Abstract
The crystal and molecular structure of tert-butyl 4-(2-tert-butoxy-2-oxoethyl)-piperazine-1-carboxylate is reported. The title compound crystallizes from a petroleum ether/ethyl acetate mixture in the monoclinic space group P 21/c with four molecules in the unit cell. The unit cell parameters are: a = 8.4007(2) Å, b = 16.4716(4) Å, c = 12.4876(3) Å; β = 90.948(1)° and V = 1727.71(7) Å3. Bond lengths and angles of this piperazine-carboxylate are typical.
1. Introduction
The piperazine moiety plays an important role and is found in various bioactive compounds. In particular, the piperazinoacetic acid motif was found in highly selective factor Xa trypsin-like protease inhibitors [1]. Furthermore, the piperazine residue was used as spacer in pleuromutilin derivatives [2] or as linker in piperazine based hydroxamic acids as histone acylase (HDAC) inhibitors [3]. Functionalized piperazine derivatives were applied in radiopharmaceutical research as starting material for spiro-compounds, which were used for the mild introduction of fluorine-18 [4]. Finally, the acetic acid-piperazine core was used for the linkage of biological active peptides [5]. Alongside to the convenient reaction of piperazine with haloacetic acid derivatives via nucleophilic substitution, several mild methods were developed using Triton B [6] or RuCl3 [7] as catalysts.
2. Results and Discussion
The preparation of the title compound tert-butyl 4-(2-tert-butoxy-2-oxoethyl)piperazine-1-carboxylate (3) in a high yield of 79% was accomplished via nucleophilic displacement of the bromine in tert-butyl bromoacetate (2) with the secondary amine of the Boc-protected piperazine 1 under basic conditions using triethylamine. (Figure 1) The reaction was performed under mild conditions at 60 °C overnight using tetrahydrofuran as solvent. Crystals of 3 were grown during the purification step from a saturated petroleum ether/ethyl acetate solution.
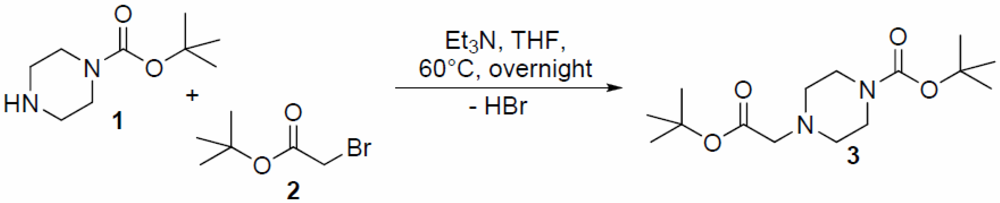
Figure 1.
The synthesis of title compound 3.
The crystal and instrumental parameters used in the unit cell determination, the data collection, and structure refinement parameters are summarized in Table 1. The molecular structure of 3 is shown in Figure 2 with the used atom-labeling scheme. The displacement thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths comprising key features of tert-butyl 4-(2-tert-butoxy-2-oxoethyl)piperazine-1-carboxylate (3), are given in Table 2. The central piperazine ring adopts a chair conformation. Whereas the carboxyl unit of the Boc residue, which is attached to N1, is almost in plane with the mean plane of the piperazine ring atoms (22.3°), the plane through the atoms of the second carboxyl unit (C11, O3 and O4) has an angle of 116.3° to the mean plane through the piperazine ring atoms. The packing of the molecules in the unit cell in a view along the crystallographic a direction is demonstrated in Figure 3. As visible from this plot, the title molecules have two different but symmetry-related orientations with respect to each other in the crystals of 3. Intermolecular contacts are limited to those of van-der-Waals type. The shortest intermolecular distances of the polar atoms are between O3 and H atoms of the neighboring molecule at 2.991 Å and between O4 and H atoms at 2.702 and 2.710 Å.
