting to remark that, when TAO is low (let say below 40 K), the cooling rate affects the achievement of the AO process for anions, such as ClO4 and NO3 [165], weakly coupled to the TMTSF, but has no real effect for the BF4 anion [149] more strongly linked to the donors (see Section 7.2.1). This means that the use of a phenomenological model of a static double well of potential, external to the anion, to describe the kinetics of ordering is oversimplified especially if the ordering process involves a sizeable interaction with the TMTSF stack able to modify substantially the shape of this potential in order to lower the potential height to overcome during the ordering process.
Let us now discuss more quantitatively the texture of the AO domains. First, superconductivity occurs in ClO4 ordered domains. Figure 12 shows that, in function of the cooling rate, the fraction of ordered ClO4 [164] is identical to the fraction of the superconducting volume determined by the measurement of Meissner effect [166].
Figure 12.
Fraction of ordered ClO4 (black symbols defined in [164]) and fraction of superconducting volume (red crosses) in (TMTSF)2ClO4 in function of the cooling rate. The fraction of superconducting volume has been determined from the data of [166] assuming that the relaxed sample is completely superconducting.
Figure 12.
Fraction of ordered ClO4 (black symbols defined in [164]) and fraction of superconducting volume (red crosses) in (TMTSF)2ClO4 in function of the cooling rate. The fraction of superconducting volume has been determined from the data of [166] assuming that the relaxed sample is completely superconducting.
Second, the profile of the (0, 1/2, 0) super-lattice reflections evolves with the cooling rate [164], which means that the topology of the AO domain pattern changes. The superlattice reflections of relaxed samples have a Lorentzian square profile (LS in Figure 13b) and those of quench samples, a Gaussian profile (G in Figure 13a). The texture of relaxed samples (right part of Figure 13b) is made of a collection of domains where the ClO4 is ordered in one of its two opposite orientations (I and II in Figure 13b). Due to the intricate pattern of the domain walls and to the random distribution of domain walls there is along a given direction (straight line in Figure 13b) a large distribution of domain size, which Fourier transform gives a LS function [167]. The texture of quenched samples (right part of Figure 13a) is made of AO domains of about the same size and shape which are embedded in a matrix of disordered ClO4 (in grey in Figure 13a). With a small (i.e., Gaussian) distribution of domain sizes, the Fourier transform of the shape of ordered domains is a G function [168]. For intermediate cooling rates the profile of the (0, 1/2, 0) superlattice reflections evolves from a LS to a G [164]. This means that non ordered ClO4 regions begin to develop between type I and II ordered domains.
Figure 13.
Left part: profile of the same AO superstructure reflection of (TMTSF)2ClO4 in the (a) quenched and (b) relaxed states (data of [164]). Note the angular broadening (larger than the experimental resolution) of the superstructure reflection when the sample is either in the quenched or in the relaxed state. Right part: texture corresponding to the (a) quenched and (b) relaxed states. In (a) L is the typical size of a domain of ordered ClO4, l is a typical inter-domain distance and the grey background represents non ordered ClO4. In (b) domains of types I and II with opposite ClO4 orientations are indicated. Along the line drawn in (b) there is a large distribution of domain sizes.
Figure 13.
Left part: profile of the same AO superstructure reflection of (TMTSF)2ClO4 in the (a) quenched and (b) relaxed states (data of [164]). Note the angular broadening (larger than the experimental resolution) of the superstructure reflection when the sample is either in the quenched or in the relaxed state. Right part: texture corresponding to the (a) quenched and (b) relaxed states. In (a) L is the typical size of a domain of ordered ClO4, l is a typical inter-domain distance and the grey background represents non ordered ClO4. In (b) domains of types I and II with opposite ClO4 orientations are indicated. Along the line drawn in (b) there is a large distribution of domain sizes.

The change of topology, and the presence of additional regions of non ordered ClO4, has to be considered to interpret the cooling rate dependence of the superconductivity critical temperature, TS. For intermediate cooling rates, let say between 0.5 K/min and 5 K/min, the reduction of the fraction of AO superconducting domains, given in Figure 12, is mostly due to the increase of the average distance, l, between the AO domains (by a factor 7) while average domain size L decreases more slowly (by a factor 2). For intermediate cooling rates TS, after a rapid drop, saturates. In presence of such a texture superconductivity is achieved through Josephson coupling between AO superconducting domains through the non ordered regions. The quantitative evolution of TS has been calculated [169] using the dependence of L and l with the cooling rate. It is found that the rate of decrease of TS saturates in agreement with the experimental finding. Finally, for larger cooling rate, when L becomes smaller than the superconductivity coherence length, ξ0, TS rapidly drops to zero
In the solid solution (TMTSF)2(ClO4)1-x(ReO4)x, the profile of the superlattice reflections remains LS in the domain of existence of the (0, 1/2, 0) local order [170]. However the probability μ to cross a domain wall per unit length [167] increases considerably with x. As μ−1 amounts to the average distance between ReO4 substituent for x = 5% and 7% [171], ReO4 appears to be very efficient to break the (0, 1/2, 0) AO spatial coherence. This can be easily understood since ReO4 develops different interactions with TMTSF (interaction 2 in Figure 9) than does ClO4 (interaction 1 in Figure 9) [172]. With this texture it is found, by the measurement [173,174] of a quasi-linear decrease of Ts with x increasing, that ReO4 acts as non magnetic point defects on the superconductivity of (TMTSF)2ClO4. This behavior, interpreted as due to the reduction of the Cooper pair lifetime with the increasing number of defects, is taken as an evidence of the presence of nodes in the superconducting gap. However a microscopic theory linking this pair breaking effect at the local perturbations of the electronic structure due to the fragmentation of the (0, 1/2, 0) AO domain pattern with the ReO4 is lacking.
An important aspect of textured (TMTSF)2ClO4 is that the electronic parameters should vary spatially because the local order around the “ordered” and “non ordered” ClO4 is different. This is illustrated by the observation of a significant change of lattice parameters with the cooling rate. In particular the c parameter increases (decreases) in the quenched (relaxed) state with respect to the dependence extrapolated from the thermal variations above TAO [175,176]. Similarly the γ angle increases (decreases) in the relaxed (quenched) state. The decrease of c, direction along which TMTSF and ClO4 layers alternate, has to be related to the contraction of the methyl group cavity when the ordered ClO4 establishes its H-bonds in the relaxed state. The opening of γ corresponds to a shear deformation of the TMTSF layer when molecule A adopts the boat shape. In addition, high resolution measurements [164] show that the growth of the AO order parameter upon cooling below TAO is accompanied by erratic angular deformations of the lattice, consisting mostly in sudden rotations of the c direction probably due to the establishment of the H-bond network with newly ordered ClO4. Each angular jump, accompanied by a narrowing of the angular distribution of c directions, realizes an improvement of the crystalline order. Differently, the decrease of the AO order parameter upon heating is accompanied by the increase of the angular distribution of c directions due to the disorder induced by the successive breaks of the H-bonds. This shows that the AO process, via the progressive setting of the H-bond network, is accompanied by considerable elastic constraints.
7.4. The AO/CO Transitions of the SCN Salts
The early discovery [94] of the 2nd order AO transition of (TMTTF)2SCN was a very important step to assess the importance of electron-electron repulsions in organic conductors. Indeed the stabilization of the (0, 1/2, 1/2) superstructure below TAO = 160 K was the first realization of a MI transition [66] not accompanied by the opening of a gap in the spin degrees of freedom. In other words the AO of (TMTTF)2SCN achieved the spin-charge decoupling expected from the stabilization of a 4kF periodicity. Recently the structural refinement of the AO structure [103] was able to prove from the variation of the intra-molecular distances that the AO transition is accompanied by a 4kF CDW of an amplitude of 0.15 electrons, with an excess of hole on the TMTTF towards which the SCN points and an excess of electron in the opposite situation. This transition is also accompanied by a reduction of the amplitude of the stack dimerization. This result is expected by the repulsive coupling between the 4kF CDW and 4kF BOW order parameters (this is exactly the opposite of the effect described in Section 6.2 and Annex A.3 where the establishment of the SP tetramerization reduced the amplitude of the CO). At low temperature (TMTTF)2SCN undergoes at TN = 7 K [94] an AF transition which stabilizes the qAF = (1/2, 1/4, ?) commensurate magnetic order [177].
(TMTSF)2SCN also undergoes a MI transition at ~90K which does not affect the spin degrees of freedom and which is also associated to a structural modulation [178]. Thus a “4kF” charge localization should accompany this MI transition as in (TMTTF)2SCN. However the structural modulation surprisingly corresponds to the establishment of a triply incommensurate short range order. The wave vector of this modulation (0.48 ± 0.015, 0.65 ± 0.01, 0.1 ± 0.02) is quite different to the one, (0, 1/2, 1/2), stabilized at the CO/AO transition of (TMTTF)2SCN. It is however interesting to remark that the wave vector components of the incommensurate modulation of (TMTTF)2SCN approximately verify the relationship qd1 = 0, i.e., qa− qb + qc = 0 (mod. 1) derived in [60], for the setting of CO via direct interactions (S…NCS) between anions and donors. As the SCN anion can only take two orientations, the incommensurability of the modulation is certainly related to the probability of occupancy of one of the two orientations of the anion. For such a wave the incommensurability observed in diffraction experiments results from a statistical average because locally the modulation must be commensurate. The perfection of such a wave is altered (i.e., the harmonic content of the modulation is reduced) if there is a broad distribution of commensurate domain sizes. In addition, if visualizing the modulation from the origin of the lattice there are increasing fluctuations on the position of successive domain walls, or equivalently cumulative fluctuations of domain size, the long range order is lost. The Fourier transform of such a modulation pattern leads to broadened satellite reflections. Similar features are observed for the incommensurate CO modulation of (DMtTTF)2ClO4 [102].
7.5. The (1/2, 0, 0) AO Transition of the NO3 Salts
Both (TMTSF)2NO3 and (TMTTF)2NO3 undergo an AO transition at TAO = 41 K [143] and 50 K [157] respectively and which stabilizes the same (1/2, 0, 0) superstructure. Even if this superstructure establishes the 2kF periodicity in stack direction, the AO transition has a weak influence on the electronic properties of the donor stack.
The (1/2, 0, 0) superstructure of (TMTSF)2NO3 has been refined [165]. No substantial stack tetramerization and differentiation between the individual TMTSF are observed below TAO (the doubling of stack periodicity should, in principle, differentiate two molecules per unit cell). This means that the NO3 anion orders without significantly perturbing its methyl group cavity. Such a result is expected because of the small size of the NO3 anion. As the organic array is not perturbed by the AO process the electronic structure remains basically unchanged below TAO and (TMTSF)2NO3 does not become semi-metallic as previously predicted [144,145]. Its FS remains quasi-1D below TAO [179] and available for a nesting instability. Indeed (TMTSF)2NO3 stabilizes a SDW ground state at TSDW = 9 K whose modulation wave vector, determined by NMR [180], is close to the wave vector of the SDW phase of (TMTSF)2PF6 [54,55].
8. Conclusions
We have reviewed the very rich panel of structural instabilities exhibited by the Bechgaard and Fabre salts. We have presented a coherent synthesis of the latest experimental developments and interpreted more quantitatively older results in a unified picture. This allows us to correlate the structural instabilities with the electronic, magnetic and superconducting properties of these salts.
The main conclusion is that structural degrees of freedom have a decisive influence on the physical properties the Bechgaard and Fabre salts; an influence which is underestimated in the present literature. Structural instabilities are not considered in the interpretation of the generic electronic phase diagram [52] giving the evolution of the crossovers and of the ground states exhibited by the octahedral anion salts in function of a generalized coordinate, which can be the size of the anion or the pressure although there is not a direct correspondence between these variables [17]. The current interpretation [51] describes the evolution of 1D instabilities and ground states by taking into account the variation of electronic interactions and couplings with pressure, nature of the donor and size of the anion. However we have shown in this review that the orientation disorder of the anions and of the methyl groups should influence the quasi-1D electronic structure in a manner which is not considered until now. The generic phase diagram accounts for the Mott-Hubbard charge localization, the crossover from a 1D Luttinger liquid to 2D and 3D Fermi liquids, the FS nesting instability leading to the density wave ground state and, with the deterioration of the nesting, the restoration of the metallic state which achieves superconductivity. However BOW, CO and ferroelectric intra-chain instabilities involving intra-stack electronic charge redistributions and inter-chain Coulomb couplings are not included in this description. In particular structural degrees of freedom which take into account the reaction of the lattice at electronic charge redistributions should directly control the development of charge instabilities and set the inter-chain coupling via a generalized electron-phonon coupling including the anion shift. Lattice degrees of freedom play also a role in SP, AF and SDW transitions via “magneto-elastic” couplings which are quite important in the Fabre and Bechgaard salts [25,128,181]).
In this review we have also shown that anions set direct and/or indirect inter-chain coupling mechanisms allowing stabilizing CO, ferroelectric and SP ground states. Also the blockade of the anions in the methyl group cavities can be taken as responsible for the suppression of the CO and SP phase transitions under pressure and of the 2kF BOW instability of (TMTSF)2PF6 at low temperature. In addition, the adiabaticity of the phonon field controlling the lattice dynamics in reaction at the charge redistribution must be considered. We provide here arguments showing that non-adiabaticity is relevant to explain the SP/AF phase diagram of Fabre salts.
Figure 14.
CO (of amplitude ±δ) and AF exchange coupling JS1S2 function of a generalized distortion parameter u of the organic stack. The drawing represents the extreme δ = 0.5 situation where the HOMO of the donor is alternatively occupied by one electron (ionic donor D+) and two electrons (neutral donor D0). In the drawing the pink dots represent the electrons. The holes, corresponding to the absence of one electron on D+, bear the spins S1 and S2. The exchange interaction J(u) is tuned by the position, u, of the neutral molecule which controls the intra-stack overlap of the HOMO’s. The figure shows also that the molecular shift u changes the orientation of the electric dipole P of the D0D+ dimer.
Figure 14.
CO (of amplitude ±δ) and AF exchange coupling JS1S2 function of a generalized distortion parameter u of the organic stack. The drawing represents the extreme δ = 0.5 situation where the HOMO of the donor is alternatively occupied by one electron (ionic donor D+) and two electrons (neutral donor D0). In the drawing the pink dots represent the electrons. The holes, corresponding to the absence of one electron on D+, bear the spins S1 and S2. The exchange interaction J(u) is tuned by the position, u, of the neutral molecule which controls the intra-stack overlap of the HOMO’s. The figure shows also that the molecular shift u changes the orientation of the electric dipole P of the D0D+ dimer.
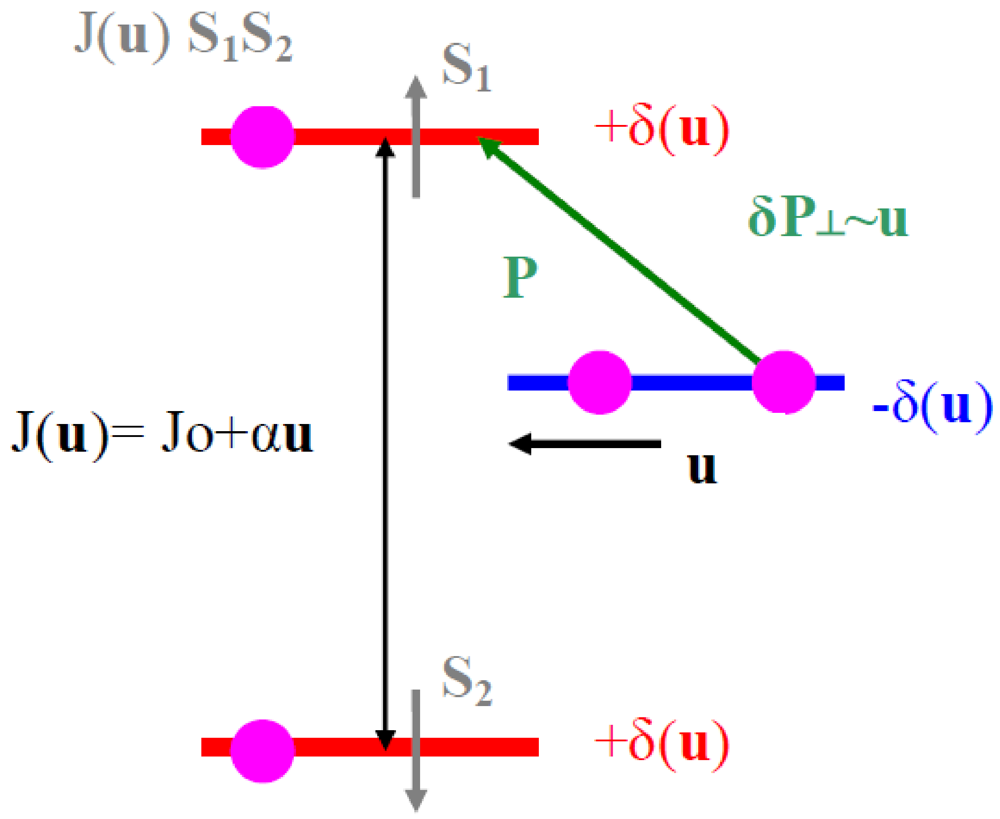
The generic phase diagram does not include salts with non centro-symmetrical anions such as those with ReO4 or FSO3 which become superconductors at larger TS than TS of salts with octahedral anions. In these salts, AO transitions, involving important charge redistributions, activate CDW and BOW instabilities of the organic stack. These charge displacements are accompanied by substantial deformations of the lattice structure. The modification of the electronic structure which thus results drives noticeable change of electronic properties. In this manner non centro-symmetrical anions can tune novel ground state competitions. Also little is known regarding the general pressure evolution of the AO ground states which, for salts with ReO4 and FSO3 anions stabilize new metallic super-structures which exhibit superconductivity, some of them being probably non-centrosymmetric when a ferroelectric dipolar anion order is achieved.
A beautiful illustration of the interplay between SDW and superconductivity is shown by (TMTSF)2ClO4. In this salt we have shown that this competition is controlled by the texture of the AO pattern. This is a simple case where superconductivity and SDW can be simply associated to structurally different regions. Texture effects are also important in the competition between SDW and superconductivity in pressurized (TMTSF)2PF6 [182]. In a general way it is interesting to remark that, similarly to pressurized (TMTSF)2PF6, the highest TS in pressurized (TMTSF)2ReO4 [151,153] and FSO3 [159,160] is achieved in the de-mixing region between different AO phases.
Finally we conclude on the possible interplay between ferroelectricity and (antiferro-) magnetism in the Fabre salts which could give rise to a multiferroic behavior [183] where the action on the dielectric polarization reacts on the magnetic polarization and vice-versa. In particular, CO and the associated lattice deformation could control via a sizeable spin-phonon coupling the magnitude of the exchange interaction and the direction of the dielectric polarization as schematically illustrated by Figure 14. In addition, dipolar anions located between donor stacks could add a new degree of freedom at the ferroelectricity. Multiferroicity is now well documented in inorganic materials (see for example [184,185]). Its manifestation in spin-charge decoupled organic materials such as the ones discussed in this review deserve special attention in the future.
Conflict of Interest
The author declares no conflict of interest.
Acknowledgments
The work reported here is due to wide collaborations which have been quoted in the references. For the most recent achievements outlined in this review, useful discussions with E. Canadell, S. Charfi-Kaddour, C. Coulon, M. de Souza, M. Dressel, G. Giovannetti, S. Haddad, P. Foury-Leylekian, M. Lang, M. Poirier, E. Rose and H. Sawa are acknowledged. The author is also very grateful to C. Bourbonnais for a general discussion on the content of the paper.
Appendix: Landau Theory with Coupled Order Parameters Applied at the Phase Diagram of the Bechgaard and Fabre Salts
A.1. The AF Phase of (TMTTF)2Br
The primary order parameter η(q) describes the AF modulation. This two components AF order parameter, where qAF = (1/2, ±1/4, ?), can be represented by the complex number ηeiθ. To the structural modulation, with qS = (1, 1/2, ?), corresponds the real order parameter ρ. The Landau development of the free energy, which is function of η, θ and ρ, contains 3 contributions corresponding respectively in expression (A1) to (a) AF coupling, (b) cost of elastic deformation and (c) magneto-elastic coupling:

The 3rd term in FAF(η,θ) is the umklapp contribution due to commensurability 4 of the modulation: 4qAF is a reciprocal lattice wave vector. θ fixes the phase of the AF modulation with respect to the lattice. The minimization of F (η,θ,ρ) with respect to ρ gives :

In (A2), ρ is proportional to the square of the magnetization and depends on the phase θ of the AF modulation. (−λ2/8d) renormalizes the 4rd order coefficients b and u, which thus become b' = b − (λ2/8e) and u’ = u − (λ2/8e). By replacing ρ by expression (A2) in (A1), F(η,θ) reads:
F(η, θ) = a η2 +b' η4 +u' η4 cos4θ (A3)
As the AF transition of (TMTTF)2Br is of 2nd order one has b' > 0 in (A3). (A3) is minimum with respect to θ for sin4θ = 0. This leads to θ = pπ/4, with p integer, from which two types of free energy F(η) can be obtained:

The minimization of F(η) with respect to η fixes the primary order parameter which is non-zero when a < 0 (a = 0 at TN). ρ is related to η and θ by the expression (A.2). The two classes of solutions of (A4) are schematically represented in Figure 6. The magneto-elastic coupling λ favours solution (a) while the umklapp term u favours solution (b). Solution (a) is the most stable if λ2 > 8 eu. The structural order parameter ρ is different from zero for solution (a) since the magneto-elastic coupling provides an energy gain only if η(q) located on one site out of two. The charge modulation which thus results corresponds to a 4kF CDW or a CO.