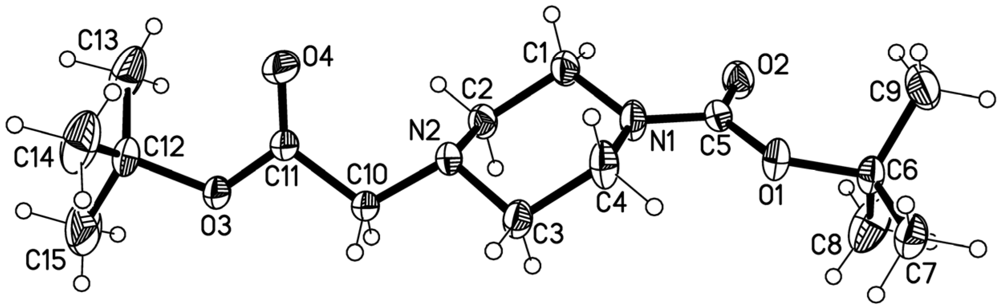
Figure 2.
A view of the structure of the title molecules in crystals of 3 showing the atom labeling scheme. Displacement ellipsoids are drawn at the 50% probability level.

Table 1.
Crystal data and structure refinement for compound 3.
| Crystal data | Refinement | ||
|---|---|---|---|
| Formula | C15H28N2O4 | Refinement method | Full-matrix least- |
| Formula weight | 300.39 g·mol−1 | squares on F2 | |
| Temperature | 173 K | Data/restraints/parameters | 7089/0/190 |
| Wavelength | 0.71073 Å | Measured reflections | 52521 |
| Crystal system | monoclinic | 2 θmax | 68.6° |
| Space group | P21/c | Rint | 2.4% |
| Unit cell dimensions | a = 8.4007(2) Å | ||
| b = 16.4716(4) Å | Goodness-of-fit on F2 | 1.04 | |
| c = 12.4876(3) Å | |||
| β = 90.948(1)° | Final R indices | R1 = 0.0629 | |
| Volume | 1727.71(7) Å3 | [I > 2σ(I)] | wR2 = 0.1791 |
| Z | 4 | R indices (all data) | R1 = 0.0801 |
| Density (calcd.) | 1155 g·cm−3 | wR2 = 0.1969 | |
| Absorption coefficient | 0.08 mm−1 | Largest diff. peak and hole | 0.97/−0.52 e·Å−3 |
| F(000) | 656 | ||
| Crystal size | 0.63 × 0.42 × 0.39 mm3 | ||
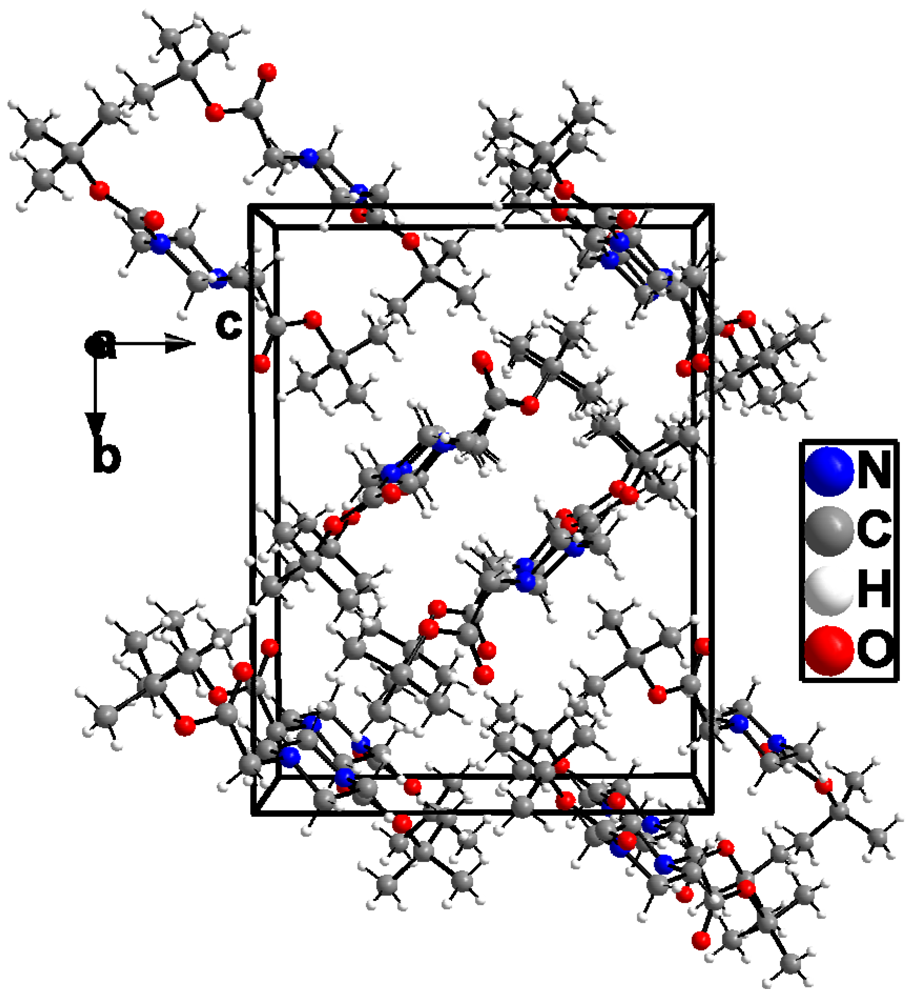
Figure 3.