This simple model ignores the stack dimerization otherwise a magnetoelastic BOW deformation could be achieved in solution (b). In this case there is an energy gain to put the parallel magnetizations on the intra-dimer bond and to shorten this bond. This shortening will favour the delocalization of the hole bearing the spin between the two sites forming the dimer.
A.2. The Density Wave Phases of (TMTSF)2PF6
According to data reported in Section 4.2, the analysis of the density wave phase transition should include 3 order parameters related to the 2kF SDW (η1(q)), 2kF CDW (η2(q)), and 4kF CDW (ρ) modulations. As in Annex A.1, the first two order parameters are complex quantities and the third one is real. As qSDW is incommensurate there is no umklapp term in the free energy. In addition because of the incommensurability only the phase difference ω between the two complex order parameters η1 and η2 is relevant. There is also a phase shift θ, already considered in the Annex A.1, between η2 and ρ. In order to perform a derivation as simple as possible we shall ignore ρ and the phase shift θ (in Annex A.1 expression (A.4) shows that the minimization of the free energy with respect to ρ and θ leads to a renormalization of the 4rd order coefficient of the primary order parameter). The Landau development of the free energy F(η1,η2,ω) is given by:

The free energy is minimum if ω=0 (=π/2) for negative (positive) ν. This leads to a coupling term:
FC (η1, η2)=cη12 η22, where c= μ − │ν│ (A6)
Figure A1.
Phase diagrams of the Landau free energy, written on top of the figure, in function of the temperature (T) and of an external parameter, h, allowing the variation of the Landau coefficients ai. In the figure both b1 and b2 are positive. The value and sign of the coupling c between the order parameters are indicated for each phase diagram. Thin (thick) lines represent 2nd (1st) order transition lines. The phases are labeled by the order parameter which differs from zero. “0” corresponds to the non modulated (η1 = η2 = 0) phase. The critical temperature achieving pure η1 or η2 phases on each side of the phase “0” trough 2nd order transitions (thin green lines) is given by a1 = 0 or a2 = 0 respectively. The arrows connected to the 2nd order transition lines (thin red (blue) lines) delimiting the (η1, η2) phase for 0 < c < 2√(b1b2) (0 > c > −2√(b1b2)) indicate the sense of displacement of the phase boundaries when c increases (c decreases). This displacement shows, when c > 2√(b1b2), that the (η1, η2) phase collapses to a first order transition (thick red line) linking the pure η1 and η2 phases. When c < −2√(b1b2), the expansion of the (η1, η2) phase renders this phase directly reachable from the “0” phase through a first order phase transition (thick blue line).
Figure A1.
Phase diagrams of the Landau free energy, written on top of the figure, in function of the temperature (T) and of an external parameter, h, allowing the variation of the Landau coefficients ai. In the figure both b1 and b2 are positive. The value and sign of the coupling c between the order parameters are indicated for each phase diagram. Thin (thick) lines represent 2nd (1st) order transition lines. The phases are labeled by the order parameter which differs from zero. “0” corresponds to the non modulated (η1 = η2 = 0) phase. The critical temperature achieving pure η1 or η2 phases on each side of the phase “0” trough 2nd order transitions (thin green lines) is given by a1 = 0 or a2 = 0 respectively. The arrows connected to the 2nd order transition lines (thin red (blue) lines) delimiting the (η1, η2) phase for 0 < c < 2√(b1b2) (0 > c > −2√(b1b2)) indicate the sense of displacement of the phase boundaries when c increases (c decreases). This displacement shows, when c > 2√(b1b2), that the (η1, η2) phase collapses to a first order transition (thick red line) linking the pure η1 and η2 phases. When c < −2√(b1b2), the expansion of the (η1, η2) phase renders this phase directly reachable from the “0” phase through a first order phase transition (thick blue line).
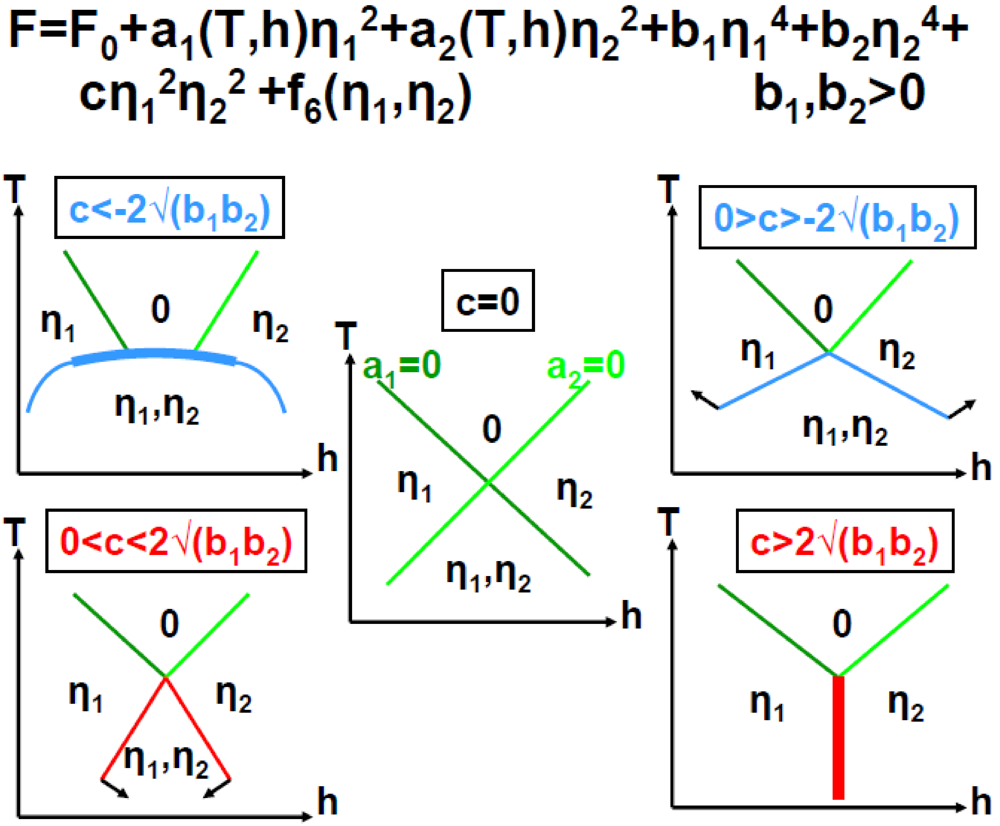
The functional (A5) with FC given by (A6), together with the presence of additional 6th order terms providing the stability of the phases, has been studied in the literature [186,187,85]. These works lead to elaborated phase diagrams depending upon the value and sign of the coefficients entering in the Landau development. The results for positive b1 and b2 are summarized in Figure A1. Figure A1 shows that for an attractive enough coupling, c < −2√(b1b2), between the 2kF SDW and 2kF CDW order parameters the SDW/CDW modulated phase can be reached trough a first order transition even if b1 and b2 coefficients of the 4th order terms are positive (see also Figure 7a). This peculiar feature was first noticed in [186]. This result requires going beyond the standard theory reported in [187], which, being restricted to c > −2√(b1b2), gives a transition to the mixed phase only through a peculiar point corresponding to the simultaneous vanishing: a1 = a2 = 0. A more general treatment considering negative bi coefficients has been developed in [85]. This theory leads to phase diagrams stabilizing the mixed (η1, η2) phases if one or two bi coefficients are negative. All these phase diagrams are summarized in Figure 7. The stabilization of the (η1, η2) phase implies that c should be negative (attractive interaction between the 2kF SDW and the 2kF CDW) and that b1 and b2 are small or even negative. c negative means 0 < μ < │ν│or μ < 0. The condition b1 and b2 small or even negative is obtained if there is an efficient renormalization of the bi’s due to a strong coupling between the 2kF SDW and/or the 2kF CDW and the secondary order parameter ρ corresponding to the 4kF CDW. This renormalization is due to the magneto-elastic coupling previously considered in Annex A.1.
A.3. Competition between CO and SP Transitions
We assume that CO develops first and that the SP transition occurs inside the CO phase. These two modulated states are characterized by the real order parameters ηCO and ηSP, characterized by the qCO = (1, 0, 0) and qSP = (1/2, 1/2, 1/2) wave vectors respectively. When ηCO is already established, the Landau development of the free energy for the SP order parameter is given by:

Note that the Landau development in ηSP implies the presence of an adiabatic phonon field. In (A7) one has aSP = αSP(T− TSP0), bSP > 0. We shall assume in the following that ηCO(T) is enough small so that its value near TSP can still be obtained by the minimization of the two first terms in the development of the CO free energy given by:
F(ηCO)= aCOηCO2 +bCOηCO4. (A8)
Then the minimization ∂F(ηCO)/∂ηCO = 0 leads to:
ηCO(T) = [αCO(T− TCO)/2bCO]1/2 (A9)
In the following we shall assume that ηCO remains small enough so that only the lowest order λ coupling term will be considered in FC(ηCO, ηSP). With these approximations ηCO renormalizes the Landau coefficient aSP, which thus becomes:
a'SP = aSP + ληCO = αSP(T− TSP) (A10)
This leads to a renormalization of the critical temperature TSP0, given by aSP = αSP(T− TSP0), to TSP defined by (A.10). With TSP much smaller than TCO one gets:
TSP = TSP0 − ληCO(TSP)/αSP (A11)
With ηCO(TSP) given by (A9), (A11) becomes:
TSP = TSP0 − A(TCO− TSP)1/2, with A = (λ/αSP)[αCO/2bCO]1/2 (A12)
The relationship (A12) between TSP and TCO is drawn in Figure 11. Note that if the μ biquadratic term of FC(ηCO,ηSP) is considered instead of the λ linear-quadratic term there is a linear relationship between TSP and TCO. This linear plot is shown in Figure 12 of [73].
The minimization of (A7) + (A8) with respect to ηCO leads to a decrease of ηCO(T) below TSP, when the repulsive coupling FC(ηCO,ηSP) develops with the growth of ηSP. The (negative) relative correction ΔηCO(T)/ηCO(TSP) at the ηCO order parameter variation given by (A9) is, in first order in λ:

The μ biquadratic term in FC(ηCO,ηSP) leads also to a negative correction proportional to ηSP(T)2.
References and Notes
- Jérome, D.; Schulz, H.J. Organic conductors and superconductors. Adv. Phys. 1982, 31, 299–490. [Google Scholar] [CrossRef]
- Ishiguro, T.; Yamaji, K.; Saito, G. Organic Super-Conductors, 2nd ed; Springer-Verlag: Berlin/Heidelberg, Germany, 1998; Volume 88. [Google Scholar]
- Toyota, N.; Lang, M.; Műller, J. Low-Dimensional Molecular Metals; Springer-Verlag: Berlin/Heidelberg, Germany, 2007; Volume 154. [Google Scholar]
- Henriques, R.T.; Alcácer, L.; Pouget, J.P.; Jérome, D. Electrical conductivity and x-ray diffuse scattering study of organic conductors (perylene)2M(mnt)2, (M=Pt, Pd, Au). J. Phys. C: Solid State Phys. 1984, 17, 5197–5208. [Google Scholar] [CrossRef]
- Rovira, C.; Veciana, J.; Ribera, E.; Tarrès, J.; Canadell, E.; Rousseau, R.; Mas, M.; Molins, E.; Almeida, M.; Henriques, R.T.; et al. An organic spin-ladder molecular material. Angew. Chem. Int. Ed. 1997, 36, 2324–2326. [Google Scholar] [CrossRef]
- Canadell, E.; Rachidi, I. E.-I.; Ravy, S.; Pouget, J.-P.; Brossard, L.; Legros, J.P. On the band electronic structure of X[M(dmit)2]2 (X = TTF, (CH3)4N; M = Ni, Pd) molecular conductors and superconductors. J. Phys. France 1989, 50, 2967–2981. [Google Scholar] [CrossRef]
- Pouget, J.-P.; Ravy, S. Structural aspects of the bechgaard salts and related compounds. J. Phys. I France 1996, 6, 1501–1525. [Google Scholar] [CrossRef]
- Pouget, J.-P. Structural features of the Bechgaard salts and related compounds. J. Phys. IV France 2000, 10, Pr3-43–Pr3-56. [Google Scholar] [CrossRef]
- Thorup, N.; Rindorf, G.; Soling, H.; Bechgaard, K. The structure of Di(2,3,6,7-tetramethyl-1,4,5,8-tetraselenafulvalenium) Hexafluorophosphate, (TMTSF)2PF6, the First Superconducting Organic Solid. Acta Cryst. B 1981, 37, 1236–1240. [Google Scholar] [CrossRef]
- Pashkin, A.; Dressel, M.; Ebbinghaus, S.G.; Hanfland, M.; Kuntscher, C.A. Pressure-induced structural phase transition in the Bechgaard-Fabre salts. Synth. Met. 2009, 159, 2097–2100. [Google Scholar] [CrossRef]
- Gallois, B.; Gaultier, J.; Hauw, C.; Lamcharfi, T.; Filhol, A. Neutron Low-Temperature (4 and 20K) and X-ray High-Pressure (6.5 × 102 and 9.8 × 102 MPa) Structures of the Organic Superconductor Di(2,3,6,7-tetramethyl-1,4,5,8-tetraselenafulvalenium) Hexafluorophosphate, (TMTSF)2PF6. Acta Cryst. B 1986, 42, 564–575. [Google Scholar] [CrossRef]
- Emery, V.J.; Bruinsma, R.; Barisic, S. Electron-electron umklapp scattering in organic superconductors. Phys. Rev. Lett. 1982, 48, 1039–1043. [Google Scholar] [CrossRef]
- Ishibashi, S.; Manuel, A.A.; Kohyama, M. Ab initio pseudopotential calculation for (TMTSF)2ClO4. J. Phys. Cond. Matt. 1999, 11, 2279–2283. [Google Scholar] [CrossRef]
- Liautard, B.; Peytavin, S.; Brun, G.; Maurin, M. Corrélations structurales dans la série (TMTTF)2X. J. Phys. (Paris) 1982, 43, 1453–1459. [Google Scholar] [CrossRef]
- Liautard, B.; Peytavin, S.; Brun, G.; Maurin, M. Structural studies and physical properties in the organic conductors series (TMTTF)2X and (TMTSF)2X. J. Phys. (Paris) 1983, 44, C3-951–C3-956. [Google Scholar] [CrossRef]
- Kistenmacher, T.J. Cavity size versus anion size in (TMTSF)2X salts: Possible implications for the uniqueness of (TMTSF)2ClO4. Solid State Commun. 1984, 50, 729–733. [Google Scholar] [CrossRef]
- This assertion is incorrect for TMTSF and TMTSF salts because the anion modifies mainly the c parameter [14,15] while pressure leads to a relative decrease of the parameter a twice larger than for parameters b and c [10,11].
- Kistenmacher, T.J. Anion-donor coupling in (TMTSF)2X salts: Symmetry considerations. Solid State Commun. 1984, 51, 931–934. [Google Scholar] [CrossRef]
- Beno, M.A.; Blackman, G.S.; Leung, P.C.W.; Williams, J.M. Hydrogen bond formation and anion ordering in superconducting (TMTSF)2ClO4 and (TMTSF)2AsF6. Solid State Commun. 1983, 48, 99–103. [Google Scholar] [CrossRef]
- Granier, T.; Gallois, B.; Fritsch, A.; Ducasse, L.; Coulon, C. 135K Crystallographic and Electronic Structure of (TMTTF)2SbF6. In Lower-Dimensional Systems and Molecular Electronics; Metzger, R.M., Day, P., Papavassiliou, G.C., Eds.; Plenum Press: New York, NY, USA, 1990; Volume 248, pp. 163–168. [Google Scholar]
- Pouget, J.-P.; Foury-Leylekian, P.; Alemany, P.; Canadell, E. Charge ordering in low dimensional organic conductors: structural aspects. Phys. Status Solidi B 2012, 249, 937–942. [Google Scholar] [CrossRef]
- Jacobsen, C.S.; Tanner, D.B.; Bechgaard, K. Optical and infrared properties of tetramethyltetraselenafulvalene [(TMTSF)2X] and tetramethyltetrathiafulvalene [(TMTTF)2X] compounds. Phys. Rev. B 1983, 28, 7019–7032. [Google Scholar] [CrossRef]
- de Souza, M.; Hofmann, D.; Foury-Leylekian, P.; Moradpour, A.; Pouget, J.-P.; Lang, M. Exploring the charge-ordering transition in (TMTTF)2X via thermal expansion measurements. Phys. B 2010, 405, S92–S94. [Google Scholar] [CrossRef]
- de Souza, M.; Foury-Leylekian, P.; Moradpour, A.; Pouget, J.-P.; Lang, M. Evidence for lattice effects at the charge-ordering transition in (TMTTF)2X. Phys. Rev. Lett. 2008, 101, 216403. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.; Műller, J.; Steglich, F.; Brűhl, A.; Wolf, B.; Dressel, M. Anomalous thermal expansion behaviour in (TMTTF)2PF6 and (TMTSF)2PF6 around 100K: evidence for a strong spin-phonon interaction? J. Phys. IV France 2004, 114, 111–112. [Google Scholar] [CrossRef]
- Bechgaard, K.; Jacobsen, C.S.; Mortensen, K.; Pedersen, H.J.; Thorup, N. The properties of five highly conducting salts: (TMTSF)2X, X=PF6−, AsF6−, SbF6−, BF4− and NO3− derived from tetramethyltetraselenafulvalene (TMTSF). Solid State Commun. 1980, 33, 1119–1125. [Google Scholar] [CrossRef]
- Yu, W.; Zhang, F.; Zamborszky, F.; Alavi, B.; Baur, A.; Merlic, C.A.; Brown, S.E. Electron-lattice coupling and broken symmetries of the molecular salt (TMTTF)2SbF6. Phys. Rev. B 2004, 70, 121101 (R). [Google Scholar]
- Furukawa, K.; Hara, T.; Nakamura, T. Deuteration effect and possible origin of the charge-ordering transition of (TMTTF)2X. J. Phys. Soc. Jpn. 2005, 74, 3288–3294. [Google Scholar] [CrossRef]
- Williams, J.M.; Beno, M.A.; Sullivan, J.C.; Banovetz, L.M.; Braam, J.M.; Blackman, G.S.; Carlson, C.D.; Greer, D.L.; Loesing, D.M.; Carneiro, K. The design of organic metals based on TMTSF and TMTTF: novel structural implications and predictions. J. Phys. (Paris) 1983, 44, C3-941–C3-949. [Google Scholar] [CrossRef]
- Scott, J.C.; Pedersen, H.J.; Bechgaard, K. Proton NMR in the organic conductor tetramethyltetraselenafulvalenium hexafluorophosphate. Phys. Rev. B 1981, 24, 475–477. [Google Scholar] [CrossRef]
- McBrierty, V.J.; Douglass, D.C.; Wudl, F.; Aharon-Shalom, E. Nuclear resonance and relaxation in ditetramethyltetraselenafulvalenium salts. Phys. Rev. B 1982, 26, 4805–4809. [Google Scholar] [CrossRef]
- Scott, J.C.; Engler, E.M.; Clark, W.G.; Murayama, C.; Bechgaard, K.; Pedersen, H.J. NMR studies of (TMTSF)2PF6. Mol. Cryst. Liq. Cryst. 1982, 79, 61–65. [Google Scholar]
- Stein, P.C.; Moradpour, A.; Jérome, D. Nuclear relaxation in tetramethyltetraselenafulvalene salts (TMTSF)2X, (X=PF6, ClO4). J. Phys. Lett. (Paris) 1985, 46, L241–L247. [Google Scholar] [CrossRef]
- Foury-Leylekian, P.; Petit, S.; André, G.; de Souza, M.; Lang, M.; Ressouche, E.; Moradpour, A.; Pouget, J.-P*. Low temperature structural ordering in the (TMTSF)2PF6 and AsF6 Bechgaard salts. In preparation, 2012.*; Laboratoire de Physique des Solides, Université Paris-sud: 91405 Orsay, France.