View of the packing of molecules in crystals of 3 along the crystallographic a axis.
Table 2.
Selected atom distances [Å] in 3.
| atoms | distance | atoms | distance |
|---|---|---|---|
| N1−C1 | 1.459(2) | C5−O2 | 1.220(1) |
| C1−C2 | 1.520(2) | O1−C6 | 1.471(1) |
| C2−N2 | 1.463(2) | N2−C10 | 1.452(1) |
| N2−C3 | 1.463(2) | C10−C11 | 1.516(2) |
| C3−C4 | 1.517(2) | C11−O3 | 1.333(1) |
| C4−N1 | 1.466(2) | C11−O4 | 1.200(2) |
| N1−C5 | 1.358(1) | O3−C12 | 1.477(1) |
| C5−O1 | 1.348(1) |
3. Experimental Section
3.1. General
NMR spectra were recorded on a Varian Inova-400 and chemical shifts of the 1H and 13C spectra are reported in parts per million (ppm) using tetramethylsilane as internal standard. The melting point was determined on a Galen III (Cambridge Instruments) melting point apparatus (Leica, Vienna, Austria) and is uncorrected. The mass spectrum (MS) was obtained on a Quattro/LC mass spectrometer (MICROMASS) by electrospray ionization.
3.2. Synthesis of tert-Butyl 4-(2-tert-butoxy-2-oxoethyl)piperazine-1-carboxylate (3)
N-Boc-piperazine (207 mg, 1.11 mmol) and Et3N (225 mg, 2.22 mmol) were dissolved in anhydrous THF (10 mL). tert-Butyl bromoacetate (434 mg, 2.22 mmol) was added dropwise at ambient temperature and the mixture was stirred at 60 °C overnight. After cooling to room temperature, saturated hydrogen carbonate solution (15 mL) was added and the aqueous layer was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were dried over Na2SO4, the solvent was removed and purification was done via column chromatography (petroleum ether/ethyl acetate = 4:1) to yield 3 as colorless solid (264 mg, 79%). m.p. 102 °C. 1H NMR (400 MHz, CDCl3): δ = 1.45 (s, 9H, tBu), 1.46 (s, 9H, tBu), 2.52 (t, 3J = 4.9 Hz, 4H, NCH2), 3.12 (s, 2H, NCH2), 3.47 (t, 3J = 4.9 Hz, 4H, NCH2). 13C NMR (101 MHz, CDCl3): δ = 28.3, 28.6 (2 × tBu), 52.8 (NCH2), 60.1 (NCH2), 79.8, 81.4 (2 × Cquart), 154.8 (C=O). MS (ESI+): m/z = 323 (11) [M+Na], 301 (100) [M++H].