- Aoyagi, B.; Kato, K.; Ota, A.; Yamochi, A.; Saito, G.; Suematsu, H.; Sakata, M.; Takata, M. Direct Observation of Bonding and Charge Ordering in (EDO-TTF)2PF6. Angew. Chem. Int. Ed. 2004, 43, 3670–3676. [Google Scholar] [CrossRef]
- Pouget, J.-P. Structural Instabilities. In Highly Conducting Quasi One Dimensional Organic Crystals; Conwell, E.M., Ed.; Academic Press: New York, NY, USA, 1988; Semiconductors and Semimetals; Volume 27, pp. 87–215. [Google Scholar]
- Shirane, G.; Shapiro, S.M.; Comès, R.; Garito, A.F.; Heeger, A.J. Phonon dispersion and Kohn anomaly in tetrathiafulvalene-tetracyanoquinodimethane (TTF-TCNQ). Phys. Rev. B 1976, 14, 2325–2334. [Google Scholar] [CrossRef]
- Pouget, J.P. Structural Instabilities of One-Dimensional Conductors. In Low-Dimensional Conductors and Superconductors; Jérome, D., Caron, L.G., Eds.; Plenum Press: New York, NY, USA, 1987; Volume 155, pp. 17–45. [Google Scholar]
- Ribault, M.; Pouget, J.-P.; Jérome, D.; Bechgaard, K. Superconductivity and Absence of a Kohn Anomaly in the Quasi One Dimensional Organic Conductor: (TMTSF)2AsF6. J. Phys. Lett. (Paris) 1980, 41, L607–L610. [Google Scholar] [CrossRef]
- Pouget, J.-P.; Moret, R.; Comès, R.; Bechgaard, K.; Fabre, J.M.; Giral, L. X-Ray Diffuse Scattering Study of Some (TMTSF)2X and (TMTTF)2X Salts. Mol. Cryst. Liq. Cryst. 1982, 79, 129–143. [Google Scholar]
- Slater, J.C. Magnetic Effect and the Hartree-Fock Equation. Phys. Rev. 1951, 82, 538–541. [Google Scholar] [CrossRef]
- Pouget, J.-P. X-ray diffuse scattering as precursor of incommensurate Peierls transitions in one-dimensional organic charge transfer conductors. Z. Kristallogr. 2004, 219, 711–718. [Google Scholar] [CrossRef]
- Cao, N.; Timusk, T.; Bechgaard, K. Unconventional Electrodynamic Response of the Quasi-One-Dimensional Organic Conductor (TMTSF)2ClO4. J. Phys. I France 1996, 6, 1719–1726. [Google Scholar] [CrossRef]
- Dressel, M.; Schwartz, A.; Grüner, G.; Degiorgi, L. Deviations from Drude Response in Low-Dimensional Metals: Electrodynamics of the Metallic State of (TMTSF)2PF6. Phys. Rev. Lett. 1996, 77, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.; Dressel, M.; Grüner, G.; Vescoli, V.; Degiorgi, L.; Giamarchi, T. On-chain electrodynamics of metallic (TMTSF)2X salts: observation of Tomonaga-Luttinger liquid response. Phys. Rev. B 1998, 58, 1261–1271. [Google Scholar] [CrossRef]
- Wzietek, P.; Creuzet, F.; Bourbonnais, C.; Jérome, D.; Bechgaard, K.; Batail, P. Nuclear-relaxation and electronic correlations in quasi-one-dimensional organic conductors. 2. Experiments. J. Phys. I France 1993, 3, 171–201. [Google Scholar] [CrossRef]
- Chaikin, P.M.; Tiedje, T.; Bloch, A.N. Sound velocity measurements in (TMTSF)2PF6. Solid State Commun. 1982, 41, 739–742. [Google Scholar] [CrossRef]
- Shi, X.D.; Chiang, L.; Upasani, R.; Chaikin, P.M. Sound velocity studies of Bechgaard salts (TMTSF)2PF6 and(TMTSF)2ClO4. In Advanced Organic Solid State Materials; Chiang, L.Y., Chaikin, P.M., Cowan, D.O., Eds.; Materials Research Society: Pittsburgh, PA, USA, 1990; pp. 239–244. [Google Scholar]
- Fertey, P.; Poirier, M.; Batail, P. Microwave transport approach to the coherence of interchain hopping in (TMTSF)2PF6. Eur. Phys. J. B 1999, 10, 305–309. [Google Scholar] [CrossRef]
- Fertey, P.; Poirier, M.; Batail, P. H-T behaviors of the transverse microwave conductivity of (TMTSF)2X crystals. Synth. Met. 1999, 103, 2076–2077. [Google Scholar] [CrossRef]
- Bourbonnais, C. Organic Superconductors: Reduced Dimensionality and Correlation Effects. Synth. Met. 1997, 84, 19–24. [Google Scholar] [CrossRef]
- Bourbonnais, C.; Jérome, D. Electronic confinement in organic metals. Science 1998, 281, 1155–1156. [Google Scholar] [CrossRef]
- Giamarchi, T. Theoretical Framework for Quasi-One Dimensional Systems. Chem. Rev. 2004, 104, 5037–5055. [Google Scholar] [CrossRef]
- Delrieu, J.M.; Roger, M.; Toffano, Z.; Moradpour, A.; Bechgaard, K. NMR proton lineshape in (TMTSF)2X- incommensurability of nesting wave vector and order parameter. J. Phys. (Paris) 1986, 47, 839–861. [Google Scholar] [CrossRef]
- Takahashi, T.; Maniva, Y.; Kawamura, H.; Saito, G. Determination of SDW characteristics in (TMTSF)2PF6 by 1H NMR analysis. J. Phys. Soc. Jpn. 1986, 55, 1364–1373. [Google Scholar] [CrossRef]
- Ducasse, L.; Abderrabba, M.; Gallois, B. Temperature dependence of the Fermi surface topography in the (TMTSF)2X and (TMTTF)2X families. J. Phys. C: Solid State Phys. 1985, 18, L947–L951. [Google Scholar] [CrossRef]
- Vaca, P.; Coulon, C. Magnetic phase-transition in Bechgaard salts and related-compounds - electronic localization and competing interchain couplings. Phase Transitions 1991, 30, 49–62. [Google Scholar] [CrossRef]
- Moret, R.; Pouget, J.P.; Comès, R.; Bechgaard, K. X-ray Scattering evidence for anion ordering and structural distortions in the low-temperature phase of Di (Tetramethyltetraselenafulvalenium) perrhenate [(TMTSF)2ReO4]. Phys. Rev. Lett. 1982, 49, 1008–1012. [Google Scholar] [CrossRef]
- Rindorf, G.; Soling, H.; Thorup, N. Di(4,4',5,5'-tetramethyl-2,2'-bi-&,3-diselenolyliden)ium Perrhenate, C20H24Se+8.ReO4, (TMTSF)2ReO4. Detailed superstructure at 120 K. Acta Cryst. C 1984, 40, 1137–1139. [Google Scholar] [CrossRef]
- By the same argument one obtains a 3D 4kF CDW or CO if the alternation of charge rich and charge poor molecules along a given stack is fixed by uniform shifts of anions separated by a and di = a ± b – c (± corresponding to the two coupling process considered in the text and in Figure 9). qa = qdi = 0 (mod. 2π) leads to qa=0 and qa ± qb + qc = 0 (mod. 1). These relationships leads to ±qb + qc =0 (mod.1) which has two solutions: qb=qc=0 and qb = qc = 1/2. They corresponds respectively to the ferroelectric qF = (0, 0, 0) and antiferroelectric qAF = (0, 1/2, 1/2) CO patterns.
- Pouget, J.-P.; Shirane, G.; Bechgaard, K.; Fabre, J.M. X-ray evidence of a structural phase transition in di-tetramethyltetraselenafulvalenium perchlorate [(TMTSF)2ClO4], pristine and slightly doped. Phys. Rev. B 1983, 27, 5203–5206. [Google Scholar] [CrossRef]
- Pouget, J.-P.; Moret, R.; Comès, R.; Shirane, G.; Bechgaard, K.; Fabre, J.M. X-ray evidence of competing orderings in (TMTSF)2ClO4 and related alloys. J. Phys. (Paris) 1983, 44, C3-969–C3-975. [Google Scholar] [CrossRef]
- Liu, Q.; Ravy, S.; Pouget, J.P.; Johannsen, I. X-ray investigation of the tetramethyldithiadiselenafulvalene (TMDTDSF)2X series of organic conductors. II. Influence of the orientational disorder on the structural instabilities. J. Phys. I France 1993, 3, 821–837. [Google Scholar] [CrossRef]
- Liu, Q.; Ravy, S.; Pouget, J.P.; Johannsen, I.; Bechgaard, K. X-ray investigation of the tetramethyldithiadiselenafulvalene (TMDTDSF)2X series of organic conductors. I. Study of the orientational disorder of the TMDTDST molecule. J. Phys. I France 1993, 3, 803–819. [Google Scholar] [CrossRef]
- Pouget, J.-P.; Ravy, S. X-Ray evidence of charge density wave modulations in the magnetic phases of (TMTSF)2PF6 and (TMTTF)2Br. Synth. Met. 1997, 85, 1523–1528. [Google Scholar] [CrossRef]
- Coulon, C.; Delhaes, P.; Flandrois, S.; Lagnier, R.; Bonjour, E.; Fabre, J.M. A new survey of the physical properties of the (TMTTF)2X series. Role of the counterion ordering. J. Phys. (Paris) 1982, 43, 1059–1067. [Google Scholar] [CrossRef]
- Coulon, C.; Delhaes, P.; Amiell, J.; Manceau, J.P.; Fabre, J.M.; Giral, L. Effet of doping (TMTSF)2ClO4 with TMTTF I. Ambient pressure result: A competition between the different possible ground states. J. Phys. (Paris) 1982, 43, 1721–1729. [Google Scholar] [CrossRef]
- Foury-Leylekian, P.; Le Bolloc’h, D.; Hennion, B.; Ravy, S.; Moradpour, A.; Pouget, J.-P. Neutron-scattering evidence for a spin-Peierls ground state in (TMTTF)2PF6. Phys. Rev. B 2004, 70, 180405 (R). [Google Scholar]
- Dumoulin, L.; Bourbonnais, C. Theory of lattice and electronic fluctuations in weakly localized spin-peierls systems. J. Phys. I France 1996, 6, 1727–1744. [Google Scholar] [CrossRef]
- Liu, Q.; Ravy, S.; Pouget, J.-P.; Coulon, C.; Bourbonnais, C. Structural fluctuations and spin-Peierls transitions revisited. Synth. Met. 1993, 56, 1840–1845. [Google Scholar] [CrossRef]
- Dumm, M.; Loidl, A.; Alavi, B.; Starkey, K.P.; Montgomery, L.K.; Dressel, M. Comprehensive ESR studies of the antiferromagnetic ground states in the one dimensional spin systems (TMTSF)2PF6, (TMTSF)2AsF6 and (TMTTF)2Br. Phys. Rev. B 2000, 62, 6512–6520, and references there in.. [Google Scholar] [CrossRef]
- Pouget, J.-P. Microscopic interactions in CuGeO3 and organic Spin-Peierls systems deduced from their pretransitional fluctuations. Eur. Phys. J. B 2001, 20, 321–333, (Erratum Eur. Phys. J. B 2001, 24, 415).. [Google Scholar] [CrossRef]
- Pouget, J.-P. Bond and charge ordering in low-dimensional organic conductors. Phys. B 2012, 407, 1762–1770. [Google Scholar] [CrossRef]
- Creuzet, F.; Bourbonnais, C.; Caron, L.G.; Jérome, D.; Bechgaard, K. A 13C NMR study of the interplay between the spin-Peierls and antiferromagnetic ground states in (TMTTF)2PF6 under pressure. Synth. Met. 1987, 19, 289–294. [Google Scholar] [CrossRef]
- Nakamura, T.; Nobutoki, T.; Kobayashi, Y.; Takahashi, T.; Saito, G. 1H-NMR investigation of the SDW wave-number in (TMTTF)2Br. Synth. Met. 1995, 70, 1293–1294. [Google Scholar] [CrossRef]
- Seo, H.; Merino, J.; Yoshioka, H.; Ogata, M. Theoretical aspects of charge ordering in molecular conductors. J. Phys. Soc. Jpn. 2006, 75, 051009. [Google Scholar] [CrossRef]
- Zorina, L.; Simonov, S.; Mézière, C.; Canadell, E.; Suh, S.; Brown, S.E.; Foury-Leylekian, P.; Fertey, P.; Pouget, J.P.; Batail, P. Charge ordering, symmetry and electronic structures issues and Wigner crystal structure of the quarter-filled band Mott insulators and high pressure metalsδ-(EDT-TTF-CONMe2)2X, X=Br and AsF6. J. Mat. Chem. 2009, 19, 6980–6994. [Google Scholar] [CrossRef] [Green Version]
- Tomic, S.; Biskup, N.; Babic, S.D.; Maki, K. Commensurate spin-density–wave state in (TMTTF)2Br- single particle and collective charge dynamics. Europhys. Lett. 1994, 26, 295–301. [Google Scholar] [CrossRef]
- Ashikawa, A.; Matsunaga, N.; Nomura, K.; Nakamura, T.; Takahashi, T.; Saito, G. Pressure and Magnetic Field Dependence of SDW transition in (TMTTF)2Br. Phys. Stat. Sol. B 2001, 223, 539–543. [Google Scholar] [CrossRef]
- Fujiyama, S.; Nakamura, T. NMR studies of the localized states of (TMTTF)2Br. J. Phys. Chem. Solids 2002, 63, 1259–1261. [Google Scholar] [CrossRef]
- Javadi, H.H.S.; Laversanne, R.; Epstein, A.J. Microwave conductivity and dielectric constant of tetramethyltetrathiafulvalene salts [(TMTTF)2X, X=SCN, ReO4, SbF6. Phys. Rev. B 1988, 37, 4280–4283. [Google Scholar] [CrossRef]
- Nad, F.; Monceau, P.; Fabre, J.M. Low frequency dielectric permittivity of quasi-one-dimensional conductor (TMTTF)2Br. Eur. Phys. J. B 1998, 3, 301–306. [Google Scholar] [CrossRef]
- Clark, W.G.; Hanson, M.E.; Wong, W.H.; Alavi, B. Evidence that the SDW transition in (TMTSF)2PF6 is first order. Phys. B 1994, 194–196, 285–286. [Google Scholar]
- Kagoshima, S.; Saso, Y.; Maesato, M.; Kondo, R.; Hasegawa, T. Low temperature diffuse X-ray studies of charge-density waves coexisting with spin-density waves in the organic conductors (TMTSF)2PF6 and (TMTSF)2AsF6. Solid State Commun. 1999, 110, 479–483. [Google Scholar] [CrossRef]
- Gufan; Yu, M.; Larin, E.S. Theory of phase transitions described by two order parameters. Sov. Phys. Solid State 1980, 22, 270–275. [Google Scholar]
- Takahashi, T.; Maniwa, Y.; Kawamura, H.; Saito, G. Determination of SDW characteristics in (TMTSF)2PF6 by 1H-NMR analysis. Phys. B 1986, 143, 417–421. [Google Scholar]
- Takahashi, T.; Maniwa, Y.; Kawamura, H.; Murata, K.; Saito, G. NMR analysis of electronic properties in organic superconductors (TMTSF)2PF6 and β-(BEDT-TTF)2I3. Synth. Met. 1987, 19, 225–230. [Google Scholar] [CrossRef]
- Nickel, J.C.; Duprat, R.; Bourbonnais, C.; Dupuis, N. Triplet Superconductivity Pairing and Density Wave Instabilities in Organic Conductors. Phys. Rev. Lett. 2005, 95, 247001. [Google Scholar] [CrossRef] [PubMed]
- Nickel, J.C.; Duprat, R.; Bourbonnais, C.; Dupuis, N. Superconductivity pairing and density wave instabilities in quasi-one-dimensional conductors. Phys. Rev. B 2006, 73, 165126. [Google Scholar] [CrossRef]
- Kuroki, K. Pairing symmetry considerations in organic superconductors. J. Phys. Soc. Jpn. 2006, 75, 051013. [Google Scholar] [CrossRef]
- Ng, H.K.; Timusk, T.; Bechgaard, K. Far infra-red studies of bis (tetramethyltetraselenafulvalene) hexafluoroantimonate [(TMTSF)2SbF6]: Coexistence of metallic and semiconducting states. Phys. Rev. B 1984, 30, 5842–5846. [Google Scholar] [CrossRef]
- Grüner, G. The dynamics of spin-density waves. Rev. Mod. Phys. 1994, 66, 1–24. [Google Scholar] [CrossRef]
- Laversanne, R.; Coulon, C.; Gallois, B.; Pouget, J.P.; Moret, R. Structural and electrical properties of (TMTTF)2MF6 salts (M=P, As, Sb). Role of the anions. J. Phys. Lett.(Paris) 1984, 45, L393–L399. [Google Scholar] [CrossRef]
- Coulon, C.; Maaroufi, A.; Amiell, J.; Dupart, E.; Flandrois, S.; Delhaes, P.; Moret, R.; Pouget, J.P.; Morand, J.P. Antiferromagnetic and structural instabilities in tetramethyltetrathiafulvalene thiocyanate [(TMTTF)2SCN]. Phys. Rev. B 1982, 26, 6322–6325. [Google Scholar] [CrossRef]
- Coulon, C.; Parkin, S.S.P.; Laversanne, R. Structureless transition and strong localization effects in bis-tetramethyltetrathiafulvalenium salts [(TMTTF)2X]. Phys. Rev. B 1985, 31, 3583–3587. [Google Scholar] [CrossRef]
- Brown, S.E.; Javadi, H.H.S.; Laversanne, R. Sound Velocity Measurements in (TMTTF)2X Salts, X= ReO4, AsF6, SbF6. In Advanced Organic Solid State Materials; Chiang, L.Y., Chaikin, P.M., Cowan, D.O., Eds.; Materials Research Society: Pittsburgh, PA, USA, 1990; pp. 245–250. [Google Scholar]
- Chow, D.S.; Zamborsky, F.; Alavi, B.; Tantillo, D.J.; Baur, A.; Merlic, C.A.; Brown, S.E. Charge ordering in the TMTTF family of molecular conductors. Phys. Rev. Lett. 2000, 85, 1698–1701. [Google Scholar] [CrossRef] [PubMed]
- Dumm, M.; Salameh, B.; Abaker, M.; Montgomery, L.K.; Dressel, M. Magnetic and optical studies of spin and charge ordering in (TMTTF)2AsF6. J. Phys. IV (France) 2004, 114, 57–60. [Google Scholar] [CrossRef]
- Dumm, M.; Abaker, M.; Dressel, M.; Montgomery, L.K. Charge order in (TMTTF)2PF6 investigated by infrared spectroscopy. J. Low. Temp. Phys. 2006, 142, 609–612. [Google Scholar] [CrossRef]
- Monceau, P.; Nad, F.; Brazovski, S. Ferroelectric Mott-Hubbard phase of organic (TMTTF)2X conductors. Phys. Rev. Lett. 2001, 86, 4080–4083. [Google Scholar] [CrossRef] [PubMed]
- Foury-Leylekian, P.; Ravy, S.; Pouget, J.-P. Stuctural properties of strongly correlated quasi-1D organic systems. Phys. B 2002, 312–313, 574–577. [Google Scholar] [CrossRef]
- Ravy, S.; Foury-Leylekian, P.; Le Bolloc’h, D.; Pouget, J.-P.; Fabre, J.M.; Prado, R.J.; Lagarde, P. Structural instability and electronic localization in the 2/1 salts: The case of the Fabre and the (DMtTTF)2ClO4 salts. J. Phys. (Paris) IV 2004, 114, 81–85. [Google Scholar]
- Nogami, Y.; Nakamura, T. X-ray observation of 2kF and 4kF charge orderings in (TMTTF)2ReO4 and (TMTTF)2SCN associated with anion orderings. J. Phys. (Paris) IV 2002, 12, Pr9-145–Pr9-148. [Google Scholar]
- Coulon, C.; Lalet, G.; Pouget, J.-P.; Foury-Leylekian, P.; Moradpour, A.; Fabre, J.M. Anisotropic conductivity and charge ordering in (TMTTF)2X salts probed by ESR. Phys. Rev. B 2007, 76, 085126. [Google Scholar] [CrossRef]
- Foury-Leylekian, P.; Petit, S.; Andre, G.; Moradpour, A.; Pouget, J.P. Neutron scattering evidence for a lattice displacement at the charge ordering transition of (TMTTF)2PF6. Phys. B 2010, 405, S95–S97. [Google Scholar] [CrossRef]
- Granier, T.; Gallois, B.; Ducasse, L.; Fritsch, A.; Filhol, A. 4K crystallographic and electronic structures of (TMTTF)2X salts (X−: PF6−; AsF6−). Synth. Met. 1988, 24, 343–356. [Google Scholar] [CrossRef]
- Sawa, H. Direct Observation of Electronic State of (TMTTF)2PF6 Using Synchrotron Radiation X-ray Analysis. In Proceedings of International Research School and Workshop on Electronic Crystals; ECRYS, Cargèse, France, 15–27 August 2011.