3.3. Data Collection and Refinement
Crystallographic data were collected with a Bruker-Nonius Apex-X8 CCD-diffractometer with monochromatic Mo–Kα radiation (λ = 0.71073 Å) and a CCD detector. Preliminary data of the unit cell dimensions were obtained from the reflection positions of 36 frames, measured in three different directions of the reciprocal space. After completion of the data measurements the reflection intensities were corrected for Lorentz, polarization, and absorption effects. The data set of 7089 reflections was averaged from 52521 reflections (up to 68.6°) with an internal R value of 2.4% in Laue group 2/m. Averaging in mmm (orthorhombic) gives an Rint larger than 50%, indicating the monoclinic crystal system to be the correct choice. The structures were solved by direct methods using SHELXS-97 and refined against F2 on all data by full-matrix least-squares methods using SHELXL-97 version 2 [8,9]. All non-hydrogen atoms were refined anisotropically; all hydrogen atoms bonded to carbon atoms were placed on geometrically calculated positions and refined using riding models. Crystallographic data has been deposited with the Cambridge Crystallographic Data Centre, CCDC-858567. It can be retrieved free of charge through deposit@ccdc.cam.ac.uk or http://www.ccdc.cam.ac.uk.
4. Conclusions
The crystal and molecular structure of tert-butyl 4-(2-tert-butoxy-2-oxoethyl)-piperazine-1-carboxylate (3) is reported. These data represent a crystallographically characterized example of a molecular compound with a piperazine building block, which found various applications in the preparation of biological active compounds in pharmaceutical research.
References
- Huang, W.; Naughton, M.A.; Yang, H.; Su, T.; Dam, S.; Wong, P.W.; Arfsten, A.; Edwards, S.; Sinha, U.; Hollenbach, S.; Scarborough, R.M.; Zhu, B.-Y. Design, synthesis, and structure-Activity relationships of unsubstituted piperazinone-Based transition state factor Xa inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 723–728. [Google Scholar]
- Hirokawa, Y.; Kinoshita, H.; Tanaka, T.; Nakamura, T.; Fujimoto, K.; Kashimoto, S.; Kojima, T.; Kato, S. Pleuromutilin derivatives having a purine ring. Part 2: Influence of the central spacer on the antibacterial activity against Gram-positive pathogens. Bioorg. Med. Chem. Lett. 2009, 19, 170–174. [Google Scholar]
- Rossi, C.; Porcelloni, M.; D’Andrea, P.; Fincham, C.I.; Ettorre, A.; Mauro, S.; Squarcia, A.; Bigioni, M.; Parlani, M.; Nardelli, F.; Binaschi, M.; Maggi, C.A.; Fattori, D. Alkyl piperidine and piperazine hydroxamic acids as HDAC inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2305–2308. [Google Scholar]
- Grosse-Gehling, P.; Wuest, F.R.; Peppel, T.; Köckerling, M.; Mamat, C. 1-(3-[18F]fluoro-propyl)piperazines as model compounds for the radiofluorination of pyrido[2,3-d]pyrimidines. Radiochim. Acta 2011, 99, 365–373. [Google Scholar] [CrossRef]
- Dutta, A.S.; Crowther, M.; Gormley, J.J.; Hassall, L.; Hayward, C.F.; Gellert, P.R.; Kittlety, R.S.; Alcock, P.J.; Jamieson, A.; Moores, J.M.; et al. Potent cyclic peptide inhibitors of VLA-4 (α4β1 integrin)-mediated cell adhesion. Discovery of compounds like cyclo(MePhe-Leu-Asp-Val-D-Arg-D-Arg) (ZD7349) compatible with depot formulation. J. Peptide Sci. 2000, 6, 321–341. [Google Scholar] [CrossRef]
- Meshram, H.M.; Chennakesava Reddy, B.; Ramesh Goud, P. Triton B-Mediated Mild, Convenient, and Efficient Method for the Selective Alkylation of Cyclic Secondary Amines and Thiols. Synth. Commun. 2009, 39, 2297–2303. [Google Scholar] [CrossRef]
- Varala, R.; Enugala, R.; Adapa, S.R. Ruthenium(III) chloride-catalyzed efficient protocol for ethyl diazoacetate insertion into the N–H bond of secondary amines. Monatsh. Chem. 2008, 139, 1369–1372. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXS/L-97, Programs for the Solution and Refinement of Crystal Structures; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).