- In this respect the analysis performed in [105] and which considers only the shift of the anion sublattice is incomplete. As the neutron scattering length is similar for all the nucleus of (TMTTF)2PF6, the 3 contributions given in the text have to be treated on equal footing in the structural refinement. Note that in deuterated samples the neutron scattering could be particularly sensitive to the deformation of the methyl group cavity at TCO
- Dressel, M.; Dumm, M.; Knoblauch, T.; Masimo, M. Comprehensive optical investigation of Charge Order in Organic chain compounds (TMTTF)2X. Crystals 2012, in press.. [Google Scholar]
- Rose, E.; Dressel, M. Coupling between molecular chains and anions in (TMTTF)2X salts. Phys. B 2012, 407, 1787–1892. [Google Scholar] [CrossRef]
- Riera, J.; Poilblanc, D. Influence of the anion potential on the charge ordering in quasi-one-dimensional charge-transfer salts. Phys. Rev. B 2001, 63, 241102(R). [Google Scholar]
- Pouget, J.-P.; Foury-Leylekian, P.; Le Bolloc’h, D.; Hennion, B.; Ravy, S.; Coulon, C.; Cardoso, V.; Moradpour, A. Neutron-Scattering Evidence for a Spin-Peierls ground State in (TMTTF)2PF6. J. Low. Temp. Phys. 2006, 142, 147–152. [Google Scholar] [CrossRef]
- Kőhler, B.; Rose, E.; Dumm, M.; Untereiner, G.; Dressel, M. Comprehensive transport study of anisotropy and ordering phenomena in quasi-one-dimensional (TMTTF)2X salts (X=PF6, AsF6, SbF6, BF4, ClO4, ReO4. Phys. Rev. B 2011, 84, 035124. [Google Scholar] [CrossRef]
- Nad, F.; Monceau, P.; Nakamura, T.; Furukawa, K. The effect of deuteration on the transition into a charge ordered state of (TMTTF)2X salts. J. Phys.: Condens. Matter 2005, 35, L399–L406. [Google Scholar]
- Moret, R.; Pouget, J.P.; Comès, R.; Bechgaard, K. X-ray study of the anion ordering transition in di(tetramethyltetraselenafulvalen)-ium perchlorate (TMTSF)2ClO4: quenching and irradiation effects. J. Phys. (Paris) 1985, 46, 1521–1732. [Google Scholar] [CrossRef]
- Zamborszky, F.; Yu, W.; Raas, W.; Brown, S.E.; Alavi, B.; Merlic, C.A.; Baur, A. Competition and coexistence of bond and charge orders in (TMTTF)2AsF6. Phys. Rev. B 2002, 66, 081103(R). [Google Scholar]
- Nad, F.; Monceau, P. Dielectric Response of the Charge Ordered State in Quasi-One Dimensional Organic Conductors. J. Phys. Soc. Jpn. 2006, 75, 051005. [Google Scholar] [CrossRef]
- Coulon, C.; Foury-Leylekian, P.; Fabre, J.M.; Pouget, J.-P. Electronic instabilities and irradiation effects in the (TMTTF)2X series. Centre de Recherche Paul Pascal, Université Bordeaux I, France, 2012; Unpublished work. [Google Scholar]
- Brazovskii, S. Ferroelectricity and Charge Ordering in Quasi-1D Organics Conductors. In The Physics of Organic Superconductors and Conductors; Lebed, A., Ed.; Springer-Verlag: Berlin, Germany, 2008; pp. 313–355. [Google Scholar]
- Kirova, N.; Brazovskii, S. Ferroelectricity: From organic conductors to electronic polymers. Phys. B 2009, 404, 382–384. [Google Scholar] [CrossRef]
- Yamamoto, K.; Iwai, S.; Boyko, S.; Kashiwazaki, A.; Hiramatsu, F.; Okabe, C.; Nishi, N.; Yakushi, K. Strong optical nonlinearity and its ultrafast response associated with electron ferroelectricity in an organic conductor. J. Phys. Soc. Jpn. 2008, 77, 074709. [Google Scholar] [CrossRef]
- Alemany, P.; Pouget, J.-P.; Canadell, E. Essential role of anions in the charge ordering transition of α-(BEDT-TTF)2I3. Phys. Rev. B 2012, 85, 195118. [Google Scholar] [CrossRef]
- Pouget, J.-P.; Khanna, S.K.; Denoyer, F.; Comès, R.; Garito, A.F.; Heeger, A.J. X-ray Observation of 2kF and 4kF Scatterings in Tetrathiafulvalene-Tetracyanoquinodimethane (TTF-TCNQ). Phys. Rev. Lett. 1976, 37, 437–440. [Google Scholar] [CrossRef]
- Nogami, Y.; Moret, R.; Pouget, J.-P.; Yamamoto, Y.; Oshima, K.; Hiraki, K.; Kanoda, K. X-ray structural study of 4kF Superlattice in (DMDCNQI)2Ag. Synth. Met. 1999, 102, 1778. [Google Scholar] [CrossRef]
- Foury-Leylekian, P.; Auban-Senzier, P.; Coulon, C.; Jeannin, O.; Fourmigué, M.; Pasquier, C.; Pouget, J.-P. Phase diagram of the correlated quarter-filled-band organic salt series (o-DMTTF)2X (X=Cl, Br, I) . Phys. Rev. B 1994, 50, 7131–7139. [Google Scholar]
- Zamborszky, F.; Yu, W.; Raas, W.; Brown, S.E.; Alavi, B.; Merlic, C.A.; Baur, A.; Lefebvre, S.; Wzieteck, P. Influence of charge order on the ground state of TMTTF conductors. J. Phys. (Paris) IV 2002, 12, Pr9-139–Pr9-144. [Google Scholar]
- In contrast 75As NMR T1−1 does not exhibit a divergence at TCO but only a change of slope. This result could be understood if AsF6 plays a weaker role in the CO transition (through process 1) than in the SP transition (through process 2).
- de Souza, M.; Brűhl, A.; Műller, J.; Foury-Leylekian, P.; Moradpour, A.; Pouget, J.-P.; Lang, M. Thermodynamic studies at the charge ordering and spin-Peierls transitions in (TMTTF)2X. Phys. B 2009, 404, 494–498. [Google Scholar] [CrossRef]
- Dumm, M.; Loidl, A.; Fravel, B.W.; Starkey, K.P.; Montgomery, L.K.; Dressel, M. Electron spin resonance studies on the organic linear chain compounds (TMTTF)2X (C= S, Se; X= PF6, AsF6, ClO4, Br). Phys. Rev. B 2001, 61, 511–521. [Google Scholar]
- Salameh, B.; Yasin, S.; Dumm, M.; Untereiner, G.; Montgomery, L.; Dressel, M. Spin dynamics of the organic linear chain compounds (TMTTF)2X (X = SbF6, AsF6, BF4, ReO4, and SCN). Phys. Rev. B 2011, 83, 205126. [Google Scholar] [CrossRef]
- Foury-Leylekian, P.; Petit, S.; Coulon, C.; Hennion, B.; Moradpour, A.; Pouget, J.-P. Inelastic neutron scattering investigation of the magnetic excitations in the spin-Peierls ground state of (TMTTF)2PF6. Phys. B 2009, 404, 537–540. [Google Scholar] [CrossRef]
- Langlois, A.; Poirier, M.; Bourbonnais, C.; Foury-Leylekian, P.; Moradpour, A.; Pouget, J.-P. Microwave dielectric study of spin-Peierls and charge ordering transitions in (TMTTF)2PF6 salts. Phys. Rev. B 2010, 81, 125101. [Google Scholar] [CrossRef]
- Brown, S.E.; Clark, W.G.; Zamborszky, F.; Klemme, B.J.; Kriza, G.; Alavi, B.; Merlic, C.; Kuhns, P.; Moulton, W. 13C NMR measurements of the high-magnetic-field, low-temperature phases of (TMTTF)2PF6. Phys. Rev. Lett. 1998, 80, 5429–5432. [Google Scholar] [CrossRef]
- Brown, S.E.; Clark, W.G.; Alavi, B.; Hall, D.; Naughton, M.J.; Tantillo, D.J.; Merlic, C.A. High–field magnetization of the spin-Peierls compound (TMTTF)2PF6. Phys. Rev. B 1999, 60, 6270–6272. [Google Scholar] [CrossRef]
- Hirsch, J.E.; Scalapino, D.J. 2pF and 4pF instabilities in the one-quarter-filled-band Hubbard model. Phys. Rev. B 1983, 27, 7169–7185. [Google Scholar] [CrossRef]
- Hirsch, J.E.; Scalapino, D.J. 2pF and 4pF instabilities in the one dimensional Hubbard model. Phys. Rev. B 1984, 29, 5554–5561. [Google Scholar] [CrossRef]
- Ilakovac, V.; Ravy, S.; Pouget, J.P.; Lenoir, C.; Boubekeur, K.; Batail, P.; Dolanski Babic, S.; Biskup, N.; Korin-Hamzic, B.; Tomic, S.; Bourbonnais, C. Enhanced charge localization in the organic alloys [(TMTSF)1-x(TMTTF)x]2ReO4. Phys. Rev. B 2011, 84, 195134. [Google Scholar] [CrossRef]
- The assertion done in [137] that the inversion symmetry is broken on the anion sites at RT above the AO transition is incorrect. Only inversion centers located on the organic stacks between the donors are removed by the chemical CO.
- Chow, D.S.; Wzietek, P.; Fogliatti, D.; Alavi, B.; Tantilo, D.J.; Merlic, C.A; Brown, S.E. Singular behavior in the pressure-tuned competition between spin-Peierls and antiferromagnetic ground states of (TMTTF)2PF6. Phys. Rev. Lett. 1998, 81, 3984–3987. [Google Scholar] [CrossRef]
- Rose, E.; Loose, C.; Kortus, J.; Pashkin, A.; Kuntscher, C.A.; Ebbinghaus, S.G.; Hanfland, M.; Lissner, F.; Schleid, Th.; Dressel, M. Pressure dependence of Crystal Structure and Electronic Band Structure of (TMTTF)2PF6. J. Phys.: Condens. Matter 2012, in press.. [Google Scholar]
- Le Pévelen, D.; Gaultier, J.; Barrens, Y.; Chasseau, D.; Castet, F.; Ducasse, L. Temperature and pressure dependencies of the crystal structure of the organic superconductor (TMTSF)2ClO4. Eur. Phys. J. B 2001, 19, 363–373. [Google Scholar] [CrossRef]
- Jacobsen, C.S.; Pedersen, H.J.; Mortensen, K.; Rindorf, G.; Thorup, N.; Torrance, J.B.; Bechgaard, K. An unusual metal-insulator transition: bis(tetramethyltetraselenafulvalenium)-perrhenate (TMTSF)2ReO4. J. Phys. C: Solid State Phys. 1982, 15, 2651–2663. [Google Scholar] [CrossRef]
- Pouget, J.-P.; Moret, R.; Comès, R.; Bechgaard, K. X-ray diffuse scattering study of superstructure formation in tetramethyltetraselenafulvalenium perrhenate (TMTSF)2ReO4 and nitrate (TMTSF)2NO3. J. Phys. Lett. (Paris) 1981, 42, L543–L546. [Google Scholar] [CrossRef]
- Grant, P.M. Broken-Symmetry Band Structure of Ditetramethyltetradelenafulvalene-X [(TMTSF)2X]. Phys. Rev. Lett. 1983, 50, 1005–1008. [Google Scholar] [CrossRef]
- Grant, P.M. Electronic structure of the 2:1 charge transfer salts of TMTCF. J. Phys. (Paris) 1983, 44, C3-847–C3-857. [Google Scholar] [CrossRef]
- Emery, V.J. Some basic questions in organic superconductivity. J. Phys. (Paris) 1983, 44, C3-977–C3-982. [Google Scholar] [CrossRef]
- Brazovslii, S.; Yakovenko, V. On the theory of phase transitions in organic superconductors. J. Phys. Lett.(Paris) 1985, 46, L111–L116. [Google Scholar] [CrossRef]
- Bruinsma, R.; Emery, V.J. Theory of anion ordering in TMTSF compounds. J. Phys. (Paris) 1983, 44, C3-1115–C3-1120. [Google Scholar] [CrossRef]
- Emge, T.J.; Wang, M.H.; Beno, M.A.; Williams, J.M.; Whangbo, M.H.; Evain, M. Effect of anion ordering on the H-anion interaction and band electronic structure of (TMTSF)2BF4 at 20K. J. Am. Chem. Soc. 1986, 108, 8215–8223. [Google Scholar] [CrossRef]
- Ducasse, L.; Abderrabba, M.; Gallois, B.; Chasseau, D. Influence of temperature on the band structures of (TMTSF)2X and (TMTTF)2X (X−= tetrahedral anion). Synth. Met. 1987, 19, 327–332. [Google Scholar] [CrossRef]
- Tomic, S.; Jérome, D.; Bechgaard, K. Influence of the cooling rate on the ground state of the organic conductor (TMTSF)2ReO4. J. Phys. C: Solid State Phys. 1984, L11–L16. [Google Scholar]
- Moret, R.; Ravy, S.; Pouget, J.P.; Comès, R.; Bechgaard, K. Anion-Ordering Phase Diagram of di(tetramethyltetraselenafulvalenium)-Perrhenate, (TMTSF)2ReO4. Phys. Rev. Lett. 1986, 57, 1915–1918. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.J.; Kang, H.; Kang, W. Review of the P-T phase diagram of the organic superconductor (TMTSF)2ReO4. Synth. Met. 2001, 120, 1043–1044. [Google Scholar] [CrossRef]
- Nakamura, T.; Furukawa, K.; Hara, T. 13C NMR Analyses of Successive Charge Ordering in (TMTTF)2ReO4. J. Phys. Soc. Jpn. 2006, 75, 013707. [Google Scholar] [CrossRef]
- Nogami, Y.; Ito, T.; Yamamoto, K.; Irie, N.; Horita, S.; Kambe, T.; Nagao, N.; Oshima, K.; Ikeda, N.; Nakamura, T. X-ray structural study of charge and anion orderings of TMTTF salts. J. Phys. (Paris) IV 2005, 131, 39–42. [Google Scholar]
- Ravy, S.; Pouget, J.-P.; Moret, R.; Wudl, F. Successive phase transitions in the organic conductor (TMTSF)2PF2O2. J. Phys. I France 1991, 1, 703–720. [Google Scholar] [CrossRef]
- Moret, R.; Pouget, J.-P.; Comès, R.; Bechgaard, K. Structural phase transitions in the (TMTSF)2X and (TMTTF)2X series: A survey with some new results. J. Phys. (Paris) 1983, 44, C3-957–C3-962. [Google Scholar] [CrossRef]
- Yamaura, J.; Jo, Y.J.; Kang, H.; Chung, O.H.; Kang, W. X-ray study at low temperature and under pressure in (TMTSF)2FSO3. 2005. Unpublished work.. [Google Scholar]
- Lacoe, R.C.; Wolf, S.A.; Chaikin, P.M.; Wudl, F.; Aharon-Shalom, E. Metal-insulator transitions and superconductivity in ditetramethyltetraselenafulvalenium fluorosulfonate [(TMTSF)2FSO3]. Phys. Rev. B 1983, 27, 1947–1950. [Google Scholar] [CrossRef]
- Jo, Y.J.; Choi, E.S.; Kang, H.; Kang, W.; Seo, I.S.; Chung, O.H. Refinement of the pressure-temperature phase diagram of the organic superconductor with asymmetric anion (TMTSF)2FSO3. Phys. Rev. B 2003, 67, 014516. [Google Scholar] [CrossRef]
- Takahashi, T. Charge fluctuation, charge ordering, and zero-gap state in organic conductors. Phys. B 2012, 407, 1757–1761. [Google Scholar] [CrossRef]
- Alemany, P.; Canadell, E.; Pouget, J.-P. Ab-initio electronic structure of (TMTSF)2ClO4. Institut de Ciència de Materials de Barcelona: Bellaterra, Spain, 2012; Unpublished work. [Google Scholar]
- Nagai, Y.; Nakamura, H.; Machida, M. Superconducting gap function in the organic superconductor (TMTSF)2ClO4 with anion ordering; First-principles calculations and quasiclassical analysis for angle-resolved heat capacity. Phys. Rev. B 2011, 83, 104523. [Google Scholar] [CrossRef]
- Pouget, J.-P.; Kagoshima, S.; Tamegai, T.; Nogami, Y.; Kubo, K.; Nakajima, T.; Bechgaard, K. High resolution x-ray scattering study of the anion ordering phase transition of (TMTSF)2ClO4. J. Phys. Soc. Jpn. 1990, 59, 2036–2053. [Google Scholar] [CrossRef]
- Barrens, Y.; Gaultier, J.; Brachetti, S.; Guionneau, P.; Chasseau, D.; Fabre, J.M. Low temperature structural properties of the (TMTSF)2NO3 salt. Synth. Met. 1999, 103, 2042–2043. [Google Scholar] [CrossRef]
- Matsunaga, N.; Ishikawa, A.; Hoshikawa, A.; Nomura, K.; Takasaki, S.; Yamada, J.; Nakatsuji, S.; Anzai, H. Anion disorder and two-dimensionality in the superconducting and SDW states of (TMTSF)2ClO4. J. Low Temp. Phys. 1999, 117, 1735–1739. [Google Scholar] [CrossRef]
- In the case of a random distribution of domain sizes, the probability that two sites separated by x belong to the same domain is given by exp-μx, where μ is the constant probability to cross a domain wall per unit length. The 3D Fourier transform of this probability function, assuming μ isotropic, is a Lorentzian squared function, whose HWHM Δq is linearly related to μ by Δq ≈ 0.64 μ. μ is connected to the linear density of domain walls along the direction of measurement of Δq. μ should also depend upon the geometry the domain pattern.
- The average domain size L in a given direction is inversely proportional to the HWHM Δq of the reflection (expressed in Å-1), measured in this direction. In a general manner the profile of the reflection is the Fourier transform of the average shape of the ordered domain. L is given by the relationship L≈C(d)/Δq, where the constant C(d) depends upon the dimension, d, of independent directions involved in the Fourier transform. For a segment of length L (or along one edge of a parallelepiped): C(1)≈2.79; for a disc of diameter L: C(2) ≈ 3.2, and for a sphere of diameter L: C(3)≈3.72. L depends weakly of the shape of the domain. In [164], L is obtained using C(1).
- Haddad, S.; Charfi-Kaddour, S.; Pouget, J.-P. Inhomogeneous superconductivity in organic conductors: the role of disorder and magnetic field. J. Phys.: Condens. Matter 2011, 23, 464205. [Google Scholar] [CrossRef]
- Ilakovac, V.; Ravy, S.; Boubekeur, K.; Lenoir, C.; Batail, P.; Pouget, J.-P. Substitutional disorder and anion ordering transition in the (TMTSF)2(ReO4)1-x(ClO4)x solid solution. Phys. Rev. B 1997, 56, 13878–13887. [Google Scholar] [CrossRef]
- μ−1, which is quasi-isotropic, amounts to 400 Å in relaxed samples and decreases rapidly to 25Å / 20 Å for x = 5% / 7% of ReO4. Interestingly for these last two concentrations, μ-1 amounts to the average distance between ReO4 substituent (24 Å and 21 Å respectively).
- In this respect one observes at low temperature for x = 7% of ReO4 a coexistence of (0, 1/2, 0) AO and (1/2, 1/2, 1/2) AO domains in close contact and whose diffraction pattern is a superimposition of LS shaped superlattice reflections [170]. As there is a random distribution of the two species of anion, (1/2, 1/2, 1/2) AO domains are nucleated in regions of excess of ReO4 (with respect to the average concentration) while (0, 1/2, 0) AO domains are nucleated in regions deficient in ReO4.
- Joo, N.; Auban-Senzier, P.; Pasquier, C.R.; Monod, P.; Jérome, D.; Bechgaard, K. Supression of superconductivity by non-magnetic disorder in the organic superconductor (TMTSF)2(ClO4)1-x(ReO4)x. Eur. Phys. J. B 2004, 40, 43–48. [Google Scholar] [CrossRef]
- Joo, N.; Auban-Senzier, P.; Pasquier, C.R.; Jérome, D.; Bechgaard, K. Impurity-controlled superconductivity/spin density wave interplay in the organic superconductor (TMTSF)2ClO4. Europhys. Lett. 2005, 72, 645–651. [Google Scholar] [CrossRef]
- Fertey, P.; Sayetat, F.; Muller, J.; Pouget, J.-P.; Lenoir, C.; Batail, P. Thermal behaviour of (TMTSF)2ClO4 lattice parameters in the range 10K-300K. Physica C 1994, 235–240, 2459–2460. [Google Scholar]
- Fertey, P.; Canadell, E.; Pouget, J.-P.; Sayetat, F.; Lenoir, C.; Batail, P; Muller, J. Cooling rate dependence of the lattice parameters of (TMTSF)2ClO4. Synth. Met. 1995, 70, 761–762. [Google Scholar] [CrossRef]
- Nakamura, T.; Kinami, R.; Takahashi, T.; Saito, G. 1H NMR Study of the Magnetic Structure in (TMTTF)2SCN. Synth. Met. 1997, 86, 2053–2054. [Google Scholar] [CrossRef]
- Parkin, S.S.P.; Coulon, C.; Moret, R.; Pouget, J.-P. Incommensurate structural modulation and electronic localization in bis-tetramethyltetraselenafulvalene thiocyanate. Phys. Rev. B 1987, 36, 2246–2250. [Google Scholar] [CrossRef]
- Kang, W.; Chung, O.H. Quasi-one-dimensional Fermi surface of (TMTSF)2NO3. Phys. Rev. B 2009, 79, 045115. [Google Scholar] [CrossRef]
- Hiraki, K.; Nemoto, T.; Takahashi, T.; Kang, K.; Jo, Y.; Kang, W.; Cung, O.-H. NMR studies of the exotic members of the Bechgaard salts, NO3 and FSO3 salts. Synth. Met. 2003, 135–136, 691–692. [Google Scholar]
- Poirier, M.; Langlois, A.; Bourbonnais, C.; Foury-Leylekian, P.; Moradpour, A.; Pouget, J.-P. Magneto-elastic coupling in the spin-Peierls ground state of hydrogenated and deuterated (TMTTF)2PF6 salts. Departement de Physique, Université de Sherbrooke: Shergrooke, Canada, 2012; Unpublished work. [Google Scholar]
- Pasquier, C.R.; Kang, N.; Salameh, B.; Auban-Senzier, P.; Jérome, D.; Brazovskii, S. Evolution of the spin-density wave-superconductivity texture in the organic superconductor (TMTSF)2PF6 under pressure. Phys. B 2012, 407, 1806–1809, and references there in.. [Google Scholar] [CrossRef]
- Giovannetti, G.; Kumar, S.; Pouget, J.-P.; Cappone, M. Magnetic and charge orderings in predicting multiferroicity in TMTTF2-PF6 organic crystals. 2012. Unpublished work.. [Google Scholar] [CrossRef]
- Khomskii, D.I. Multiferroics: Different ways to combine magnetism and ferroelectricity. J. Magn. Magn. Mater. 2006, 306, 1–8. [Google Scholar] [CrossRef]
- van den Brink, J.; Khomskii, D.I. Multiferroicity due to charge ordering. J. Phys.: Condens. Matter 2008, 20, 434217. [Google Scholar] [CrossRef]
- Holakovský, J. A New Type of Ferroelectric Phase Transition. Phys. Stat. Sol. B 1973, 56, 615–619. [Google Scholar] [CrossRef]
- Imry, Y. On the statistical mechanics of coupled order parameters. J. Phys. C: Solid State Phys. 1975, 8, 567–577. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Pouget, J.-P. Structural Aspects of the Bechgaard and Fabre Salts: An Update. Crystals 2012, 2, 466-520. https://doi.org/10.3390/cryst2020466
AMA Style
Pouget J-P. Structural Aspects of the Bechgaard and Fabre Salts: An Update. Crystals. 2012; 2(2):466-520. https://doi.org/10.3390/cryst2020466
Chicago/Turabian StylePouget, Jean-Paul. 2012. "Structural Aspects of the Bechgaard and Fabre Salts: An Update" Crystals 2, no. 2: 466-520. https://doi.org/10.3390/cryst2020466